

Abstract
BACKGROUND
Esophageal adenocarcinoma (EAC) is an aggressive disease with high mortality and an overall 5-year survival rate of less than 20%. Barrett’s esophagus (BE) is the only known precursor of EAC, and patients with BE have a persistent and excessive risk of EAC over time. Individuals with BE are up to 30-125 times more likely to develop EAC than the general population. Thus, early detection of EAC and BE could significantly improve the 5-year survival rate of EAC. Due to the limitations of endoscopic surveillance and the lack of clinical risk stratification strategies, molecular biomarkers should be considered and thoroughly investigated.
AIM
To explore the transcriptome changes in the progression from normal esophagus (NE) to BE and EAC.
METHODS
Two datasets from the Gene Expression Omnibus (GEO) in NCBI Database (https://www.ncbi.nlm.nih.gov/geo/) were retrieved and used as a training and a test dataset separately, since NE, BE, and EAC samples were included and the sample sizes were adequate. This study identified differentially expressed genes (DEGs) using the R/Bioconductor project and constructed trans-regulatory networks based on the Transcriptional Regulatory Element Database and Cytoscape software. Enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) terms was identified using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources. The diagnostic potential of certain DEGs was assessed in both datasets.
RESULTS
In the GSE1420 dataset, the number of up-regulated DEGs was larger than that of down-regulated DEGs when comparing EAC vs NE and BE vs NE. Among these DEGs, five differentially expressed transcription factors (DETFs) displayed the same trend in expression across all the comparison groups. Of these five DETFs, E2F3, FOXA2, and HOXB7 were up-regulated, while PAX9 and TFAP2C were down-regulated. Additionally, the majority of the DEGs in trans-regulatory networks were up-regulated. The intersection of these potential DEGs displayed the same direction of changes in expression when comparing the DEGs in the GSE26886 dataset to the DEGs in trans-regulatory networks above. The receiver operating characteristic curve analysis was performed for both datasets and found that TIMP1 and COL1A1 could discriminate EAC from NE tissue, while REG1A, MMP1, and CA2 could distinguish BE from NE tissue. DAVID annotation indicated that COL1A1 and MMP1 could be potent biomarkers for EAC and BE, respectively, since they participate in the majority of the enriched KEGG and GO terms that are important for inflammation and cancer.
CONCLUSION
After the construction and analyses of the trans-regulatory networks in EAC and BE, the results indicate that COL1A1 and MMP1 could be potential biomarkers for EAC and BE, respectively.
Keywords: Esophageal adenocarcinoma, Differentially expressed genes, Barrett’s esophagus, Transcription factors, Microarray
Core tip: We did comprehensive bioinformatics analyses to identify the differentially expressed genes in Barrett’s esophagus (BE) and esophageal adenocarcinoma (EAC) samples from two Gene Expression Omnibus datasets, and certain potential biomarkers were revealed in the progression of BE and EAC. After trans-regulatory network prediction, receiver operating characteristic curve evaluation and Database for Annotation, Visualization, and Integrated Discovery annotation, an association between COL1A1 and EAC was found. Similarly, an association between MMP1 and BE was also predicted. Our study provided a novel perspective on the molecular mechanisms involved in the development of BE and EAC.
INTRODUCTION
According to the Montreal definition and classification[1], Barrett’s esophagus (BE) is defined as a specialized intestinal metaplasia condition in the distal esophagus and may progress to low-grade dysplasia, high-grade dysplasia, and esophageal adenocarcinoma (EAC), which is a chronic inflammatory process[2,3]. BE is the only recognized precursor of EAC, and BE patients have a persistent and excessive risk of EAC over time[4]. Individuals with BE are 30-125 times more likely to develop EAC than the general population, and almost 50% of EAC patients progressed from BE[5,6]. The prevalence of EAC has continuously increased over the past few decades with 450000 new cases in 2008 alone[7,8]. EAC is an aggressive disease with high mortality and an overall 5-year survival rate of less than 20%[9]. Thus, early detection of EAC and BE could significantly improve the 5-year survival rate of EAC, and additional studies of pathogenesis and carcinogenesis of EAC and BE are urgently required[2,6].
Due to the limitations of endoscopic surveillance and the lack of clinical risk stratification strategies available to identify BE patients who are at a higher risk of progressing to EAC, more attention has been paid to improving endoscopic techniques and to the discovery of molecular biomarkers for EAC and BE[10,11]. Panels of markers have been developed and used to explore the relationships among normal esophagus (NE), BE, and EAC, yet the molecular drivers are still not clear, and no biomarkers are currently utilized for clinical application[11]. Recently, gene profiling analysis has been introduced to explore the molecular changes in EAC and BE , however, there are no consistent conclusions among these different studies[12-14].
In this study, we aimed to explore the changes in the transcriptome, and to determine the differentially expressed genes and their roles in the multistep morphological progression of BE and EAC based on two Gene Expression Omnibus (GEO) datasets. We screened for potential genes and determined their diagnostic value using bioinformatics analyses that were based on several tools. Using this approach, a number of potent genes and their underlying mechanisms could be identified and clarified.
