

Abstract
H2S is an endogenous gasotransmitter that increases cerebral blood flow. In the cerebral vascular endothelium, H2S is produced by cystathionine δ-lyase (CSE). Endothelin-1 (ET-1) has constrictor and dilator influences on the cerebral circulation. The mechanism of the vasodilation caused by ET-1 may involve endothelium-derived factors. We hypothesize that ET-1-elicited dilation of pial arterioles requires an elevation of H2S production in the cerebral vascular endothelium. We investigated the effects of ET-1 on CSE-catalyzed brain H2S production and pial arteriolar diameter using cranial windows in newborn pigs in vivo. H2S was measured in periarachnoid cerebrospinal fluid. ET-1 (10−12–10−8 M) caused an elevation of H2S that was reduced by the CSE inhibitors propargylglycine (PPG) and β-cyano-l-alanine (BCA). Low doses of ET-1 (10−12–10−11 M) produced vasodilation of pial arterioles that was blocked PPG and BCA, suggesting the importance of H2S influences. The vasodilator effects of H2S may require activation of smooth muscle cell membrane ATP-sensitive K+ (KATP) channels and large-conductance Ca2+-activated K+ (BK) channels. The KATP inhibitor glibenclamide and the BK inhibitor paxilline blocked CSE/H2S-dependent dilation of pial arterioles to ET-1. In contrast, the vasoconstrictor response of pial arterioles to 10−8 M ET-1 was not modulated by PPG, BCA, glibenclamide, or paxilline and, therefore, was independent of CSE/H2S influences. Pial arteriolar constriction response to higher levels of ET-1 was independent of CSE/H2S and KATP/BKCa channel activation. These data suggest that H2S is an endothelium-derived factor that mediates the vasodilator effects of ET-1 in the cerebral circulation via a mechanism that involves activation of KATP and BK channels in vascular smooth muscle.
NEW & NOTEWORTHY Disorders of the cerebral circulation in newborn infants may lead to lifelong neurological disabilities. We report that vasoactive peptide endothelin-1 exhibits vasodilator properties in the neonatal cerebral circulation by stimulating production of H2S, an endothelium-derived messenger with vasodilator properties. The ability of endothelin-1 to stimulate brain production of H2S may counteract the reduction in cerebral blood flow and prevent the cerebral vascular dysfunction caused by stroke, asphyxia, cerebral hypoxia, ischemia, and vasospasm.
Keywords: ATP-sensitive K+ channels, cerebral blood flow regulation, cranial window, endothelium-derived vasoactive factors, large-conductance Ca2+-activated K+ channels, newborn pigs, pial arterioles
INTRODUCTION
Disorders of the cerebral circulation in newborns are among the leading causes of mortality and of severe morbidities that cause lifelong disabilities (6, 30). The neonatal brain is more susceptible than the adult brain to injury from fluctuations in blood flow because of the rapid development and proliferation of new neurons and vessels (6). Encephalopathy can result from repeated insults after asphyxia and hypoxia-ischemia, to which vascular dysfunction makes an important contribution (5, 6, 30). It is essential to understand the basic physiology of the cerebral circulation to understand these devastating disorders and to find potential solutions.
H2S has been known for centuries to be a toxic substance and environmental hazard. Over the last decade, with the identification of endogenous sulfides in the mammalian brain, H2S has become appreciated as a physiological mediator (10, 12, 14, 15). H2S is endogenously produced in various body systems, including the cardiovascular, neuronal, immune, renal, respiratory, gastrointestinal, reproductive, liver, and endocrine systems (10, 12, 15, 25, 37). H2S affects blood flow, blood pressure, hormone secretion, muscle relaxation, immune responses, and neurotransmission (19, 23–25, 32, 34, 36, 37).
In the brain and cerebral vasculature, H2S is produced from l-cysteine by three major enzymes, including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfur transferase (10, 12, 14). CSE is mainly expressed in the cerebral vascular endothelium, whereas CBS is localized to astrocytes and neurons (12). Both enzymes contribute to the endogenous production of H2S by the brain under physiological and pathological conditions. H2S plays an important role in the regulation of vascular tone. It dilates the cerebral vasculature along with other vascular beds (10, 12, 14, 16, 32, 34, 37). Vascular smooth muscle ATP-sensitive K+ (KATP) channels and large-conductance Ca+-activated K+ (BK) channels play an important role in mediating the dilation response of cerebral arterioles to H2S and hypercapnia (12, 17, 20, 32). Endogenously produced H2S also has cytoprotective properties and could contribute to reduction of brain injury from various causes, including asphyxia and cerebral ischemia (10, 15, 32).
