

Abstract
Myocardial ischemia-reperfusion injury induces a sterile inflammatory response, leading to further injury that contributes to the final infarct size. Locally released danger-associated molecular patterns lead to priming and triggering of the NOD-like receptor protein 3 inflammasome and amplification of the inflammatory response and cell death by activation of caspase-1. We review strategies inhibiting priming, triggering, or caspase-1 activity or blockade of the inflammasome-related cytokines interleukin-1β and interleukin-18, focusing on the beneficial effects in experimental models of acute myocardial infarction in animals and the initial results of clinical translational research trials.
Keywords: inflammasome, interleukin-1β, interleukin-18, ischemia-reperfusion injury, nucleotide-binding oligomerization domain-like receptor protein 3
THE INFLAMMASOME
The term inflammasome refers to a class of macromolecular structures that are formed from the activation of intracellular receptors for danger- or pathogen-associated molecular patterns (DAMPs or PAMPs, respectively) (15, 130), part of nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I-like receptors (130), and absent in melanoma 2 (AIM2)-like receptor (123, 130). The evolutionary role of inflammasomes is to link detection of a “danger signal,” in the form of a plethora of molecular structures (microbial, cellular, and organic or inorganic simple molecules), to a specific cellular response that implies activation of the proinflammatory caspase-1 and release of the proinflammatory cytokines IL-1β and IL-18 (Fig. 1) (123, 130). The various inflammasomes have different ligand specificity and, together, can recognize a wide range of DAMPs and PAMPs (106). As such, inflammasomes are central to innate immunity, a nonspecific first line of defense promptly activated during the course of infections, in immune and nonimmune cells, based on the chemical properties of the antigen. Innate immunity is different from adaptive immunity, which requires activity of specialized cells and produces an antigen-specific response. DAMPs are a class of endogenous (self) molecules, including nucleic acids, nucleotides, proteins, fatty acids, and inorganic molecules, that, when in the extracellular space, in endocytotic vesicles, or free in the cytoplasm, trigger activation of the innate immune response independently from the presence of the infectious pathogen (112). The response to DAMPs in the absence of a microbial agent is, therefore, defined as “sterile immunity” (112). Congruent activation of the innate immune response in the presence or absence of a pathogen is due to the virtually indistinguishable chemical nature of DAMPs from PAMPs. The most studied inflammasome receptor is NACHT, leucine-rich repeat, and pyrin domain (PYD)-containing protein 3 [NLR protein 3 (NLRP3)], a pattern recognition receptor (PRR) that recognizes bacterial and viral PAMPs as well as numerous DAMPs involved in tissue injury (29, 93, 130). Several intracellular and extracellular stimuli that originate from disruption of cell homeostasis trigger NLRP3 activation, making this protein central in control of the inflammatory response to tissue injury (130). The focus of this review is to describe the activation of the NLRP3 inflammasome, its associated cytokines, and their consequences in response to myocardial ischemia-reperfusion (I/R) injury.
Fig. 1.

Inflammasomes react to the presence of danger-associated molecule and activate a specific cytokine pathway. A plethora of molecular structures, stemming from pathogen invasion or host tissue/cell damage, result in a streamlined initiation and activation of the innate immune response. During pathogen invasion, bacterial and viral products, such as lipopolysaccharide, DNA, or RNA, serve as pathogen-associated molecular patterns (PAMPs) to the inflammasome. During tissue damage, molecules of intracellular origin serve as danger-associated molecular patterns (DAMPs) to also activate the subsequent immune response. Activation of the inflammasome induces production and release of active IL-1β and/or IL-18.
INJURY AND INFLAMMATION IN ACUTE MYOCARDIAL INFARCTION
Acute myocardial infarction (AMI) is one of commonest causes of morbidity, hospitalization, and mortality on a global scale (9). Sudden occlusion of a coronary artery due to rupture of the fibrous cap that overlays a lipid plaque and subsequent thrombus formation results in immediate downstream reduction of oxygen delivery to the myocardium and intracellular energy that culminates in cardiomyocyte necrosis (26, 50). Restoration of blood flow to the ischemic myocardium, although a standard of care in AMI, can paradoxically compound injury to the myocardium through multiple mechanisms, deemed reperfusion injury (153). AMI represents a prototypical example of sterile injury, in which the NLRP3 inflammasome coordinates an inflammatory response in the absence of pathogens (130). While this response is essential to guarantee healing of the wounded tissue, uncontrolled or exuberant inflammation becomes a mechanism of disease (49). Prompted by local and systemic signals associated with cell damage and necrosis, leukocytes are recruited first to the region of ischemia and infarction (117). After AMI, there is no restitutio ad integrum. In fact, the resultant clearance of cellular debris, together with immune cell proliferation and fibrotic scar formation, leads to an incomplete functional recovery of the affected tissue, compounded by negligible cell cycle entry and regeneration of adult mammalian cardiomyocytes (48, 70, 117). Although the inflammatory response is needed to coordinate these healing phases, a chronic and unresolved inflammatory response leads to continual loss of functional myocytes, deleterious geometrical abnormalities, and aneurysm formation, worsening the consequences of AMI (23, 52, 117). The activity of the NLRP3 inflammasome is a decisive component of the innate immune response to ischemia, repair, and perpetuation of injury (125, 130).
Danger-Associated Molecular Patterns
Prolonged ischemia induces a loss of membrane integrity, leading to necrosis, and is associated with the release of intracellular contents not found in the normal interstitium (21, 40). Several intracellular proteins with different biological functions are collectively named “alarmins” for their ability to become alarm or danger signals for nearby cells. Identified alarmins include IL-1α (see below), high-mobility group box 1, S100 family proteins, some heat shock proteins (HSPs), and glucose-regulated proteins (GRPs) (10, 14, 77, 152). A common function of these alarmins is to activate NF-κB (14). As a protein regulating transcription of pro- and anti-inflammatory genes, NF-κB is a central orchestrator and key regulator of the inflammatory response and inflammasome pathway after AMI (137). One of the best-characterized alarmins is high-mobility group box 1. This nuclear protein is passively released during necrosis, leading to paracrine diffusion and binding to Toll-like receptor (TLR)-4, a PRR of the TLR family, or the receptor for advanced glycation end products, increasing NF-κB activation and perpetuating reperfusion injury (14, 42, 78). The S100 family of proteins also diffuse after necrosis, binding to the receptor for advanced glycation end products and TLR-4 and eliciting NF-κB activation (41). A similar function has been observed for GRPs (e.g., GRP94/gp96 and GRP170) and HSPs (e.g., hsp70 and hsp90) (12, 83). Among other known DAMPs, extracellular ATP serves as a trigger for aggregation of the components of the NLRP3 inflammasome, leading to the active macromolecular structure (15, 21, 40, 130).
NLRP3: a Sensor That Links Injury to Inflammation
NLRP3 is a member of a family of 14 NLRPs that includes PRRs and proteins not involved in the inflammatory response. NLRP3 is structurally characterized by a domain of leucine-rich repeats assembled at the COOH terminus, a central NATCH domain (also known as NOD), and an NH2-terminal effector PYD (130). The function of NLRP3 is mostly associated with the inflammasome; however, there have been reports of NLRP3 activity independent of the inflammasome pathway (146, 149). Upon ligand sensing, NLRP3 oligomerizes and, through its NH2-terminal PYD, interacts with the PYD of adaptor protein apoptosis-associated speck-like protein containing a COOH-terminus caspase activation and recruitment domain (ASC) (Fig. 2) (112, 130). This PYD-PYD interaction initiates polymerization of ASC into filamentous, insoluble structures that macroscopically distinguish the presence of the inflammasome as a large perinuclear speck (17). ASC is a scaffold protein that is necessary for recruitment of the effector enzyme pro-caspase-1 into the NLRP3 inflammasome (123, 130).
Fig. 2.

Stages of NOD-like receptor protein 3 (NLRP3) inflammasome activation: priming, triggering, and downstream effects. NLRP3 inflammasome activation requires two independent steps. During tissue damage, the presence of damage-associated molecular patterns (DAMPs) stimulates pattern recognition receptors (PRRs) like Toll-like receptors (TLRs) or the IL-1 receptor, leading to translocation of NF-κB into the nucleus. This event induces transcription of hundreds of proinflammatory genes, notably all components of the inflammasome (inflammasome priming). Sufficient translation of the inflammasome components NLRP3, apoptosis-associated speck-like protein containing a COOH-terminal caspase activation and recruitment domain (ASC), and pro-caspase-1 is a necessary step toward inflammasome formation but does not directly result in its activation. Extracellular DAMPs, like extracellular ATP (eATP), or intracellular DAMPs can induce NLRP3 activation (inflammasome trigger) through different mechanisms that involve K+ efflux. For example, eATP activates the purinergic P2X7 receptor, resulting in K+ efflux and activation of NLRP3. Once active, NLRP3 oligomerizes into a platform for pyrin domain (PYD) interactions between itself and ASC. Subsequent polymerization of ASC into filamentous insoluble structures serves as a platform for pro-caspase-1 recruitment. Formation of the macromolecular structure is followed by autocatalytic activation of pro-caspase-1 into caspase-1 and cleavage of the pro-IL-1β and pro-IL-18 into their active forms. Caspase-1-mediated cleavage of gasdermin-D (GSDMD), culminating in oligomerization of the NH2-terminal fragment of GSDMD into a plasma membrane pore, mediates release of IL-1β and IL-18 into the extracellular space for further autocrine, paracrine, and endocrine immune responses. Caspase-1 activity also mediates a form of regulated cell death termed pyroptosis.
Initially named IL-1β-converting enzyme for its recognized role in IL-1β activation, caspase-1 is a cysteine proteinase synthesized as a zymogen (45). Although similar to other caspases, caspase-1 is not part of the apoptotic pathway, inasmuch as caspase-1-deficient mice remain sensitive to apoptotic stimuli and progress through normal development (72). Predominantly associated with the inflammasome and functioning to process cytokines, caspase-1 cleaves the inactive cytosolic pro-IL-1β and pro-IL-18 into their respective active forms (45). Caspase-1 is also associated with a regulated form of cell death, termed pyroptosis (13). Morphologically and functionally distinct from apoptosis, which cleanly packages intracellular contents for immune cell phagocytosis, pyroptosis features plasma membrane rupture and release of intracellular and proinflammatory contents (13). This inflammation-linked cell death is a driving factor of reperfusion injury (Figs. 2 and 3). NLRP3 can also serve as a platform for noncanonical activation on two other caspases, caspase-8 and caspase-11. However, it is not known whether these two caspases have a role in the response to tissue injury (57, 69).