MATERIALS AND METHODS
Dataset retrieval from the GEO database
The gene expression profile datasets of EAC and BE were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), and the GSE1420 and GSE26886 datasets were selected for further analyses. The GSE1420 dataset consisted of 8 NE, 8 BE, and 8 EAC samples[3]. The samples were obtained from 8 EAC patients after transhiatal esophagectomy. The GSE26886 test dataset included 19 NE, 20 BE, and 21 EAC samples.
Identification of differentially expressed genes (DEGs)
Data analysis was conducted using the R/Bioconductor project[15]. The AffyPLM package and the gcRMA algorithm were used for data preprocessing including background correction, normalization, and summarization. The Limma package was used to identify the DEGs between any two groups. Genes with a fold change (FC) of at least 1.5 and a P-value < 0.05 were considered DEGs.
Prediction and analysis of differentially expressed transcription factors (DETFs)
The transcription factor (TF)-gene regulation modes of 36 cancer-related TF families were obtained from the Transcriptional Regulatory Element Database (TRED) (http://rulai.cshl.edu/TRED)[16], including more than 170 cancer-related TFs. We then integrated the DEGs with the TF-gene regulation modes and constructed the dys-regulated TF-DEGs networks. The trans-regulatory networks were visualized with the help of the Cytoscape software (v3.6.0, National Institute of General Medical Science, Bethesda, MD, United States)[17].
Functional annotation of DEGs
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources (https://david.ncifcrf.gov/) was used to provide functional annotations for the selected DEGs[18], including the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway analysis and Gene Ontology (GO) analysis. The enriched terms identified by the KEGG Pathway and GO analysis [including Gene Ontology_Biological Process (GO_BP), Cellular Components (GO_CC) and Molecular Function (GO_MF)] were collected, and those with a P-value < 0.05 were considered to be significantly enriched terms.
Statistical analysis
Statistical analyses were performed using the SPSS software (v20.0, SPSS Inc., Chicago, IL, United States) and the GraphPad Prism software (v6, GraphPad Software, San Diego, CA, United States). The receiver operating characteristic curve (ROC) analysis was conducted using the Med-Calc statistical software (version 15.2.2.2, MedCalc Software Bvba, Ostend, Belgium), and the sensitivity, specificity, and area under the curve (AUC) were calculated. The results with P-values < 0.05 were considered statistically significant.
RESULTS
Identification of DEGs in EAC and BE
In this study, we first identified DEGs in groups of EAC compared to NE (EAC vs NE), BE compared to NE (BE vs NE), and EAC compared to BE (EAC vs BE) in the GSE1420 dataset. The numbers of DEGs in the three different comparison groups are shown in Figure 1 (|FC| > 1.5, P-values < 0.05). Refer to Supplementary Tables 1-3 for details.
Figure 1.
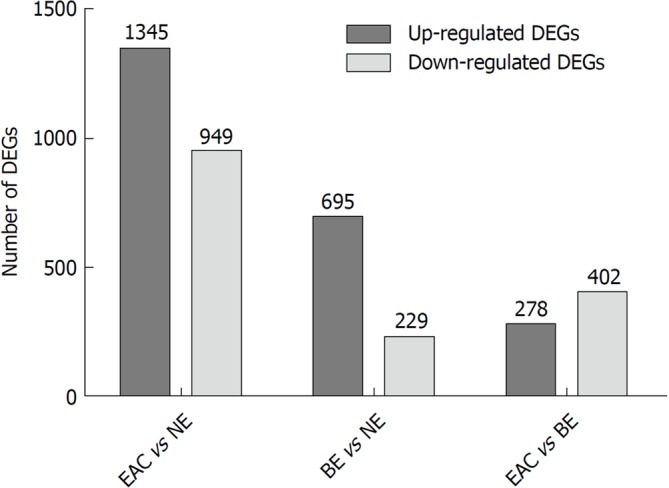
The numbers of differentially expressed genes in different comparison groups (esophageal adenocarcinoma vs normal esophagus, Barrett's esophagus vs normal esophagus, and esophageal adenocarcinoma vs Barrett's esophagus) in the GSE1420 dataset. The GSE1420 dataset consists of three different groups [normal esophagus (NE), Barrett's esophagus (BE), and esophageal adenocarcinoma (EAC)]. The differentially expressed genes (DEGs) in different comparison groups (EAC vs NE, BE vs NE, and EAC vs BE) were identified using the R/Bioconductor software (|FC| > 1.5, P-values < 0.05), and the numbers of DEGs in different comparison groups were summarized.