Endothelin-1 (ET-1) is an endogenous 21-amino acid vasoactive peptide produced by the cerebral vascular endothelium (29, 33), but it is also found in neurons, astrocytes, and the choroid plexus (7, 18, 25, 26). ET-1 elicits dose-dependent vasodilator and vasoconstrictor effects via two receptor subtypes (ETA and ETB receptors) expressed in vascular smooth muscle and endothelial cells (4, 9, 11, 18, 21, 24–28, 31) as well as in astrocytes (7). ET-1 is best known as the most potent vasoconstrictor in systemic and cerebral circulation (1, 2, 5, 11, 18, 26, 31). The mechanisms responsible for the vasoconstrictor properties of ET-1 are largely directed at ETA receptors in the vascular smooth muscle and involve a plethora of pathways, including PKC, MAPK, and phosphatidylinositol 3-kinase/PKB-dependent signaling, elevation in intracellular Ca2+, and Ca2+-dependent activation of calmodulin and myosin light chain kinase as well as modulation of ion channels (4, 9, 29, 31, 33, 35). In contrast, the mechanisms by which ET-1 elicits vasodilator effects in the cerebral vasculature and other vascular beds remain largely unexplored. The mechanism of the vasodilation caused by ET-1 may involve endothelium-derived vasodilator factors, prostacyclin, and nitric oxide (NO) during physiological and pathological conditions (1, 2, 5, 9, 11, 28).
We pursued the novel hypothesis that H2S is an endothelium-derived vasoactive factor that mediates the vasodilator effects of ET-1 in the cerebral circulation. We propose that ET-1-elicited dilation of pial arterioles involves an elevation of H2S production in the cerebral vasculature, leading to activation of KATP and BK channels. To test this hypothesis, we investigated the effects of inhibitors of CSE activity and K+ channels on vasodilator and vasoconstrictor responses of pial arterioles to ET-1 in newborn pigs. We concluded that CSE/H2S-mediated activation of vascular KATP and BK channels contributes to the vasodilator but not vasoconstrictor effects of ET-1 in the neonatal cerebral circulation.
MATERIALS AND METHODS
The Animal Care and Use Committee of the University of Tennessee Health Science Center reviewed and approved all procedures involving animals.
Animal study protocol.
Newborn pigs at 1–5 days old and weighing 1.5–3.5 kg were used (n = 50). Pigs were anesthetized with ketamine (33 mg/kg im) and acepromazine (3.3 mg/kg im), intubated via tracheostomies, and placed on mechanical ventilation to maintain blood gases at the normal range for newborn piglets (arterial Pco2: 30–40 mmHg, arterial Po2: 70–90 mmHg, and pH 7.3–7.4), as previously described (12, 13, 22). Femoral artery catheterization was performed for continuous blood pressure monitoring and arterial blood gas sampling. Long-term anesthesia was achieved with α-chloralose (50 mg/kg iv) once femoral vein catheterization was established. Arterial blood gases, blood pressure, and body temperature were measured regularly and maintained within the normal range.
Cranial window installation.
The piglet’s scalp was incised and retracted. The fascia over the skull was scraped off. A 2-cm circular opening was created in the skull over the parietal cortex. The dura was cut and retracted over the cut bone edges. A stainless steel frame with a glass window pane was placed into the hole. The cranial window was sealed with bone wax and cemented into place with dental acrylic. The space under the window was filled with artificial cerebrospinal fluid (aCSF) through needle ports on the sides of the window frame. aCSF was equilibrated with 6% CO2 and 6% O2 to maintain pH (7.35–7.40), arterial Po2 (42–46 mmHg), and arterial Pco2 (42–46 mmHg) in the normal range for CSF. The cranial window allowed direct observation of the cerebral arterioles leading to the capillaries of the brain parenchyma. Arteriolar diameters were measured using a video micrometer connected to a television camera mounted on the microscope and a video monitor. Two to three pial arterioles with diameters of 50 to 70 µm were observed in each piglet. Because the sensitivity of different pial arterioles to various vasoactive stimuli may vary, the data from the different arterioles were pooled for each group.
Pharmacological agents were administered topically to the brain surface through the cranial window ports. aCSF was used as the control agent at the beginning of the experiments and between treatments with different agents to allow the arterioles to return to baseline. Sodium nitroprusside (SNP) and isoproterenol were used as endothelium-independent vasodilators to assess cerebral arteriolar reactivity. SNP (10−6 M), a cGMP-dependent vasodilator, and isoproterenol (10−6 M), a cAMP-dependent vasodilator, act directly on cerebral vascular smooth muscle. Ascending concentrations of ET-1 from 10−12 to 10−8 M were used. ET-1 was stored in ethanol (10−6 M, −20°C) and diluted to concentrations of 10−12−10−8 M in aCSF. SNP and isoproterenol were dissolved in distilled water and diluted to 10−6 M with aCSF. The concentrations of the K+ channel blockers glibenclamide and paxilline were selected on the basis of our previous publications (12, 16, 17, 22). Glibenclamide stock solution in DMSO was diluted with aCSF to 10−7 M. Paxilline stock solution in ethanol was diluted with aCSF to 4 × 10−5 M. dl-propargylglycine (PPG), a selective inhibitor of CSE-catalyzed H2S formation, was dissolved in water and diluted to 5 mM with aCSF. The concentration of PPG was selected on the basis of previous publications by us and other investigators (3, 10, 12, 15, 23, 24). β-Cyano-l-alanine (BCA), another selective CSE inhibitor, was dissolved in ethanol and diluted with aCSF to 10−4 M (3, 15, 24).
Experimental approach.
