

Abstract
Angiotensin-converting enzyme (ACE) inhibitors reduce body weight, lower blood pressure (BP), and improve insulin sensitivity in animal models of cardiometabolic syndrome. These effects are generally attributed to reduced angiotensin (ANG) II formation; however, these therapies also increase levels of ANG-(1–7), a beneficial hormone opposing ANG II actions. We hypothesized that this ANG-(1–7) generation contributes to the insulin-sensitizing effects of ACE inhibition in obese mice. Adult male C57BL/6J mice were placed on a 60% high-fat diet for 11 wk. During the last 3 wk of diet, mice received normal water or water containing the ACE inhibitor captopril (50 mg/l) as well as the ANG-(1–7) mas receptor antagonist A779 (400 or 800 ng·kg−1·min−1) or saline vehicle via subcutaneous osmotic minipumps. At the end of treatment, arterial BP was measured, and hyperinsulinemic-euglycemic clamps were performed in conscious obese mice receiving vehicle, captopril, captopril plus A779, or A779 (n = 6–13/group). Captopril reduced body weight (28 ± 2 vs. 41 ± 2 g saline; P = 0.001), lowered systolic BP (109 ± 6 vs. 144 ± 7 mmHg saline; P = 0.041), and improved whole-body insulin sensitivity (steady-state glucose infusion rate: 31 ± 4 vs. 16 ± 2 mg·kg−1·min−1 saline; P = 0.001) in obese mice. A779 attenuated captopril-mediated improvements in insulin sensitivity (23 ± 2 mg·kg−1·min−1; P = 0.042), with no effect on body weight (32 ± 2 g; P = 0.441) or BP (111 ± 7 mmHg; P = 0.788). There was no effect of A779 alone on cardiometabolic outcomes. These data suggest that insulin-sensitizing effects of ACE inhibition are in part due to activation of ANG-(1–7)/mas receptor pathways and provide new insight into mechanisms underlying the positive metabolic effects of these therapies.
Keywords: animal models, hypertension, insulin resistance, obesity, renin-angiotensin system
INTRODUCTION
Overactivation of the renin-angiotensin (ANG) system (RAS) is an important hormonal mechanism contributing to hypertension and insulin resistance (27, 44). In this system, the precursor angiotensinogen is cleaved by the enzyme renin to form ANG I, which is subsequently cleaved by ANG-converting enzyme (ACE) to form ANG II. The actions of ANG II at AT1 receptors elevate blood pressure to promote hypertension through multiple mechanisms including vasoconstriction, aldosterone release, oxidative stress, inflammation, and sympathetic nervous system activation (44). In addition, circulating and adipose ANG II levels are increased in obesity and can increase hepatic glucose output and contribute to insulin resistance via uncoupling intracellular insulin signaling pathways and inhibiting insulin-stimulated glucose uptake in peripheral tissues (29, 33, 35). Pharmacological blockade of ANG II formation with ACE inhibitors is widely used for hypertension treatment. These therapies are often used in obese patients because of their positive metabolic profile and have been shown to reduce incidence of new-onset type II diabetes mellitus in large randomized clinical trials (1). In obese rodents, ACE inhibitors lower body weight and improve hypertension, insulin resistance, and glucose intolerance (9, 10, 32).
The beneficial cardiovascular and metabolic effects of ACE inhibition are generally attributed to reduced ANG II formation; however, these therapies also increase circulating levels of ANG-(1–7). (7, 24, 26, 28) ANG-(1–7) is a more recently discovered bioactive peptide of the RAS that generally opposes ANG II actions (38). ANG-(1–7) is formed from cleavage of ANG I by various endopeptidases, such as neprilysin, or from cleavage of ANG II by ACE2. The basis for the potent rise in ANG-(1–7) with ACE inhibition is primarily due to decreased degradation of ANG-(1–7) to ANG-(1–5) by ACE (8, 34). ANG-(1–7) binds mas G protein-coupled receptors distributed in numerous tissues to lower blood pressure in rodent models of hypertension through vascular, neural, renal, and cardiac mechanisms (36, 38). Accumulating evidence from our laboratory and others have shown that ANG-(1–7) also has positive effects on lipid metabolism, insulin action, glucose homeostasis, and energy balance in rodent models of cardiometabolic syndrome independent of effects on blood pressure (16, 48). For example, our laboratory recently showed that ANG-(1–7) produces blood pressure-independent improvement in whole-body insulin sensitivity in obese mice by enhancing insulin-stimulated skeletal muscle glucose uptake (48).
Emerging studies have shown a role for endogenous ANG-(1–7) generation in the cardiovascular, antithrombotic, and renal-protective effects of ACE inhibition in animal models of hypertension and type I diabetes (2, 4, 19, 26, 50). There are, however, limited studies examining the importance of ANG-(1–7) to the metabolic effects of ACE inhibitors (12, 31). In this study, we tested the hypothesis that ANG-(1–7) mas receptor-mediated pathways contribute to the insulin-sensitizing effects of chronic ACE inhibition in obese mice. To test this, we performed hyperinsulinemic-euglycemic clamps to measure whole-body insulin sensitivity in conscious obese mice following chronic treatment with saline vehicle, the ACE inhibitor captopril, the ANG-(1–7) mas receptor antagonist A779, or captopril plus A779. We also measured blood pressure in these animals to provide information on the role of ANG-(1–7) in integrated cardiometabolic function in obesity.
MATERIALS AND METHODS
General.
All procedures conformed to the NIH Guide for Care and Use of Laboratory Animals and were approved by the Penn State College of Medicine Institutional Animal Care and Use Committee. Mice were housed in a humidity- and temperature-controlled room (23°C) on a 12-h:12-h light/dark cycle. Mice had ad libitum access to food and water throughout the study.
Diet and drug delivery.