Fig. 3.

Wavefront of reperfusion injury. Prolonged ischemia inevitably leads to death of cardiomyocytes mainly through necrosis. This initial area of necrotic cells represents only a part of the mature infarct measured hours after reperfusion. In the initial phases (<3 h) of reperfusion, activity of the inflammasome is negligible, but damage-associated molecular patterns (DAMPs) released by necrotic cells contribute to increase expression of inflammasome components. When levels of inflammasome proteins reach a threshold for activation, the high intensity of inflammasome activity becomes associated with a rapid growth of the infarct through pyroptosis, the inflammatory cell death mediated by the inflammasome. The first hours before the increase in this inflammasome activity represent a time window for successful therapeutic intervention.
NLRP3 INFLAMMASOME ACTIVATION CONTRIBUTES TO INFARCT SIZE
In the first report of inflammasome activation during AMI, Kawaguchi et al. (68) noted visible ASC aggregates in the human heart at autopsies. They subsequently showed smaller infarcts, reduced IL-1β synthesis, and preservation of cardiac function in mice lacking ASC or caspase-1 (68). This is consistent with prior reports of the cardioprotective effect of caspase-1 deletion, preceding the notion of the inflammasome (53). Targeted deletion of caspase-1 reduces early mortality and left ventricular (LV) dilation after myocardial infarction (MI) (53, 105). Further evidence in support of a role of the NLRP3 inflammasome in AMI derived from gene silencing of NLRP3 in a different in vivo model of MI in the mouse by Mezzaroma et al. (94). These reports were followed by the work of Sandanger et al. (114), who found reduced infarct size in Nlrp3−/− mice in an ex vivo Langendorff model of I/R.
Priming of the NLRP3 Inflammasome
Activation of the NLRP3 inflammasome involves two distinct and independent phases: inflammasome priming and triggering of NLRP3 (Fig. 2) (130, 131). NF-κB drives transcription of hundreds of proinflammatory genes, including all the components of the NLRP3 inflammasome in leukocytes and cardiac resident cells, such as cardiomyocytes, fibroblasts, and endothelial cells (78, 123, 130). This step mediated by NF-κB, termed priming, is necessary to produce a critical mass of inflammasome components to suffice formation of the macromolecular complex (130, 131). The TLR signaling induced by DAMPs, including alarmins, and the activation of other cytokine receptors are the main factors that mediate the priming step (50, 130, 131).
Triggering of the NLRP3 Inflammasome
The presence of extracellular or intracellular triggers then leads to activation of NLRP3 (Fig. 2) (123). Many of these signals (see below) are associated with K+ efflux. Common trigger signals include extracellular ATP, reactive oxygen species (ROS), and impairment of the autophagy process, which has been shown to exacerbate the NLRP3 inflammasome-mediated response (150). When present in high extracellular concentration, ATP binds to the purinergic P2X7 receptor, inducing K+ efflux and triggering the NLRP3 activation cascade (Fig. 2) (130).
In some instances, as in monocytes, the priming signal induced by lipopolysaccharide (LPS) is sufficient to induce release of IL-1β (101). However, in the heart, the combination of the priming and triggering signals is necessary to elicit NLRP3 inflammasome activation (130, 131). The requirement for two signals broadens the therapeutic possibilities aimed at blocking inflammasome activation. The complexity of NLRP3 signaling and converging cellular processes continues to grow. Some of the mechanisms relevant to the sterile inflammatory response are described below.
ROLE OF THE INFLAMMASOME AND PYROPTOTIC CELL DEATH IN THE WAVEFRONT OF REPERFUSION INJURY
In the absence of priming, the concentration of inflammasome components in the heart is insufficient to respond to a trigger signal (131). This has been demonstrated using the mouse model of constitutively active mutant NLRP3, which fails to activate caspase-1 in the heart in the absence of priming (131).
I/R injury offers a combination of priming and triggering that activates the inflammasome pathway in the heart. We and others have shown that, after reperfusion, the size of the infarct increases in a time-dependent fashion (99, 127). Even if reperfusion reduces the total amount of myocardial necrosis, reperfusion fails to salvage all the myocardium that could be salvaged. Indeed, during the early phases of reperfusion, most of the infarct is represented by the necrotic core that is generated during ischemia plus a bordering portion that is part of the area at risk that is rescued from the hypoxic necrosis but has been exposed to sublethal injury and proinflammatory stimuli by DAMPs generated by the neighboring necrotic cells. Similar to the wavefront of ischemic injury during prolonged ischemia, this phenomenon represents a “wavefront of reperfusion injury,” whereby the size of the infarct continues to expand over the 3–6 h after reperfusion (Fig. 3) (109). The wavefront of reperfusion injury may, at least in part, be due to a propagation of cell damage, of an inflammatory nature, to cells that were previously harmed but not killed. Expression of NLRP3 and the inflammasome activation indeed increase over time after reperfusion, resulting in cell death (82, 127). In accordance with these dynamics, we have shown, in the mouse, a therapeutic intervention window of a few hours after reperfusion, characterized by a reduction in infarct size with inhibition of the NLRP3 inflammasome, that is lost if treatment is given 3 h after reperfusion (127). These results indicate that, in the mouse heart, a sustained NLRP3 inflammasome activation occurs between 1 and 3 h after reperfusion, and a therapeutic intervention before 3 h may improve the reperfusion outcome. As explained above, the mechanism by which the NLRP3 inflammasome induces cardiomyocyte death and infarct expansion is likely through pyroptosis and not through production of IL-1β (94). Deletion or inhibition of the inflammasome components (NLRP3, ASC, or caspase-1) reduces infarct size, whereas IL-1β blockade (see below) at the time of reperfusion fails to reduce infarct size (68, 84, 86, 113, 114, 127, 138). However, the mouse lacking IL-1 receptor type I (IL-1RI) or the mouse pretreated with an IL-1 blocker is protected, and this may be related to a further downregulation of the sensors of the inflammasome in the heart (133).
MECHANISMS OF REGULATION OF THE NLRP3 INFLAMMASOME
As described above, NLRP3 responds to a variety of different stimuli. The extensive research of the past few years has pointed out some key pathways and events that lead to the activation of NLRP3.
ROS and Mitochondrial Dysfunction
ROS, among the most common cellular byproducts produced during I/R injury, lead to oxidative stress and activation of the inflammasome cascade via multiple mechanisms (52, 108). Oxidative stress leads to an unfolded protein response that can cause detachment of thioredoxin from thioredoxin-interacting protein, which interacts with NLRP3 to promote activation of the inflammasome (82). siRNA against thioredoxin-interacting protein was shown to reduce inflammasome formation in the heart and reduce infarct size after I/R injury (82). Mitochondria are responsible for cellular energy production and are actively involved in the induction of necrotic, apoptotic, and pyroptotic cell death (50). These organelles constitute one of the major sources of ROS after reperfusion, due to damage to the segment between complex III and cytochrome oxidase of the mitochondrial respiratory chain (22). ROS represent a potent trigger of NLRP3 and induce NLRP3-dependent lysosomal damage and inflammasome activation (62, 130). Another activator of NLRP3 of mitochondrial origin is cardiolipin, a phospholipid that is located in the inner membrane of the mitochondria and is responsible for several bioenergetic processes (65). In ROS-mediated mitochondrial dysfunction, exposed cardiolipin binds to and activates NLRP3 (65). Released cardiolipin, in combination with an ineffective autophagic clearance of damaged mitochondria, leads to NLRP3-mediated activation of caspase-1 and subsequent production of IL-1β. Additionally, impaired mitochondrial fission can contribute to activation of NLRP3. This has been shown by deletion of dynamin-related protein 1, which leads to an increase in NLRP3-dependent caspase-1 activation and IL-1β secretion (103).
Autophagy
Autophagy is an additional important pathway that is linked to myocardial ischemia and inflammasome activation (122, 150). The primary function of autophagy is removal and recycling of misfolded or damaged proteins and organelles (97). Autophagy is, therefore, essential to maintain tissue homeostasis and becomes fundamental in limiting cellular damage (43, 97). Recent evidence has shown that autophagy is necessary to reduce myocardial damage after AMI (150). The autophagic process can limit activation of the NLRP3 inflammasome by removing damaged mitochondria (122). It has also been shown that physiological autophagy limits secretion of mature IL-1β in macrophages in a mouse model of acute liver toxicity; however, this has not been shown in the heart (38, 124). The overall effect exerted on the inflammasome pathway by functional, physiological autophagy would be to minimize activation of the inflammasome and reduce secretion of cytokines. However, impairment of the autophagic pathways, as a consequence of extensive cell damage, would lead to ineffective or incomplete autophagy (and mitophagy), inducing activation of NLRP3 and exacerbation of myocardial damage.
Posttranslational Regulation of NLRP3 Inflammasome Activity
Activation of the inflammasome is also regulated by posttranslational modifications of its components (Fig. 4). In a model of stroke, Bruton’s tyrosine kinase has been shown to interact with both NLRP3 and ASC, suggesting a potential role of Bruton’s tyrosine kinase in the phosphorylation of ASC (64). Two other important kinases, spleen tyrosine kinase and c-Jun NH2-terminal kinase, are involved in the response to oxidative stress and enhance ASC oligomerization through posttranslational modification of ASC (151). An additional kinase, NEK7, a member of the never in mitosis gene A-related kinase family, acts downstream of the P2X7 receptor and binds to NLRP3, regulating its activation and oligomerization (61). NEK7 is highly expressed in heart muscle and might be a target for intervention in the heart (61).
Fig. 4.