To investigate dose-dependent effects of ET-1 on pial arteriolar diameter and brain H2S production via a CSE-catalyzed reaction, we used the following three groups of animals: 1) a control group (ET-1 was diluted with aCSF, n = 9), 2) a BCA-treated group (ET-1 was diluted with 0.1 mM BCA, n = 95), and 3) a PPG-treated group (ET-1 was diluted with 5 mM PPG, n = 7). For control measurements of baseline arteriolar diameter and H2S production, the brain surface was exposed for 15 min to aCSF, 0.1 mM BCA, or 5 mM PPG, respectively. ET-1 at ascending concentrations (10−12–10−8 M) dissolved in aCSF, BCA, or PPG, respectively, was placed under the cranial window. After a 10-min exposure to each concentration, the diameter of the pial arterioles was recorded, and the sample of periarachnoid CSF (pCSF) (0.5 ml) was collected through the port of the cranial window for further H2S measurement.
The effects of K+ channel inhibitors on cerebral vascular responses to ET-1 were investigated using a repeated-measures protocol. After control pial arteriolar responses to ET-1 (10−12–10−8 M) were recorded as described above, the brain surface under the cranial window was flushed with aCSF for 30 min to allow pial arterioles to return to baseline diameter. Paxilline (4 × 10−5 M) and/or glibenclamide (10−7 M) were topically applied to the brain for 30 min, and the responses to ET-1 (10−12–10−8 M), combined with the corresponding inhibitor, were recorded. Each animal received one control treatment and one experimental treatment with ET-1 and the channel inhibitor (n = 5 pigs/treatment).
Localization of CSE and CBS in the cerebral cortex.
Immunohistochemistry was performed on slices of the formalin-fixed/paraffin-embedded newborn pig brain cortex using the avidin-biotin-peroxidase complex technique with monoclonal antihuman antibodies highly selective to CSE or CBS (Novus Biologicals), as we have previously described (13). Brain sections were counterstained with hematoxylin and viewed with an Olympus BX50 light microscope.
Measurement of H2S.
H2S levels in pCSF were measured using a H2S-selective electrode (Lazar Research Laboratories) on a Jenko model 6230 microcomputer-based pH/mV/Temp meter (Jenko Electronics), as previously described elsewhere (8, 19, 37). pCSF samples were collected in 500-µl tubes after being under the cranial window for 10 min. Collections of samples from under the cranial windows was accomplished as we have previously described (12, 13, 17). NaOH was added to pCSF samples (final concentration: 40 mM) to keep samples under alkali conditions and enable measurements of all biological forms of unbound H2S-derived species in solution. Na2S solution in distilled water (0.5–10 µM) was used for a calibration curve, as suggested by the manufacturer (Jenko). Using the standard calibration curve, H2S sample readings in millivolts were normalized to baseline and expressed in micromoles. The H2S detection limit was 0.4 ± 0.2 µM.
Statistical analysis.
Data are presented as means ± SE. Comparisons among populations within each experimental group were performed with repeated-measures ANOVA. P values of <0.05 were considered statistically significant.
Materials.
Endothelin-1, dl-propargylglycine, BCA, SNP, isoproterenol, Na2S, and glibenclamide were purchased from Sigma. Paxilline was obtained from Tocris Bioscience. H2S gas (99.5%) was obtained from NexAir.
RESULTS
Immunohistochemistry of H2S-producing enzymes in the newborn brain cortex showed high expression of CSE in pial arterioles and penetrating vessels, whereas CBS was detected mainly in cortical astrocytes and neurons (Fig. 1A). Under baseline conditions, the H2S level in pCSF was 1.1 ± 0.2 µM (n = 9 pigs). Topical ET-1 (10−12–10−8 M) caused an increase in the H2S level three- to fourfold above the baseline level (P < 0.05; Fig. 1B). To investigate whether the ET-1-evoked H2S response occurs via CSE activation, we used the selective CSE inhibitors BCA (0.1 mM) and PPG (5 mM). When placed topically under the cranial window, both inhibitors greatly attenuated (BCA) or completely blocked (PPG) the H2S elevation in response to ET-1 (P < 0.05; Fig. 1B). These data suggest that CSE activation is a main contributor to the ET-1-evoked H2S production in the cerebral microcirculation.
Fig. 1.