There were four groups of obese mice in this study: 1) saline (n = 8, saline infusion plus normal water); 2) CAP (n = 6, saline infusion plus captopril water); 3) A779 (n = 6, A779 infusion plus normal water); and 4) CAP + A779 (n = 13, A779 infusion plus captopril water). We did not include chow-fed mice, as we previously shown no effect of chronic ANG-(1–7) administration on cardiovascular or metabolic function under normal conditions (48). Male C57BL/6J mice (Jackson Laboratory) were placed on a diet containing 60% kcal from fat (high-fat diet, HFD; Bioserv F3282) starting at 5 wk of age. After 8 wk of HFD, mice were implanted with a subcutaneous osmotic minipump (Alzet Model 2004) for chronic 3-wk infusion of saline or the ANG-(1–7) mas receptor antagonist A779 (Bachem). To examine potential dose-response relationships, we administered A779 at 400 ng·kg−1·min−1 (n = 6) and 800 ng·kg−1·min−1 (n = 7) doses. Data were pooled, as there were no significant differences in any of the outcomes measured between these two doses of A779. Similar doses, duration, and route of administration for A779 were shown to prevent protective effects of ANG-(1–7) on atherogenesis in obese ApoE-knockout mice as well as on weight gain in obese rats (31, 49). Immediately following minipump implantation, mice received either normal tap water or water containing the ACE inhibitor captopril (50 mg/l; Tocris Bioscience). We chose captopril, as it is known to elevate circulating ANG-(1–7) levels in several species, including rodents (7, 24, 26, 28, 31). Captopril was provided daily at a dose shown to reduce body weight and improve glucose tolerance in obese mice (32). There were no differences in daily water intake between mice receiving normal water and water containing captopril (2.9 ± 0.1 and 3.1 ± 0.2 ml, respectively; P = 0.409). Mice receiving captopril consumed ~0.16 mg of drug per day, resulting in a dose of 3.7 to 6.7 mg·kg−1·day−1 depending on the amount of fluid consumed and body mass for an individual animal. This dosing regimen in mice has been previously shown to be equivalent to captopril doses used for treatment of hypertensive patients (32). We chose to employ HFD-induced obese mice, as this animal model closely mimics the human condition, exhibits ANG-(1–7) deficiency, and allows for integrated study of insulin resistance and hypertension (45, 48).
Body composition.
At the end of treatment, body mass was measured, and body composition was determined using an mq10 nuclear magnetic resonance analyzer (Bruker Optics).
Blood pressure.
As previously reported (48), blood pressure and heart rate were measured in conscious mice on the morning of insulin clamps at t = −150 min via an indwelling carotid artery catheter connected to a strain-gauge transducer and BP analyzer (BPA-400; Digi-Med). This method was used because direct arterial measurement is more sensitive and accurate compared with tail cuff measurement, and it is not possible to measure blood pressure by gold standard radiotelemetry methods in insulin-clamped animals because of the need for carotid artery catheterization for both procedures.
Hyperinsulinemic-euglycemic clamps (insulin clamp).
Carotid artery and jugular vein catheters were implanted ~5 days before experiments. Insulin clamps were performed in conscious, unrestrained, and 5-h-fasted mice as previously reported (20, 48). A primed, continuous intravenous infusion of [3-3H]glucose (0.04 μCi/min; Perkin Elmer) was started at t = −90 min to determine rates of plasma glucose appearance (Ra) and disappearance (Rd). Arterial blood was drawn from the carotid catheter at baseline to measure glucose-specific activity (t = −15 and −5 min) and insulin (t = −5 min). At t = 0 min, continuous insulin (4 mU·kg−1·min−1; Humulin R; Eli Lilly) and variable glucose (D50 + 50 μCi [3-3H]glucose) infusions were started. Donor erythrocytes were infused to prevent a fall in hematocrit. Arterial glucose was measured every 10 min from t = 0–120 min. The exogenous glucose infusion rate (GIR) was adjusted to maintain euglycemia at 110–130 mg/dl during the steady-state period from t = 80–120 min. GIR and [3-3H]glucose kinetics were determined during steady state from t = 80–120 min. Clamp insulin levels were measured at t = 100 and 120 min, with the average value reported. At t = 120 min, mice received an intravenous bolus of 2[14C]deoxyglucose (2[14C]DG; 13 μCi; Perkin Elmer) to determine tissue-specific glucose uptake (Rg). After the bolus, blood samples were obtained at 2, 7, 15, 25, and 35 min to measure plasma 2[14C]DG disappearance. At t = 155 min, mice were killed with tissues freeze clamped and stored at −80°C for biochemical analysis. Insulin was measured by double antibody radioimmunoassay. Radioactivity of 3[3H]glucose, 2[14C]2DG, and 2[14C]2DG-6-phosphate was measured using liquid scintillation counting.
Insulin clamp calculations.
Ra and Rd were calculated using non-steady-state equations. Endogenous glucose production (EndoRa) was calculated by subtracting GIR from total Ra. Tissue Rg was calculated as previously described in skeletal muscle (gastrocnemius, vastus lateralis, and soleus), adipose tissue (visceral epididymal, subcutaneous inguinal, and interscapular brown), heart, and brain (25). To account for potential differences among groups in arterial glucose and insulin levels during the insulin clamp, an insulin sensitivity index (SI) was calculated using the formula SI index = GIR/(G × ΔI), where GIR is the steady-state GIR (mg·kg−1·min−1), G is steady-state blood glucose levels (mg/ml), and ΔI is the difference between steady-state and basal insulin levels (μU/ml).
Plasma ANG peptides.
Plasma ANG II and ANG-(1–7) levels were measured in a separate cohort of obese mice that did not undergo insulin clamps (n = 6/group). Blood was collected in a peptidase inhibitor cocktail to prevent in vitro metabolism, with plasma sent to the Biomarker Analytical Core Laboratory at Wake Forest University for radioimmunoassay analysis (3, 40).
Statistical analysis.