Transcription factors in DEGs
We then focused on the changes in the transcriptome during the progression of BE and EAC, and used the TRED database to perform the transcriptome analysis. Table 1 demonstrates the DETFs in the above three comparison groups. The table shows that a great number of DETFs was present in the EAC vs NE group compared to that in the other groups. Moreover, five DETFs displayed the same trend in expression among all three comparison groups, with up-regulation of E2F3, FOXA2, and HOXB7, and down-regulation of PAX9 and TFAP2C. Additionally, we analyzed the trans-regulatory information to further elucidate the interactions between DETFs and their regulatory DEGs. The EAC vs NE network consisted of 146 regulation modes that involved 129 nodes. Among these nodes, 10 were TFs. The majority of the genes in the network were up-regulated, including 68 up-regulated and 51 down-regulated DEGs (Figure 2A). The network also shows that one TF could target multiple genes, and TFAP2A is the TF with the highest number of interactions (79 connections). Additionally, the CLO1A1 gene could be regulated by three different TFs (SP3, MYBL2, and TFAP2A). Similarly, the trans-regulatory networks of BE vs NE and EAC vs BE were constructed and are shown in Figure 2B and 2C.
Table 1.
Differentially expressed transcription factors in the comparison groups in the GSE1420 dataset
| Comparison group | Up-regulated TFs | Down-regulated TFs |
| EAC vs NE | ATF3, ATF5, CEBPB, E2F31, EGR1, EGR3, EGR4, FOXA21, HOXA10, HOXB71, MYBL2, NFIB, PPARG, RELB, SMAD6, SP3, STAT1 | PAX91, RARG, SMAD2, TFAP2A, TFAP2B, TFAP2C1, FOXE1, HLF, HOXA4, HOXB2 |
| BE vs NE | ATF3, E2F31, EPAS1, ETS2, FOS, FOSB, FOXA21, HOXB71, NFIB, PPARG, STAT1 | PAX91, TFAP2B, TFAP2C1, HLF, HOXA1, HOXA4 |
| EAC vs BE | E2F31, EGR4, FOXA21, HOXA10, HOXB71, MYBL2 | PAX91, PPARD, RARG, TFAP2C1, ELK3, ETS2, FOXE1 |
Differentially expressed transcription factors, which displayed the same expression tendency among all the three comparison groups. TF: Transcription factor; EAC: Esophageal adenocarcinoma; NE: Normal esophagus; BE: Barrett's esophagus.
Figure 2.

The trans-regulatory networks of differentially expressed transcription factors and their regulatory differentially expressed genes. Circles represent the differentially expressed genes (DEGs) (red for up-regulated DEGs and green for down-regulated DEGs) regulated by the predicted transcription factor (TFs) (yellow for TFs). The direction of arrows is from the predicted TFs to their target DEGs. A: The trans-regulatory network of differentially expressed TFs and their regulatory DEGs in the comparison group of esophageal adenocarcinoma (EAC) vs normal esophagus (NE); B: The trans-regulatory network of differentially expressed TFs and their regulatory DEGs in the comparison group of Barrett's esophagus (BE) vs NE; C: The trans-regulatory network of differentially expressed TFs and their regulatory DEGs in the comparison group of EAC vs BE.
Bioinformatics analyses of DEGs in the trans-regulatory networks
To explore the value of the DEGs in the trans-regulatory networks described above, we performed similar bioinformatics analyses in the GSE26886 dataset. As a test dataset, the GSE26886 dataset contained a higher number of samples in each group. After the DEGs were identified in each comparison group (BE vs NE, EAC vs NE, and EAC vs BE) in GSE26886, we obtained the intersections between the DEGs in the trans-regulatory networks from the GSE1420 dataset and the DEGs from the GSE26886 dataset for each comparison group, respectively. Unsurprisingly, the majority of the genes in each network were included in the intersections. Moreover, all of the DEGs in each intersection exhibited the same direction of expression changes, indicating that our data are reliable and suitable for further investigation. The detailed gene lists are shown in Table 2 for the EAC vs NE group and the BE vs NE group, both of which are more relevant to the pathogenesis of EAC and BE. Additionally, the bi-cluster analysis showed that the different kinds of tissues could be distinguished (Figure 3).
Table 2.