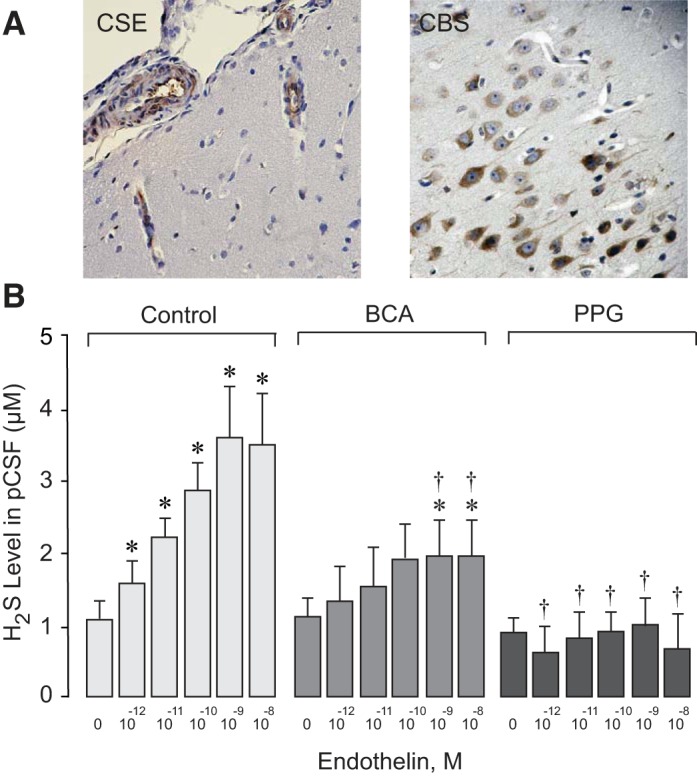
A: immunohistochemistry of cystathionine δ-lyase (CSE) and cystathionine β-synthase (CBS) in the newborn pig cerebral cortex by the avidin-biotin-peroxidase complex technique. CSE-positive staining (brown) was revealed in pial arterioles and in penetrating cerebral vessels. CBS-positive staining (brown) was revealed in cortical astrocytes and neurons. Hematoxylin was used as a counterstain. B: effects of endothelin-1 (ET-1) on CSE-catalyzed H2S production by the brain. ET-1 in increasing concentrations (10−12–10−8 M) was applied to the brain surface in the three following groups of newborn pigs: 1) control group (n = 9 piglets), 2) β-cyano-l-alanine-treated group (BCA; 0.1 mM, n = 9 piglets), and 3) propargylglycine-treated group (PPG; 5 mM, n = 7 piglets). pCSF was sampled after a 10-min exposure to each concentration of ET-1, and the H2S concentration was measured by a sulfide-sensitive electrode. Values are expressed as means ± SE. *P < 0.05 compared with baseline; †P < 0.05 compared with the corresponding control values.
Topical ET-1 caused concentration-dependent changes in pial arteriolar diameters (Fig. 2). Pial arterioles dilated 12–15% above the baseline diameter in response to lower concentrations of ET-1 (10−12–10−11 M), whereas a higher concentration of ET-1 (10−8 M) caused 25–30% vasoconstriction. H2S has a potent vasodilator effect in the newborn cerebral circulation (12, 16, 17). We investigated whether H2S produced via CSE activity contributes to cerebral vasodilation in response to ET-1. After topical treatment with BCA and PPG, the pial arteriolar dilation response to ET-1 (10−12–10−11 M) was completely abolished (Fig. 2). In contrast, BCA and PPG had no effect on the vasoconstrictor effects of ET-1 (10−8 M). These data suggest that CSE-catalyzed H2S generation selectively contributes to the vasodilation of pial arterioles in response to low doses of ET-1. In contrast, we found no evidence that CSE-derived H2S contributes to the vasoconstriction of pial arterioles caused by high doses of ET-1.
Fig. 2.
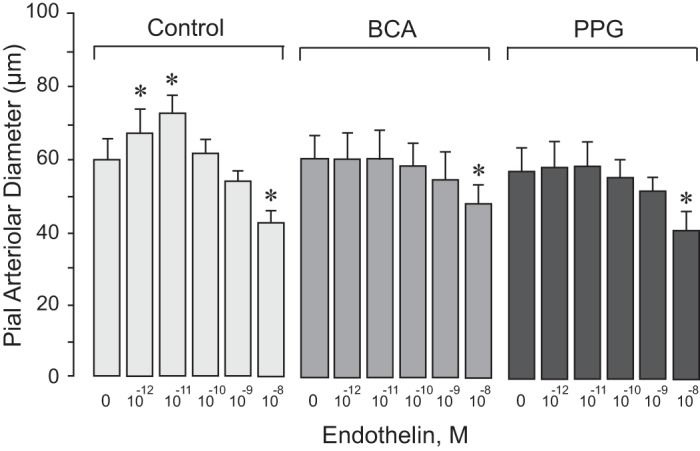
Cystathionine δ-lyase (CSE) inhibitors selectively block the vasodilator response of pial arterioles to endothelin-1 (ET-1). ET-1 in increasing concentrations (10−12–10−8 M) was applied to the brain surface in the following three groups of newborn pigs: 1) control group (n = 9 piglets); 2) β-cyano-l-alanine-treated group (BCA; 0.1 mM, n = 9 piglets); and 3) propargylglycine-treated group (PPG; 5 mM, n = 7 piglets). Values are expressed as means ± SE. *P < 0.05 compared with baseline.
Previously, we demonstrated that the vasodilator effects of H2S require activation of smooth muscle cell membrane KATP and BK channels. We addressed the hypothesis that K+ channels are important mediators of the vasoactive properties of ET-1 in the newborn cerebral circulation. Glibenclamide, a KATP channel inhibitor, and paxilline, a BK channel inhibitor, given alone or together, completely blocked the vasodilator response of pial arterioles to ET-1, whereas the vasoconstrictor response to high doses of ET-1 was not affected by the inhibitors (Fig. 3). Glibenclamide and paxilline had no inhibitory effect on endothelium-independent vasodilator responses of pial arterioles to SNP and isoproterenol (Fig. 4), suggesting that K+ channel inhibitors have selective effects only on endothelium-mediated responses to ET-1. These data demonstrate that both KATP and BK channels are selectively involved in vasodilator responses of pial arterioles to ET-1 that are mediated by endogenously produced H2S.