Data are presented as means ± SE. Analyses were performed using GraphPad Prism (Version 7.0) with a two-tailed P value <0.05 defined as statistically significant. Differences in outcomes were compared using two-way ANOVA with Tukey’s multiple-comparisons post hoc tests. This study was powered to detect differences in insulin sensitivity (GIR needed to maintain euglycemia), the primary outcome, and included similar numbers of mice as our previous study using these methods (48). On the basis of the present data showing a mean difference in steady-state GIR means of 9.3 between CAP and CAP + A779 groups with a within-group standard deviation of 4.4 and 0.05 α-level, this study has 91% power to detect a difference among these treatment groups using independent t-test analysis (PS Dupont software, Version 3.1.2) (11).
RESULTS
Effect of ACE inhibition on circulating angiotensin peptides.
Chronic ACE inhibition with captopril resulted in an ~50% reduction in ANG II levels (Table 1). In addition, there was a main effect for captopril to increase ANG-(1–7) levels approximately twofold in the circulation of obese mice (Table 1). There was no effect of A779, either alone or in combination with captopril, on circulating ANG II or ANG-(1–7) levels.
Table 1.
Contribution of ANG-(1–7) to hormonal and metabolic effects of ACE inhibition with captopril in obese mice
| Parameter, unit | Saline | CAP | CAP + A779 | A779 | PCAP | PA779 | PINT |
|---|---|---|---|---|---|---|---|
| N | 8 | 6 | 13 | 6 | |||
| ANG II, pg/ml | 167 ± 34 | 82 ± 17*† | 86 ± 28*† | 190 ± 20 | 0.002 | 0.619 | 0.724 |
| ANG-(1–7), pg/ml | 54 ± 18 | 91 ± 15 | 116 ± 15* | 69 ± 8 | 0.016 | 0.916 | 0.409 |
| Body composition | |||||||
| Body mass, g | 41 ± 2 | 28 ± 2* | 32 ± 2* | 43 ± 2 | 0.001 | 0.142 | 0.595 |
| Adiposity, % | 35 ± 3 | 25 ± 2* | 29 ± 2 | 30 ± 4 | 0.045 | 0.879 | 0.129 |
| Lean mass, % | 46 ± 3 | 54 ± 2† | 49 ± 2 | 42 ± 2 | 0.002 | 0.024 | 0.759 |
| Fluid mass, % | 7 ± 1 | 6 ± 1 | 6 ± 1 | 6 ± 1 | 0.123 | 0.127 | 0.103 |
| Arterial glucose | |||||||
| Basal, mg/dl | 127 ± 7 | 112 ± 10 | 116 ± 4 | 118 ± 5 | 0.212 | 0.733 | 0.299 |
| Clamp, mg/dl | 122 ± 3 | 114 ± 1 | 120 ± 2 | 119 ± 4 | 0.164 | 0.594 | 0.093 |
| Arterial insulin | |||||||
| Basal, μU/ml | 81 ± 14 | 20 ± 4* | 41 ± 9 | 49 ± 13 | 0.007 | 0.658 | 0.030 |
| Clamp, μU/ml | 153 ± 27 | 76 ± 5 | 100 ± 12 | 148 ± 29 | 0.006 | 0.640 | 0.477 |
| SI, ml2⋅μU1⋅kg−1⋅min−1 | 0.27 ± 0.08 | 0.53 ± 0.05 | 0.31 ± 0.05 | 0.20 ± 0.06 | 0.012 | 0.264 | 0.042 |
Data are means ± SE and were analyzed by 2-way ANOVA for main effects of captopril (CAP) treatment via drinking water (PCAP) and A779 treatment via osmotic minipump (PA779) and their interaction (PINT) in high-fat diet-induced obese mice. Arterial glucose and insulin were measured at baseline (basal) and during the steady state of the hyperinsulinemic-euglycemic clamp (clamp). ACE, angiotensin-converting enzyme; A779, angiotensin (ANG)-(1–7) mas receptor antagonist; SI, insulin sensitivity index.
P < 0.05 vs. saline and
P < 0.01 vs. A779 from post hoc pairwise comparisons with Tukey’s correction for multiple comparisons.
Contribution of ANG-(1–7) to blood pressure effects of ACE inhibition.
As shown in Fig. 1, obese mice treated with saline exhibited modest hypertension, as evidenced by an average arterial blood pressure level of 142/105 mmHg. Captopril significantly lowered systolic (PCAP = 0.002), diastolic (PCAP = 0.001), and mean (PCAP = 0.001) blood pressure in obese mice, with no effect on heart rate (PCAP = 0.530). There was no significant main effect of A779 on blood pressure or heart rate in obese mice (PA779 = 0.672 systolic, 0.842 diastolic, 0.135 mean, 0.152 heart rate), as well as no interaction between captopril and A779 treatments (PINT = 0.435 systolic, 0.846 diastolic, 0.614 mean, 0.600 heart rate).
Fig. 1.

Angiotensin (ANG)-(1–7) does not contribute to blood pressure-lowering effects of ANG-converting enzyme (ACE) inhibition in obese mice. A–C: arterial blood pressure was measured in conscious obese mice treated with saline (n = 8), captopril (n = 6) (CAP), captopril plus A779 (n = 13), or A779 (n = 6). Chronic ACE inhibition with captopril significantly lowered blood pressure in obese mice (PCAP: 0.002 systolic, 0.001 diastolic, and 0.001 mean). There was no effect of the ANG-(1–7) mas receptor antagonist A779 alone on blood pressure in obese mice (PA779: 0.672 systolic, 0.842 diastolic, and 0.135 mean) and no interaction of A779 and captopril treatments (PINT: 0.435 systolic, 0.846 diastolic, and 0.614 mean). These data suggest that ANG-(1–7) mas receptor pathways do not play a major role in the blood pressure-lowering effects of ACE inhibition in obese mice. D: there were no main effects of treatments on heart rate (PCAP: 530, PA779: 0.152, PINT: 0.600). Data are means ± SE and analyzed by 2-way ANOVA for main effects of captopril (PCAP) and A779 (PA779) treatments and their interaction (PINT). P values on graphs represent post hoc pairwise comparisons with Tukey’s correction for multiple comparisons. bpm, beats per minute.