Molecular signaling of regulation of the NOD-like receptor protein 3 (NLRP3) inflammasome during tissue injury. Several stimuli regulate activation of NLRP3 [and apoptosis-associated speck-like protein containing a COOH-terminal caspase activation and recruitment domain (ASC)], controlling formation of the inflammasome. Mitochondria can stimulate NLRP3 activation though generation of reactive oxygen species (ROS). Although there are alternative sources of ROS, the mitochondrial origin of ROS is predominant during the reperfusion phase of injury. ROS also cause dissociation of thioredoxin-interacting protein (TNXIP) from thioredoxin. Free TNXIP mediates activation of NLRP3. Damaged mitochondria also release cardiolipin, a phospholipid present only in the inner mitochondrial membrane, but can be released in the cytoplasm and activate NLRP3 during damage. The mitochondrial antiviral signaling protein (MAVS) is also an important regulator of NLRP3 during viral infection and tissue damage. Interaction of MAVS with the nucleotide-binding oligomerization domain, leucine-rich repeat containing X1 (NLRX1) protein prevents NLRP3 activation. Damaged mitochondria enter autophagic vesicles for effective removal of the potentially dangerous organelle. Effective autophagy and mitophagy, leading to complete removal of defective proteins and intracellular organelles, prevent activity of the inflammasome. On the other hand, defective autophagy and mitophagy lead to the leak of the proteolytic enzyme cathepsin B inside the cytoplasm, leading to NLRP3 activation. NEK7, a member of the never in mitosis gene A (NIMA)-related kinases family, promotes NLRP3 activation after K+ efflux. Bruton’s tyrosine kinase (BTK) also activates the inflammasome by interacting with NLRP3 and ASC. Two other kinases, spleen tyrosine kinase (SYK) and c-jun NH2-terminal kinase (JNK), enhance oligomerization of ASC.
Recently, a member of the NLR family has been found to act as a negative regulator of the inflammatory response during AMI. In an in vitro model of hypoxia-induced myocardial injury, NLRX1 has been found to inhibit NLRP3 activation mediated by mitochondrial antiviral signaling protein (121). Mitochondrial antiviral signaling protein is a mitochondrion-associated adaptor protein that is essential for recruitment of NLRP3 to the mitochondrial outer membrane (Fig. 4) (80, 121).
INFLAMMASOME-ASSOCIATED CYTOKINES
Inhibition of the NLRP3 inflammasome limits the inflammatory injury after myocardial I/R in the mouse. As described above, caspase-1 has a central role in the function of the inflammasome. This enzyme is fundamental for induction of pyroptotic cell death and is also necessary for processing and release of the inflammasome-associated cytokines IL-1β and IL-18 (Fig. 5) (45, 46). The mechanism by which caspase-1 accomplishes these two functions is through cleavage of the protein gasdermin D (GSDMD) followed by oligomerization of the NH2-terminal domain of GSDMD, to form a pore in the plasma membrane (36, 38, 61, 101). This pore disrupts the ionic gradients, causing Na+ and water influx, osmotic swelling, and membrane rupture (46, 119). Recent data hypothesized an intermediate phase between inflammasome activation and pyroptotic cell rupture, deemed “hyperactivated,” where GSDMD pores permit IL-1β (and IL-18) release in macrophages with activated inflammasomes and intact lipid bilayers (44, 74).
Fig. 5.
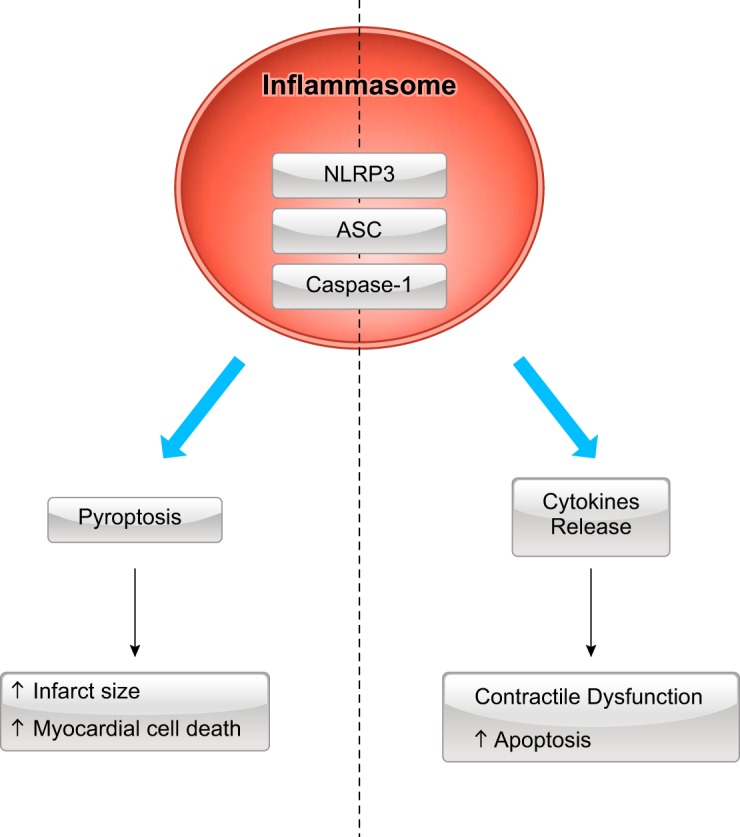
Dual function of caspase-1 activation during myocardial infarction. Activation of the inflammasome leads to caspase-1 activation, which has two associated, but different, effects: 1) inflammatory cell death (pyroptosis), which fosters increased infarct size and myocardial cell death, and 2) release of cytokines IL-1β and IL-18, which spur contractile dysfunction and regulated cell death through apoptosis. NLRP3, NOD-like receptor protein 3; ASC, apoptosis-associated speck-like protein containing a COOH-terminal caspase activation and recruitment domain.
Interleukin-1β
IL-1β, the original member of the IL-1 family of cytokines, has been extensively studied because of its integral role in initiating and perpetuating physiological and pathological inflammation (31–33). IL-1β is normally transcribed and translated in the cytosol at low basal levels in its inactive pro form; however, upon stimulation of TLRs or other cytokine receptors, transcription and translation are dramatically increased within 15 min (33). As previously reported, IL-1β secretion in its active form requires the active form of caspase-1 (33). Therefore, the rate-limiting step in IL-1β processing and secretion is inflammasome activation. Lacking a signal peptide for the classic endoplasmic reticulum-Golgi apparatus protein secretion pathway, IL-1β secretion requires formation of a GSDMD pore (13, 44). Once released in the extracellular space, IL-1β acts in an autocrine, paracrine, and endocrine fashion to initiate and sustain proinflammatory activity (33, 44).
The heterodimerization of two plasma membrane receptors, IL-1RI and IL-1 receptor accessory protein (IL-1RAcP), mediates signal transduction of the two isoforms of IL-1 (IL-1β and IL-1α) (16). This signaling involves interaction of the Toll-IL-1 receptor (TIR) domain of IL-1RI with the TIR domain of the myeloid differentiation factor 88 that activates a kinase-dependent signaling cascade (35). A second type II receptor, IL-1RII, is membrane bound but lacks the intracellular TIR domain and acts as a scavenger receptor (30, 35). The intracellular cascades [reviewed in detail elsewhere (123)] result in transcription of a multitude of chemokines, cytokines, and adhesion molecules that drive immune cell recruitment, activation, and differentiation (94). IL-1 signaling also alters intracellular protein expression, changing cellular function and metabolism (31). Sharing ~26% amino acid homology with IL-1β is IL-1 receptor antagonist (IL-1Ra), an endogenous protein that tightly binds to and occupies IL-1RI, inhibits the binding of IL-1RAcP and subsequent receptor heterodimerization, and prevents the intracellular transduction of signals (130). Production of this high-affinity competitor is induced by several proinflammatory stimuli, including LPS, a complex molecule common on the wall of gram-negative bacteria that activates TLR4, and IL-1β, presumably to balance a perpetual cycle of proinflammatory signaling (120, 126). Intracellular functions, specifically antiapoptotic, of IL-1Ra have also been noted in cardiomyocytes (144).
Interleukin-18
IL-18, also a member of the IL-1 family of cytokines, requires caspase-1-mediated cleavage to the active form (45). IL-18 is produced and stored in the cytoplasm, also in the absence of TLR or other proinflammatory stimulation, and is readily available for cleavage and secretion in case of inflammasome activation or by extracellular proteases in case of passive diffusion due to cell necrosis (102, 130). Receptor binding and signaling of IL-18 resemble receptor binding and signaling of IL-1, whereby a heterodimeric complex of IL-18Rα and IL-18Rβ combine to evoke intracellular cascades, culminating in proinflammatory gene transcription (67, 102). A second IL-18 receptor, Na+-Cl− cotransporter (NCC) was identified in 2015. NCC is a solute carrier symporter that is mainly present in the distal tubule of the glomeruli in the kidney (89). NCC becomes expressed in the atherosclerotic plaques of apolipoprotein E-deficient mice and mediates IL-18 signaling in cytokine-primed macrophages (147). An endogenous circulating binding protein of IL-18 (IL-18BP) in healthy humans is normally present at levels 25–50 times higher than IL-18, with significantly higher affinity for membrane-bound IL-18Rα (37).
Differences and Similarities Between IL-1β and IL-18
The production and signaling of IL-1β and IL-18 are similar, because both cytokines require caspase-1 and GSDMD for their secretion, bind a heterodimeric receptor of a similar structure, and share some intracellular downstream cascades involving myeloid differentiation factor 88 activation and convergence on NF-κB (18, 45, 102). The specificity of the signal depends on the receptor and cell type; therefore, depending on the target, these two cytokines promote different biological effects (79). As previously mentioned, basal gene expression for IL-1β in several cell types (e.g., blood mononuclear cells, hematopoietic cells, and endothelial cells) is extremely low (107). In contrast, the IL-18 precursor is present in blood monocytes from healthy subjects, unstimulated macrophages, and epithelial and endothelial cells (37). In addition, IL-1 induces fever and IL-18 does not. [IL-18 does not affect cyclooxygenase-2 or PGE2 production (37, 79).] IL-1 (α or β) is active at concentrations severalfold lower than IL-18 (37). In addition, in some diseases, the activity of IL-1 and IL-18 is different. IL-1 is more involved in rheumatoid arthritis, whereas IL-18 is more involved in inflammatory bowel disease (34, 66). IL-18 is also involved in homeostasis. When fed a standard diet, mice lacking IL-18 develop obesity and hyperglycemia, a phenotype exacerbated by a high-fat diet (19, 100). Finally, in the same cells, IL-1 and IL-18 can activate different responses: IL-1β strongly induces NF-κB, whereas IL-18 may promote a p38 MAPK-specific response (79).
