

Abstract
Primary aldosteronism is characterized by excess aldosterone secretion by the adrenal gland independent of the renin-angiotensin system and accounts for ~10% of hypertensive patients. Excess aldosterone causes cardiac hypertrophy, fibrosis, inflammation, and hypertension. The molecular mechanisms that trigger the onset and progression of aldosterone-mediated cardiac injury remain incompletely understood. MicroRNAs (miRNAs) are endogenous, small, noncoding RNAs that have been implicated in multiple cardiac pathologies; however, their regulation and role in aldosterone-mediated cardiac injury and dysfunction remains mostly unknown. We previously reported that microRNA-21 (miR-21) is the most upregulated miRNA by excess aldosterone in the left ventricle in a rat experimental model of primary aldosteronism. To elucidate the role of miR-21 in aldosterone-mediated cardiac injury and dysfunction, miR-21 knockout mice and their wild-type littermates were treated with aldosterone infusion and salt in the drinking water for 2 or 8 wk. miR-21 genetic ablation exacerbated aldosterone/salt-mediated cardiac hypertrophy and cardiomyocyte cross-sectional area. Furthermore, miR-21 genetic ablation increased the cardiac expression of fibrosis and inflammation markers and fetal gene program. miR-21 genetic ablation increased aldosterone/salt-mediated cardiac dysfunction but did not affect aldosterone/salt-mediated hypertension. miR-21 target gene Sprouty 2 may be implicated in the cardiac effects of miR-21 genetic ablation. Our study shows that miR-21 genetic ablation exacerbates aldosterone/salt-mediated cardiac hypertrophy, injury, and dysfunction blood pressure independently. These results suggest that miR-21 plays a protective role in the cardiac pathology triggered by excess aldosterone. Furthermore, miR-21 supplementation may be a novel therapeutic approach to abolish or mitigate excess aldosterone-mediated cardiovascular deleterious effects in primary aldosteronism.
Keywords: aldosterone, cardiac injury, gene expression, microRNAs, mineralocorticoids
INTRODUCTION
Primary aldosteronism is characterized by excess autonomous secretion of aldosterone (ALDO) by the adrenal gland independent of the renin-angiotensin system and accounts for ~10% of hypertensive patients (13, 30, 57, 60, 81, 107). Excess ALDO causes hypertension and cardiac hypertrophy, inflammation, and fibrosis that lead to cardiac dysfunction (10, 51, 69, 70, 97, 105, 106). Despite the societal and individual burden of the target organ injury associated with primary aldosteronism, the molecular mechanism responsible for excess ALDO-mediated cardiac injury and dysfunction are poorly understood.
MicroRNAs (miRNAs) are short, endogenous, single-strand, noncoding RNAs that modulate gene expression protein levels post-transcriptionally, mainly by inducing translational repression or mRNA degradation, or both, of specific target gene mRNAs (2, 6, 27, 80, 99). miRNAs have been implicated in a variety of physiological functions, including development, proliferation, apoptosis, metabolism, and immune response (12, 29, 74, 82). Furthermore, miRNAs have been implicated in multiple pathologies, including cardiovascular ones, where they exert driving, permissive, or protective roles (7, 20, 22, 35, 42, 74, 79, 80, 87). Overall, there is increasing evidence that miRNAs alongside transcription factors are critical regulators that modulate the expression levels of potentially all genes in most, if not all, pathophysiological processes (15, 37).
We previously reported that ALDO/SALT treatment modulates the expression of multiple miRNAs in the left ventricle (LV) in vivo (4). Among the multiple miRNAs regulated, microRNA-21 (miR-21) stands as the most upregulated, with its expression rising almost fourfold after 2 wk of ALDO/SALT treatment and remaining elevated for up to 8 wk after treatment initiation. Furthermore, we reported that miR-21 downregulation, using chemically modified, nuclease-resistant, cholesterol-conjugated antisense oligonucleotides, exacerbates ALDO/SALT-mediated cardiac hypertrophy, inflammation, fibrosis, and dysfunction (4). These findings strongly suggest that miR-21 plays a protective role in ALDO/SALT-mediated cardiac injury. Despite these exciting findings, the use of antisense oligonucleotides always carries the concern of off-target effects. The rules of miRNA-target mRNA binding are poorly understood, and that is reflected in the low degree of agreement between different algorithms to predict miRNA targets in silico (23, 36). Furthermore, recent studies performing unbiased transcriptome-wide miRNA binding site analysis in vivo have shown that noncanonical miRNA:target mRNA interactions are widespread and physiologically relevant (19, 31, 47, 59). Until we have a better knowledge of the miRNA:target mRNA interaction rules, off-target effects of miRNA inhibitors are a significant concern when predicted in silico. Although theoretically possible, analyzing off-target effects of miRNAs expressed in multiple cell types and tissues, as is the case of miR-21, is impractical at either the mRNA or protein level. To overcome these limitations, we make use of genetically engineered mouse models that allow precise ablation of a particular miRNA. We used miR-21 knockout (miR21KO) mice to perform a comprehensive analysis of the role of miR-21 in ALDO/SALT-mediated cardiac injury and dysfunction.
MATERIALS AND METHODS
Animals.
miR21KO mice generation has been previously reported (49). Mice were backcrossed for more than 10 generations on a C57BL/6N genetic background (Charles Rivers, Wilmington, MA). Animals were maintained under specific pathogen-free conditions in a temperature-controlled environment with a 12:12-h light-dark cycle and provided with standard chow (Teklad diet no. 2018) and water ad libitum.
Experimental protocol.
Eight-week-old male miR21KO or their wild-type (WT) littermate controls were uninephrectomized, allowed to recover overnight, and then randomly assigned to Control and ALDO/SALT groups and treated for 2 or 8 wk unless otherwise stated. ALDO (0.15 µg/h; approximate dose: 137 µg·kg body wt−1·day−1) was administered in polyethylene glycol 300 (vehicle, Fluka) by subcutaneously implanted osmotic minipumps (Alzet 1002 or 1004, Durect Corp.). SALT (1.0% NaCl + 0.3% KCl) was administered in tap water. KCl was administered to avoid ALDO-mediated hypokalemia. At the end of the experiment, blood was collected via cardiac puncture, animals were perfused with saline, and tissues were harvested, weighed, snap-frozen in liquid nitrogen, and stored at −80°C for further analysis.
For histological analysis, animals were injected intraperitoneally with heparin (100 U) 10 min before euthanasia. Under isoflurane anesthesia, blood was collected via cardiac puncture, animals were perfused with cardioplegic solution (50) followed by 10% neutral buffered formalin, and tissues were harvested and processed for histological analysis.
All protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center, and studies were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, 8th Edition, 2011.
Histological analysis.
Formalin-fixed, paraffin-embedded 5-µm cardiac sections were stained with hematoxylin and eosin and Picro Sirius Red using standard techniques. Interstitial and perivascular fibrosis volumetric areas were blindly analyzed using ImageJ software as reported (4).
Cardiomyocyte cross-sectional area.
Formalin-fixed, paraffin-embedded 5-µm cardiac sections were stained with Alexa Fluor 488-labeled wheat germ agglutinin (Molecular Probes), images were acquired by confocal microscopy, and pictures were blindly analyzed with ImageJ to determine cardiomyocyte cross-sectional area (24, 101). At least 100 cardiomyocytes in 3 different regions of the right, left, and interventricular septum were quantified, and cardiomyocyte cross-sectional area was expressed as μm2.
RNA extraction and gene expression quantification.
Total RNA was extracted with TRI-Reagent (MRC Gene) following manufacturer’s suggested protocols, resuspended in nuclease-free water, DNase treated with Turbo DNA-free kit (Ambion), and quantified by UV absorbance using the Nanodrop spectrophotometer (Nanodrop Technologies). Gene and miRNA expression were quantified by RT-qPCR using SyBR Green I technology as previously reported (102). Primer pairs were designed with Primer3 software (72) (Tables 1 and 2). Gene and miRNA PCR product quantification was performed by the relative quantification method and expressed as arbitrary units standardized against β2-microglobulin or let-7f, respectively (46, 78).