Contribution of ANG-(1–7) to metabolic effects of ACE inhibition.
Chronic captopril treatment significantly reduced body mass in obese mice (Table 1; PCAP = 0.001). This weight-reducing effect was associated with decreased adiposity and increased lean mass (PCAP = 0.002 and 0.002, respectively), with no effect on fluid mass (PCAP = 0.123). There was no effect of ANG-(1–7) mas receptor antagonism with A779, either alone or in combination with captopril, on body mass, or adiposity (Table 1). There was, however, a main effect for A779 to reduce lean mass (PA779 = 0.024). There were no significant differences in basal arterial glucose levels among treatment groups (Table 1). During steady-state hyperinsulinemia, glucose was similarly clamped in the predefined range of 110–130 mg/dl among groups (Table 1; Fig. 2A). Despite similar glucose levels, arterial insulin was reduced at baseline (PCAP = 0.034) and similarly during the insulin clamp (PCAP = 0.005) following chronic captopril treatment, suggesting improved insulin sensitivity. There was no main effect of A779 on glucose or insulin levels (Table 1); however, A779 prevented the lowering of basal insulin levels produced by captopril (PINT = 0.034).
Fig. 2.
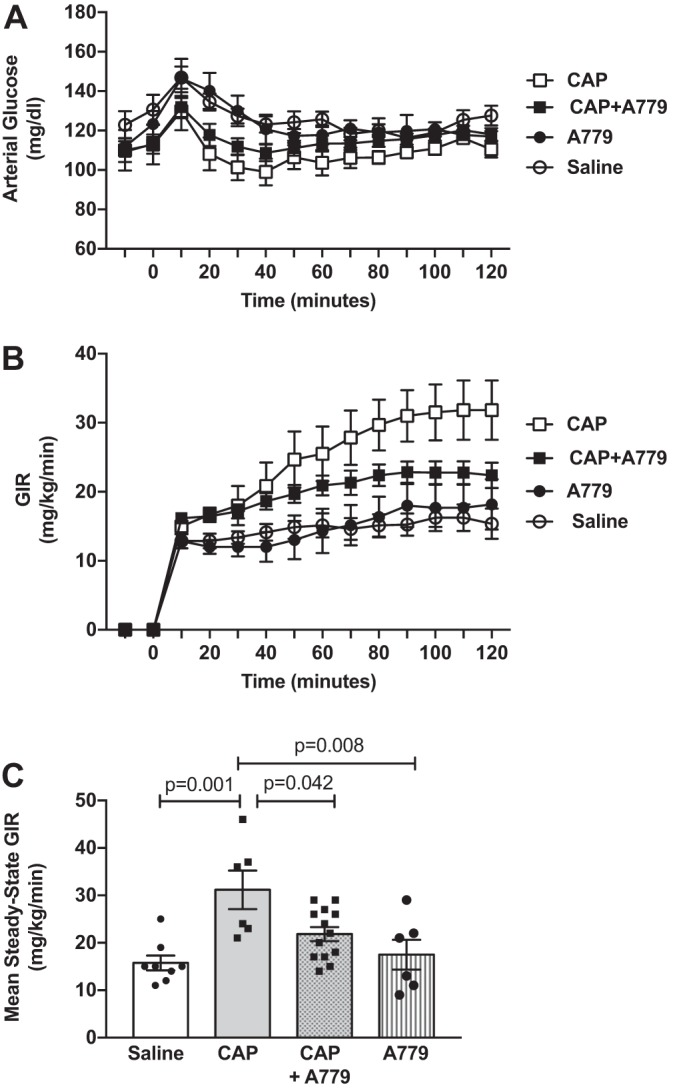
Angiotensin (ANG)-(1–7) contributes to the improvement in whole-body insulin sensitivity produced by ANG-converting enzyme (ACE) inhibition in obese mice. A: arterial glucose levels were maintained at ~120 mg/dl during a steady-state period of insulin clamps (t = 80–120 min) by variable intravenous infusion of 50% glucose. B: glucose infusion rate (GIR) needed to maintain euglycemia, a measure of whole-body insulin sensitivity, during the 120-min insulin clamp period. C: ACE inhibition with captopril significantly improved the mean steady-state GIR in obese mice, an effect that was attenuated in the presence of the ANG-(1–7) mas receptor antagonist A779 (PCAP: 0.001, PA779: 0.181, PINT: 0.047; 2-way ANOVA). P values on graphs are post hoc pairwise comparisons with Tukey’s correction. Data are means ± SE in obese mice treated with saline (n = 8), captopril (n = 6) (CAP), captopril plus A779 (n = 13), or A779 (n = 6).
Contribution of ANG-(1–7) to ACE inhibitor-mediated insulin sensitization.
Despite lower clamp insulin levels, captopril significantly increased whole-body insulin sensitivity in obese mice, as measured by both the GIR needed to maintain euglycemia during steady state of the insulin clamp (Fig. 2, B and C; PCAP = 0.001) and the SI (Table 1; PCAP = 0.012). This enhancement in insulin sensitivity was due to increased insulin-stimulated peripheral glucose disposal (Rd; Fig. 3A; PCAP = 0.041), as well as enhanced insulin-mediated suppression of endogenous glucose production (EndoRa; Fig. 3B; PCAP = 0.008). Consistent with effects on peripheral glucose disposal, captopril increased insulin-stimulated glucose uptake (Rg) in soleus and gastrocnemius skeletal muscles, brown fat, and heart in obese mice, with no effects in vastus skeletal muscle, epididymal fat, subcutaneous fat, or brain (Fig. 3, C and D). There was no effect of A779 alone on insulin sensitivity, Rd, EndoRa, or glucose uptake in vastus skeletal muscle, fat, heart, and brain (Table 1, Figs. 2 and 3); however, there was a main effect for A779 to increase glucose uptake in soleus and gastrocnemius muscles (PA779 = 0.040 and 0.016, respectively). Importantly, chronic mas receptor antagonism with A779 attenuated captopril-mediated improvements in insulin sensitivity (mean steady-state GIR: PINT = 0.047; SI: PINT = 0.042), clamp Rd (PINT = 0.009), and glucose uptake in soleus and gastrocnemius muscles (PINT = 0.008 and 0.006, respectively). There was no interaction between captopril and A779 treatments on basal Rd, EndoRa, or glucose uptake in vastus, fat, heart, and brain.