Fig. 3.
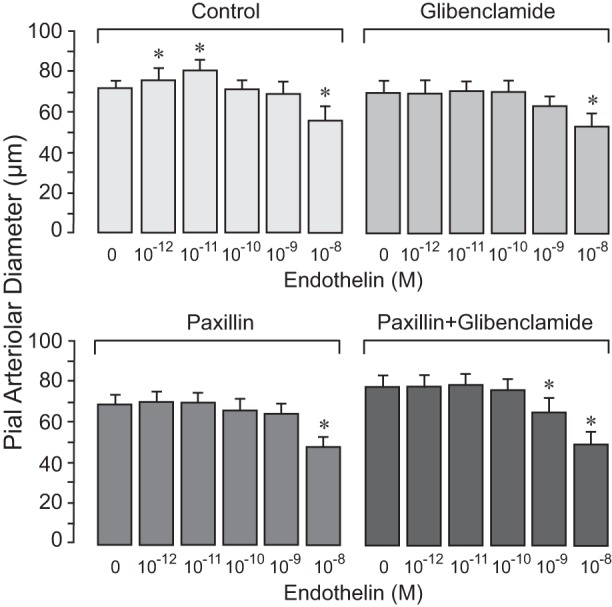
K+ channel inhibitors selectively block the vasodilator response of pial arterioles to endothelin-1 (ET-1). Responses of pial arterioles to ET-1 (10−12–10−8 M) were repeatedly tested in the same animals before (control; n = 10 piglets) and after treatment with the following inhibitors: the ATP-sensitive K+ channel inhibitor glibenclamide (10−7 M) and the large-conductance Ca+-activated K+ channel inhibitor paxilline (4 × 10−5 M), applied alone or in combination (n = 5 piglets/treatment). Values are expressed as means ± SE. *P < 0.05 compared with the corresponding baseline diameter.
Fig. 4.
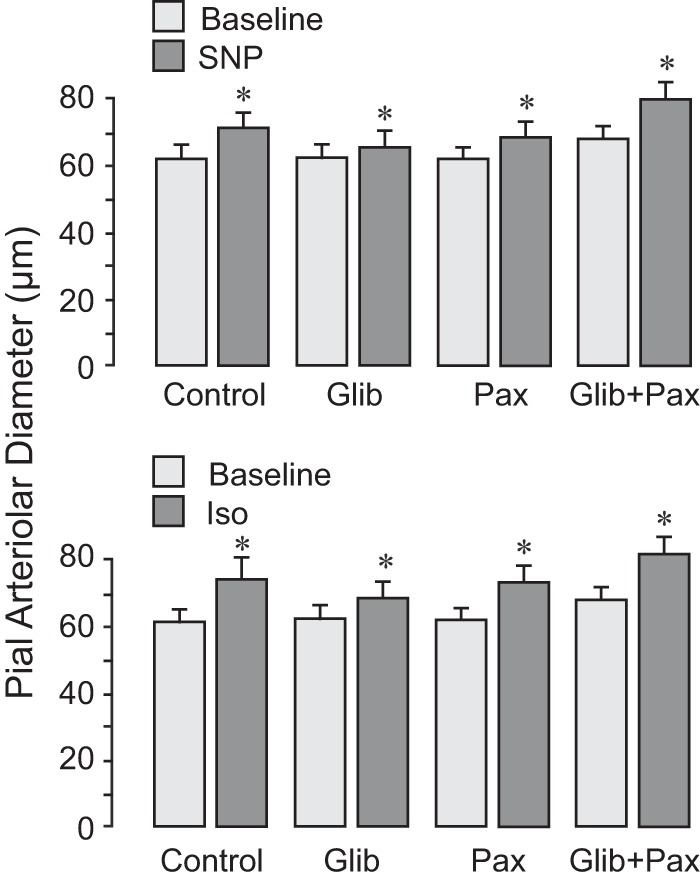
K+ channel inhibitors do not alter vasodilator responses of pial arterioles to the smooth muscle-targeting vasodilators sodium nitroprusside (SNP) and isoproterenol (Iso). Responses of pial arterioles to SNP (10−6 M; top) and Iso (10−6 M; bottom) were repeatedly tested in the same animals before (control; n = 10 piglets) and after treatment with the following inhibitors: the ATP-sensitive K+ channel inhibitor glibenclamide (10−7 M) and the large-conductance Ca+-activated K+ channel inhibitor paxilline (4 × 10−5 M), applied alone or in combination (n = 5 piglets/treatment). Values are expressed as means ± SE. *P < 0.05 compared with the corresponding baseline diameter.
DISCUSSION
We provide evidence that H2S produced via CSE activity is an endothelium-derived factor that mediates vasodilator effects of ET-1 in the cerebral circulation via a mechanism that involves activation of vascular smooth muscle KATP and BK channels. This is supported by the following new findings in newborn pigs: 1) ET-1 increases H2S production by the brain via CSE activation, 2) the vasodilator effect of ET-1 requires CSE-catalyzed production of endothelium-derived H2S and involvement of vascular smooth muscle KATP and BK channels, and 3) vasoconstrictor effects of higher ET-1 concentrations are independent of H2S production via CSE and do not involve KATP and BK channels.