Table 1.
qPCR primers
| Gene | Accession No. | Direction | Sequence | Annealing Temp, °C | Amplicon Size, bp |
|---|---|---|---|---|---|
| Ctgf | NM_010217 | Forward | GGGCCTCTTCTGCGATTTC | 60.0 | 151 |
| Reverse | ATCCAGGCAAGTGCATTGGTA | ||||
| Lox | NM_010728 | Forward | GATGCGCTGCGGAAGAAAAC | 60.0 | 75 |
| Reverse | GTACCCTGTGGTCATAGTCTCT | ||||
| Fn1 | NM_010233 | Forward | AATGGAAAAGGGGAATGGAC | 65.0 | 244 |
| Reverse | CTCGGTTGTCCTTCTTGCTC | ||||
| Col1a1 | NM_007742 | Forward | GCCTCCCAGAACATCACCTA | 65.0 | 76 |
| Reverse | CCTTCTTGAGGTTGCCAGTC | ||||
| Col3a1 | NM_009930 | Forward | AGGCTGAAGGAAACAGCAAA | 65.0 | 73 |
| Reverse | CTCCATTCCCCAGTGTGTTT | ||||
| PAI-1 (Serpine1) | NM_008871 | Forward | AGTCTTTCCGACCAAGAGCA | 65.0 | 169 |
| Reverse | GCCGAACCACAAAGAGAAAG | ||||
| MCP-1 (Ccl2) | NM_011333 | Forward | AGGTCCCTGTCATGCTTCTG | 65.0 | 84 |
| Reverse | CGTTAACTGCATCTGGCTGA | ||||
| Spp1 (OPN) | NM_001204203 | Forward | AGCAAGAAACTCTTCCAAGCAA | 60.0 | 134 |
| Reverse | GTGAGATTCGTCAGATTCATCCG | ||||
| IL-6 | NM_031168 | Forward | CCAAGAGGTGAGTGCTTCCC | 60.0 | 118 |
| Reverse | CTGTTGTTCAGACTCTCTCCCT | ||||
| TGF-β (Tgfb1) | NM_011577 | Forward | CTCCCGTGGCTTCTAGTGC | 60.0 | 133 |
| Reverse | GCCTTAGTTTGGACAGGATCTG | ||||
| TNF-α (Tnf) | NM_013693 | Forward | CCCTCACACTCAGATCATCTTCT | 60.0 | 61 |
| Reverse | GCTACGACGTGGGCTACAG | ||||
| Nppa | NM_008725 | Forward | GCTTCCAGGCCATATTGGAG | 60.0 | 126 |
| Reverse | GGGGGCATGACCTCATCTT | ||||
| Nppb | NM_008726 | Forward | GAGGTCACTCCTATCCTCTGG | 60.0 | 100 |
| Reverse | GCCATTTCCTCCGACTTTTCTC | ||||
| Myh6 (α-MHC) | NM_010856 | Forward | GCCCAGTACCTCCGAAAGTC | 60.0 | 110 |
| Reverse | GCCTTAACATACTCCTCCTTGTC | ||||
| Myh7 (β-MHC) | NM_080728 | Forward | ACTGTCAACACTAAGAGGGTCA | 60.0 | 114 |
| Reverse | TTGGATGATTTGATCTTCCAGGG | ||||
| Pten | NM_008960 | Forward | CAATCATGTTGCAGCAATTCACT | 60.0 | 122 |
| Reverse | CCCCATAAAAATCTAGGGCCTCT | ||||
| Spry1 | NM_011896 | Forward | GGTCATAGGTCAGATCGGGTC | 60.0 | 120 |
| Reverse | CTTGCCACACTGTTCGCAG | ||||
| Spry2 | NM_011897 | Forward | TCCAAGAGATGCCCTTACCCA | 60.0 | 170 |
| Reverse | GCAGACCGTGGAGTCTTTCA | ||||
| Bcl2 | NM_177410 | Forward | GTCGCTACCGTCGTGACTTC | 60.0 | 284 |
| Reverse | CAGACATGCACCTACCCAGC | ||||
| Pdcd4 | NM_011050 | Forward | GGAAGTGAAGCGGTTAGAAGTG | 60.0 | 204 |
| Reverse | CTCCTGGTCGTCATCATAGTTTG | ||||
| Β2m | NM_009735 | Forward | GCTCGGTGACCCTGGTCTTT | 65.0 | 117 |
| Reverse | TGTTCGGCTTCCCATTCTCC |
Col1a1, collagen I; Col3a1, collagen III; Ctgf, connective tissue growth factor; Fn1, fibronectin; IL-6, interleukin 6; Lox, lysyl oxidase; MCP-1 (Ccl2), monocyte chemoattractant protein-1; Myh6 (α-MHC), α-myosin heavy chain; Myh7 (β-MHC), β-myosin heavy chain; PAI-1 (Serpine1), plasminogen activator inhibitor type 1; Pdcd4, programmed cell death 4; Pten, phosphatase and tensin homolog; qPCR, quantitative polymerase chain reaction; Spp1 (OPN), osteopontin; Spry1, Sprouty 1; Spry2, Sprouty 2; TGF-β (Tgfb1), transforming growth factor β; TNF-α (Tnf), tumor necrosis factor α; Nppa, natriuretic peptide A; Nppb, natriuretic peptide B; B2m, beta-2-microglobulin; Bcl2, Bcl2, apoptosis regulator.
Table 2.
miRNA qPCR primers
| miRNA | Target Sequence | Exiqon Product No. |
|---|---|---|
| mmu-let-7f-5p | UGAGGUAGUAGAUUGUAUAGUU | 204359 |
| mmu-miR-21–5p | UAGCUUAUCAGACUGAUGUUGA | 204230 |
miRNA, microRNA; qPCR, quantitative polymerase chain reaction.
Western blot analysis.
Tissues were homogenized in radioimmunoprecipitation assay buffer containing Halt protease and phosphatase inhibitor cocktail (Thermo Scientific), and total protein concentration was quantified with the bicinchoninic acid protein assay kit (Thermo Scientific). One hundred micrograms of total protein were resolved by SDS-PAGE and transferred to PVDF membranes. The PVDF membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature and then incubated overnight at 4°C with the following primary antibodies at the indicated dilutions: connective tissue growth factor (Ctgf; 1:10,000; Abcam ab-209780), plasminogen activator inhibitor type 1 (PAI-1; 1:30,000; Abcam ab-182973), Nppa (1:30,000; Abcam ab-126149), Sprouty 1 (Spry1; 1:30,000; Cell Signaling Technology 13013), Sprouty 2 (Spry2; 1:1,000,000; Cell Signaling Technology 14954), phosphatase and tensin homolog (Pten; 1:30,000; Abcam ab-170941), programmed cell death 4 (Pdcd4; 1:30,000; Abcam ab-8448), and Bcl2 (1:3,000; Abcam ab-692). Membranes were probed with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG secondary antibodies (Jackson Immuno Research 111–035–003 and 115–035–003) for 1 h at room temperature. Detection by chemiluminescence was performed with SuperSignal West Pico PLUS kit (Thermo Scientific). The membranes were exposed to X-ray film, developed, scanned, and quantified with ImageJ (National Institutes of Health). For normalization, membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific), incubated overnight at 4°C with anti-GAPDH antibody (1:3,000,000; Cell Signaling Technology 5174), and processed as described above.
Echocardiography.
Echocardiographic assessment of cardiac function was conducted using a Vevo 770 High Resolution In Vivo Imaging System (VisualSonics) as previously described (4, 34) according to the American Society of Echocardiography Guidelines using the leading-edge technique (41).
Mice were anesthetized with 1.5% isoflurane gas and placed on a prewarmed pad, and body temperature was monitored with a rectal probe. Heart rate was monitored via an EKG transducer pad throughout each echocardiography session. Two-dimensional B-Mode parasternal long axis view was obtained first to visualize the aortic and mitral valves. The transducer was then rotated clockwise 90° to obtain the parasternal short axis view, and M-Mode images were obtained at the mid-papillary muscle level from this view. Fractional shortening (%), ejection fraction (%), and cardiac output (ml/min) were analyzed and calculated using the VisualSonics Advanced Cardiovascular Measurements Package using the mean of 12–15 cardiac cycles derived from 3 separate M-Mode images during each session. The operator was unaware of the treatments and genotypes of the animals during image acquisition and analysis.
Blood pressure determination.
Mean arterial pressure was measured in conscious, freely moving mice by means of an indwelling carotid artery catheter at the end of a 2- or 8-wk treatment period, as we previously reported (54, 55).
Statistical analysis.
All results were expressed as means ± SE. Multiple experimental groups were analyzed by one- or two-way ANOVA followed by Newman-Keuls post hoc contrasts. Statistical calculations were performed with GraphPad Prism package version 7.04 (GraphPad Software). Differences were considered significant at P < 0.05.
RESULTS
ALDO upregulates cardiac miR-21 expression.
We previously reported that cardiac miR-21 is upregulated by ALDO/SALT treatment in Sprague-Dawley rats (4). To study the role of miR-21 in ALDO/SALT-mediated cardiac injury using genetically engineered mice models, we first validated that ALDO/SALT-mediated LV miR-21 expression is subjected to a similar regulation. miR-21 was upregulated ~2.5-fold in the LV after 2 wk of treatment and remained elevated for up to 8 wk (Fig. 1). ALDO/SALT-mediated LV miR-21 regulation showed a similar pattern and magnitude of regulation in mice and rats (4).
Fig. 1.