Fig. 3.

Angiotensin (ANG)-(1–7) contributes to the improved insulin-stimulated peripheral glucose disposal and skeletal muscle glucose uptake produced by ANG-converting enzyme (ACE) inhibition (ACEi) in obese mice. A: there was no effect of the ACE inhibitor captopril (CAP) or ANG-(1–7) mas receptor antagonist A779 on basal peripheral glucose disposal (Rd; PCAP: 0.911, PA779: 0.105, PINT: 0.935; 2-way ANOVA). Captopril increased insulin-stimulated Rd, and this effect was significantly attenuated in the presence of A779 treatment (PCAP: 0.041, PA779: 0.641, PINT: 0.009). B: there was no effect of captopril or A779 on basal rates of endogenous glucose production (EndoRa) (PCAP: 0.945, PA779: 0.205, PINT: 0.837). Captopril reduced insulin-stimulated EndoRa with no effect of A779 (PCAP: 0.008, PA779: 0.156, PINT: 0.759). C and D: captopril significantly increased the glucose metabolic index (Rg) determined by 2[14C] deoxyglucose accumulation in soleus and gastrocnemius skeletal muscles, an effect attenuated by cotreatment with A779 (PCAP: 0.024, PA779: 0.040, PINT: 0.008; PCAP: 0.040, PA779: 0.016, PINT: 0.006, respectively). Captopril also increased Rg in heart and brown fat, with no significant effect of A779 (PCAP: 0.021, PA779: 0.057, PINT: 0.446; PCAP: 0.009, PA779: 0.197, PINT: 0.088, respectively). There were no effects of captopril or A779 on Rg in vastus muscle (PCAP: 0.298, PA779: 0.089, PINT: 0.057), epididymal fat (PCAP: 0.148, PA779: 0.242, PINT: 0.531), subcutaneous fat (PCAP: 0.072, PA779: 0.076, PINT: 0.771), or brain (PCAP: 0.384, PA779: 0.798, PINT: 0.274). Data are means ± SE in obese mice treated with saline (n = 8), captopril (n = 6), captopril plus A779 (n = 13), or A779 (n = 6). P values on graphs are post hoc pairwise comparisons with Tukey’s correction. HFD, high-fat diet.
DISCUSSION
The main finding of this study is that the insulin-sensitizing effects produced by chronic ACE inhibition with captopril in obese mice appear to predominantly stem from activation of ANG-(1–7) mas receptor-mediated pathways. The contribution of ANG-(1–7) pathways to captopril-mediated insulin sensitization includes enhancement of insulin-mediated peripheral glucose disposal with glucose uptake specifically into gastrocnemius and soleus skeletal muscles. Despite these insulin-sensitizing effects, our data suggest that weight-attenuating and blood pressure-lowering effects of captopril are independent of ANG-(1–7) in obese mice and therefore may reflect blockade of ANG II formation or other pathways. These overall findings provide new insight into mechanisms underlying the insulin-sensitizing effects of ACE inhibition and support the rationale of direct targeting of ANG-(1–7) pathways to improve insulin sensitivity in obesity.
Precise mechanisms underlying beneficial cardiovascular and metabolic effects of ACE inhibition remain unclear, as these therapies concomitantly decrease ANG II and increase ANG-(1–7) levels (7, 24, 26, 28, 31). In this study, we observed an ~50% reduction in ANG II, as well as a twofold increase in ANG-(1–7) levels in obese mice treated with the ACE inhibitor captopril. Our findings are consistent with previous studies showing two- to fourfold increases in ANG-(1–7) following ACE inhibition in rats, canines, and humans (13, 23, 24, 28). A few studies have shown up to 25-fold increases in ANG-(1–7) with ACE inhibition in rats (4, 7), perhaps attributable to differences in the specific ACE inhibitor used as well as dose and length of treatment. Although not measured in this study, plasma renin activity and ANG I levels are reported to increase following captopril treatment in obese rats (10).
ACE inhibition improves whole-body insulin sensitivity by increasing peripheral glucose disposal and glucose uptake in skeletal muscle and heart (18). Consistent with these findings, we used gold standard hyperinsulinemic-euglycemic clamp methods combined with isotopic tracers to show that captopril improves whole-body insulin sensitivity in obese mice by enhancing insulin-stimulated peripheral glucose disposal and glucose uptake in skeletal muscle, brown fat, and heart. Although not examined in this study, ACE inhibitors improve skeletal muscle insulin sensitivity by increasing Glut4 translocation to the sarcolemma either by stimulating bradykinin-nitric oxide pathways or by inhibiting deleterious actions of ANG II on insulin signaling (18). We also found that captopril lowered basal insulin levels without affecting basal endogenous glucose production and increased insulin-mediated suppression of endogenous glucose production, suggesting improved hepatic insulin sensitivity. This finding is consistent with literature suggesting that ANG II blockade with ACE inhibitors or ANG receptor blockers suppresses hepatic endogenous glucose production (33, 43).