Endogenous H2S production from l-cysteine is catalyzed by two major enzymes: CSE and CBS. In the newborn brain, CSE is expressed in pial arterioles and penetrating cerebral vessels and is localized to the vascular endothelium. CBS has been detected in the brain parenchyma, including astrocytes and neurons (10, 12, 15). Both CSE and CBS contribute to brain H2S production that leads to H2S accumulation in pCSF that bathes the brain surface. The concentration of H2S detected in tissues and fluids depends upon the methods used for measurements. We have experience using two alternative approaches for the detection of H2S in biological liquids. The H2S-selective electrode technique is accepted by experts in H2S physiology as an efficient approach to measurements of all biological forms of unbound H2S in biological fluids (19, 34). Using this approach, Yang et al. (34) detected a basal H2S concentration of 4 μM in serum. We measured a mean baseline H2S concentration of 1.1 ± 0.2 μM in piglet pCSF using an electrochemical detection method. This value is comparable with the baseline pCSF H2S concentration of 0.5–1.0 μM detected by a GC-MS method (12). GC-MS detects gaseous H2S, whereas electrochemical methods measure H2S-derived sulfur ions in liquid.
At low concentrations (10−12–10−11 M), ET-1 caused vasodilation of cerebral arterioles that was concomitant with activation of CSE-dependent H2S production in the cerebral vasculature. BCA and PPG, selective inhibitors of CSE, largely reduced brain H2S production and completely blocked the vasodilator response to ET-1. Previously, we demonstrated that H2S dilates piglet cerebral arterioles via the activation of both BK and KATP channels, activating Ca2+ sparks in cerebral arteriole vascular smooth muscle and producing membrane hyperpolarization (17). Now, we present data suggesting that KATP and BK channels are involved in the CSE/H2S-mediated mechanism of cerebral vasodilation in response to low doses of ET-1. Inhibitors of BK and KATP channels (paxilline and glibenclamide), alone or combined, blocked the dilation of pial arterioles to ET-1. Overall, ET-1 stimulates the activity of endothelial CSE and upregulates the production of an endothelium-derived mediator H2S that activates BK and KATP channels, thus producing vasodilation. As we have previously demonstrated, the interaction between CSE (but not CBS)-derived H2S and BK/KATP channels is also involved in the mechanism of the cerebral vasodilation response to hypercapnia (12). It appears that ET-1 produces vasodilation of cerebral arterioles by activating a functional cerebrovascular unit composed of endothelial CSE localized in proximity to H2S-sensitive BK and/or KATP channels in vascular smooth muscle. Current evidence demonstrates that pleiotropic physiological properties of H2S are based on its ability to modify various proteins via S-sulfhydration on cysteine-SH residues, thus affecting a variety of biological pathways (19, 20, 25). The mechanism of H2S-mediated posttranslational modulation of ion channel properties may involve eliciting S-sulfhydration of cysteine-rich K+ channels (20).
The vasoconstrictor effect of ET-1 has been implicated in the cerebrovascular dysfunction occurring in stroke, subarachnoid hemorrhage, chronic intermittent hypoxia, and brain trauma (1, 5, 9, 11, 18, 23, 26, 27, 31). It is generally accepted that the mechanisms of vasoconstrictor effects of ET-1 involves smooth muscle ETA receptors that initiate phosphorylation-dependent signaling, including PKC, MAPK, and phosphatidylinositol 3-kinase/PKB pathways, and also Ca2+-dependent activation of calmodulin and myosin light chain kinase (4, 9, 29, 31, 33, 35). ET-1 also acts on BK channels via a dual mechanism that produces the channel opening via initiating Ca2+ sparks and BK-mediated vasodilation (17) or BK channel blockade leading to vasoconstriction (35). A recently described mechanism of the vasoconstrictor effect of ET-1 in rat cerebral arterioles involves PKC activation, leading to phosphorylation-mediated inhibition of myocyte BK channels (35). The PKC-mediated vasoconstrictor response to ET-1 involves inhibition of surface trafficking of the BK channel β1-subunits and a reduction in the Ca2+ sensitivity of the channel (35). We provide evidence that the pial arteriolar constriction response to ET-1 is independent of CSE/H2S and KATP/BK channel activation. Although ET-1 at high doses continuously increased brain H2S production, pial arteriolar constriction was not affected by the CSE inhibitors glibenclamide and paxilline, indicating no functional relationships between CSE activation, K+ channels, and vascular contraction.
We cannot exclude the possibility that ET-1 also stimulates CBS activity that is highly expressed in cortical astrocytes and neurons. The insufficient selectivity of currently available CBS inhibitors (3) does not allow testing this hypothesis in vivo. We conducted preliminary experiments (data not shown) using the CBS inhibitor aminooxyacetic acid. Aminooxyacetic acid effectively reduced ET-1-elicited H2S elevation in pCSF but did not block vasodilator or vasoconstrictor influences of ET-1. These findings suggest that CBS-derived H2S is not involved in vasoactive effects of ET-1 in the cerebral circulation.