Differentially expressed genes in the trans-regulatory networks existed in both GSE1420 and GSE26886 datasets
| Comparison group | Up-regulated DEGs | Down-regulated DEGs |
| EAC vs NE | UCP2, TOP1, TIMP1, TAP1, STAT11, ST6GAL1, SOCS1, SMURF2, SLC19A1, SKIL, SERPINE1, SDC4, RELB1, RARRES3, PTPN6, PSMB9, PRKCA, PPARG1, PLAUR, PLAU, PECAM1, PDGFA, ODC1, NT5E, NR1H3, NQO1, MYBL21, MUC5AC, MTRR, MMP9, MMP7, MET, MAN2B1, LAMB1, ITGB1, ITGAV, IRF3, IRF1, IGFBP3, ICAM1, HSD17B2, HOXB71, HMGA2, HFE, HEXB, HEXA, GUSB, GBP1, GALC, F2R, ENG, EGR11, DSG2, CXCL1, COL5A2, COL1A2, COL1A1, CFTR, CD14, ATP2A3, APOE, ADAM28, A2M | TOM1, TGM1, TGFA, TFAP2C1, TFAP2A1, TALDO1, SRPX2, SNAI2, SMARCA2, SMAD21, SLC6A4, SLC13A4, SERPINB2, RORA1, RARG1, PRNP, PPL, PIM1, PEX3, PBX1, OAT, MAPT, MAPK1, LMO2, KRT5, KRT4, KRT16, KRT14, IMPA2, HSPB1, HRAS, HOXA41, HMOX1, HLF1, GSTP1, GM2A, GLUD1, FLG, FGFR3, FCER1A, ETF1, DSG3, DSG1, DBI, CYP11A1, CSTA, CRABP2, CEACAM1, CALB2, BCKDHA, ATP12A, ALDH3A2, AKR1C1, AGA, ADIPOR1, ACOX1, ACAA1 |
| BE vs NE | UCP2, TIMP1, TGFBR2, SOCS3, REG1A, PPARG1, PLAUR, NR1H3, NQO1, MMP1, KRT18, GDF15, GBP1, FOS1, CA2, ANPEP, A2M | RORA, HOXA11, FCER1A, CYP11A1, BMP7 |
Differentially expressed transcription factors. DEGs: Differentially expressed genes; EAC: Esophageal adenocarcinoma; NE: Normal esophagus; BE: Barrett's esophagus.
Figure 3.

Bi-cluster analysis of the differentially expressed genes in the trans-regulatory networks existing in both the GSE1420 and GSE26886 datasets. Each row represents one of the differentially expressed genes (DEGs) in the trans-regulatory networks, and each column represents a tissue sample from the two datasets (GSE1420 and GSE26886). The column “normal esophagus (NE)” represents normal esophagus tissue, the column “Barrett's esophagus (BE)” represents Barrett's esophagus tissue, and the column “esophageal adenocarcinoma (EAC)” represents tissue from esophageal adenocarcinoma. The heat map was constructed using each DEG expression value in every tissue sample. A: The heat map for the comparison group of EAC vs. NE in the GSE1420 dataset; B: The heat map for the comparison group of EAC vs NE in the GSE26886 dataset; C: The heat map for the comparison group of BE vs NE in the GSE1420 dataset; D: The heat map for the comparison group of BE vs NE in the GSE26886 dataset.
We suspected that genes with higher FCs in expression might be more important to pathogenesis and carcinogenesis. Thus, we reviewed the DEGs with an expression FC of at least 5 in each intersection of the EAC vs NE group and the BE vs NE group to identify the biomarkers. TIMP1 and COL1A1 were selected for EAC, and MMP1, REG1A, CA2, and ANPEP were selected for BE. We then performed ROC analysis for these genes in both datasets. The ROC curves indicated that the expression of TIMP1 and COL1A1 could discriminate EAC from NE tissue, while the expression of MMP1, REG1A, and CA2 could discriminate BE from NE tissue (Figures 4 and 5).
Figure 4.

Receiver operating characteristic curve analysis for tissue discrimination between esophageal adenocarcinoma vs normal esophagus and Barrett's esophagus vs normal esophagus in the GSE1420 dataset. A: Receiver operating characteristic (ROC) curve of TIMP1; B: ROC curve of COL1A1; C: ROC curve of MMP1; D: ROC curve of REG1A; E: ROC curve of CA2; F: ROC curve of ANPEP.
Figure 5.

Receiver operating characteristic analysis for tissue discrimination between esophageal adenocarcinoma vs normal esophagus and Barrett's esophagus vs normal esophagus in the GSE26886 dataset. A: Receiver operating characteristic (ROC) curve of TIMP1; B: ROC curve of COL1A1; C: ROC curve of MMP1; D: ROC curve of REG1A; E: ROC curve of CA2; F: ROC curve of ANPEP.
To investigate the biological functions of the DEGs in Table 2, we used DAVID to provide the annotation of up- and down-regulated DEGs in each comparison group, and generated a series of enriched KEGG pathway and GO terms for each separately. In the EAC vs NE group (Supplementary Table 4), COL1A1 participated in nearly a third of the enriched KEGG pathway terms such as extracellular matrix (ECM)-receptor interaction and the PI3K-Akt signaling pathway. COL1A1 was also enriched in 20 GO_BP terms, and the total number of enriched GO_BP terms was 78. Interestingly, pathways in cancer was up-regulated and enriched in the BE vs NE group, and the enrichment included the genes MMP1, FOS, TGFBR2, and PPARG. These four genes participated in almost all the GO_BP terms, including negative regulation of interferon-gamma-mediated signaling pathway, organ regeneration, extracellular matrix disassembly, and transforming growth factor beta receptor signaling pathway. Refer to Supplementary Table 5 for detailed information.