We further demonstrate that blocking ANG-(1–7) mas receptors with A779 in obese mice reversed the ability of captopril to lower basal insulin levels, improve whole-body insulin sensitivity, and enhance insulin-mediated plasma glucose disposal via skeletal muscle glucose uptake. In support of this concept, A779 blocks the ability of acute captopril to enhance insulin-stimulated skeletal muscle glucose uptake in anesthetized Wistar rats (12). Furthermore, mas receptor deficiency in mice results in hyperinsulinemia and worsened insulin sensitivity (37). The underlying mechanism by which ANG-(1–7) mediates ameliorative effects on skeletal muscle insulin sensitivity has been defined by our group and others. Our recent study showed that, in obese mice, chronic systemic ANG-(1–7) administration produced direct insulin-sensitizing effects on skeletal muscle by reducing protein levels of AS160, a negative regulator of Glut4 trafficking, to allow Glut4 translocation to the sarcolemma (48). Similarly, others have shown that ANG-(1–7) increases AS160 phosphorylation and Glut4 protein levels in skeletal muscle in rodents and that A779 decreases Glut4 expression in mice (30, 42). There was no effect of A779 alone on whole-body insulin sensitivity in obese mice, perhaps reflecting the known ANG-(1–7) deficiency in this model (48). Despite contributing to skeletal muscle insulin sensitization, there was no effect of A779 either alone or in combination with captopril on basal or insulin-stimulated endogenous glucose production. This is consistent with our previous study showing lack of effect of direct ANG-(1–7) administration on hepatic insulin sensitivity in obese mice (48). This finding may reflect the absence of hepatic insulin resistance in HFD-induced obese mice as well as the higher physiological dose of insulin infused during clamps, which can fully suppress endogenous glucose production and mask more subtle effects on hepatic insulin sensitivity (6, 22, 48).
The circulating ANG II/ACE pathway chronically contributes to positive energy balance to promote weight gain. Genetic or pharmacological inhibition of ACE lowers body weight and adiposity by increasing energy expenditure (10, 21, 31, 32, 46); although these findings do not readily translate to clinical populations. In this study, captopril lowered body mass by ~32% and decreased adiposity and increased lean mass in obese mice. Other similar studies did not report lean mass following captopril treatment or reported no change in grams of lean mass in the face of reduced body mass, suggesting an increased percentage of lean mass (10, 31, 32, 47). The weight-reducing effects of ACE inhibitors are thought to reflect reduced food intake in obese rats and mice; however, varying effects are observed depending on brain penetrance of the drug used (10, 31). Because captopril does not readily cross the blood-brain barrier, effects on food intake are thought to be mediated by increased central ANG II levels (10). Previous studies have shown that A779 reverses weight-attenuating effects of ANG II blockade in normal and obese rats (31, 39). In this study, A779 was not able to reverse effects of captopril on body composition, suggesting minor contributions of ANG-(1–7) mas receptor pathways to ACE inhibitor-mediated effects on energy balance in obese mice. This is consistent with studies showing no effect of chronic ANG-(1–7) administration on body composition in obese mice (5, 48). Furthermore, global knockout of mas receptors in mice increases adiposity without changing total body mass, perhaps suggesting decreased lean mass (37).
Finally, ANG-(1–7) is known to lower blood pressure in hypertensive rat models (38), and several studies have shown that A779 attenuates ACE inhibitor-mediated cardiovascular effects in rodents (2, 4, 14, 26, 50). In this study, A779 did not produce any effect on blood pressure either alone or in combination with captopril, suggesting that depressor effects of ACE inhibition in obese mice are due to inhibition of ANG II formation or potentially other mechanisms, such as bradykinin release. We and others have similarly shown that ANG-(1–7) does not lower blood pressure in obese mice despite protective effects on vascular function and insulin sensitivity (5, 48). These disparate findings may reflect differences in species, the degree of hypertension, diets, and length of treatment. Importantly, these findings support growing evidence that the antihypertensive effects of ANG-(1–7) are not evident in all research models.
In summary, we extend the present knowledge of hormonal mechanisms involved in ACE inhibition and highlight potentially divergent RAS mechanisms for effects of ACE inhibition on insulin action, blood pressure, and energy balance in obesity. We studied male obese mice, as there is risk for teratogenic effects with ACE inhibitors in premenopausal females, as well as evidence for reduced effectiveness in female clinical populations (41). Because there is known sexual dimorphism in the expression of RAS components and the cardiometabolic phenotype in obesity (17), however, there could be important sex differences that were not explored. These findings also support direct targeting of ANG-(1–7) to improve insulin sensitivity in obesity, which may prove advantageous over ACE inhibitors, which produce limiting adverse effects in some patients because of accumulation of bradykinin (15). It would be of interest to determine in future research whether ANG-(1–7) in combination with an ACE inhibitor provides further metabolic benefits compared with ACE inhibition alone.
GRANTS
This study was supported by NIH Grants HL-122507 and U24-DK-059637.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.S. and A.C.A. conceived and designed research; J.L., A.J.M., S.S.B., and A.C.A. performed experiments; J.L., A.J.M., S.S.B., and A.C.A. analyzed data; J.L., Y.S., and A.C.A. interpreted results of experiments; A.C.A. prepared figures; J.L., Y.S., and A.C.A. drafted manuscript; A.J.M. and S.S.B. edited and revised manuscript; J.L., A.J.M., S.S.B., Y.S., and A.C.A. approved final version of manuscript.
ACKNOWLEDGMENTS
Technical support was provided by the Vanderbilt Mouse Metabolic Phenotyping Center.