Vasoactive effects of ET-1 in cerebral circulation are mediated via two main subtypes of receptors, ETA and ETB, localized in neurovascular cells. ETA receptors localized in vascular smooth muscle mediate vascular contraction and may contribute to ET-1-induced cerebral vasospasm caused by subarachnoid hemorrhage and traumatic brain injury (1, 9, 18). The contribution of ETA receptors to cerebrovascular dysfunction has also been described in the model of chronic intermittent hypoxia (5). Vasodilator effects of ET-1 appear to involve ETB receptors in endothelial cells that trigger enhanced production of the endothelium-derived vasorelaxants nitric oxide and prostacyclin (1, 2, 9, 11, 28). We speculate that the H2S-mediated vasodilator effect of ET-1 in the neonatal cerebral circulation may involve targeting ETB receptors in endothelial cells. An interplay between distinct endothelium-derived vasorelaxants H2S, prostanoids, and nitric oxide as mediators of the vasodilator response to ET-1 cannot be excluded.
Overall, we provide evidence that H2S is an endothelium-derived factor that mediates vasodilator effects of ET-1 in the cerebral circulation via a mechanism that involves activation of vascular smooth muscle KATP and BK channels. ET-1 as a promising endogenous activator of cerebrovascular H2S may be important for neuroprotection in certain pathological conditions in neonates. The ability of ET-1 to stimulate brain production of H2S, a potent endogenous vasodilator and cytoprotective mediator, may counteract reduction in cerebral blood flow and prevent cerebral vascular dysfunction and encephalopathy caused by stroke, asphyxia, cerebral hypoxia, ischemia, and vasospasm. Further studies evaluating the role of ET-1 in the setting of hypoxic-ischemic injury and other cerebrovascular disease will be important.
GRANTS
This work was supported by National Institutes of Health Grants RO1-HL-034059 (to C. W. Leffler), RO1-HL-42851 (to C. W. Leffler), and RO1-NS-101717 (to H. Parfenova).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.P., M.A.W., M.P., M.H., H.P., and C.W.L. conceived and designed research; S.P., A.L.F., J.L., and M.A.W. performed experiments; S.P., J.L., M.P., M.H., H.P., and C.W.L. analyzed data; S.P., H.P., and C.W.L. drafted manuscript; A.L.F. and H.P. prepared figures; J.L., M.A.W., M.P., M.H., H.P., and C.W.L. approved final version of manuscript; M.A.W. and C.W.L. interpreted results of experiments; H.P. and C.W.L. edited and revised manuscript.
REFERENCES
- 1.Armstead WM. Role of endothelin in pial artery vasoconstriction and altered responses to vasopressin after brain injury. J Neurosurg 85: 901–907, 1996. doi: 10.3171/jns.1996.85.5.0901. [DOI] [PubMed] [Google Scholar]
- 2.Armstead WM, Mirro R, Leffler CW, Busija DW. Influence of endothelin on piglet cerebral microcirculation. Am J Physiol Heart Circ Physiol 257: H707–H710, 1989. doi: 10.1152/ajpheart.1989.257.2.H707. [DOI] [PubMed] [Google Scholar]
- 3.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol 169: 922–932, 2013. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouallegue A, Daou GB, Srivastava AK. Endothelin-1-induced signaling pathways in vascular smooth muscle cells. Curr Vasc Pharmacol 5: 45–52, 2007. doi: 10.2174/157016107779317161. [DOI] [PubMed] [Google Scholar]
- 5.Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J, Davisson RL, Iadecola C. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension 60: 106–113, 2012. doi: 10.1161/HYPERTENSIONAHA.112.193672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groenendaal F, De Vries LS. Hypoxic-ischemic Encephalopathy. In: Fanaroff & Martin’s Neonatal-Perinatal Medicine: Diseases of the Fetus and Infant, edited by Martin RJ, Fanaroff AA, Walsh MC. Philadelphia, PA: Saunders, 2015, chapt. 6, p. 907–921. [Google Scholar]
- 7.Hostenbach S, D’haeseleer M, Kooijman R, De Keyser J. The pathophysiological role of astrocytic endothelin-1. Prog Neurobiol 144: 88–102, 2016. doi: 10.1016/j.pneurobio.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Hughes MN, Centelles MN, Moore KP. Making and working with hydrogen sulfide: The chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic Biol Med 47: 1346–1353, 2009. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Kasemsri T, Armstead WM. Endothelin impairs ATP-sensitive K+ channel function after brain injury. Am J Physiol Heart Circ Physiol 273: H2639–H2647, 1997. doi: 10.1152/ajpheart.1997.273.6.H2639. [DOI] [PubMed] [Google Scholar]
- 10.Kimura H. The physiological role of hydrogen sulfide and beyond. Nitric Oxide 41: 4–10, 2014. doi: 10.1016/j.niox.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Kitazono T, Heistad DD, Faraci FM. Dilatation of the basilar artery in response to selective activation of endothelin B receptors in vivo. J Pharmacol Exp Ther 273: 1–6, 1995. [PubMed] [Google Scholar]