IL-1α, a Cytokine and an Alarmin
IL-1α is also produced and stored in the cell (39). Different from IL-1β and IL-18, IL-1α translocates to the nucleus to influence transcription and is functionally active in its precursor form (14, 32). Therefore, pro-IL-1α is capable of acting as an alarmin, initiating proinflammatory paracrine responses immediately after release from the cytosol after cellular necrosis (14, 36, 63), before IL-1β becomes active and exerts its function (14, 36). IL-1α is not recognized by caspase-1 and thus is not activated through the inflammasome (36), although it has been reported that active release of IL-1α also necessitates inflammasome activation, likely through GSDM pore formation (14). The initial inflammatory signal that follows necrosis and recruitment of neutrophils has been shown to be due to the precursor of IL-1α, whereas recruitment of macrophages to potentiate and maintain the inflammatory process was characteristic of IL-1β (110).
CELL-SPECIFIC EFFECTS OF THE INFLAMMASOME AND ITS CYTOKINES ON AMI
Cell-Specific Effects of the Inflammasome
After AMI, ASC specks can be detected in most of the resident cells (endothelial cells, cardiomyocytes, and fibroblasts) and in cells infiltrating the infarct. In the subacute phase, the majority of the specks are observed in cells of the granulation tissue. However, during healing and maturation of the scar, as the infiltrate resolves, ASC positivity is detected mostly in isolated cardiomyocytes or fibroblasts (94, 130). The effects of NLRP3 inflammasome formation in the different cardiac resident cells are cell specific (125). IL-1β production has been documented in cardiac fibroblasts and endothelial cells but not in cardiomyocytes, despite an increase in NLRP3 and caspase-1 activity in the latter (68, 82, 94, 114). Inflammasome and caspase-1 activation in cardiomyocytes, instead, induces loss of cell membrane integrity and death (94, 125).
Cell-Specific Effects of IL-1β
After AMI, locally produced active IL-1β also has regulatory and detrimental effects on cardiac resident and infiltrating cells. The initial release of active IL-1β in the area of infarction creates homing signals for leukocytes and creates a “sticky” endothelium (48, 56). Abrupt chemokine release after ischemia creates a gradient that attracts leukocytes to the region (11, 51). Proinflammatory cytokines, including IL-1α and IL-1β, increase endothelial cell surface expression of adhesion molecules that bind to the recruited leukocytes and begin the process of leukocyte extravasation from the vessel into the injured area (52). IL-1β is also capable of increasing chemokines, such as monocyte chemoattractant protein-1, responsible for mobilizing proinflammatory monocytes to the area (104). Resident cardiac fibroblasts are also important players in the post-AMI inflammatory response. In the initial phase after ischemic injury, IL-1β has been shown to increase fibroblast expression of collagenases and inhibit transforming growth factor-β signaling, possibly to ensure that the injured matrix area has been effectively broken down and cleared of additional damage signals before tissue reparative engines are fully engaged and fibroblast populations are converted to myofibroblasts (48, 115). Cardiomyocytes are also sensitive to IL-1β. In fact, IL-β depresses contractility, in vitro and in vivo, via uncoupling of the L-type Ca2+ channel from the β-adrenergic receptor (81, 116), decreasing expression of phospholamban and the sarco(endo)plasmic reticulum Ca2+-ATPase and increasing local nitric oxide, which affects ATP production within the mitochondria (25). Moreover, IL-1β induces apoptotic cell death of cardiomyocytes in the subacute and chronic phases after AMI (5, 132).
Cell-Specific Effects of IL-18
The cell-specific effects of IL-18 on myocardial cells post-AMI have been investigated to a lesser extent. IL-18 appears to induce angiogenesis but, at the same time, inhibits angiogenesis after ischemic damage (148). In vitro experiments using recombinant IL-18 proved that it induces apoptosis of cardiac endothelial cells in vitro (20). Cardiac fibroblasts exposed to IL-18 proliferate, produce collagen, and remodel the extracellular matrix (47). IL-18 depresses the contractility of cardiomyocytes in vitro and in vivo, and in an ex vivo model of hypoxia, IL-18 impairs the contractility of human cardiac strips (105).
PHARMACOLOGICAL INHIBITION OF THE NLRP3 INFLAMMASOME
Several NLRP3 inflammasome inhibitors have been identified; they include molecules already known and synthetized and newly designed compounds derived in attempts to increase specificity and successful clinical application (Tables 1 and 2).
Table 1.
NLRP3 inflammasome inhibitors in experimental AMI
| Name(s) | Method(s) | Finding(s) | Reference(s) |
|---|---|---|---|
| Colchicine | 0.1 mg/kg ip daily for 7 days to the mouse starting before ischemia in the nonreperfused AMI model | Smaller scar, reduced adverse remodeling, and heart failure and improved survival | 8, 54 |
| N-[(7S)-1,2,3,10-tetramethoxy-9-oxo-6,7-dihydro-5H-benzo[a ]heptalen-7yl] acetamide | 0.4 mg/kg ip 25 min before reperfusion in a model of transient I/R | Reduced infarct size and fibrosis in the chronic setting | |
| Glyburide derivative; | 100 mg/kg ip before surgery in a reperfused AMI model | Reduced infarct size and improved LV function | 84, 86, 127 |
| 16673-34-0 | 100 mg/kg ip after ischemia in a model of nonreperfused AMI | Improved cardiac remodeling and LV dysfunction | |
| 5-chloro-2-methoxy-N-[2-(4-sulfamoyl-phenyl) ethyl] benzamide | 100 mg/kg ip up to 60 min after reperfusion | Reduced infarct size at 24 h | |
| MCC950 | 3–6 mg/kg iv 15 min before reperfusion and again after 24 h and following 5 days in the pig balloon angioplasty inflation ischemia model | Reduced infarct size; improved cardiac remodeling and prevention of LV dysfunction | 138 |
| N-((1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)-4-(2-hydroxypropan-2-yl)furan-2-sulfonamide; N-[[(1,2,3,5,6,7-hexahydro-S-indacen-4-yl)amino]carbonyl]-4-(1-hydroxy-1-methylethyl)-2-furansulfonamide | |||
| BAY 11-7082 | Treatment (dose not specified) before ischemia or before reperfusion in I/R models in mouse and rat | Reduced infarct size; preserved cardiac function | 71 |
| 3-[(4-methyl- phenyl)-sulfonyl]-(2E)-propenenitrile | |||
| INF4E | 50 μM in an ex vivo myocardial ischemia model 20 min before ischemia | Reduced infarct size at 60 minReduced infarct size | 88 |
| ethyl 2-((2-chlorophenyl)(hydroxy)methyl)acrylate | |||
| OLT1177 (dapansutrile) | 6–600 mg/kg ip before, or up to 60 min after, reperfusion in the mouse surgical coronary artery ligation model | 128 | |
| 3-(methanesulfonyl) propanenitrile |
AMI, acute myocardial infarction; I/R, ischemia-reperfusion; LV, left ventricular; NLRP3, NOD-like receptor protein 3.
Table 2.
Clinical trials of colchicine and IL-1 blockers in AMI and heart failure
| Disease | Clinical Trial Name | Study Design and Drug Regimen | Main Finding(s) | Reference(s) |
|---|---|---|---|---|
| ST-segment elevation AMI | Anti-Inflammatory Treatment With Colchicine in AMI | Randomization 1:1 | Reduced infarct size (cardiac magnetic resonance and cardiac biomarkers); reduced peak CRP | 28 |
| Colchicine or placebo | ||||
| (n = 151) | Loading dose of 2 mg (1.5 mg initially followed by 0.5 mg 1 h later) and continuing with 0.5 mg twice daily, or placebo, for 5 days | |||
| ST-segment elevation AMI | VCU-ART | Randomization 1:1 | Reduced peak CRP; trend toward reduced incidence of heart failure at 3 mo and at long-term followup with anakinra | 2, 3, 6 |
| (n = 10) | VCU-ART2 | Anakinra or placebo | ||
| (n = 30) | Anakinra 100 mg once daily for 14 days | |||
| Non-ST-segment elevation AMI | MRC-ILA Heart Study | Randomization 1:1 | Reduced peak CRP; no differences in major adverse cardiac events at 30 days and 3 mo, but more events after 6 mo in the anakinra-treated group | 98 |
| (n = 182) | Anakinra or placebo | |||
| Anakinra 100 mg once daily for 14 days |
AMI, acute myocardial infarction; CRP, C-reactive protein.
Colchicine
Colchicine is one of the oldest anti-inflammatory drugs and, recently (2009), was found to inhibit NLRP3 activity by preventing its assembly and activation (87). Although historically used for treatment of Mediterranean fever, colchicine has been recently approved by the United States Food and Drug Administration for treatment of gout (27). Besides its effect on microtubule polymerization, colchicine inhibits NLRP3 activation, preventing the pore formation induced by the P2X7 receptor and, therefore, limiting K+ efflux (87). Because of the integral role of microtubules in providing a platform for mediating intracellular trafficking and the known translocation and aggregation of NLRP3, ASC, and caspase-1, colchicine may inhibit the inflammasome by decreasing aggregation of its components. Colchicine suppressed the colocalization of ASC to NLRP3 and decreased amounts of mature IL-1β in the peritoneal cavity of mice during acute gout (95). The novel understanding of the mechanism of action has opened the way to the investigation in the field of cardiology (27, 91). In a nonreperfused AMI mouse model, a high dose (0.1 mg·kg−1·day−1) of colchicine administered for 7 days significantly inhibited the increase in NLRP3 mRNA and caspase-1 activity at 24 h post-MI, inhibited expansion of LV scar size at 1 wk, attenuated ventricular remodeling at 7 days and 4 wk after MI, and improved 7-day survival (54). These findings show that colchicine inhibits the inflammasome pathway in the in vivo AMI model. Similarly, in a model of reperfused AMI induced by 45 min of transient ischemia, a single dose (0.4 mg/kg ip) of colchicine administered 25 min before reperfusion significantly reduced infarct size, troponin T levels, and inflammatory markers 24 h post-MI and significantly reduced fibrosis 10 wk after AMI (8). Recently, in a clinical trial, patients presenting with ST-segment elevation MI (STEMI) undergoing primary percutaneous coronary intervention were treated with colchicine (initial dose of 1.5 mg followed 1 h later by 0.5 mg and 0.5 mg twice daily for 5 days). The clinical trial results revealed a reduction in infarct size with colchicine, measured as area under the curve for creatine kinase myocardial band (28).