REFERENCES
- 1.Abuissa H, Jones PG, Marso SP, O’Keefe JH Jr. Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for prevention of type 2 diabetes: a meta-analysis of randomized clinical trials. J Am Coll Cardiol 46: 821–826, 2005. doi: 10.1016/j.jacc.2005.05.051. [DOI] [PubMed] [Google Scholar]
- 2.Al-Maghrebi M, Benter IF, Diz DI. Endogenous angiotensin-(1-7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol Res 59: 263–268, 2009. doi: 10.1016/j.phrs.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold AC, Okamoto LE, Gamboa A, Shibao C, Raj SR, Robertson D, Biaggioni I. Angiotensin II, independent of plasma renin activity, contributes to the hypertension of autonomic failure. Hypertension 61: 701–706, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benter IF, Yousif MH, Al-Saleh FM, Raghupathy R, Chappell MC, Diz DI. Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J Cardiovasc Pharmacol 57: 559–567, 2011. doi: 10.1097/FJC.0b013e31821324b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beyer AM, Guo DF, Rahmouni K. Prolonged treatment with angiotensin 1-7 improves endothelial function in diet-induced obesity. J Hypertens 31: 730–738, 2013. doi: 10.1097/HJH.0b013e32835ecbe5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonner JS, Lantier L, Hocking KM, Kang L, Owolabi M, James FD, Bracy DP, Brophy CM, Wasserman DH. Relaxin treatment reverses insulin resistance in mice fed a high-fat diet. Diabetes 62: 3251–3260, 2013. doi: 10.2337/db13-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijn AJ. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension 18: 763–773, 1991. doi: 10.1161/01.HYP.18.6.763. [DOI] [PubMed] [Google Scholar]
- 8.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 31: 362–367, 1998. doi: 10.1161/01.HYP.31.1.362. [DOI] [PubMed] [Google Scholar]
- 9.Coppey L, Lu B, Gerard C, Yorek MA. Effect of inhibition of angiotensin-converting enzyme and/or neutral endopeptidase on neuropathy in high-fat-fed C57Bl/6J mice. J Obes 2012: 326806, 2012. doi: 10.1155/2012/326806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Kloet AD, Krause EG, Kim DH, Sakai RR, Seeley RJ, Woods SC. The effect of angiotensin-converting enzyme inhibition using captopril on energy balance and glucose homeostasis. Endocrinology 150: 4114–4123, 2009. doi: 10.1210/en.2009-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dupont WD, Plummer WD Jr. Power and sample size calculations. A review and computer program. Control Clin Trials 11: 116–128, 1990. doi: 10.1016/0197-2456(90)90005-M. [DOI] [PubMed] [Google Scholar]
- 12.Echeverría-Rodríguez O, Del Valle-Mondragón L, Hong E. Angiotensin 1-7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo. Peptides 51: 26–30, 2014. doi: 10.1016/j.peptides.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 13.Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Ann Tallant E, Smith RD, Chappell MC. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int 68: 2189–2196, 2005. doi: 10.1111/j.1523-1755.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- 14.Flores-Monroy J, Valencia-Hernández I, Martínez-Aguilar L. Ang (1-7) is a modulator of the vasoconstrictor actions of Ang I and Ang II. J Renin Angiotensin Aldosterone Syst 16: 254–259, 2015. doi: 10.1177/1470320314563560. [DOI] [PubMed] [Google Scholar]
- 15.Gavras H, Gavras I. Angiotensin converting enzyme inhibitors. Properties and side effects. Hypertension 11: II37–II41, 1988. doi: 10.1161/01.HYP.11.3_Pt_2.II37. [DOI] [PubMed] [Google Scholar]
- 16.Guimaraes PS, Oliveira MF, Braga JF, Nadu AP, Schreihofer A, Santos RA, Campagnole-Santos MJ. Increasing angiotensin-(1-7) levels in the brain attenuates metabolic syndrome-related risks in fructose-fed rats. Hypertension 63: 1078–1085, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01847. [DOI] [PubMed] [Google Scholar]
- 17.Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, Karounos M, Cassis LA. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol 32: 1392–1399, 2012. doi: 10.1161/ATVBAHA.112.248559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henriksen EJ, Jacob S. Modulation of metabolic control by angiotensin converting enzyme (ACE) inhibition. J Cell Physiol 196: 171–179, 2003. doi: 10.1002/jcp.10294. [DOI] [PubMed] [Google Scholar]
- 19.Höcht C, Gironacci MM, Mayer MA, Schuman M, Bertera FM, Taira CA. Involvement of angiotensin-(1-7) in the hypothalamic hypotensive effect of captopril in sinoaortic denervated rats. Regul Pept 146: 58–66, 2008. doi: 10.1016/j.regpep.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Hughey CC, Wasserman DH, Lee-Young RS, Lantier L. Approach to assessing determinants of glucose homeostasis in the conscious mouse. Mamm Genome 25: 522–538, 2014. doi: 10.1007/s00335-014-9533-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayasooriya AP, Mathai ML, Walker LL, Begg DP, Denton DA, Cameron-Smith D, Egan GF, McKinley MJ, Rodger PD, Sinclair AJ, Wark JD, Weisinger HS, Jois M, Weisinger RS. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc Natl Acad Sci USA 105: 6531–6536, 2008. doi: 10.1073/pnas.0802690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang L, Mayes WH, James FD, Bracy DP, Wasserman DH. Matrix metalloproteinase 9 opposes diet-induced muscle insulin resistance in mice. Diabetologia 57: 603–613, 2014. doi: 10.1007/s00125-013-3128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohara K, Brosnihan KB, Chappell MC, Khosla MC, Ferrario CM. Angiotensin-(1-7). A member of circulating angiotensin peptides. Hypertension 17: 131–138, 1991. doi: 10.1161/01.HYP.17.2.131. [DOI] [PubMed] [Google Scholar]
- 24.Kohara K, Brosnihan KB, Ferrario CM. Angiotensin(1-7) in the spontaneously hypertensive rat. Peptides 14: 883–891, 1993. doi: 10.1016/0196-9781(93)90063-M. [DOI] [PubMed] [Google Scholar]
- 25.Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol Endocrinol Metab 248: E353–E362, 1985. doi: 10.1152/ajpendo.1985.248.3.E353. [DOI] [PubMed] [Google Scholar]
