

Abstract
A drug design for safer phenylbutazone was been explored by reactivity and docking studies involving single electron transfer mechanism, as well as toxicological predictions. Several approaches about its structural properties were performed through quantum chemistry calculations at the B3LYP level of theory, together with the 6-31+G(d,p) basis sets. Molecular orbital and ionization potential were associated to electron donation capacity. The spin densities contribution showed a preferential hydroxylation at the para-positions of phenyl ring when compared to other positions. In addition, on electron abstractions the aromatic hydroxylation has more impact than alkyl hydroxylation. Docking studies indicate that six structures 1, 7, 8 and 13–15 have potential for inhibiting human as well as murine COX-2, due to regions showing similar intermolecular interactions to the observed for the control compounds (indomethacin and refecoxib). Toxicity can be related to aromatic hydroxylation. In accordance to our calculations, the derivatives here proposed are potentially more active as well safer than phenylbutazone and only structures 8 and 13–15 were the most promising. Such results can explain the biological properties of phenylbutazone and support the design of potentially safer candidates.
Keywords: phenylbutazone, DFT, electron transfer, metabolism, toxicity
1. Introduction
Phenylbutazone is a non-steroidal anti-inflammatory drug that has analgesic and antipyretic actions and can be used in some situations related do acute pain and muscular-skeletal disorders, such as in cases of ankylosing spondylitis and rheumatoid arthritis [1]. This medicine belongs to the group of pyrazolones, which are derived from enolic acid associated to its hydrogen α, α-dicabonylic [2].
It was synthesized in 1946 and introduced into clinical practice in 1949. However, in 1980 were attributed severe restrictions on the use of phenylbutazone in humans, because of the recognition that such a drug induced blood dyscrasias, which includes aplastic anemia, leukopenia, agranulocytosis and thrombocytopenia in some cases leading to death [3].
The toxicity of phenylbutazone is directly related to its mechanism of action, which shows non-selective effect for COX-1 isoform and may act on COX-2, thus the side effects are more evident in the organs and systems where COX-1 is found. The most known adverse reactions are diffused gastritis, gastric ulcers, venous thrombosis, nephritis and chronic renal injury [4,5]. As their toxicity depends on of metabolism and kinetics is a dose-dependent type, dosages up to two times above the recommended can result in fatal protein loss enteropathy, within 10 to 14 days [6]. Also, their main metabolites found were oxyphenbutazone and γ-hydroxy-phenylbutazone, where the first is active and second inactive. Only 1% of the administered dose is excreted in the urine as the parent drug while the majority (within 72%) is excreted in urine as metabolites and 10% is excreted in bile as well as metabolites [7].
Recently we have developed a theoretical prediction model of toxicity for pyrazolone drugs for agranulocytosis. Our study shows that this toxicity is mainly related to their ionization potential and nucleophilicity values, on electron transfer process [8]. Nevertheless, at moment the mechanism to phenylbutazone was not proposed. Thus, the study aimed to examine the molecular structure of phenylbutazone derivatives in a particular biological system, obtaining means for predicting the safer potential candidates.
2. Results and Discussion
2.1. Tautomerism Study
The relative barrier is the energy difference of the most stable derivative to the less stable, that is, how much energy was needed to obtain the protonated form. In this study, we have investigated the phenylbutazone (1a) and phenylbutazone tautomers (1b) shown in Figure 1, depicting the keto-enol equilibrium for phenylbutazone. Results were obtained from quantum chemistry calculations at the theory level B3LYP and 6-31G+(d,p) basis set for the equilibrium structures, demonstrating that the conformation of the phenylbutazone molecule (1a) in the keto form is approximately 9.36 kcal/mol more stable than the other one. For the anionic form of phenylbutazone we observed greater planarity and rigidity of the five-membered ring (1b), on contrary to the proton form. In a qualitative analysis, we can infer a greater degree of electron delocalization, which could result in the appearance of a greater amount of resonance forms and consequent relocation of charges on the oxygen atoms.
Figure 1.

Tautomerism keto-enolic of phenylbutazone (1a) and phenylbutazone tautomers (1b).