Glyburide Derivative
Glyburide, a sulfonylurea used to increase pancreatic β-cell insulin secretion to treat type 2 diabetes mellitus, was shown to inhibit IL-1β release in vitro by acting upstream of NLRP3 and downstream of the P2X7 receptor (76). The use of glyburide at a dose proven to inhibit the NLRP3 inflammasome would, however, cause lethal hypoglycemia in the mouse (84). A derivative of glyburide, 4-[2-(5-chloro-2-methoxybenzamido)ethyl]benzenesulfamide, which lacks the moiety responsible for inducing insulin secretion, was developed to maintain the inflammasome inhibitory effect and also to abolish the hypoglycemic effect of glyburide (84). This compound, known also as 16673-34-0, has been studied in a murine model of acute transient (30 or 75 min) coronary ligation and permanent ligation as well as in a model of cardiotoxicity induced by a single dose (10 mg/kg) of doxorubicin (86). Single-dose (100 mg/kg) glyburide analog treatment at reperfusion significantly decreased macromolecular aggregation of ASC, caspase-1 activity, plasma levels of cardiac troponin I, and infarct size, without effects on glycemia (84). Furthermore, when administered within 1 h of reperfusion injury, 16673-34-0 is as effective as when it is given at the time of reperfusion (127). This finding is particularly important, because it confirms the timing of activation of NLRP3 after I/R in AMI. Drugs that necessitate prompt administration after reperfusion are at higher risk of failure during testing in the clinical setting (136). Therefore, identification of a therapeutic target that can be blocked during an extended time window of intervention may lead to increased success during the clinical translation phase. After permanent ligation in the mouse, daily treatment with 16673-34-0 (100 mg/kg) for 7 days significantly preserved LV fractional shortening and LV end-diastolic and end-systolic diameters without affecting infarct size, indicating that some of the benefits for cardiac function are independent of infarct reduction, possibly due to reduced IL-1β and IL-18 cytokine secretion (see below) (86). In the cardiotoxicity model, 16673-34-0 reduced myocardial fibrosis and preserved LV fractional shortening (84). The mechanism of action seems directed toward NLRP3 oligomerization downstream of activation, since 16673-34-0 inhibits macrophages with constitutively active mutant NLRP3 (86).
Recently, a compound derived from 16673-34-0, termed JC124, was tested in a setting of longer-term (75 min) ischemia; it was given after reperfusion and significantly reduced infarct size, indicating benefits also after more dramatic ischemic damage (55).
MCC950
A potent inhibitor of NLRP3, MCC950, is part of a class of drugs previously shown to inhibit NLRP3-induced ASC oligomerization, preventing formation of the specks (24, 75). MCC950 reduced IL-1β production in vivo when given 1 h before LPS treatment and blocked ASC oligomerization in vitro without inhibiting NLRP3 ATPase activity (24). MCC950 has been shown to be effective also in a mouse model of experimental autoimmune encephalomyelitis in which NLRP3 is required for development of the disease (58). Administration of MCC950 suppressed T cell responses and attenuated the severity of the disease (58). Additionally, MCC950 has been effective in treating mice displaying Muckle-Wells syndrome, a cryopyrin-associated periodic syndrome. These mice, carrying a gain-of-function mutation of Nlrp3, are contradistinguished by neonatal mortality due to high IL-1β and IL-18 plasma levels. Treatment for 28 days with MCC950 rescued these mice with constitutively active mutant NLRP3 (24).
In a porcine model of 75-min transluminal coronary artery balloon occlusion, MCC950 treatment (6 or 3 mg/kg) 15 min before ischemia and once daily after ischemia decreased infarct size, preserved LV ejection fraction, decreased myocardial IL-1β levels, and attenuated myocardial neutrophil influx, validating the therapeutic target in the large-animal model (138).
BAY 11-7082
BAY 11-7082, previously known to inhibit NF-κB, has been shown to directly inhibit NLRP3 ATPase activity in response to NLRP3-specific stimuli in vitro (71). In a rat model of I/R, pretreatment with BAY 11-7082 (130 µg/kg) 30 min before reperfusion significantly reduced infarct size and cardiac fibrosis and improved LV fractional shortening (71).
INF4E
Acrylamide derivatives were developed by an Italian cohort as covalent inhibitors of NLRP3 and its ATPase activity required for inflammasome activation (88). An ex vivo model of I/R including pretreatment with the NLRP3 inhibitor ethyl-2-{[(2-chlorophenyl)hydroxyl]methyl}acrylate (INF4E) showed a significant reduction of infarct size and an improvement in LV developed pressure 60 min after ischemia (88).
Dapansutrile/OLT1177
Perhaps the NLRP3 inhibitor most advanced into the clinical testing realm is dapansutrile (or OLT1177). Preclinical models of 30 min of transient coronary ligation followed by OLT1177 administration (60 mg/kg) within 60 min at reperfusion in the mouse have shown significant reduction of infarct size, reduction of plasma cardiac troponin I, and preservation of LV fractional shortening (128). OLT1177 was tested as an orally active β-sulfonyl nitrile molecule in humans, and its safety has been shown at oral doses up to 1,000 mg/day for 8 days, with no clinical, hematological, or organ toxicities (85). Marchetti and colleagues (85) described an effect of OLT1177 on NLRP3 ATPase activity, leading to decreased NLRP-ASC oligomerization, but no effect on NLRC4 or AIM2 inflammasomes, K+ efflux, gene expression, or synthesis of IL-1β precursor or on other kinases on a broad-range screen. The potential for OLT1177 as a treatment for acute gouty arthritis and osteoarthritis is under investigation in a phase II clinical trial (136a, 136b).
BLOCKADE OF IL-1α AND IL-1β IN AMI
The benefit of an anti-inflammatory strategy aimed at the inflammasome appears to extend beyond inhibition of inflammasome formation and to be, at least in part, related to reduced synthesis and secretion of proinflammatory cytokines.
Genetic deletion or pharmacological inhibition of IL-1 activity, via direct antibody neutralization/modulation of IL-1β or IL-1α, IL-1 receptor antagonism, or IL-1β/IL-1α sequestration via a chimeric protein made by the ectodomains of IL-1RI and IL-1RAcP (IL-1Trap), has been shown to lead to significantly reduced cardiomyocyte apoptosis and prevent LV dilation and systolic dysfunction (142, 143). One of the first reports of the use of anakinra, a recombinant IL-1Ra, given at different doses in an experimental AMI in mice and in rats, reduced the adverse remodeling process and myocardial apoptotic rate and preserved LV function (5). These data were supported by another study in which IL-1R1−/− mice exhibited, after I/R injury, an attenuated proinflammatory and profibrotic response followed by a reduction of infarcted area and an attenuation of adverse remodeling of the LV (4). As shown in an elegant study from Sager et al. (113) using parabiotic bone marrow transplantation, release of IL-1β after AMI fosters mobilization of inflammatory cells from the hematopoietic system, leading to granulation tissue and scar formation in the heart after AMI. Moreover, neutralization of the systemically released IL-1β with an anti–IL-1β treatment lowered monocyte and neutrophil infiltration and ameliorated adverse ventricular remodeling (113). As previously described, IL-1Ra prevents binding of both IL-1α and IL-1β to IL-1RI, making it impossible to distinguish the role of the two isoforms (32).
Anakinra is approved by the United States Food and Drug Administration for the treatment of rheumatoid arthritis and cryopyrin-associated periodic syndromes, diseases driven by enhanced IL-1 activity (73). Pilot clinical trials completed for the use of anakinra (100 mg sc daily) in patients with reperfused STEMI and primary percutaneous coronary intervention (n = 40) noted that IL-1 blockade was safe, significantly limited the C-reactive protein (CRP) increase at 72 h, and lowered the rate of new onset heart failure after STEMI (Table 2) (2, 3, 6). The VCU-ART3 trial evaluating a higher dose of anakinra (100 mg twice daily) versus the standard dose (100 mg daily) versus placebo is ongoing (136c).
In the phase II clinical trial MRC-ILA heart study involving 182 patients with non-STEMI who were treated with anakinra (100 mg daily for 14 days), a similar reduction in the acute inflammatory response was observed (98). At 30 days and 3 mo, there was no change in clinical events, but by 12 mo, ischemic events were significantly increased in the anakinra- compared with placebo-treated patients.
Phase II clinical trials in patients with heart failure also support a beneficial role of IL-1 blockade with anakinra (Table 2) (139–141).
In additional studies conducted in mice after AMI, selective blockade of IL-1β was explored using a monoclonal IgG2a antibody directed against IL-1β, a mouse equivalent of canakinumab (7, 129). The antibody, given at reperfusion and repeated 1 wk later improved long-term survival after AMI; it inhibited myocardial apoptosis, limiting ventricular enlargement (7), independent of infarct size reduction (113, 132). In a recent study conducted on mice, administration of a polyclonal antibody against IL-1α after I/R injury, on the other hand, limited inflammasome activation, decreased infarct size, and preserved LV function (90). This points to a different role and timing of action of the two isoforms of IL-1, IL-1α and IL-1β. In a recent preclinical study in diabetic rats, administration of gevokizumab, a monoclonal antibody against IL-1β, limited the progression of HF after MI and reduced oxidative stress, ventricular remodeling, scar size, and coronary endothelium-dependent relaxation, independent of infarct size (60, 134).
There have been no clinical trials of selective IL-1β or IL-1α blockade in AMI. In a randomized, double-blind study involving 10,061 patients who had experienced AMI >30 days before enrollment and had serum CRP ≥2 mg/l, canakinumab, a human monoclonal antibody that inhibits IL-1β, given subcutaneously once every 3 mo, decreased the incidence of nonfatal MI, nonfatal stroke, or cardiovascular death at a median followup of 3.7 yr. Administration of 150 mg of canakinumab every 3 mo significantly reduced the incidence of the primary end point (cardiac death, nonfatal MI, and nonfatal stroke) with a hazard ratio of 0.85. This outcome was largely due to a 24% reduction in MI. Interestingly, there was also a 36% reduction in urgent revascularization and a 37% reduction in cardiac arrest (1). In the patients who had a high-sensitivity CRP of <2.0 mg/l within 3 mo after the first dose of canakinumab, incidence of the primary end point was significantly decreased over time, whereas patients who had a high-sensitivity CRP of >2.0 mg/l did not benefit from the treatment. This signifies the importance of identifying specific responders to the anti-inflammatory intervention within the heterogeneous group of patients. Additionally, the decreased incidence of cardiovascular events was independent of lipid levels (111). The effects on AMI-specific outcomes or HF have not been reported. A small single-center substudy of the Canakinumab Anti-Inflammatory Thrombosis Outcome Study in patients with systolic heart failure showed an improvement in peak aerobic exercise capacity in patients treated with canakinumab (135).