DISCUSSION
Numerous studies related to EAC and BE have been conducted over the last decades, and more attention should be paid to the pathogenesis and the molecular mechanisms involved in the progression of BE and EAC, which could help in prevention and improve prognosis[19]. Currently, histological examinations are the golden standard for the diagnosis of EAC and BE; however, the inter- and intra-observer variabilities are not ideal[20]. Additionally, histology depends on a biopsy taken during an endoscopic procedure, and this introduces additional inter- and intra-observer variability[21]. Due to the limitations of endoscopic surveillance and histological examination, developing biomarkers that could identfify the neoplastic progression is crucial[10]. The discovery of molecular changes during BE and EAC progression may identify biomarkers that can be used for early diagnosis and prognostic prediction[22]. Advances in biological ‘omics’-based techniques have been used to explore the changes in gene expression during disease progression[3], and these new technologies could identify a series of abnormally expressed genes that may be new targets for EAC and BE[12-14]. However, there are no consistent conclusions among these different studies, and many of the critical genes and pathways have not been thoroughly investigated.
Krishnamoorthi et al[23] proposed that the persistence of non-dysplastic BE is not protective against the progression to adenocarcinoma. Therefore, we focused on the three conditions (NE, BE, and EAC) to provide a broad view of their underlying mechanisms, and explored changes in the transcriptome to address the complexity of BE and EAC while evaluating potential biomarkers. We first identified the DEGs and predicted their roles in the multistep morphological progression of BE and EAC. Since DEGs with higher expression may play more important roles in pathogenesis and carcinogenesis, the DEGs with an FC in expression of at least 5 in the trans-regulatory networks were reviewed and included in the ROC analysis. After using various analytical approaches that are based on several bioinformatics tools, the results showed that COL1A1 was associated with EAC, and MMP1 was predicted to be associated with BE. These two respective genes were evaluated as potent biomarkers for EAC and BE, because they might participate in the majority of the important and enriched KEGG or GO terms that were related to inflammation and cancer. It is well known that the tissue micro-environment can promote esophageal carcinogenesis at its earliest stage[24], and that inflammation plays an important role in this process. Additionally, inflammatory cells release various molecules that activate or inhibit signal transduction. In this study, many inflammatory pathways were enriched, which is consistent with the previous data and warrants further investigation.
COL1A1, which is located on chromosome 17, encodes the pro-alpha1 chain of the type I collagen[25]. Type I collagen is highly expressed in embryonic and connective tissues[26]. Previous studies found that COL1A1 participates in the development of several cancers[27,28] such as non-small cell lung cancer and hepatocellular carcinoma. This gene may take part in the suppression of apoptosis induced by radiation in cervical cancer cells, and may also regulate the pathogenesis of several other cancers[29,30]. However, studies on COL1A1 in EAC are rare. In this study, the sensitivity, specificity, and AUC of COL1A1 were evaluated and our data indicated that COL1A1 could be a potential biomarker for distinguishing EAC and might play an important role in EAC pathogenesis.
MMP1[31], which is located on chromosome 11, encodes for a member of the peptidase M10 family of matrix metalloproteinases (MMPs). Proteins in this family participate in the breakdown of the ECM in pathophysiological processes such as embryonic development and tissue remodeling. Multiple studies indicated that MMP1 participates in the progression of various cancers and is associated with an increased cancer risk[32,33]. Up-regulation of MMP1 has already been observed in EAC and BE samples[34], but has not been fully investigated. In our study, the ROC analysis found that the sensitivity, specificity, and AUC were relatively high in both datasets. Moreover, MMP1 participated in cancer pathways and most of the enriched GO_BP terms, including numerous inflammatory processes. Since BE is recognized as the only precursor of EAC to date, MMP1 could be an active protein in the progression of BE and EAC, suggesting that the gene is a potent biomarker for BE.
Some limitations undoubtedly existed in the present study. The genes identified could not be further discussed. And future studies should be performed to validate and evaluate the potential genes and biomarkers. Importantly, biomarker development could influence the regulatory guidelines, and the translation of biomarker discoveries into the clinic was included in the Precision Medicine Initiative[35]. As studies have previously shown, modern ‘omics’-based technologies may provide new perspectives on the molecular mechanisms and the pathways involved in neoplasms[22]. The combination of genetics, gene-expression profiling of esophageal tissue, and pathway signatures might constitute a risk-prediction model for EAC and BE[22]. If the clinical and environmental factors could be included, the personalized risk-prediction model for EAC and BE may be established and implemented in the clinic[22]. Additionally, the associations between risk factors and molecular subtypes of EAC should be investigated further, and screening and surveillance trials for high-risk individuals are necessary[36].
In conclusion, comprehensive bioinformatics analyses of the DEGs from two datasets were performed, and the genes and biomarkers potentially involved in the progression of BE and EAC were identified. After trans-regulatory network prediction, ROC evaluation, and DAVID annotation, an association between COL1A1 and EAC was found. Similarly, an association between MMP1 and BE was also predicted. This study provides a novel perspective on the molecular mechanisms involved in the development of BE and EAC.
ARTICLE HIGHLIGHTS
Research background
Barrett’s esophagus (BE) is the only known precursor of esophageal adenocarcinoma (EAC), and patients with BE have a persistent and excessive risk of EAC over time. As an aggressive disease with high mortality, the overall 5-year survival rate of EAC is less than 20%. Therefore, early detection of BE and EAC could significantly improve the 5-year survival rate of EAC. Due to the limitations of endoscopic surveillance and the lack of clinical risk stratification strategies, more attention has been paid to improving endoscopic techniques and to the discovery of molecular biomarkers for EAC and BE.