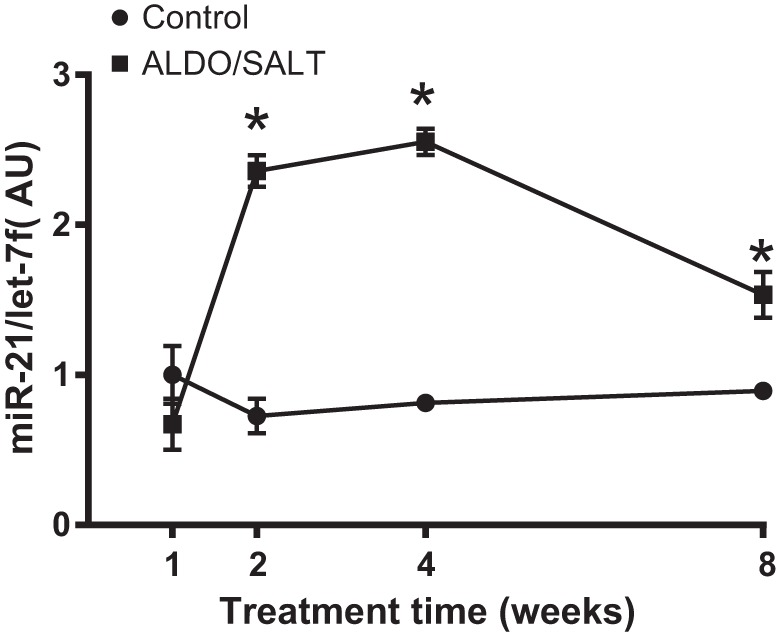
miR-21 is upregulated by ALDO/SALT in the left ventricle. Wild type C57BL/6 mice were treated with ALDO/SALT or vehicle for up to 8 wk. miRNAs were quantified by RT-qPCR. Results are expressed as means ± SE (n = 6). *P < 0.05 vs. Control. ALDO, aldosterone; AU, arbitrary unit; MiR-21, microRNA-21.
miR-21 genetic ablation exacerbates ALDO/SALT-mediated cardiac hypertrophy.
To elucidate the role of miR-21 on ALDO/SALT-mediated cardiac hypertrophy, we used genetically engineered mice with genetic miR-21 ablation (miR21KO) (49). miR21KO mice are viable, thrive and reproduce normally, and do not exhibit any overt phenotype under basal conditions. miR21KO and WT littermate control mice were treated with ALDO/SALT or vehicle for 2 or 8 wk. ALDO/SALT caused cardiac and LV hypertrophy after 2 wk of treatment only in WT animals (Fig. 2, A and B). However, after 8 wk of treatment, miR21KO mice not only catch up but further present an exacerbated cardiac and LV hypertrophy when compared with WT animals (Fig. 2, A and B). ALDO/SALT-mediated cardiac hypertrophy exacerbation in miR21KO animals was also observed at the cellular level in cardiomyocyte cross-sectional area analysis. ALDO/SALT increased cardiomyocyte cross-sectional area only in miR21KO but not WT animals (Fig. 2C). Frequency distribution analysis of ALDO/SALT-treated animals clearly showed a shift to increased cardiomyocyte cross-sectional area in miR21KO mice compared with WT animals (Fig. 2D), also evidenced in microscopy images (Fig. 2E). Furthermore, an in-depth analysis of cardiomyocyte hypertrophy showed that ALDO/SALT-mediated effect in miR21KO mice was restricted to the LV and the interventricular septum walls (data not shown).
Fig. 2.

miR-21 genetic ablation exacerbates ALDO/SALT-mediated cardiac and cardiomyocyte hypertrophy. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 2 or 8 wk. Heart (A) or left ventricle (B) weights were determined by gravimetry and corrected by body weight. Results are means ± SE (n = 6–8). *P < 0.05. WT or miR21KO mice were treated with ALDO/SALT or vehicle for 8 wk, and cardiomyocyte cross-sectional areas were quantified by WGA staining in FFPE cardiac sections (C and D). Representative images of cardiomyocyte cross-sectional area (E). At least 100 cardiomyocytes in 3 different regions of the right, left, and interventricular septum were quantified, and cardiomyocyte cross-sectional areas were expressed as μm2 (n = 4–5 mice per treatment). Cumulative distributions were analyzed by the non-parametric Kolmogorov-Smirnov test. *P < 0.05. ALDO, aldosterone; BW, body weight; FFPE, formalin-fixed, paraffin-embedded; LV, left ventricle; miR-21, microRNA-21; miR21KO, miR-21 knockout; WGA, wheat germ agglutinin; WT, wild type; NS, not significant.
miR-21 genetic ablation increased ALDO/SALT-mediated cardiac injury.
To analyze the role of miR-21 on ALDO/SALT-mediated cardiac injury, we analyzed the expression of fibrosis and inflammation markers in the LV of WT and miR21KO animals subjected to ALDO/SALT treatment for 2 or 8 wk (Figs. 3 and 4). When we analyzed fibrosis marker expression, the most striking findings were the significant upregulation of Ctgf and lysyl oxidase (Lox) in the LV of ALDO/SALT-treated miR21KO animals (Fig. 3, A and B). Fibronectin (Fn1) and collagen I (Col1a1) were also upregulated, although to a lower degree, by ALDO/SALT treatment only in miR21KO animals (Fig. 3, C and D). Collagen III (Col3a1) expression was 30% lower in miR21KO mice compared with WT animals under control conditions but was not affected by ALDO/SALT treatment (Fig. 3E). Histological analysis showed that the increase in fibrosis markers expression translates into an exacerbation of interstitial (Fig. 3F), but not perivascular (data not shown), ALDO/SALT-mediated fibrosis in miR21KO animals.
Fig. 3.

miR-21 genetic ablation exacerbates ALDO/SALT-mediated LV fibrosis. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 2 or 8 wk, and LV gene expression was quantified by RT-qPCR (A–E). miR21KO or WT mice were treated with ALDO/SALT or vehicle for 8 wk, and cardiac interstitial fibrosis was quantified in Picro Sirius Red-stained sections (F). Results are means ± SE (n = 6–8). *P < 0.05. ALDO, aldosterone; AU, arbitrary unit; Col1a1, collagen I; Col3a1, collagen III; Ctgf, connective tissue growth factor; Fn1, fibronectin 1; Lox, lysyl oxidase; LV, left ventricle; miR-21, microRNA-21; miR21KO, miR-21 knockout; WT, wild type.
Fig. 4.

miR-21 genetic ablation exacerbates ALDO/SALT-mediated LV inflammation marker gene expression. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 2 or 8 wk and left ventricle gene expression quantified by RT-qPCR (A–D). Results are means ± SE (n = 6–8). *P < 0.05. ALDO, aldosterone; AU, arbitrary unit; IL6, interleukin 6; LV, left ventricle; MCP-1, monocyte chemoattractant protein-1; miR-21, microRNA-21; miR21KO, miR-21 knockout; PAI-1, plasminogen activator inhibitor type 1; Spp1, osteopontin; WT, wild type.
Analysis of inflammation markers indicated that the most significant findings were that ALDO/SALT treatment upregulated the expression of PAI-1 (Serpine1), monocyte chemoattractant protein-1 (MCP-1, Ccl2), osteopontin (OPN, Spp1), and interleukin-6 (IL6) only in miR21KO animals after 8 wk of treatment (Fig. 4, A–D). On the other hand, there was not a genotype effect on transforming growth factor β (Tgfb) or tumor necrosis factor α (Tnf) LV expression (data not shown).
To validate mRNA expression studies at the protein level, we analyzed expression levels of selected fibrosis and inflammatory markers by Western blot (Fig. 5, A and B). Fibrosis marker Ctgf and inflammatory marker PAI-1 (Serpine1) protein expression were significantly upregulated only in miR21KO mice after 8 wk of ALDO/SALT treatment, following a pattern similar to the mRNA expression.
Fig. 5.

miR-21 genetic ablation exacerbates ALDO/SALT-mediated LV fibrosis and inflammation markers protein expression. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 8 wk, and LV protein levels of Ctgf (A), PAI-1 (B), and Nppa (C) were quantified by Western blot. Results are means ± SE (n = 4). *P < 0.05. ALDO, aldosterone; AU, arbitrary unit; Ctgf, connective tissue growth factor; LV, left ventricle; miR-21, microRNA-21; miR21KO, miR-21 knockout; PAI-1, plasminogen activator inhibitor type 1; WT, wild type.
miR-21 genetic ablation decreased cardiac function in ALDO/SALT-treated animals.
To analyze the role of miR-21 on ALDO/SALT-mediated cardiac dysfunction, we assessed cardiac function by echocardiography in WT and miR21KO animals subjected to ALDO/SALT treatment for 8 wk (Fig. 6). ALDO/SALT-treated miR21KO animals showed a significant decrease in ejection fraction, fractional shortening, and cardiac output when compared either to control miR21KO or ALDO/SALT-treated WT animals (Fig. 6, A–C). The deleterious effect of miR-21 ablation on ALDO/SALT-mediated cardiac dysfunction was blood pressure independent, since 8 wk of ALDO/SALT treatment raised blood pressure to similar levels in WT and miR21KO animals (Fig. 6D). No effect of genotype or treatment was observed in blood pressure after 2 wk of treatments (data not shown), suggesting similar time course of blood pressure rise in both genotypes.
Fig. 6.

miR-21 genetic ablation exacerbates ALDO/SALT-mediated cardiac dysfunction without affecting hypertension. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 8 wk. Ejection fraction (A), fractional shortening (B), and cardiac output (C) were measured by echocardiography. Each determination is the mean of 12–15 cardiac cycles derived from 3 separate M-Mode images during each session. Blood pressure was measured by means of an indwelling catheter in freely moving conscious animals (D). Results are means ± SE (n = 5). *P < 0.05. ALDO, aldosterone; miR-21, microRNA-21; miR21KO, miR-21 knockout; NS, not significant; WT, wild type.