BLOCKADE OF IL-18 IN AMI
The role and blockade of IL-18 in myocardial ischemia and MI are less established. IL-18 expression and plasma levels increase after reperfusion in animal models, with myocardial levels peaking at 3 h after reperfusion and serum levels peaking at 6 h (145). Treatment with an IL-18-neutralizing antibody 1 h before I/R significantly decreased infarct size (145). The role of IL-18 has also been investigated in I/R injury in an experimental heart transplantation model. Using syngeneic heterotopic heart transplantation after myocardial ischemia, Gu et al. (59) explored the cardioprotective effects of IL-18BP that prevented IL-18 from binding its receptor. Pretreatment with IL-18BP before ischemia increased graft survival, reduced myocardial damage and leukocyte infiltration, and lowered expression of proinflammatory cytokines (59).
Clinical trials have been completed or are underway to prove the safety of intravenous infusion of GSK1070806, a humanized monoclonal antibody to IL-18, in patients with diabetes mellitus or inflammatory bowel disease (92, 96). A separate recombinant human IL-18BP (tadekinig alfa) phase III trial is underway in patients with NLRC4 macrophage activation syndrome (NLRC4-MAS) mutation or X-linked inhibitor of apoptosis deficiency (136d).
CONCLUSIONS
Myocardial I/R induces a sterile injury and inflammatory response, characterized by the priming and triggering of the NLRP3 inflammasome by locally released DAMPs. Strategies inhibiting the priming, triggering, or activity of the inflammasome or blockade of the inflammasome-related cytokines IL-1β and IL-18 have shown beneficial effects in experimental models of AMI in animals (118, 134). None of the inflammasome inhibitors are approved for clinical use in myocardial I/R syndromes. IL-1 blockers have shown promising results in small studies of patients with STEMI and heart failure (anakinra) and in a large secondary prevention study in patients with prior AMI (canakinumab). NLRP3 inflammasome inhibitors are under development in phase I−II clinical programs (136a, 136b).
DISCLOSURES
S. Toldo has received research grants from Olatec. A. Abbate has received research grants from Novartis and Swedish Orphan Biovitrum and has served as a scientific adviser to Olatec.
AUTHOR CONTRIBUTIONS
S.T., A.G.M., Z.S.C., and A.A. drafted manuscript; S.T., A.G.M., Z.S.C., and A.A. edited and revised manuscript; S.T., A.G.M., Z.S.C., and A.A. approved final version of manuscript; A.G.M. and Z.S.C. prepared figures.
REFERENCES
- 1.Abbate A. Why the CANTOS is a game changer in cardiovascular medicine. J Cardiovasc Pharmacol 70: 353–355, 2017. doi: 10.1097/FJC.0000000000000546. [DOI] [PubMed] [Google Scholar]
- 2.Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW, Oddi C, Carbone S, Trankle CR, Roberts CS, Mueller GH, Gambill ML, Christopher S, Markley R, Vetrovec GW, Dinarello CA, Biondi-Zoccai G. Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). Am J Cardiol 115: 288–292, 2015. doi: 10.1016/j.amjcard.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GGL, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW; VCU-ART Investigators . Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] pilot study). Am J Cardiol 105: 1371–1377.e1, 2010. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 4.Abbate A, Salloum FN, Van Tassell BW, Vecile E, Toldo S, Seropian I, Mezzaroma E, Dobrina A. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PLoS One 6: e27923, 2011. doi: 10.1371/journal.pone.0027923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117: 2670–2683, 2008. doi: 10.1161/CIRCULATIONAHA.107.740233. [DOI] [PubMed] [Google Scholar]
- 6.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol 111: 1394–1400, 2013. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, Salloum FN, Smithson L, Dinarello CA. Interleukin-1β modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail 12: 319–322, 2010. doi: 10.1093/eurjhf/hfq017. [DOI] [PubMed] [Google Scholar]
- 8.Akodad M, Fauconnier J, Sicard P, Huet F, Blandel F, Bourret A, de Santa Barbara P, Aguilhon S, LeGall M, Hugon G, Lacampagne A, Roubille F. Interest of colchicine in the treatment of acute myocardial infarct responsible for heart failure in a mouse model. Int J Cardiol 240: 347–353, 2017. doi: 10.1016/j.ijcard.2017.03.126. [DOI] [PubMed] [Google Scholar]
- 9.Anderson JL, Morrow DA. Acute myocardial infarction. N Engl J Med 376: 2053–2064, 2017. doi: 10.1056/NEJMra1606915. [DOI] [PubMed] [Google Scholar]
- 10.Asea A. Heat shock proteins and Toll-like receptors. Handb Exp Pharmacol 111–127, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Barreiro O, Martín P, González-Amaro R, Sánchez-Madrid F. Molecular cues guiding inflammatory responses. Cardiovasc Res 86: 174–182, 2010. doi: 10.1093/cvr/cvq001. [DOI] [PubMed] [Google Scholar]
- 12.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-κB pathway. Int Immunol 12: 1539–1546, 2000. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 13.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertheloot D, Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol 14: 43–64, 2017. doi: 10.1038/cmi.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81: 1–5, 2007. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 16.Boiselle PM. The Journal of Thoracic Imaging welcomes the European Society of Thoracic Imaging. J Thorac Imaging 26: 2, 2011. doi: 10.1097/RTI.0b013e31820325a0. [DOI] [Google Scholar]
- 17.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182: 3173–3182, 2009. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem 273: 12203–12209, 1998. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 19.Carbone S, Lee PJH, Mauro AG, Mezzaroma E, Buzzetti R, Van Tassell B, Abbate A, Toldo S. Interleukin-18 mediates cardiac dysfunction induced by Western diet independent of obesity and hyperglycemia in the mouse. Nutr Diabetes 7: e258, 2017. doi: 10.1038/nutd.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, Mummidi S. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem 279: 20221–20233, 2004. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 21.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837, 2010. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 278: 36027–36031, 2003. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 23.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling−concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35: 569–582, 2000. doi: 10.1016/S0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 24.Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KHG, Masters SL, Schroder K, Cooper MA, O’Neill LAJ. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255, 2015. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Combes A, Frye CS, Lemster BH, Brooks SS, Watkins SC, Feldman AM, McTiernan CF. Chronic exposure to interleukin 1β induces a delayed and reversible alteration in excitation-contraction coupling of cultured cardiomyocytes. Pflugers Arch 445: 246–256, 2002. doi: 10.1007/s00424-002-0921-y. [DOI] [PubMed] [Google Scholar]
- 26.Crea F, Libby P. Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation 136: 1155–1166, 2017. doi: 10.1161/CIRCULATIONAHA.117.029870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dasgeb B, Kornreich D, McGuinn K, Okon L, Brownell I, Sackett DL. Colchicine: an ancient drug with novel applications. Br J Dermatol 178: 350–356, 2018. doi: 10.1111/bjd.15896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deftereos S, Giannopoulos G, Angelidis C, Alexopoulos N, Filippatos G, Papoutsidakis N, Sianos G, Goudevenos J, Alexopoulos D, Pyrgakis V, Cleman MW, Manolis AS, Tousoulis D, Lekakis J. Anti-inflammatory treatment with colchicine in acute myocardial infarction: a pilot study. Circulation 132: 1395–1403, 2015. doi: 10.1161/CIRCULATIONAHA.115.017611. [DOI] [PubMed] [Google Scholar]
- 29.de Torre-Minguela C, Mesa Del Castillo P, Pelegrín P. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol 8: 43, 2017. doi: 10.3389/fimmu.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood 77: 1627–1652, 1991. [PubMed] [Google Scholar]
- 31.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood 87: 2095–2147, 1996. [PubMed] [Google Scholar]
- 32.Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev 8: 253–265, 1997. doi: 10.1016/S1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- 33.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550, 2009. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 34.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117: 3720–3732, 2011. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Semin Immunol 25: 389–393, 2013. doi: 10.1016/j.smim.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Dinarello CA, van der Meer JWM. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol 25: 469–484, 2013. doi: 10.1016/j.smim.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol 4: 289, 2013. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding WX, Jaeschke H. Autophagy in macrophages regulates the inflammasome and protects against liver injury. J Hepatol 64: 16–18, 2016. doi: 10.1016/j.jhep.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Paolo NC, Shayakhmetov DM. Interleukin 1α and the inflammatory process. Nat Immunol 17: 906–913, 2016. doi: 10.1038/ni.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48: 504–511, 2010. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 proteins. Curr Mol Med 13: 24–57, 2013. doi: 10.2174/156652413804486214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol 174: 7506–7515, 2005. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 43.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16: 663–669, 2004. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 44.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48: 35–44.e6, 2018. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1β: two cytokine substrates for ICE (caspase-1). J Clin Immunol 19: 1–11, 1999. doi: 10.1023/A:1020506300324. [DOI] [PubMed] [Google Scholar]
- 46.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8: 1812–1825, 2006. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 47.Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine 53: 19–28, 2011. doi: 10.1016/j.cyto.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11: 255–265, 2014. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frangogiannis NG. Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol 30: 240–245, 2015. doi: 10.1097/HCO.0000000000000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol 5: 1841–1875, 2015. doi: 10.1002/cphy.c150006. [DOI] [PubMed] [Google Scholar]
- 51.Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med 15: 163–169, 2005. doi: 10.1016/j.tcm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 52.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res 53: 31–47, 2002. doi: 10.1016/S0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 53.Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, Tracey D, Allen H, Lee RT, Kelly RA. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol 35: 685–694, 2003. doi: 10.1016/S0022-2828(03)00113-5. [DOI] [PubMed] [Google Scholar]