- 12.Leffler CW, Parfenova H, Basuroy S, Jaggar JH, Umstot ES, Fedinec AL. Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol 300: H440–H447, 2011. doi: 10.1152/ajpheart.00722.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leffler CW, Parfenova H, Fedinec AL, Basuroy S, Tcheranova D. Contributions of astrocytes and CO to pial arteriolar dilation to glutamate in newborn pigs. Am J Physiol Heart Circ Physiol 291: H2897–H2904, 2006. doi: 10.1152/ajpheart.00722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: gaseous messengers in cerebrovascular circulation. J Appl Physiol 100: 1065–1076, 2006. doi: 10.1152/japplphysiol.00793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci 29: 84–90, 2008. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Liang GH, Adebiyi A, Leo MD, McNally EM, Leffler CW, Jaggar JH. Hydrogen sulfide dilates cerebral arterioles by activating smooth muscle cell plasma membrane KATP channels. Am J Physiol Heart Circ Physiol 300: H2088–H2095, 2011. doi: 10.1152/ajpheart.01290.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang GH, Xi Q, Leffler CW, Jaggar JH. Hydrogen sulfide activates Ca2+ sparks to induce cerebral arteriole dilatation. J Physiol 590: 2709–2720, 2012. doi: 10.1113/jphysiol.2011.225128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maguire JJ, Davenport AP. Endothelin@25 - new agonists, antagonists, inhibitors and emerging research frontiers: IUPHAR Review 12. Br J Pharmacol 171: 5555–5572, 2014. doi: 10.1111/bph.12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal 2: ra72, 2009. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268, 2011. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson D, Wackenfors A, Gustafsson L, Ugander M, Paulsson P, Ingemansson R, Edvinsson L, Malmsjö M. Endothelin receptor-mediated vasodilatation: effects of organ culture. Eur J Pharmacol 579: 233–240, 2008. doi: 10.1016/j.ejphar.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 22.Nnorom CC, Davis C, Fedinec AL, Howell K, Jaggar JH, Parfenova H, Pourcyrous M, Leffler CW. Contributions of KATP and KCa channels to cerebral arteriolar dilation to hypercapnia in neonatal brain. Physiol Rep 2: e12127, 2014. doi: 10.14814/phy2.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, Madden JA. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol 209: 4011–4023, 2006. doi: 10.1242/jeb.02480. [DOI] [PubMed] [Google Scholar]
- 24.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabó C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA 106: 21972–21977, 2009. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paul BD, Snyder SH. H2S: A novel gasotransmitter that signals by sulfhydration. Trends Biochem Sci 40: 687–700, 2015. doi: 10.1016/j.tibs.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodríguez-Pascual F, Busnadiego O, Lagares D, Lamas S. Role of endothelin in the cardiovascular system. Pharmacol Res 63: 463–472, 2011. doi: 10.1016/j.phrs.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Silpanisong J, Kim D, Williams JM, Adeoye OO, Thorpe RB, Pearce WJ. Chronic hypoxia alters fetal cerebrovascular responses to endothelin-1. Am J Physiol Cell Physiol 313: C207–C218, 2017. doi: 10.1152/ajpcell.00241.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tirapelli CR, Casolari DA, Yogi A, Montezano AC, Tostes RC, Legros E, D’Orléans-Juste P, de Oliveira AM. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K+ channels in ETB-induced relaxation. Br J Pharmacol 146: 903–912, 2005. doi: 10.1038/sj.bjp.0706388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorin E, Webb DJ. Endothelium-derived endothelin-1. Pflügers Arch 459: 951–958, 2010. doi: 10.1007/s00424-009-0763-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuor UI, Del Bigio MR, Chumas PD. Brain damage due to cerebral hypoxia/ischemia in the neonate: pathology and pharmacological modification. Cerebrovasc Brain Metab Rev 8: 159–193, 1996. [PubMed] [Google Scholar]
- 31.Vignon-Zellweger N, Heiden S, Miyauchi T, Emoto N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci 91: 490–500, 2012. doi: 10.1016/j.lfs.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 32.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92: 791–896, 2012. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 33.Yakubu MA, Leffler CW. Regulation of ET-1 biosynthesis in cerebral microvascular endothelial cells by vasoactive agents and PKC. Am J Physiol Cell Physiol 276: C300–C305, 1999. doi: 10.1152/ajpcell.1999.276.2.C300. [DOI] [PubMed] [Google Scholar]
- 34.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322: 587–590, 2008. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhai X, Leo MD, Jaggar JH. Endothelin-1 stimulates vasoconstriction through Rab11A serine 177 phosphorylation. Circ Res 121: 650–661, 2017. doi: 10.1161/CIRCRESAHA.117.311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang LM, Jiang CX, Liu DW. Hydrogen sulfide attenuates neuronal injury induced by vascular dementia via inhibiting apoptosis in rats. Neurochem Res 34: 1984–1992, 2009. doi: 10.1007/s11064-009-0006-9. [DOI] [PubMed] [Google Scholar]
- 37.Zhao W, Wang R. H2S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 283: H474–H480, 2002. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]