Thus, the keto tautomer is more stable than the enol tautomer. Since phenylbutazone anion is reported as the active form in inflammation, it could act as free radical by a one-electron transfer mechanism, possibly via enol pathway [9]. However, despite this form to be unstable, any changes on pH medium should induce modification of the equilibrium relative to this tautomerism. The inflammation results in marked reductions of tissue pH by generation of acid derivatives as well as protons [10].
In addition, other pyrazole system showed the same behavior [11]. Hence for each calculation of phenylbutazone derivatives was realized from the keto tautomer. These conformations were used for electronic study of chemical reactivity and theoretical metabolism.
2.2. Molecular Simplification
The electron donation capacity for the simplified phenylbutazone derivatives (Figure 2) were theoretically measured using HOMO, LUMO, GapL-H and ionization potential (IP) values. For the phenylbutazone compound, simplification of the phenyl and alkyl groups was carried out in order to investigate the structure-toxicity relationships at the pharmacophoric region: In compound 4, the two phenyl rings were withdrawn due to the symmetry of the original molecule (phenylbutazone). The structures shown in Figure 3 are derived from oxidative metabolism, originating two main metabolites, through hydroxylation.
Figure 2.

Molecular simplification of phenylbutazone.
Figure 3.

Molecular metabolism of phenylbutazone.
The HOMO and LUMO values are an important parameter of molecular structure related to their reactivity. They show all nucleophilic and electrophilic regions, respectively, around the molecule. The molecule which has the higher HOMO energy has strong donating electron ability. On the contrary, the higher LUMO value implies that the molecule is a good electron accepting compound [12]. The GapL-H values of these structures can indicate their chemical reactivity on electron transfers. The lowest GapL-H value is related to high chemical reactivity. Phenylbutazone showed HOMO value of −6.24 eV and GapL-H of 4.98 eV. Previous works have demonstrated a good tendency between HOMO and IP [13]. The IP value for the phenylbutazone is 177.46 kcal/mol. Additionally, the alkyl and phenyl moieties had been great influence on electron donation capacity for phenylbutazone derivatives. Indeed, our results showed that phenyl moiety has more impact than alkyl group due to HOMO decreases and by the increase of the GapL-H and IP values (Table 1).
Table 1.
Theoretical properties of phenylbutazone derivatives.
| Derivatives | HOMO (eV) | LUMO (eV) | GAPL-H (eV) | IP (kcal/mol) |
|---|---|---|---|---|
| 1 | −6.24 | −1.25 | 4.98 | 177.46 |
| 2 | −6.31 | −1.31 | 5.01 | 180.32 |
| 3 | −6.35 | −1.30 | 5.05 | 185.11 |
| 4 | −7.15 | −1.21 | 5.94 | 215.45 |
| 5 | −6.04 | −1.20 | 4.83 | 166.43 |
| 6 | −6.20 | −1.22 | 4.99 | 170.12 |
Therefore, alkyl moieties and phenyl rings had a remarkable effect on electron donation capacity. In fact, the inflammation prostaglandin-mediated can to occur from an oxidation process by electron or hydrogen transfers followed for an oxygen addition. So, structures that have high electron donation capacity would have more inhibitory effect due to its antioxidant capacity [14].
2.3. Theoretical Metabolism
The oxidative metabolism of phenylbutazone (1) gives two main metabolites by means of hydroxylation, called oxyphenbutazone (5) and γ-hydroxyphenbutazone (6)—see Figure 3. The former is active and the second one is inactive [7]. Our calculations show that any hydroxylation can be related to changes on electronic behavior of phenylbutazone, such as electron donation capacity. However, the alkyl hydroxylation at N-buthyl position can be related to changes on solubility due to the loss of lipophilic interaction at the pocket of enzyme responsible for arachidonic acid oxidation.
In fact, oxyphenbutazone (5) has demonstrated more electron donation capacity than phenylbutazone. As we can see the best values for HOMO of −6.04 eV, GapL-H of 4.83 eV and IP of 166.43 kcal/mol for 5. These values would be related to its biological activity [15]. Likewise, these properties change for hydroxylated alkyl derivative 6 is few when compared to oxyphenbutazone (5). In addition, the high electron donation capacity of oxyphenbutazone can be related to their toxicity increases [15]. The aromatic rings are the best nucleophilic moieties especially due to nitrogen atoms. As a result, after an electron transfer the most reactive positions can be localized by spin densities calculations. It may be used to prediction a regioselective hydroxylation.