Research motivation
The exact molecular mechanisms of EAC and BE are controversial, so a top priority is to explore their basic molecular mechanisms and pathways. The discovery of molecular changes during their progression may identify biomarkers that can be used for early diagnosis and prognostic prediction. Advances in biological ‘omics’-based techniques have been used to explore the changes in gene expression during disease progression, including gene chips, serial analysis of gene expression, and proteomics. These new technologies could identify a series of abnormally expressed genes that may be new targets for EAC and BE, especially as more researchers focus on this topic. Recently, gene profiling analysis has been used to explore the molecular changes in many disorders, including EAC and BE. However there are no consistent conclusions among these different studies, and many of the potential genes and pathways have not been thoroughly investigated.
Research objectives
In this study, we focused on the three conditions [normal esophagus (NE), BE, and EAC] to provide a broad view of their underlying mechanisms, and explored changes in the transcriptome to address the complexity of EAC and BE while evaluating potential biomarkers. After using various analytical approaches that are based on several bioinformatics tools, the results showed that COL1A1 was associated with EAC, and that MMP1 was predicted to be associated with BE. These two respective genes were evaluated as potent biomarkers for EAC and BE, which could play certain roles in the pathogenesis and might be utilized for clinical application.
Research methods
The gene expression profile datasets of EAC and BE were retrieved from the Gene Expression Omnibus database. The expression profiling analysis was conducted using the R/Bioconductor project. The AffyPLM package and the gcRMA algorithm were used for data preprocessing, and the Limma package was used to identify the differentially expressed genes (DEGs) between any two groups. Genes with a fold change of at least 1.5 and a P-value < 0.05 were considered DEGs. The transcription factor (TF)-gene regulation modes of 36 cancer-related TF families were obtained from the Transcriptional Regulatory Element Database. We then integrated the DEGs with the TF-gene regulation modes and constructed the dys-regulated TF-DEGs networks. The trans-regulatory networks were visualized with the help of the Cytoscape software. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources was used to provide functional annotations. Statistical analyses were performed using the SPSS software and the GraphPad Prism software. The receiver operating characteristic curve (ROC) analysis was conducted using the Med-Calc statistical software.
Research results
In this study, we found that the number of up-regulated DEGs was larger than that of down-regulated DEGs when comparing EAC vs. NE and BE vs. NE. And the majority of the DEGs in trans-regulatory networks were up-regulated. The intersection of these potential DEGs displayed the same direction of changes in expression when comparing the DEGs in the GSE26886 dataset to the DEGs in trans-regulatory networks in the GSE1420 dataset. The ROC analysis and DAVID annotation indicated that COL1A1 and MMP1 could be potent biomarkers for EAC and BE, respectively, since they participate in the majority of the enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) terms that are important for inflammation and cancer.
Research conclusions
Our data indicated that there is an association between COL1A1 and EAC after trans-regulatory network prediction, ROC evaluation, and DAVID annotation. Similarly, an association between MMP1 and BE was also predicted. This study provides potential genes which could be applied to clinical practice, and a novel perspective on the molecular mechanisms involved in the underlying development of BE and EAC.
Research perspectives
The discovery of molecular changes during disease progression may identify biomarkers that might be used for early diagnosis and prognostic prediction. And this study also provide a potential perspective on the molecular mechanisms to the clinical gastroenterologists. In order to confirm the value of these potent genes, the validation groups will be introduced in our future study, which might provide more detailed information.
ACKNOWLEDGMENTS
The authors would like to thank Professor Feng Zhang from Health Science Center, Xi’an Jiaotong University, for the biostatistics review.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors declare that they have no conflicts of interest.
Data sharing statement: Datasets analyzed during the current study are available from the Gene Expression Omnibus (GEO) DataSets in the National Center for Biotechnology Information (NBCI) Database (http://www.ncbi.nlm.nih.gov/gds/). All data generated or analysed during this study are included in this article and its supplementary information files.
Peer-review started: October 14, 2018
First decision: November 29, 2018
Article in press: December 15, 2018
P- Reviewer: Hashimoto N, Yamada T S- Editor: Wang XJ L- Editor: Wang TQ E- Editor: Yin SY
Contributor Information
Jing Lv, Department of Clinical Laboratory, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China.
Lei Guo, Department of Spinal Surgery, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China.
Ji-Han Wang, Department of Clinical Laboratory, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China.
Yu-Zhu Yan, Department of Clinical Laboratory, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China.
Jun Zhang, Department of Gastroenterology, Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710004, Shaanxi Province, China.
Yang-Yang Wang, The Tenth Research Institute of Telecommunications Technology, Xi’an 710000, Shaanxi Province, China.
Yan Yu, Department of Clinical Laboratory, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China.
Yun-Fei Huang, Department of Spinal Surgery, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China.