Furthermore, we analyzed the expression levels of cardiac stretch markers atrial and brain natriuretic peptides (Nppa and Nppb, respectively). Nppa and Nppb LV expression was upregulated after ALDO/SALT treatment for 8 or 2 wk, respectively, in the miR21KO mice (Fig. 7, A and B). Moreover, β-myosin heavy chain (Myh7) was upregulated after 8 wk of ALDO/SALT treatment in miR21KO mice, whereas there was no effect on α-myosin heavy chain (Myh6) LV expression (Fig. 7, C and D). Analysis of Nppa protein expression levels failed to show upregulation by ALDO/SALT treatment; instead Nppa protein levels showed a decrease in miR21KO compared with WT in 8 wk-treated ALDO/SALT animals (Fig. 5C).
Fig. 7.

miR-21 genetic ablation increases fetal program gene expression in ALDO/SALT-treated mice. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 2 or 8 wk and left ventricle gene expression quantified by RT-qPCR (A–D). Results are means ± SE (n = 6–8). *P < 0.05. ALDO, aldosterone; AU, arbitrary unit; miR-21, microRNA-21; miR21KO, miR-21 knockout; Myh6, α-myosin heavy chain; Myh7, β-myosin heavy chain; WT, wild type.
miR-21 genetic ablation modulates miR-21 target genes.
To elucidate the molecular mechanisms by which miR-21 genetic ablation exacerbates ALDO/SALT-mediated cardiac injury and dysfunction, we analyzed the mRNA and protein levels of several miR-21 targets that have been reported to mediate miR-21 cardiac phenotype changes in other experimental models. We focused our analysis on Spry1, Spry2, Pten, Pdcd4, and apoptosis regulator Bcl2 (Bcl2). At the mRNA expression level, there were not significant changes with the exception of Spry1, which was modestly but significantly upregulated by ALDO/SALT treatment exclusively in miR21KO animals after 2 and 8 wk of treatment (Fig. 8, left and center). At the protein level, we observed a modest but significant increase in Spry1 protein levels exclusively in miR21KO mice treated with ALDO/SALT (Fig. 8A, right). However, the largest change in protein levels was observed in Spry2 (Fig. 8B, right). Spry2 protein levels were higher in miR21KO mice and further exacerbated by ALDO/SALT treatment in both mouse genotypes.
Fig. 8.

miR-21 genetic ablation exacerbates miR-21 target gene Sprouty 2 (Spry2) protein levels in ALDO/SALT-mediated cardiac injury. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 2 (left) or 8 wk (center), and left ventricle gene expression was quantified by RT-qPCR for Spry1 (A), Spry2 (B), Pten (C), Pdcd4 (D), and Bcl2 (E). Results are means ± SE (n = 6–8). *P < 0.05. miR21KO or WT mice were treated with ALDO/SALT or vehicle for 8 wk, and left ventricle protein levels were quantified by Western blot (right). Results are means ± SE (n = 4). *P < 0.05. ALDO, aldosterone; AU, arbitrary unit; miR-21, microRNA-21; miR21KO, miR-21 knockout; Pdcd4, programmed cell death 4; Pten, phosphatase and tensin homolog; Spry1, Sprouty 1, WT, wild type.
In summary, miR-21 genetic ablation exacerbated ALDO/SALT-mediated cardiac hypertrophy, injury, and dysfunction, despite no significant effect in blood pressure, and those effects may be mediated by the miR-21 target gene Sprouty 2.
DISCUSSION
Our results show that miR-21 genetic ablation exacerbates ALDO/SALT-mediated cardiac hypertrophy, injury, and dysfunction in an animal experimental model of primary aldosteronism. These cardiac deleterious effects may be mediated, at least partially, by an upregulation of the miR-21 target gene Sprouty 2. Moreover, these results strongly suggest that miR-21 plays a protective role on the cardiac pathology triggered by excess ALDO. These findings suggest that miR-21 supplementation may be a potential therapeutic approach to abolish or mitigate excess ALDO-mediated cardiovascular deleterious effects such as the ones observed in patients with primary aldosteronism.
We previously reported that miR-21 is the most upregulated miRNA in the LV of ALDO/SALT-treated adult male rats (4). Furthermore, we reported that miR-21 downregulation by means of antagomirs exacerbates ALDO/SALT-mediated cardiac deleterious phenotype as evidenced by an increase in cardiac hypertrophy, fibrosis, and dysfunction (4). In the present report, we confirm those findings in a genetic model of miR-21 ablation that is devoid of antagomir off-target confounding effects. Importantly, we expand on our previous findings to demonstrate that the miR-21 cardioprotective effect in the context of ALDO/SALT-mediated cardiac injury and dysfunction is blood pressure independent.
Primary aldosteronism was first described as a case report series in the early 1950s by J. W. Conn (21) and is long considered a rare disease mainly because of technical problems in the screening of the disease. In the last 2 decades, there has been overwhelming evidence that primary aldosteronism is a fairly common disease representing ~10% of hypertensive patients and ranking as the most common cause of secondary hypertension (11, 60). Moreover, a recent meta-analysis (58) and an impressive retrospective cross-sectional study comprising 2,582 patients with primary aldosteronism (61) have put to rest the notion that primary aldosteronism is just another form of hypertension and its main consequence is a rise in blood pressure. Those studies compared primary aldosteronism patients with matched essential hypertensive patients with similar blood pressure and have unequivocally shown that patients with primary aldosteronism have increased risk of stroke, coronary artery disease, atrial fibrillation, and heart failure compared with essential hypertensive patients (58, 61).
Surgical (adrenalectomy) or medical (mineralocorticoid receptor blockers) therapy are the usual options for unilateral or bilateral adrenal gland excess ALDO production, respectively. Both therapeutic approaches are highly effective in correcting the biochemical abnormalities of primary aldosteronism (52, 98). However, both surgical and medical therapies show a significant variability (up to 32%–37%) in the degree of clinical success (52, 98). A recent retrospective cohort study analyzed the cardiometabolic outcomes and mortality in 602 patients with primary aldosteronism on mineralocorticoid receptor blockade therapy and more than 40,000 patients with essential hypertension (39). The medically treated patients with primary aldosteronism present an increased hazard ratio in the primary outcome of incident cardiovascular event, defined as a composite of incident myocardial infarction or coronary revascularization, hospital admission with congestive heart failure, or stroke. The increase in cardiometabolic events in patients with primary aldosteronism receiving mineralocorticoid receptor blockade therapy was evident despite blood pressure control at the time of study entry. Thus, those studies clearly show that patients with primary aldosteronism present a worse cardiovascular phenotype than essential hypertensive patients with similar blood pressure. Moreover, medical treatment of primary aldosteronism patients with mineralocorticoid receptor blockers, the only option for patients with bilateral adrenal hyperplasia, is suboptimal for cardiometabolic outcome correction despite achieving adequate blood pressure control. The limitations of current medical strategies for optimal treatment of primary aldosteronism support the need for developing add-on therapies to mineralocorticoid receptor blockade that can benefit the cardiovascular outcome in patients with primary aldosteronism.
miR-21 is weakly expressed in normal human hearts, but it is consistently upregulated in human end-stage heart failure (56, 85, 86, 103). In animal experimental models, cardiac miR-21 has been shown to be upregulated by thoracic aortic banding (16, 75–77, 84, 89), ischemia/reperfusion (71), ischemic preconditioning (18, 28), and heat shock (104) as well as in transgenic mice expressing activated calcineurin A (CnA), in the heart (89) and in the border zone of myocardial infarct (25, 64, 90, 108), in a model of pulmonary insufficiency and stenosis (67) and in a model of diabetic nephropathy (95). Despite multiple studies, the role of miR-21 in cardiac pathophysiology remains highly controversial, with a plethora of studies suggesting that this particular miRNA can be either deleterious (1, 5, 14, 16, 32, 38, 44, 48, 63, 66, 86, 95, 108) or beneficial (17, 18, 25, 43, 62, 63, 65, 75, 77, 84, 88, 100, 104, 109) in cardiac injury.
Our results show that miR-21 genetic ablation exacerbated ALDO/SALT-mediated cardiac hypertrophy at the tissue and cellular level. These changes were blood pressure independent, as the lack of miR-21 does not modify ALDO/SALT-mediated hypertension. These results suggest that direct cardiac actions of ALDO, probably in cardiomyocytes, are partially mediated by miR-21 with this miRNA being upregulated and playing a protective role in ALDO/SALT-mediated cardiac remodeling. Rickard and colleagues (68) reported that mineralocorticoid receptor ablation in a mouse model of deoxycorticosterone (DOC)/SALT-induced cardiac injury attenuates cardiac fibrosis and inflammation. Unfortunately, the DOC/SALT mice experimental model does not fully recapitulate the observations in patients with primary aldosteronism or the ALDO/SALT rodent experimental model, as no cardiac or cardiomyocyte cross-sectional area hypertrophy is observed in WT DOC/SALT-treated animals even after 8 wk of treatment (68). DOC, or its acetate, has been used as a surrogate of ALDO for in vivo studies and its lower mineralocorticoid activity compensated with higher doses (3.6 μg ALDO/day vs. 333 μg DOC/day). However, DOC is not only a weak mineralocorticoid but also a steroid with a unique combination of biological mineralocorticoid and glucocorticoid activities that not only may not fully replicate ALDO biological actions, but that may also be unique ones that can potentially generate a different cardiac phenotype (93).