Therefore, the calculated spin density to initial electron abstraction on phenylbutazone (Figure 4) shows the main contributions from the nitrogen atoms (0.21 and 0.22), carbon atoms at the para-positions (0.14) and oxygen atom at the carbonyl groups (0.08). The contribution from the other atoms is almost an order of magnitude minor. These results showed that the reactivity increase at the para-positions (0.14) and explain the preferential regioselective radical attack of hydroxyl group to form aromatic hydroxylated-type derivatives such as oxyphenbutazone.
Figure 4.
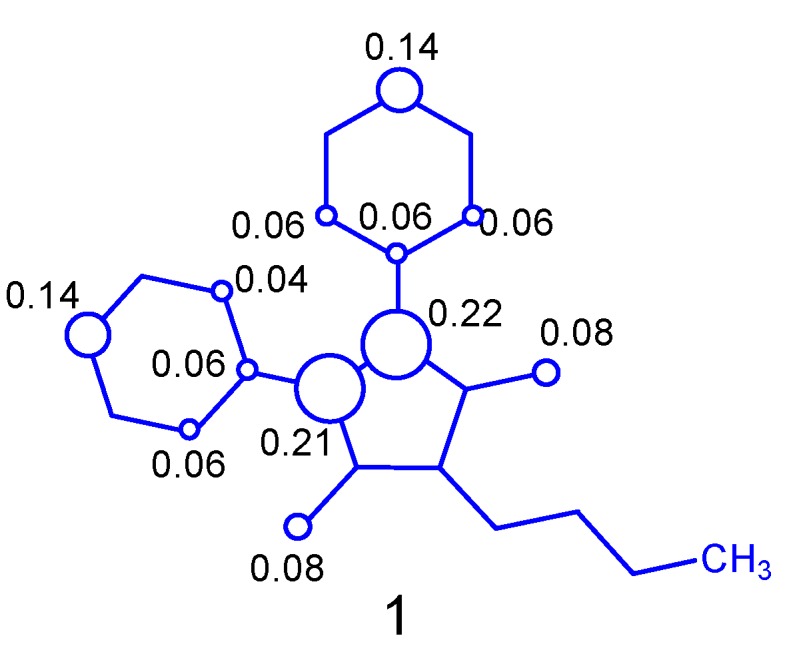
Spin densities contribution of phenylbutazone cation free radical.
2.4. Proposed Phenylbutazone Derivatives
Among the theoretical properties of phenylbutazone, the most remarkable are their high nucleophility, electron donation capacity, lipophilicity and molecular volume. These values can be observed through electronic, solubility and structural parameters in Table 2.
Table 2.
Theoretical properties of proposed phenylbutazone derivatives.
| Derivatives | HOMO (eV) | LUMO (eV) | GAPL-H (eV) | IP (kcal/mol) | ΔEiso (kcal/mol) | Log P | Vol (Å3) |
|---|---|---|---|---|---|---|---|
| 1 | −6.24 | −1.25 | 4.98 | 177.46 | 0 | 4.22 | 941.89 |
| 7 | −6.44 | −1.55 | 4.88 | 174.44 | −3.01 | 5.26 | 1027.15 |
| 8 | −5.83 | −0.94 | 4.89 | 161.07 | −16.38 | 3.94 | 979.66 |
| 9 | −5.83 | −0.99 | 4.83 | 159.79 | −17.66 | 4.81 | 1051.55 |
| 10 | −5.80 | −0.97 | 4.83 | 159.54 | −17.91 | 5.20 | 1119.93 |
| 11 | −5.91 | −1.46 | 4.44 | 159.69 | −17.76 | 5.37 | 1030.56 |
| 12 | −6.11 | −2.21 | 3.90 | 168.09 | −9.37 | 3.39 | 1010.77 |
| 13 | −6.02 | −2.10 | 3.91 | 167.21 | −10.25 | 5.37 | 960.47 |
| 14 | −6.02 | −2.02 | 4.00 | 167.15 | −10.30 | 5.37 | 963.55 |
| 15 | −5.93 | −1.90 | 4.02 | 166.03 | −11.42 | 4.85 | 920.66 |
Phenylbutazone showed HOMO value of −6.24 eV and GapL-H of 4.98 eV. It is IP, Log P and Vol values are 177.46 kcal/mol, 4.22 and 941.89 Å3, respectively. Based on them, nine derivatives were proposed by the combination among phenylbutazone, edaravone [16] and antipirine (Figure 5). The chloro moiety was added as metabolism inhibitor at para-position at the phenyl ring hydroxylation reactions [17].