- 54.Fujisue K, Sugamura K, Kurokawa H, Matsubara J, Ishii M, Izumiya Y, Kaikita K, Sugiyama S. Colchicine improves survival, left ventricular remodeling, and chronic cardiac function after acute myocardial infarction. Circ J 81: 1174–1182, 2017. doi: 10.1253/circj.CJ-16-0949. [DOI] [PubMed] [Google Scholar]
- 55.Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, Li X, Rolfe A, Sun D, Abbate A, Wang X-Y, Zhang S. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem 61: 5412–5423, 2018. doi: 10.1021/acs.jmedchem.8b00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1β in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol 16: 1000–1006, 1996. doi: 10.1161/01.ATV.16.8.1000. [DOI] [PubMed] [Google Scholar]
- 57.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TBH. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol 13: 246–254, 2012. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 58.Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP-Y. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185: 974–981, 2010. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gu H, Xie M, Xu L, Zheng X, Yang Y, Lv X. The protective role of interleukin-18 binding protein in a murine model of cardiac ischemia/reperfusion injury. Transpl Int 28: 1436–1444, 2015. doi: 10.1111/tri.12683. [DOI] [PubMed] [Google Scholar]
- 60.Harouki N, Nicol L, Remy-Jouet I, Henry JP, Dumesnil A, Lejeune A, Renet S, Golding F, Djerada Z, Wecker D, Bolduc V, Bouly M, Roussel J, Richard V, Mulder P. The IL-1β antibody gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure. JACC Basic Transl Sci 2: 418–430, 2017. doi: 10.1016/j.jacbts.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357, 2016. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191: 5230–5238, 2013. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cohen I, Rider P, Vornov E, Tomas M, Tudor C, Wegner M, Brondani L, Freudenberg M, Mittler G, Ferrando-May E, Dinarello CA, Apte RN, Schneider R. IL-1α is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci Rep 5: 14756, 2015. [Erratum in: Sci Rep 6: 19100, 2016.] doi: 10.1038/srep14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 6: 7360, 2015. doi: 10.1038/ncomms8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39: 311–323, 2013. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanai T, Kamada N, Hisamatsu T. Clinical strategies for the blockade of IL-18 in inflammatory bowel diseases. Curr Drug Targets 14: 1392–1399, 2013. doi: 10.2174/13894501113149990006. [DOI] [PubMed] [Google Scholar]
- 67.Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev 281: 138–153, 2018. doi: 10.1111/imr.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123: 594–604, 2011. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 69.Kenneth NS, Younger JM, Hughes ED, Marcotte D, Barker PA, Saunders TL, Duckett CS. An inactivating caspase 11 passenger mutation originating from the 129 murine strain in mice targeted for c-IAP1. Biochem J 443: 355–359, 2012. doi: 10.1042/BJ20120249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kikuchi K, Poss KD. Cardiac regenerative capacity and mechanisms. Annu Rev Cell Dev Biol 28: 719–741, 2012. doi: 10.1146/annurev-cellbio-101011-155739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim YS, Kim JS, Kwon JS, Jeong MH, Cho JG, Park JC, Kang JC, Ahn Y. BAY 11-7082, a nuclear factor-κB inhibitor, reduces inflammation and apoptosis in a rat cardiac ischemia-reperfusion injury model. Int Heart J 51: 348–353, 2010. doi: 10.1536/ihj.51.348. [DOI] [PubMed] [Google Scholar]
- 72.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science 267: 2000–2003, 1995. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 73.Kullenberg T, Löfqvist M, Leinonen M, Goldbach-Mansky R, Olivecrona H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology (Oxford) 55: 1499–1506, 2016. doi: 10.1093/rheumatology/kew208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuriakose T, Kanneganti TD. Gasdermin D flashes an exit signal for IL-1. Immunity 48: 1–3, 2018. doi: 10.1016/j.immuni.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 75.Laliberte RE, Perregaux DG, Hoth LR, Rosner PJ, Jordan CK, Peese KM, Eggler JF, Dombroski MA, Geoghegan KF, Gabel CA. Glutathione S-transferase-ω1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1β posttranslational processing. J Biol Chem 278: 16567–16578, 2003. doi: 10.1074/jbc.M211596200. [DOI] [PubMed] [Google Scholar]
- 76.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol 187: 61–70, 2009. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411, 2013. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol 1: a001651, 2009. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee J-K, Kim S-H, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL-1β and IL-18. Proc Natl Acad Sci USA 101: 8815–8820, 2004. doi: 10.1073/pnas.0402800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, Zhang S, Li F, Qin L. NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol Immunol 76: 90–97, 2016. doi: 10.1016/j.molimm.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 81.Liu S, Schreur KD. G protein-mediated suppression of L-type Ca2+ current by interleukin-1β in cultured rat ventricular myocytes. Am J Physiol Cell Physiol 268: C339–C349, 1995. doi: 10.1152/ajpcell.1995.268.2.C339. [DOI] [PubMed] [Google Scholar]
- 82.Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, Xin C, Zhu D, Li Y, Yan W, Xiong L, Gao E, Wang H, Tao L. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol 109: 415, 2014. doi: 10.1007/s00395-014-0415-z. [DOI] [PubMed] [Google Scholar]
- 83.Manjili MH, Park JE, Facciponte JG, Wang XY, Subjeck JR. Immunoadjuvant chaperone, GRP170, induces “danger signals” upon interaction with dendritic cells. Immunol Cell Biol 84: 203–208, 2006. doi: 10.1111/j.1440-1711.2006.01418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol 63: 316–322, 2014. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, Carta S, Tengesdal I, Nemkov T, D’Alessandro A, Henry C, Jones GS, Goodrich SA, St. Laurent JP, Jones TM, Scribner CL, Barrow RB, Altman RD, Skouras DB, Gattorno M, Grau V, Janciauskiene S, Rubartelli A, Joosten LAB, Dinarello CA. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA 115: E1530–E1539, 2018. doi: 10.1073/pnas.1716095115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN, Nordio A, Carbone S, Mauro AG, Das A, Zalavadia AA, Halquist MS, Federici M, Van Tassell BW, Zhang S, Abbate A. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J Cardiovasc Pharmacol 66: 1–8, 2015. doi: 10.1097/FJC.0000000000000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241, 2006. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 88.Mastrocola R, Penna C, Tullio F, Femminò S, Nigro D, Chiazza F, Serpe L, Collotta D, Alloatti G, Cocco M, Bertinaria M, Pagliaro P, Aragno M, Collino M. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid Med Cell Longev 2016: 5271251, 2016. doi: 10.1155/2016/5271251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mastroianni N, De Fusco M, Zollo M, Arrigo G, Zuffardi O, Bettinelli A, Ballabio A, Casari G. Molecular cloning, expression pattern, and chromosomal localization of the human Na-Cl thiazide-sensitive cotransporter (SLC12A3). Genomics 35: 486–493, 1996. doi: 10.1006/geno.1996.0388. [DOI] [PubMed] [Google Scholar]
- 90.Mauro AG, Mezzaroma E, Torrado J, Kundur P, Joshi P, Stroud K, Quaini F, Lagrasta CA, Abbate A, Toldo S. Reduction of myocardial ischemia-reperfusion injury by inhibiting interleukin-1α. J Cardiovasc Pharmacol 69: 156–160, 2017. doi: 10.1097/FJC.0000000000000452. [DOI] [PubMed] [Google Scholar]
- 91.Mauro AG, Thurber C, Abbate A. Colchicine in acute myocardial infarction: “teaching new tricks to an old dog”. Transl Med 5: 1000e133, 2015. [Google Scholar]
- 92.McKie EA, Reid JL, Mistry PC, DeWall SL, Abberley L, Ambery PD, Gil-Extremera B. A study to investigate the efficacy and safety of an anti-interleukin-18 monoclonal antibody in the treatment of type 2 diabetes mellitus. PLoS One 11: e0150018, 2016. doi: 10.1371/journal.pone.0150018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mezzaroma E, Toldo S, Abbate A. Role of NLRP3 (cryopyrin) in acute myocardial infarction. Cardiovasc Res 99: 225–226, 2013. doi: 10.1093/cvr/cvt123. [DOI] [PubMed] [Google Scholar]
- 94.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA 108: 19725–19730, 2011. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14: 454–460, 2013. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 96.Mistry P, Reid J, Pouliquen I, McHugh S, Abberley L, DeWall S, Taylor A, Tong X, Rocha Del Cura M, McKie E. Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose antiinterleukin-18 mAb GSK1070806 in healthy and obese subjects. Int J Clin Pharmacol Ther 52: 867–879, 2014. doi: 10.5414/CP202087. [DOI] [PubMed] [Google Scholar]
- 97.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075, 2008. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morton AC, Rothman AMK, Greenwood JP, Gunn J, Chase A, Clarke B, Hall AS, Fox K, Foley C, Banya W, Wang D, Flather MD, Crossman DC. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J 36: 377–384, 2015. doi: 10.1093/eurheartj/ehu272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, Manoharan J, Ranjan S, Bock F, Braun-Dullaeus RC, Esmon CT, Huber TB, Camerer E, Dockendorff C, Griffin JH, Isermann B, Shahzad K. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 130: 2664–2677, 2017. doi: 10.1182/blood-2017-05-782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Netea MG, Joosten LAB, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, Tack CJ, van Krieken H, Kim SH, Stalenhoef AF, van de Loo FA, Verschueren I, Pulawa L, Akira S, Eckel RH, Dinarello CA, van den Berg W, van der Meer JWM. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med 12: 650–656, 2006. doi: 10.1038/nm1415. [DOI] [PubMed] [Google Scholar]
- 101.Netea MG, Nold-Petry CA, Nold MF, Joosten LAB, Opitz B, van der Meer JHM, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JWM, Dinarello CA. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood 113: 2324–2335, 2009. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O’Brien LC, Mezzaroma E, Van Tassell BW, Marchetti C, Carbone S, Abbate A, Toldo S. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol Med 20: 221–229, 2014. doi: 10.2119/molmed.2014.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Park S, Won JH, Hwang I, Hong S, Lee HK, Yu JW. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci Rep 5: 15489, 2015. doi: 10.1038/srep15489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parry GC, Martin T, Felts KA, Cobb RR. IL-1β-induced monocyte chemoattractant protein-1 gene expression in endothelial cells is blocked by proteasome inhibitors. Arterioscler Thromb Vasc Biol 18: 934–940, 1998. doi: 10.1161/01.ATV.18.6.934. [DOI] [PubMed] [Google Scholar]