We previously reported that miR-21 downregulation exacerbated ALDO/SALT-mediated cardiac fibrosis and dysfunction but had a protective role in cardiac inflammation after 2 wk of treatment (4). In the present study, we report that miR-21 genetic ablation increased ALDO/SALT-mediated cardiac inflammation as evidenced by an increase in LV PAI-1, MCP1, Spp1, and Il6 mRNA expression after 8 wk of treatment. These differences may be explained by a different course of the disease in different animal species. C57BL/6 is the most popular mouse strain for transgenic studies, but that does not imply that it is the best one for all disease models. C57BL/6 mice are notoriously resistant to chronic cardiac and renal injury compared with other mouse strains (40, 94). The resistance of C57BL/6 mice to cardiorenal injury may explain some of the differences observed at the molecular and physiological level when the ALDO/SALT-induced cardiac phenotype is compared with other species. The species-specific differences in the course of the disease may also explain why miR-21 genetic ablation in mice partially protected them from ALDO/SALT-mediated cardiac hypertrophy after 2 wk of treatment, but later on, miR21KO mice showed a significant increase in cardiac and LV hypertrophy. A possible explanation for this delayed effect of miR-21 ablation in cardiac injury may be that most of the significant changes in fibrosis (Ctgf, Lox) and inflammation (PAI-1, MCP-1, Spp1, IL6) markers were only observed after 8 wk of treatment. On the other hand, discrepancy between genetic and pharmacological approaches to ablate or downregulate miR-21, respectively, can be explained by off-target effects of the antagomir antisense oligonucleotides used in our previous studies (4). In the present report, we present a refined animal experimental model that clearly shows that miR-21 plays a cardioprotective role in the setting of excess ALDO-mediated cardiac injury and dysfunction.
We observed a similar pattern at the mRNA and protein level for Ctgf and PAI-1. However, we did not observe parallel changes at the mRNA and protein level for Nppa. Other mRNA/protein pairs have been reported to have dissociated expression patterns. For example, whereas cardiac total collagen content continuously increased for 12 wk after permanent coronary artery ligation, an experimental model of myocardial infarction, collagens I and III mRNA levels peak 1 wk after the surgical procedure and return to basal levels after 2 wk (110). This classical example of cardiac collagen expression may suggest that Nppa and Nppb protein levels increase later in time and not parallel with their mRNA expression pattern.
We analyzed the expression of some of the most prominent validated candidate miR-21 target genes (Spry1, Spry2, Pten, Pdcd4, and Bcl2) that have been previously implicated in cardiac injury and dysfunction in other animal experimental models (18, 26, 71, 77, 86). Our study showed that Sprouty 2 protein levels were exacerbated by ALDO/SALT treatment in the LV of miR21KO mice. Sprouty family proteins (Spry1, Spry2, Spry3, and Spry4) are negative regulators of the MAPK/ERK intracellular signaling pathway and modulate key processes including proliferation, differentiation, motility, and survival (53). The miR-21/Sprouty proteins pathway has been linked to cardiomyocyte remodeling in cardiac hypertrophy and fibroblast activation in heart failure (73, 77, 86).
miR-21 overexpression in neonatal rat cardiomyocytes causes extensive cellular outgrowths (77). This biological effect of miR-21 is reversed by Sprouty 2 overexpression and mimicked by Sprouty 2 knockdown, suggesting that Sprouty 2 is a mediator of miR-21-induced cellular outgrowths. Moreover, miR-21-mediated Sprouty 2 downregulation translated into ERK1/2 activation as evidenced by increased ERK1/2 phosphorylation. In vitro studies using a reporter plasmid expressing an mRNA containing the predicted miR-21 target sequences in Sprouty 2 mRNA demonstrated that Sprouty 2 is indeed a direct target of miR-21. Those studies strongly suggest that Sprouty 2 is a direct miR-21 target that, by modulating MAPK/ERK intracellular signaling pathway, regulates cardiomyocyte biology.
miR-21 is upregulated in animal experimental models of heart failure as well as in samples from human failing hearts (86). miR-21 upregulation in failing hearts is restricted to cardiac fibroblasts where Sprouty 1 is co-expressed, suggesting that Sprouty 1 could be a potential target of miR-21. In vitro studies using a reporter plasmid expressing a fusion mRNA containing the 3′ untranslated region of Sprouty 1 mRNA demonstrated that Sprouty 1 is indeed a direct target of miR-21. miR-21 downregulation in rat cardiac fibroblasts decreased apoptosis and activated the MAPK/ERK intracellular signaling pathway. As expected, if Sprouty 1 was mediating miR-21 effects, Sprouty 1 knockdown led to an increase in MAPK/ERK signaling and cell survival. Overall, these studies suggest that Sprouty 1 is a direct target of miR-21 that, by modulating MAPK/ERK intracellular signaling pathway, regulates cardiac fibroblast activation and survival.
Those and several other studies have shown that the Sprouty protein family is a target of miR-21 and mediates many of its biological effects. Chronic hypoxia is a trigger of pulmonary vascular remodeling resulting in pulmonary hypertension, which is associated with smooth muscle cell (SMC) proliferation. Experimental hypoxia stimulates human pulmonary artery SMC (HPASMC) proliferation, upregulation of miR-21, and downregulation of miR-21 target Sprouty 2 (73). Moreover, miR-21 overexpression in HPASMC under normoxia conditions decreased Spry2 protein levels. On the other hand, miR-21 downregulation in HPASMC under hypoxia increased Spry2 protein levels. Taken together, those studies suggest that Sprouty 2, as well as other miR-21 targets, mediates the miR-21 effects on hypoxia-induced proliferation and migration associated with pulmonary hypertension. In summary, there is strong experimental evidence that Sprouty proteins mediate many of the biological effects triggered by miR-21.
It is important to highlight that in the above mentioned studies related to the miR-21/Sprouty axis, miR-21plays a cardiac deleterious role, whereas in our experimental model as in many other ones (17, 18, 25, 43, 62, 63, 65, 75, 77, 84, 88, 100, 104, 109) miR-21 plays a protective role. Consequently, the interpretation of the role of Sprouty proteins as mediators of miR-21 actions is more difficult and would require further studies.
Several miRNAs, such as miR-22, miR-125b, miR-150, miR-214, miR-221/222, and miR-532, have been shown to be cardioprotective in vivo in cardiac injury models of myocardial infarct, myocardial ischemia/reperfusion, and pressure overload (3, 8, 9, 33, 45, 83, 92, 96). The findings of the current report strongly suggest that miR-21, at least in the context of excess ALDO-mediated cardiac injury, should join the larger family of cardioprotective miRNAs also called “protectomiRs” (91).
A limitation of the current study is the use of a global miR21KO mice. miR-21 is expressed in multiple tissues including, but not limited to, the heart, kidney, liver, brain, and vasculature. All of these tissues can directly or indirectly affect cardiac function and are involved in blood pressure regulation. We cannot exclude that some of the biological effects of miR-21 ablation observed in the heart could be modulated or even be a direct consequence of miR-21 ablation in extra-cardiac tissues. Only the use of cell- or tissue-specific transgenic animals would allow answering the question of whether the cardiac effects of miR-21 genetic ablation reported in the current study are independent of the effects of miR-21 in other tissues or organs.
Also, it is important to note that the genetically engineered mice used have an ablation of pre-miR-21 which leads to a lack of both miR-21 (miR-21–5p) and miR-21 passenger strand (miR-21–3p or miR-21*). miR-21* has been reported to be enriched, compared with miR-21, in exosomes secreted by rat cardiac fibroblasts, in contrast to the intracellular milieu that is enriched in miR-21 (5). Cardiac fibroblast-conditioned media or cardiac fibroblast-cardiomyocyte coculture leads to increased levels of miR-21* and hypertrophy in cardiomyocytes. Moreover, thoracic aortic constriction in mice leads to increase of miR-21* in pericardial fluid. Furthermore, angiotensin II-induced cardiac hypertrophy in mice is attenuated by miR-21* downregulation at the organ and cellular level. Those findings suggest that miR-21* is not merely a bystander but has important biological functions in cardiac biology. However, it is important to state that in the two above-mentioned animal experimental models of cardiac injury and remodeling, both miR-21 and miR-21* seem to play a deleterious role. On the other hand, in our animal experimental model of excess ALDO-mediated cardiac injury and dysfunction, miR-21, and putatively miR-21*, play a protective role decreasing ALDO/SALT-mediated cardiac injury and dysfunction. Moreover, we previously reported that miR-21* levels are more than 500 times less than miR-21 in the LV of rats treated with ALDO/SALT for 2 wk (4). Nevertheless, we cannot exclude that miR-21*, in an intracrine or paracrine fashion, regulates cardiac pathophysiology in primary aldosteronism. Further studies with miR-21 strand-specific knockdown or supplementation will be needed to answer this exciting cardiac biology question.