He-Ping Zhao, Department of Clinical Laboratory, Honghui Hospital, Xi’an Jiaotong University, Xi’an 710054, Shaanxi Province, China. redcrossjyk@163.com.
References
- 1.Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol. 2006;101:1900–1920; quiz 1943. doi: 10.1111/j.1572-0241.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 2.Shaheen NJ, Falk GW, Iyer PG, Gerson LB American College of Gastroenterology. ACG Clinical Guideline: Diagnosis and Management of Barrett’s Esophagus. Am J Gastroenterol. 2016;111:30–50; quiz 51. doi: 10.1038/ajg.2015.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kimchi ET, Posner MC, Park JO, Darga TE, Kocherginsky M, Karrison T, Hart J, Smith KD, Mezhir JJ, Weichselbaum RR, Khodarev NN. Progression of Barrett’s metaplasia to adenocarcinoma is associated with the suppression of the transcriptional programs of epidermal differentiation. Cancer Res. 2005;65:3146–3154. doi: 10.1158/0008-5472.CAN-04-2490. [DOI] [PubMed] [Google Scholar]
- 4.Cook MB, Coburn SB, Lam JR, Taylor PR, Schneider JL, Corley DA. Cancer incidence and mortality risks in a large US Barrett’s oesophagus cohort. Gut. 2018;67:418–529. doi: 10.1136/gutjnl-2016-312223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desai TK, Krishnan K, Samala N, Singh J, Cluley J, Perla S, Howden CW. The incidence of oesophageal adenocarcinoma in non-dysplastic Barrett’s oesophagus: a meta-analysis. Gut. 2012;61:970–976. doi: 10.1136/gutjnl-2011-300730. [DOI] [PubMed] [Google Scholar]
- 6.Sharma P. Clinical practice. Barrett’s esophagus. N Engl J Med. 2009;361:2548–2556. doi: 10.1056/NEJMcp0902173. [DOI] [PubMed] [Google Scholar]
- 7.Thrift AP, Whiteman DC. The incidence of esophageal adenocarcinoma continues to rise: analysis of period and birth cohort effects on recent trends. Ann Oncol. 2012;23:3155–3162. doi: 10.1093/annonc/mds181. [DOI] [PubMed] [Google Scholar]
- 8.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 9.Hur C, Miller M, Kong CY, Dowling EC, Nattinger KJ, Dunn M, Feuer EJ. Trends in esophageal adenocarcinoma incidence and mortality. Cancer. 2013;119:1149–1158. doi: 10.1002/cncr.27834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wani S, Gaddam S. Editorial: Best Practices in Surveillance of Barrett’s Esophagus. Am J Gastroenterol. 2017;112:1056–1060. doi: 10.1038/ajg.2017.117. [DOI] [PubMed] [Google Scholar]
- 11.Eluri S, Shaheen NJ. Barrett’s esophagus: diagnosis and management. Gastrointest Endosc. 2017;85:889–903. doi: 10.1016/j.gie.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Baal JW, Verbeek RE, Bus P, Fassan M, Souza RF, Rugge M, ten Kate FJ, Vleggaar FP, Siersema PD. microRNA-145 in Barrett’s oesophagus: regulating BMP4 signalling via GATA6. Gut. 2013;62:664–675. doi: 10.1136/gutjnl-2011-301061. [DOI] [PubMed] [Google Scholar]
- 13.Bus P, Kestens C, Ten Kate FJ, Peters W, Drenth JP, Roodhart JM, Siersema PD, van Baal JW. Profiling of circulating microRNAs in patients with Barrett’s esophagus and esophageal adenocarcinoma. J Gastroenterol. 2016;51:560–570. doi: 10.1007/s00535-015-1133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiam K, Wang T, Watson DI, Mayne GC, Irvine TS, Bright T, Smith L, White IA, Bowen JM, Keefe D, Thompson SK, Jones ME, Hussey DJ. Circulating Serum Exosomal miRNAs As Potential Biomarkers for Esophageal Adenocarcinoma. J Gastrointest Surg. 2015;19:1208–1215. doi: 10.1007/s11605-015-2829-9. [DOI] [PubMed] [Google Scholar]
- 15.Gregory Alvord W, Roayaei JA, Quiñones OA, Schneider KT. A microarray analysis for differential gene expression in the soybean genome using Bioconductor and R. Brief Bioinform. 2007;8:415–431. doi: 10.1093/bib/bbm043. [DOI] [PubMed] [Google Scholar]
- 16.Jiang C, Xuan Z, Zhao F, Zhang MQ. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 2007;35:D137–D140. doi: 10.1093/nar/gkl1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Lv J, Liu J, Guo L, Zhang J, Cheng Y, Chen C, Zhao H, Wang J. Bioinformatic analyses of microRNA-targeted genes and microarray-identified genes correlated with Barrett’s esophagus. Cell Cycle. 2018;17:792–800. doi: 10.1080/15384101.2018.1431597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerkhof M, van Dekken H, Steyerberg EW, Meijer GA, Mulder AH, de Bruïne A, Driessen A, ten Kate FJ, Kusters JG, Kuipers EJ, Siersema PD CYBAR study group. Grading of dysplasia in Barrett’s oesophagus: substantial interobserver variation between general and gastrointestinal pathologists. Histopathology. 2007;50:920–927. doi: 10.1111/j.1365-2559.2007.02706.x. [DOI] [PubMed] [Google Scholar]