- 105.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1β. Proc Natl Acad Sci USA 98: 2871–2876, 2001. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One 3: e2119, 2008. doi: 10.1371/journal.pone.0002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1β are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA 96: 2256–2261, 1999. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Raedschelders K, Ansley DM, Chen DD. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol Ther 133: 230–255, 2012. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 109.Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs. duration of coronary occlusion in dogs. Circulation 56: 786–794, 1977. doi: 10.1161/01.CIR.56.5.786. [DOI] [PubMed] [Google Scholar]
- 110.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 187: 4835–4843, 2011. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 111.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group . Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131, 2017. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 112.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol 28: 321–342, 2010. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 132: 1880–1890, 2015. doi: 10.1161/CIRCULATIONAHA.115.016160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res 99: 164–174, 2013. doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 115.Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol 191: 4838–4848, 2013. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schreur KD, Liu S. Involvement of ceramide in inhibitory effect of IL-1β on L-type Ca2+ current in adult rat ventricular myocytes. Am J Physiol Heart Circ Physiol 272: H2591–H2598, 1997. [DOI] [PubMed] [Google Scholar]
- 117.Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol 63: 1593–1603, 2014. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 118.Seta Y, Kanda T, Tanaka T, Arai M, Sekiguchi K, Yokoyama T, Kurimoto M, Tamura J, Kurabayashi M. Interleukin 18 in acute myocardial infarction. Heart 84: 668–669, 2000. doi: 10.1136/heart.84.6.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665, 2015. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 120.Smith MF Jr, Eidlen D, Arend WP, Gutierrez-Hartmann A. LPS-induced expression of the human IL-1 receptor antagonist gene is controlled by multiple interacting promoter elements. J Immunol 153: 3584–3593, 1994. [PubMed] [Google Scholar]
- 121.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153: 348–361, 2013. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation—a two-way street. Mol Med 23: 188–195, 2017. doi: 10.2119/molmed.2017.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann NY Acad Sci 1319: 82–95, 2014. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev 281: 62–73, 2018. doi: 10.1111/imr.12613. [DOI] [PubMed] [Google Scholar]
- 125.Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J 55: 101–105, 2014. doi: 10.1536/ihj.13-388. [DOI] [PubMed] [Google Scholar]
- 126.Tilg H, Mier JW, Vogel W, Aulitzky WE, Wiedermann CJ, Vannier E, Huber C, Dinarello CA. Induction of circulating IL-1 receptor antagonist by IFN treatment. J Immunol 150: 4687–4692, 1993. [PubMed] [Google Scholar]
- 127.Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, Zhang S, Van Tassell B, Salloum FN, Abbate A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol 209: 215–220, 2016. doi: 10.1016/j.ijcard.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 128.Toldo S, Mauro AG, Marchetti C, Mezzaroma E, Van Tassell B, Skouras D, La Neve F, Dinarello C, Abbate A. Novel NLRP3 inflammasome inhibitor OLT1177 reduces infarct size in a mouse model of myocardial ischemia reperfusion injury (Abstract). Circulation 136: 18066, 2017. [Google Scholar]
- 129.Toldo S, Mezzaroma E, Bressi E, Marchetti C, Carbone S, Sonnino C, Van Tassell BW, Abbate A. Interleukin-1β blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J Cardiovasc Pharmacol 64: 1–6, 2014. doi: 10.1097/FJC.0000000000000106. [DOI] [PubMed] [Google Scholar]
- 130.Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal 22: 1146–1161, 2015. doi: 10.1089/ars.2014.5989. [DOI] [PubMed] [Google Scholar]
- 131.Toldo S, Mezzaroma E, McGeough MD, Peña CA, Marchetti C, Sonnino C, Van Tassell BW, Salloum FN, Voelkel NF, Hoffman HM, Abbate A,. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc Res 105: 203–212, 2015. doi: 10.1093/cvr/cvu259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, Abbate A. Interleukin-1β blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol 98: 734–745, 2013. doi: 10.1113/expphysiol.2012.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Toldo S, Schatz AM, Mezzaroma E, Chawla R, Stallard TW, Stallard WC, Jahangiri A, Van Tassell BW, Abbate A. Recombinant human interleukin-1 receptor antagonist provides cardioprotection during myocardial ischemia reperfusion in the mouse. Cardiovasc Drugs Ther 26: 273–276, 2012. doi: 10.1007/s10557-012-6389-x. [DOI] [PubMed] [Google Scholar]
- 134.Toldo S, Van Tassell BW, Abbate A. Interleukin-1 blockade in acute myocardial infarction and heart failure: getting closer and closer. JACC Basic Transl Sci 2: 431–433, 2017. doi: 10.1016/j.jacbts.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Trankle CR, Canada JM, Cei L, Abouzaki N, Oddi-Erdle C, Kadariya D, Christopher S, Viscusi M, Del Buono M, Kontos MC, Arena R, Van Tassell B, Abbate A. Usefulness of canakinumab to improve exercise capacity in patients with long-term systolic heart failure and elevated C-reactive protein. Am J Cardiol 122: 1366–1370, 2018. doi: 10.1016/j.amjcard.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Trankle C, Thurber CJ, Toldo S, Abbate A. Mitochondrial membrane permeability inhibitors in acute myocardial infarction: still awaiting translation. JACC Basic Transl Sci 1: 524–535, 2016. doi: 10.1016/j.jacbts.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136a.United States National Library of Medicine Phase 1, Randomized, Dose Escalation, Single Center, Safety and Pharmacokinetic Study of Single and Multi-Dose, Orally Administered OLT1177 Capsules in Healthy Subjects (Online) https://clinicaltrials.gov/ct2/show/NCT02134964?term=olt1177&rank=2 [18 August 2018].
- 136b.United States National Library of Medicine Phase 2 Efficacy Trial of OLT1177 Gel in Subjects With Moderate to Severe Pain Associated With OA of the Knee (Online) https://clinicaltrials.gov/ct2/show/NCT01768975?term=olt1177&rank=1 [18 August 2018].
- 136c.United States National Library of Medicine Interleukin-1 (IL-1) Blockade in Acute Myocardial Infarction (VCU-ART3) (VCU-ART3) (Online) https://clinicaltrials.gov/ct2/show/NCT01950299 [18 August 2018].
- 136d.United States National Library of Medicine Therapeutic Use of Tadekinig Alfa in NLRC4 Mutation and XIAP Deficiency (Online) https://clinicaltrials.gov/ct2/show/NCT03113760?term=IL-18BP&rank=2 [18 August 2018].
- 137.Valen G, Yan ZQ, Hansson GK. Nuclear factor κB and the heart. J Am Coll Cardiol 38: 307–314, 2001. doi: 10.1016/S0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 138.van Hout GPJ, Bosch L, Ellenbroek GHJM, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SCA, Robertson AAB, Pasterkamp G, Hoefer IE. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J 38: 828–836, 2017. [DOI] [PubMed] [Google Scholar]
- 139.Van Tassell BW, Abouzaki NA, Oddi Erdle C, Carbone S, Trankle CR, Melchior RD, Turlington JS, Thurber CJ, Christopher S, Dixon DL, Fronk DT, Thomas CS, Rose SW, Buckley LF, Dinarello CA, Biondi-Zoccai G, Abbate A. Interleukin-1 blockade in acute decompensated heart failure: a randomized, double-blinded, placebo-controlled pilot study. J Cardiovasc Pharmacol 67: 544–551, 2016. doi: 10.1097/FJC.0000000000000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, Shah K, Canada J, Voelkel NF, Dinarello CA, Abbate A. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One 7: e33438, 2012. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, Abouzaki NA, Dixon D, Kadariya D, Christopher S, Schatz A, Regan J, Viscusi M, Del Buono M, Melchior R, Mankad P, Lu J, Sculthorpe R, Biondi-Zoccai G, Lesnefsky E, Arena R, Abbate A. Interleukin-1 blockade in recently decompensated systolic heart failure: results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail 10: e004373, 2017. doi: 10.1161/CIRCHEARTFAILURE.117.004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Van Tassell BW, Raleigh JM, Abbate A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep 12: 33–41, 2015. doi: 10.1007/s11897-014-0231-7. [DOI] [PubMed] [Google Scholar]
- 143.Van Tassell BW, Varma A, Salloum FN, Das A, Seropian IM, Toldo S, Smithson L, Hoke NN, Chau VQ, Robati R, Abbate A. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J Cardiovasc Pharmacol 55: 117–122, 2010. doi: 10.1097/FJC.0b013e3181c87e53. [DOI] [PubMed] [Google Scholar]
- 144.Vecile E, Dobrina A, Salloum FN, Van Tassell BW, Falcione A, Gustini E, Secchiero S, Crovella S, Sinagra G, Finato N, Nicklin MJ, Abbate A. Intracellular function of interleukin-1 receptor antagonist in ischemic cardiomyocytes. PLoS One 8: e53265, 2013. doi: 10.1371/journal.pone.0053265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem 284: 7853–7865, 2009. doi: 10.1074/jbc.M808824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J, Hu R. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp Cell Res 342: 184–192, 2016. doi: 10.1016/j.yexcr.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 147.Wang J, Sun C, Gerdes N, Liu C, Liao M, Liu J, Shi MA, He A, Zhou Y, Sukhova GK, Chen H, Cheng XW, Kuzuya M, Murohara T, Zhang J, Cheng X, Jiang M, Shull GE, Rogers S, Yang CL, Ke Q, Jelen S, Bindels R, Ellison DH, Jarolim P, Libby P, Shi GP. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat Med 21: 820–826, 2015. doi: 10.1038/nm.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang M, Markel TA, Meldrum DR. Interleukin 18 in the heart. Shock 30: 3–10, 2008. [DOI] [PubMed] [Google Scholar]
- 149.Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, Beck PL, Muruve DA. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol 190: 1239–1249, 2013. doi: 10.4049/jimmunol.1201959. [DOI] [PubMed] [Google Scholar]
- 150.Wu X, He L, Chen F, He X, Cai Y, Zhang G, Yi Q, He M, Luo J. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS One 9: e112891, 2014. doi: 10.1371/journal.pone.0112891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang J, Liu Z, Xiao TS. Post-translational regulation of inflammasomes. Cell Mol Immunol 14: 65–79, 2017. doi: 10.1038/cmi.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrançois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 26: 215–226, 2007. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 357: 1121–1135, 2007. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]