In summary, our study shows that miR-21 is upregulated by excess ALDO in the LV, and miR-21 genetic ablation worsens the excess ALDO-mediated cardiac deleterious phenotype. However, despite ALDO-mediated cardiac miR-21 upregulation, the increase in miR-21 levels is not sufficient to counteract ALDO-induced cardiac injury and dysfunction. We speculate that cell-specific and miRNA strand-specific miR-21 supplementation therapies may be a novel add-on therapeutic approach to abolish or further mitigate the cardiac ALDO-mediated deleterious phenotype observed in patients with primary aldosteronism.
GRANTS
This work was supported by American Heart Association Grants 12SDG8980032 (D. G. Romero) and 0830239N (L. L. Yanes Cardozo), Endocrine Fellows Foundation Endocrine Research Grant (L. L. Yanes Cardozo), VA Merit Award BX002604 (M. J. Ryan) and National Institutes of Health Grants R21 DK113500 (D. G. Romero), P20 GM121334 (L. L. Yanes Cardozo), and K08 K099415 (M. E. Hall).
The University of Mississippi Medical Center (UMMC) Center for Psychiatric Neuroscience Imaging Core is supported by NIH Centers of Biomedical Research Excellence (COBRE) P30GM103328, and the UMMC Molecular and Genomics Facility is supported, in part, by the NIH, including Mississippi IDeA Networks of Biomedical Research Excellence P20GM103476; Center for Psychiatric Neuroscience COBRE P30GM103328; Obesity, Cardiorenal and Metabolic Diseases COBRE P20GM104357; and Mississippi Center for Excellence in Perinatal Research COBRE P20GM121334.
DISCLOSURES
M. E. Rothenberg is a consultant for Pulm One, Spoon Guru, ClostraBio, Celgene, Shire, Astra Zeneca, GlaxoSmithKline, Allakos, Adare, Regeneron, and Novartis and has an equity interest in the first four listed and Immune Pharmaceuticals and royalties from reslizumab (Teva Pharmaceuticals) and UpToDate. M. E. Rothenberg is an inventor of patents owned by Cincinnati Children’s Hospital Medical Center. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
M.S., M.E.H., M.J.R., M.E.R., L.L.Y.C., and D.G.R. conceived and designed research; M.S., J.P.B., K.W.M., M.E.H., L.L.Y.C., and D.G.R. performed experiments; M.S., J.P.B., K.W.M., M.E.H., L.L.Y.C., and D.G.R. analyzed data; M.S., J.P.B., M.J.R., M.E.R., L.L.Y.C., and D.G.R. interpreted results of experiments; M.S., J.P.B., L.L.Y.C., and D.G.R. prepared figures; M.S., L.L.Y.C., and D.G.R. drafted manuscript; M.S., J.P.B., K.W.M., M.E.H., M.J.R., M.E.R., L.L.Y.C., and D.G.R. edited and revised manuscript; M.S., J.P.B., K.W.M., M.E.H., M.J.R., M.E.R., and D.G.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the University of Mississippi Medical Center (UMMC) Center for Psychiatric Neuroscience Imaging Core, the UMMC Molecular and Genomics Facility, and UMMC Pathology Research Histology Core for outstanding service.
REFERENCES
- 1.Adam O, Löhfelm B, Thum T, Gupta SK, Puhl SL, Schäfers HJ, Böhm M, Laufs U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol 107: 278, 2012. doi: 10.1007/s00395-012-0278-0. [DOI] [PubMed] [Google Scholar]
- 2.Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14: 475–488, 2013. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 3.Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J Clin Invest 122: 1222–1232, 2012. doi: 10.1172/JCI59327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ball JP, Syed M, Marañon RO, Hall ME, Kc R, Reckelhoff JF, Yanes Cardozo LL, Romero DG. Role and regulation of microRNAs in aldosterone-mediated cardiac injury and dysfunction in male rats. Endocrinology 158: 1859–1874, 2017. doi: 10.1210/en.2016-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest 124: 2136–2146, 2014. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauersachs J. Regulation of myocardial fibrosis by MicroRNAs. J Cardiovasc Pharmacol 56: 454–459, 2010. doi: 10.1097/FJC.0b013e3181ee81df. [DOI] [PubMed] [Google Scholar]
- 8.Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y, Su H, Weintraub NL, Kim IM. A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol 114: 72–82, 2018. doi: 10.1016/j.yjmcc.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayoumi AS, Teoh JP, Aonuma T, Yuan Z, Ruan X, Tang Y, Su H, Weintraub NL, Kim IM. MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc Res 113: 1603–1614, 2017. doi: 10.1093/cvr/cvx132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 67: 1355–1364, 1990. doi: 10.1161/01.RES.67.6.1355. [DOI] [PubMed] [Google Scholar]
- 11.Buffolo F, Monticone S, Burrello J, Tetti M, Veglio F, Williams TA, Mulatero P. Is primary aldosteronism still largely unrecognized? Horm Metab Res 49: 908–914, 2017. doi: 10.1055/s-0043-119755. [DOI] [PubMed] [Google Scholar]
- 12.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol 23: 175–205, 2007. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 13.Calhoun DA. Is there an unrecognized epidemic of primary aldosteronism? Pro. Hypertension 50: 447–453, 2007. doi: 10.1161/HYPERTENSIONAHA.106.086116. [DOI] [PubMed] [Google Scholar]
- 14.Cardin S, Guasch E, Luo X, Naud P, Le Quang K, Shi Y, Tardif JC, Comtois P, Nattel S. Role for MicroRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ Arrhythm Electrophysiol 5: 1027–1035, 2012. doi: 10.1161/CIRCEP.112.973214. [DOI] [PubMed] [Google Scholar]
- 15.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8: 93–103, 2007. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol 170: 1831–1840, 2007. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol 47: 5–14, 2009. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res 87: 431–439, 2010. doi: 10.1093/cvr/cvq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19: 321–327, 2012. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Condorelli G, Latronico MV, Cavarretta E. microRNAs in cardiovascular diseases: current knowledge and the road ahead. J Am Coll Cardiol 63: 2177–2187, 2014. doi: 10.1016/j.jacc.2014.01.050. [DOI] [PubMed] [Google Scholar]
- 21.Conn JW, Louis LH. Primary aldosteronism, a new clinical entity. Ann Intern Med 44: 1–15, 1956. doi: 10.7326/0003-4819-44-1-1. [DOI] [PubMed] [Google Scholar]
- 22.Dai Y, Khaidakov M, Wang X, Ding Z, Su W, Price E, Palade P, Chen M, Mehta JL. MicroRNAs involved in the regulation of postischemic cardiac fibrosis. Hypertension 61: 751–756, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00654. [DOI] [PubMed] [Google Scholar]
- 23.Dangwal S, Bang C, Thum T. Novel techniques and targets in cardiovascular microRNA research. Cardiovasc Res 93: 545–554, 2012. doi: 10.1093/cvr/cvr297. [DOI] [PubMed] [Google Scholar]
- 24.Dolber PC, Bauman RP, Rembert JC, Greenfield JC Jr. Regional changes in myocyte structure in model of canine right atrial hypertrophy. Am J Physiol 267: H1279–H1287, 1994. doi: 10.1152/ajpheart.1994.267.4.H1279. [DOI] [PubMed] [Google Scholar]
- 25.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem 284: 29514–29525, 2009. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong S, Ma W, Hao B, Hu F, Yan L, Yan X, Wang Y, Chen Z, Wang Z. microRNA-21 promotes cardiac fibrosis and development of heart failure with preserved left ventricular ejection fraction by up-regulating Bcl-2. Int J Clin Exp Pathol 7: 565–574, 2014. [PMC free article] [PubMed] [Google Scholar]
- 27.Du T, Zamore PD. microPrimer: the biogenesis and function of microRNA. Development 132: 4645–4652, 2005. doi: 10.1242/dev.02070. [DOI] [PubMed] [Google Scholar]
- 28.Duan X, Ji B, Wang X, Liu J, Zheng Z, Long C, Tang Y, Hu S. Expression of microRNA-1 and microRNA-21 in different protocols of ischemic conditioning in an isolated rat heart model. Cardiology 122: 36–43, 2012. doi: 10.1159/000338149. [DOI] [PubMed] [Google Scholar]
- 29.Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet 9: 831–842, 2008. doi: 10.1038/nrg2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young WF Jr, Montori VM; Endocrine Society . Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 93: 3266–3281, 2008. doi: 10.1210/jc.2008-0104. [DOI] [PubMed] [Google Scholar]
- 31.Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, Rajewsky N. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell 54: 1042–1054, 2014. doi: 10.1016/j.molcel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta SK, Itagaki R, Zheng X, Batkai S, Thum S, Ahmad F, Van Aelst LN, Sharma A, Piccoli MT, Weinberger F, Fiedler J, Heuser M, Heymans S, Falk CS, Förster R, Schrepfer S, Thum T. miR-21 promotes fibrosis in an acute cardiac allograft transplantation model. Cardiovasc Res 110: 215–226, 2016. doi: 10.1093/cvr/cvw030. [DOI] [PubMed] [Google Scholar]