- 21.Gordon LG, Mayne GC, Hirst NG, Bright T, Whiteman DC Australian Cancer Study Clinical Follow-Up Study, Watson DI. Cost-effectiveness of endoscopic surveillance of non-dysplastic Barrett’s esophagus. Gastrointest Endosc. 2014;79:242–256.e6. doi: 10.1016/j.gie.2013.07.046. [DOI] [PubMed] [Google Scholar]
- 22.Konda VJA, Souza RF. Biomarkers of Barrett’s Esophagus: From the Laboratory to Clinical Practice. Dig Dis Sci. 2018;63:2070–2080. doi: 10.1007/s10620-018-5088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnamoorthi R, Ramos GP, Crews N, Johnson M, Dierkhising R, Shi Q, Ginos B, Wang K, Katzka D, Buttar N, Iyer PG. Persistence of Nondysplastic Barrett’s Esophagus Is Not Protective Against Progression to Adenocarcinoma. Clin Gastroenterol Hepatol. 2017;15:950–952. doi: 10.1016/j.cgh.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 24.Quante M, Graham TA, Jansen M. Insights Into the Pathophysiology of Esophageal Adenocarcinoma. Gastroenterology. 2018;154:406–420. doi: 10.1053/j.gastro.2017.09.046. [DOI] [PubMed] [Google Scholar]
- 25.Exposito JY, Valcourt U, Cluzel C, Lethias C. The fibrillar collagen family. Int J Mol Sci. 2010;11:407–426. doi: 10.3390/ijms11020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole WG. Collagen genes: mutations affecting collagen structure and expression. Prog Nucleic Acid Res Mol Biol. 1994;47:29–80. doi: 10.1016/s0079-6603(08)60249-4. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi M, Nomoto S, Hishida M, Inokawa Y, Kanda M, Okamura Y, Nishikawa Y, Tanaka C, Kobayashi D, Yamada S, Nakayama G, Fujii T, Sugimoto H, Koike M, Fujiwara M, Takeda S, Kodera Y. Identification of the collagen type 1 α 1 gene (COL1A1) as a candidate survival-related factor associated with hepatocellular carcinoma. BMC Cancer. 2014;14:108. doi: 10.1186/1471-2407-14-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oleksiewicz U, Liloglou T, Tasopoulou KM, Daskoulidou N, Gosney JR, Field JK, Xinarianos G. COL1A1, PRPF40A, and UCP2 correlate with hypoxia markers in non-small cell lung cancer. J Cancer Res Clin Oncol. 2017;143:1133–1141. doi: 10.1007/s00432-017-2381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S, Liao G, Li G. Regulatory effects of COL1A1 on apoptosis induced by radiation in cervical cancer cells. Cancer Cell Int. 2017;17:73. doi: 10.1186/s12935-017-0443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y, Duan Z, Zhang X, Zhang X, Wang G, Li F. Down-regulation of the let-7i facilitates gastric cancer invasion and metastasis by targeting COL1A1. Protein Cell. 2018 doi: 10.1007/s13238-018-0550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray GI. Matrix metalloproteinases: a multifunctional group of molecules. J Pathol. 2001;195:135–137. doi: 10.1002/1096-9896(200109)195:2<135::AID-PATH939>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 32.Wong JC, Chan SK, Schaeffer DF, Sagaert X, Lim HJ, Kennecke H, Owen DA, Suh KW, Kim YB, Tai IT. Absence of MMP2 expression correlates with poor clinical outcomes in rectal cancer, and is distinct from MMP1-related outcomes in colon cancer. Clin Cancer Res. 2011;17:4167–4176. doi: 10.1158/1078-0432.CCR-10-1224. [DOI] [PubMed] [Google Scholar]
- 33.Lu L, Sun Y, Li Y, Wan P. The polymorphism MMP1 -1607 (1G>2G) is associated with a significantly increased risk of cancers from a meta-analysis. Tumour Biol. 2015;36:1685–1693. doi: 10.1007/s13277-014-2769-0. [DOI] [PubMed] [Google Scholar]
- 34.Dai Y, Wang Q, Gonzalez Lopez A, Anders M, Malfertheiner P, Vieth M, Kemmner W. Genome-Wide Analysis of Barrett’s Adenocarcinoma. A First Step Towards Identifying Patients at Risk and Developing Therapeutic Paths. Transl Oncol. 2018;11:116–124. doi: 10.1016/j.tranon.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coleman HG, Xie SH, Lagergren J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology. 2018;154:390–405. doi: 10.1053/j.gastro.2017.07.046. [DOI] [PubMed] [Google Scholar]