- 26.Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7). Hypertension 40: 774–779, 2002. doi: 10.1161/01.HYP.0000035396.27909.40. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z. The renin-angiotensin system and insulin resistance. Curr Diab Rep 7: 34–42, 2007. doi: 10.1007/s11892-007-0007-5. [DOI] [PubMed] [Google Scholar]
- 28.Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens 14: 799–805, 1996. doi: 10.1097/00004872-199606000-00017. [DOI] [PubMed] [Google Scholar]
- 29.Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci 32: 734–739, 2011. doi: 10.1016/j.tips.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muñoz MC, Giani JF, Burghi V, Mayer MA, Carranza A, Taira CA, Dominici FP. The Mas receptor mediates modulation of insulin signaling by angiotensin-(1-7). Regul Pept 177: 1–11, 2012. doi: 10.1016/j.regpep.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Oh YB, Kim JH, Park BM, Park BH, Kim SH. Captopril intake decreases body weight gain via angiotensin-(1-7). Peptides 37: 79–85, 2012. doi: 10.1016/j.peptides.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Premaratna SD, Manickam E, Begg DP, Rayment DJ, Hafandi A, Jois M, Cameron-Smith D, Weisinger RS. Angiotensin-converting enzyme inhibition reverses diet-induced obesity, insulin resistance and inflammation in C57BL/6J mice. Int J Obes 36: 233–243, 2012. doi: 10.1038/ijo.2011.95. [DOI] [PubMed] [Google Scholar]
- 33.Rao RH. Pressor doses of angiotensin II increase hepatic glucose output and decrease insulin sensitivity in rats. J Endocrinol 148: 311–318, 1996. doi: 10.1677/joe.0.1480311. [DOI] [PubMed] [Google Scholar]
- 34.Roks AJ, van Geel PP, Pinto YM, Buikema H, Henning RH, de Zeeuw D, van Gilst WH. Angiotensin-(1-7) is a modulator of the human renin-angiotensin system. Hypertension 34: 296–301, 1999. doi: 10.1161/01.HYP.34.2.296. [DOI] [PubMed] [Google Scholar]
- 35.Saiki A, Ohira M, Endo K, Koide N, Oyama T, Murano T, Watanabe H, Miyashita Y, Shirai K. Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metabolism 58: 708–713, 2009. doi: 10.1016/j.metabol.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 36.Santos RA. Angiotensin-(1-7). Hypertension 63: 1138–1147, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- 37.Santos SH, Fernandes LR, Mario EG, Ferreira AV, Pôrto LC, Alvarez-Leite JI, Botion LM, Bader M, Alenina N, Santos RA. Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes 57: 340–347, 2008. doi: 10.2337/db07-0953. [DOI] [PubMed] [Google Scholar]
- 38.Schindler C, Bramlage P, Kirch W, Ferrario CM. Role of the vasodilator peptide angiotensin-(1-7) in cardiovascular drug therapy. Vasc Health Risk Manag 3: 125–137, 2007. [PMC free article] [PubMed] [Google Scholar]
- 39.Schuchard J, Winkler M, Stölting I, Schuster F, Vogt FM, Barkhausen J, Thorns C, Santos RA, Bader M, Raasch W. Lack of weight gain after angiotensin AT1 receptor blockade in diet-induced obesity is partly mediated by an angiotensin-(1-7)/Mas-dependent pathway. Br J Pharmacol 172: 3764–3778, 2015. doi: 10.1111/bph.13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senanayake PD, Moriguchi A, Kumagai H, Ganten D, Ferrario CM, Brosnihan KB. Increased expression of angiotensin peptides in the brain of transgenic hypertensive rats. Peptides 15: 919–926, 1994. doi: 10.1016/0196-9781(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 41.Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol 294: R1220–R1226, 2008. doi: 10.1152/ajpregu.00864.2007. [DOI] [PubMed] [Google Scholar]
- 42.Takeda M, Yamamoto K, Takemura Y, Takeshita H, Hongyo K, Kawai T, Hanasaki-Yamamoto H, Oguro R, Takami Y, Tatara Y, Takeya Y, Sugimoto K, Kamide K, Ohishi M, Rakugi H. Loss of ACE2 exaggerates high-calorie diet-induced insulin resistance by reduction of GLUT4 in mice. Diabetes 62: 223–233, 2013. doi: 10.2337/db12-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torlone E, Rambotti AM, Perriello G, Botta G, Santeusanio F, Brunetti P, Bolli GB. ACE-inhibition increases hepatic and extrahepatic sensitivity to insulin in patients with type 2 (non-insulin-dependent) diabetes mellitus and arterial hypertension. Diabetologia 34: 119–125, 1991. doi: 10.1007/BF00500383. [DOI] [PubMed] [Google Scholar]
- 44.Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol 89: 3A–10A, 2002. doi: 10.1016/S0002-9149(01)02321-9. [DOI] [PubMed] [Google Scholar]
- 45.Wang CY, Liao JK. A mouse model of diet-induced obesity and insulin resistance. Methods Mol Biol 821: 421–433, 2012. doi: 10.1007/978-1-61779-430-8_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weisinger HS, Begg DP, Egan GF, Jayasooriya AP, Lie F, Mathai ML, Sinclair AJ, Wark JD, Weisinger RS. Angiotensin converting enzyme inhibition from birth reduces body weight and body fat in Sprague-Dawley rats. Physiol Behav 93: 820–825, 2008. doi: 10.1016/j.physbeh.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 47.Weisinger RS, Stanley TK, Begg DP, Weisinger HS, Spark KJ, Jois M. Angiotensin converting enzyme inhibition lowers body weight and improves glucose tolerance in C57BL/6J mice maintained on a high fat diet. Physiol Behav 98: 192–197, 2009. doi: 10.1016/j.physbeh.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 48.Williams IM, Otero YF, Bracy DP, Wasserman DH, Biaggioni I, Arnold AC. Chronic angiotensin-(1-7) improves insulin sensitivity in high-fat fed mice independent of blood pressure. Hypertension 67: 983–991, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Yang X, Meng X, Dong M, Guo T, Kong J, Zhang K, Zhang Y, Zhang C. Endogenous activated angiotensin-(1-7) plays a protective effect against atherosclerotic plaques unstability in high fat diet fed ApoE knockout mice. Int J Cardiol 184: 645–652, 2015. doi: 10.1016/j.ijcard.2015.03.059. [DOI] [PubMed] [Google Scholar]
- 50.Yousif MH, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF. Characterization of angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol Res 66: 269–275, 2012. doi: 10.1016/j.phrs.2012.05.001. [DOI] [PubMed] [Google Scholar]