- 33.Gurha P, Abreu-Goodger C, Wang T, Ramirez MO, Drumond AL, van Dongen S, Chen Y, Bartonicek N, Enright AJ, Lee B, Kelm RJ Jr, Reddy AK, Taffet GE, Bradley A, Wehrens XH, Entman ML, Rodriguez A. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 125: 2751–2761, 2012. doi: 10.1161/CIRCULATIONAHA.111.044354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall ME, Maready MW, Hall JE, Stec DE. Rescue of cardiac leptin receptors in db/db mice prevents myocardial triglyceride accumulation. Am J Physiol Endocrinol Metab 307: E316–E325, 2014. doi: 10.1152/ajpendo.00005.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han M, Toli J, Abdellatif M. MicroRNAs in the cardiovascular system. Curr Opin Cardiol 26: 181–189, 2011. doi: 10.1097/HCO.0b013e328345983d. [DOI] [PubMed] [Google Scholar]
- 36.Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions beyond repression of gene expression. Nat Rev Genet 15: 599–612, 2014. [Erratum in: Nat Rev Genet 15: 702, 2014]. doi: 10.1038/nrg3765. [DOI] [PubMed] [Google Scholar]
- 37.Hobert O. Gene regulation by transcription factors and microRNAs. Science 319: 1785–1786, 2008. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- 38.Huang Z, Chen XJ, Qian C, Dong Q, Ding D, Wu QF, Li J, Wang HF, Li WH, Xie Q, Cheng X, Zhao N, Du YM, Liao YH. Signal transducer and activator of transcription 3/microRNA-21 feedback loop contributes to atrial fibrillation by promoting atrial fibrosis in a rat sterile pericarditis model. Circ Arrhythm Electrophysiol 9: e003396, 2016. doi: 10.1161/CIRCEP.115.003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol 6: 51–59, 2018. doi: 10.1016/S2213-8587(17)30367-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirchhoff F, Krebs C, Abdulhag UN, Meyer-Schwesinger C, Maas R, Helmchen U, Hilgers KF, Wolf G, Stahl RA, Wenzel U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int 73: 643–650, 2008. doi: 10.1038/sj.ki.5002689. [DOI] [PubMed] [Google Scholar]
- 41.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ; Chamber Quantification Writing Group; American Society of Echocardiography’s Guidelines and Standards Committee; European Association of Echocardiography . Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 42.Latronico MV, Catalucci D, Condorelli G. MicroRNA and cardiac pathologies. Physiol Genomics 34: 239–242, 2008. doi: 10.1152/physiolgenomics.90254.2008. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Zhang X, Wang F, Zhou L, Yin Z, Fan J, Nie X, Wang P, Fu XD, Chen C, Wang DW. MicroRNA-21 lowers blood pressure in spontaneous hypertensive rats by upregulating mitochondrial translation. Circulation 134: 734–751, 2016. doi: 10.1161/CIRCULATIONAHA.116.023926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang H, Zhang C, Ban T, Liu Y, Mei L, Piao X, Zhao D, Lu Y, Chu W, Yang B. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol 44: 2152–2160, 2012. doi: 10.1016/j.biocel.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z, Ye P, Wang S, Wu J, Sun Y, Zhang A, Ren L, Cheng C, Huang X, Wang K, Deng P, Wu C, Yue Z, Xia J. MicroRNA-150 protects the heart from injury by inhibiting monocyte accumulation in a mouse model of acute myocardial infarction. Circ Cardiovasc Genet 8: 11–20, 2015. doi: 10.1161/CIRCGENETICS.114.000598. [DOI] [PubMed] [Google Scholar]
- 46.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 47.Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell 48: 760–770, 2012. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenzen JM, Schauerte C, Hübner A, Kölling M, Martino F, Scherf K, Batkai S, Zimmer K, Foinquinos A, Kaucsar T, Fiedler J, Kumarswamy R, Bang C, Hartmann D, Gupta SK, Kielstein J, Jungmann A, Katus HA, Weidemann F, Müller OJ, Haller H, Thum T. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur Heart J 36: 2184–2196, 2015. doi: 10.1093/eurheartj/ehv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu TX, Hartner J, Lim EJ, Fabry V, Mingler MK, Cole ET, Orkin SH, Aronow BJ, Rothenberg ME. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol 187: 3362–3373, 2011. doi: 10.4049/jimmunol.1101235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Y, Chiao YA, Zhang J, Manicone AM, Jin YF, Lindsey ML. Matrix metalloproteinase-28 deletion amplifies inflammatory and extracellular matrix responses to cardiac aging. Microsc Microanal 18: 81–90, 2012. doi: 10.1017/S1431927611012220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marney AM, Brown NJ. Aldosterone and end-organ damage. Clin Sci (Lond) 113: 267–278, 2007. doi: 10.1042/CS20070123. [DOI] [PubMed] [Google Scholar]
- 52.Marzano L, Colussi G, Sechi LA, Catena C. Adrenalectomy is comparable with medical treatment for reduction of left ventricular mass in primary aldosteronism: meta-analysis of long-term studies. Am J Hypertens 28: 312–318, 2015. doi: 10.1093/ajh/hpu154. [DOI] [PubMed] [Google Scholar]
- 53.Masoumi-Moghaddam S, Amini A, Morris DL. The developing story of Sprouty and cancer. Cancer Metastasis Rev 33: 695–720, 2014. doi: 10.1007/s10555-014-9497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension 59: 673–679, 2012. doi: 10.1161/HYPERTENSIONAHA.111.190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathis KW, Venegas-Pont M, Masterson CW, Wasson KL, Ryan MJ. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am J Physiol Regul Integr Comp Physiol 301: R1281–R1285, 2011. doi: 10.1152/ajpregu.00386.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW II. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation 119: 1263–1271, 2009. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mattsson C, Young WF Jr. Primary aldosteronism: diagnostic and treatment strategies. Nat Clin Pract Nephrol 2: 198–208, 2006. doi: 10.1038/ncpneph0151. [DOI] [PubMed] [Google Scholar]
- 58.Monticone S, D’Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, Mulatero P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol 6: 41–50, 2018. doi: 10.1016/S2213-8587(17)30319-4. [DOI] [PubMed] [Google Scholar]
- 59.Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protoc 9: 263–293, 2014. doi: 10.1038/nprot.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mulatero P, Stowasser M, Loh KC, Fardella CE, Gordon RD, Mosso L, Gomez-Sanchez CE, Veglio F, Young WF Jr. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 89: 1045–1050, 2004. doi: 10.1210/jc.2003-031337. [DOI] [PubMed] [Google Scholar]
- 61.Ohno Y, Sone M, Inagaki N, Yamasaki T, Ogawa O, Takeda Y, Kurihara I, Itoh H, Umakoshi H, Tsuiki M, Ichijo T, Katabami T, Tanaka Y, Wada N, Shibayama Y, Yoshimoto T, Ogawa Y, Kawashima J, Takahashi K, Fujita M, Watanabe M, Matsuda Y, Kobayashi H, Shibata H, Kamemura K, Otsuki M, Fujii Y, Yamamoto K, Ogo A, Okamura S, Miyauchi S, Fukuoka T, Izawa S, Yoneda T, Hashimoto S, Yanase T, Suzuki T, Kawamura T, Tabara Y, Matsuda F, Naruse M, Kawaguchi T, Setoh K, Matsuda F, Takahashi Y, Nakayama T, Kosugi S; Nagahama Study; JPAS Study Group . Prevalence of Cardiovascular Disease and Its Risk Factors in Primary Aldosteronism: a multicenter study in Japan. Hypertension 71: 530–537, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10263. [DOI] [PubMed] [Google Scholar]
- 62.Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, Shaik RS, Waxman AB, Zhang YY, Maron BA, Hartner JC, Fujiwara Y, Orkin SH, Haley KJ, Barabási AL, Loscalzo J, Chan SY. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 125: 1520–1532, 2012. doi: 10.1161/CIRCULATIONAHA.111.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E, Olson EN. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest 120: 3912–3916, 2010. doi: 10.1172/JCI43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Port JD, Walker LA, Polk J, Nunley K, Buttrick PM, Sucharov CC. Temporal expression of miRNAs and mRNAs in a mouse model of myocardial infarction. Physiol Genomics 43: 1087–1095, 2011. doi: 10.1152/physiolgenomics.00074.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qin Y, Yu Y, Dong H, Bian X, Guo X, Dong S. MicroRNA 21 inhibits left ventricular remodeling in the early phase of rat model with ischemia-reperfusion injury by suppressing cell apoptosis. Int J Med Sci 9: 413–423, 2012. doi: 10.7150/ijms.4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramanujam D, Sassi Y, Laggerbauer B, Engelhardt S. Viral vector-based targeting of miR-21 in cardiac nonmyocyte cells reduces pathologic remodeling of the heart. Mol Ther 24: 1939–1948, 2016. doi: 10.1038/mt.2016.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reddy S, Hu DQ, Zhao M, Blay E Jr, Sandeep N, Ong SG, Jung G, Kooiker KB, Coronado M, Fajardo G, Bernstein D. miR-21 is associated with fibrosis and right ventricular failure. JCI Insight 2: e91625, 2017. doi: 10.1172/jci.insight.91625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, Reichelt ME, Delbridge LM, Young MJ. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 60: 1443–1450, 2012. doi: 10.1161/HYPERTENSIONAHA.112.203158. [DOI] [PubMed] [Google Scholar]
- 69.Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, McMahon EG, Delyani JA. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol 283: H1802–H1810, 2002. doi: 10.1152/ajpheart.01096.2001. [DOI] [PubMed] [Google Scholar]
- 70.Rocha R, Stier CT Jr, Kifor I, Ochoa-Maya MR, Rennke HG, Williams GH, Adler GK. Aldosterone: a mediator of myocardial necrosis and renal arteriopathy. Endocrinology 141: 3871–3878, 2000. doi: 10.1210/endo.141.10.7711. [DOI] [PubMed] [Google Scholar]
- 71.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res 82: 21–29, 2009. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132: 365–386, 2000. [DOI] [PubMed] [Google Scholar]
- 73.Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 299: L861–L871, 2010. doi: 10.1152/ajplung.00201.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev 91: 827–887, 2011. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 75.Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, Abdellatif M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem 285: 20281–20290, 2010. doi: 10.1074/jbc.M110.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100: 416–424, 2007. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 77.Sayed D, Rane S, Lypowy J, He M, Chen IY, Vashistha H, Yan L, Malhotra A, Vatner D, Abdellatif M. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol Biol Cell 19: 3272–3282, 2008. doi: 10.1091/mbc.e08-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 79.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation 121: 1022–1032, 2010. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature 469: 336–342, 2011. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab 94: 3623–3630, 2009. doi: 10.1210/jc.2009-1399. [DOI] [PubMed] [Google Scholar]
- 82.Sun K, Lai EC. Adult-specific functions of animal microRNAs. Nat Rev Genet 14: 535–548, 2013. doi: 10.1038/nrg3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang Y, Wang Y, Park KM, Hu Q, Teoh JP, Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, Tang Y, Ramesh G, Kim IM. MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc Res 106: 387–397, 2015. doi: 10.1093/cvr/cvv121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 42: 1137–1141, 2007. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116: 258–267, 2007. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 86.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456: 980–984, 2008. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 87.Tijsen AJ, Pinto YM, Creemers EE. Non-cardiomyocyte microRNAs in heart failure. Cardiovasc Res 93: 573–582, 2012. doi: 10.1093/cvr/cvr344. [DOI] [PubMed] [Google Scholar]
- 88.Toldo S, Das A, Mezzaroma E, Chau VQ, Marchetti C, Durrant D, Samidurai A, Van Tassell BW, Yin C, Ockaili RA, Vigneshwar N, Mukhopadhyay ND, Kukreja RC, Abbate A, Salloum FN. Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ Cardiovasc Genet 7: 311–320, 2014. doi: 10.1161/CIRCGENETICS.113.000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 103: 18255–18260, 2006. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 105: 13027–13032, 2008. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Varga ZV, Zvara A, Faragó N, Kocsis GF, Pipicz M, Gáspár R, Bencsik P, Görbe A, Csonka C, Puskás LG, Thum T, Csont T, Ferdinandy P. MicroRNAs associated with ischemia-reperfusion injury and cardioprotection by ischemic pre- and postconditioning: protectomiRs. Am J Physiol Heart Circ Physiol 307: H216–H227, 2014. doi: 10.1152/ajpheart.00812.2013. [DOI] [PubMed] [Google Scholar]
- 92.Verjans R, Peters T, Beaumont FJ, van Leeuwen R, van Herwaarden T, Verhesen W, Munts C, Bijnen M, Henkens M, Diez J, de Windt LJ, van Nieuwenhoven FA, van Bilsen M, Goumans MJ, Heymans S, González A, Schroen B. MicroRNA-221/222 family counteracts myocardial fibrosis in pressure overload-induced heart failure. Hypertension 71: 280–288, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10094. [DOI] [PubMed] [Google Scholar]
- 93.Vinson GP. The mislabelling of deoxycorticosterone: making sense of corticosteroid structure and function. J Endocrinol 211: 3–16, 2011. doi: 10.1530/JOE-11-0178. [DOI] [PubMed] [Google Scholar]
- 94.Walkin L, Herrick SE, Summers A, Brenchley PE, Hoff CM, Korstanje R, Margetts PJ. The role of mouse strain differences in the susceptibility to fibrosis: a systematic review. Fibrogenesis Tissue Repair 6: 18, 2013. doi: 10.1186/1755-1536-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang J, Duan L, Gao Y, Zhou S, Liu Y, Wei S, An S, Liu J, Tian L, Wang S. Angiotensin II receptor blocker valsartan ameliorates cardiac fibrosis partly by inhibiting miR-21 expression in diabetic nephropathy mice. Mol Cell Endocrinol 472: 149–158, 2018. doi: 10.1016/j.mce.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 96.Wang X, Ha T, Zou J, Ren D, Liu L, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. MicroRNA-125b protects against myocardial ischaemia/reperfusion injury via targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc Res 102: 385–395, 2014. doi: 10.1093/cvr/cvu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weber KT. Aldosterone in congestive heart failure. N Engl J Med 345: 1689–1697, 2001. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- 98.Williams TA, Lenders JWM, Mulatero P, Burrello J, Rottenkolber M, Adolf C, Satoh F, Amar L, Quinkler M, Deinum J, Beuschlein F, Kitamoto KK, Pham U, Morimoto R, Umakoshi H, Prejbisz A, Kocjan T, Naruse M, Stowasser M, Nishikawa T, Young WF Jr, Gomez-Sanchez CE, Funder JW, Reincke M, Williams TA, Gomez-Sanchez CE, Funder JW, Reincke M, Adolf C, Amar L, Auchus RJ, Bartsch DK, Baudrand R, Beuschlein F, Björklund P, Brown MJ, Burrello J, Carey RM, Catena C, Connell JM, Deinum J, Dekkers T, Fahey TJ III, Fallo F, Fardella CE, Giacchetti G, Giraudo G, Hellman P, Januszewicz A, Kitamoto KK, Kline GA, Kocjan T, Lenders JWM, Mantero F, Miller BS, Morimoto R, Mulatero P, Naruse M, Nishikawa T, Pham U, Plouin P-F, Prejbisz A, Quinkler M, Rottenkolber M, Rump CL, Satoh F, Sechi LA, Stowasser M, Umakoshi H, Veglio F, Widimský J Jr, Willenberg HS, Young WF Jr; Primary Aldosteronism Surgery Outcome (PASO) investigators . Outcomes after adrenalectomy for unilateral primary aldosteronism: an international consensus on outcome measures and analysis of remission rates in an international cohort. Lancet Diabetes Endocrinol 5: 689–699, 2017. doi: 10.1016/S2213-8587(17)30135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11: 228–234, 2009. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 100.Xiao J, Pan Y, Li XH, Yang XY, Feng YL, Tan HH, Jiang L, Feng J, Yu XY. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis 7: e2277, 2016. doi: 10.1038/cddis.2016.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest 117: 1344–1353, 2007. doi: 10.1172/JCI29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yanes Cardozo LL, Romero DG, Reckelhoff JF. Cardiometabolic features of polycystic ovary syndrome: role of androgens. Physiology (Bethesda) 32: 357–366, 2017. doi: 10.1152/physiol.00030.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 129: 1009–1021, 2014. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yin C, Wang X, Kukreja RC. Endogenous microRNAs induced by heat-shock reduce myocardial infarction following ischemia-reperfusion in mice. FEBS Lett 582: 4137–4142, 2008. doi: 10.1016/j.febslet.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Young M, Fullerton M, Dilley R, Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 93: 2578–2583, 1994. doi: 10.1172/JCI117269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Young MJ, Lam EY, Rickard AJ. Mineralocorticoid receptor activation and cardiac fibrosis. Clin Sci (Lond) 112: 467–475, 2007. doi: 10.1042/CS20060275. [DOI] [PubMed] [Google Scholar]
- 107.Young WF., Jr Minireview: primary aldosteronism changing concepts in diagnosis and treatment. Endocrinology 144: 2208–2213, 2003. doi: 10.1210/en.2003-0279. [DOI] [PubMed] [Google Scholar]
- 108.Yuan J, Chen H, Ge D, Xu Y, Xu H, Yang Y, Gu M, Zhou Y, Zhu J, Ge T, Chen Q, Gao Y, Wang Y, Li X, Zhao Y. Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem 42: 2207–2219, 2017. doi: 10.1159/000479995. [DOI] [PubMed] [Google Scholar]
- 109.Zhou Q, Sun Q, Zhang Y, Teng F, Sun J. Up-regulation of miRNA-21 expression promotes migration and proliferation of Sca-1+ cardiac stem cells in mice. Med Sci Monit 22: 1724–1732, 2016. doi: 10.12659/MSM.895753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zimmerman SD, Thomas DP, Velleman SG, Li X, Hansen TR, McCormick RJ. Time course of collagen and decorin changes in rat cardiac and skeletal muscle post-MI. Am J Physiol Heart Circ Physiol 281: H1816–H1822, 2001. doi: 10.1152/ajpheart.2001.281.4.H1816. [DOI] [PubMed] [Google Scholar]