Figure 5.

Molecular structure of proposed phenylbutazone derivatives.
Results obtained indicate that all proposed structures have more electron donation capacity than phenylbutazone, in accordance to the HOMO, GapL-H, IP and ΔEiso values. Likewise, all structures have similar lipophilicity and high molecular volume, when compared to phenylbutazone. Unfortunately, despite of all the proposed structures to be like actives and safer, in potential, other properties need to be considered, such as bioavailability and other pharmacokinetic parameters. However, validations will be completed only after synthesis and the respective biological evaluations. In addition, all the theoretical values obtained for our derivatives are very close of the observed for phenylbutazone and they do not violate the Lipinski RO5 rule [18].
2.5. Molecular Docking Study
For docking method validation, the indomethacin and rofecoxib structures with crystallographic information were submitted to docking until the folding found by software that was similar to the crystallographic information.
The comparison between crystallographic ligands IMN and RCX (red color) and best-predicted docking conformation (green), can be seen in Figure 6 that show a great pose (orientation + conformation) where the RMSD values were 0.75 and 1.04 Å for indomethacin and rofecoxib, respectively.
Figure 6.

AutoDock validation of (A) COX-2 (Mus musculus, PDB 4COX) and (B) COX-2 (Homo sapiens, PDB 5KIR) with respective crystallographic ligands.
According to Gowthaman et al. (2008) [19] and Hevener et al. (2009) [20], the binding mode prediction using the docking, affirm that when the RMSD is less than 2.0 Å on the crystallographic pose of ligand is considered successful. Therefore, our results with the methodological proposal using these parameters are optimal and satisfactory.
Using the docking methodology here selected, we have identified a potential binding mode for the compound able to interact with the active site of COX-2, similar to the crystallographic pose observed for indomethacin (PDB ID 4COX), that is, around the α-helix located between the amino acid residues 118–121, 348–352 and 522–526, as well as around the β-sheet located between the amino acid residues 353–354 and 527. For the ligand, it is possible to see common hydrogen bonds formed with Tyr355 and Arg120 residues. There is also a hydrophobic interaction with Val349, Ser353, Leu352, Val523 and Ala527 residues, according to studies found in the literature [21].
Also, the interaction sites described for rofecoxib ((PDB ID 5KIR) are located around the α-helix between the amino acid residues 88–91, as well as the amino acids residues located between 352–354 and 515–519 around the β-sheet, according to studies carried out by Orlando et al. (2016) [22]. The validation was accepted despite of the minor deviation observed between the poses, because the two crystallographic poses are possible.
In order to evaluate whether the changes made would lead to a higher binding affinity than the inhibitor respective to each targeted COX-2 structure, that is, from the Mus musculus and Homo sapiens, we docked each COX-2 structure, its specific ligand with phenylbutazone (structure 1) and phenylbutazone derivatives (structures 7–15) in Auto-Dock/Vina software.
We observed that indomethacin (IMN) as ligand showed higher binding affinity (−9.5 kcal/mol) to COX-2 (organism Mus musculus) than phenylbutazone (structure 1) and phenylbutazone derivatives (structures 7–15). However, when comparing the structures donated in Mus musculus organism, rofecoxib had a lower affinity value (−7.8 kcal/mol) when compared to phenylbutazone and their derivatives.
However, the structure indicates a theoretical higher binding affinity of −9.2 kcal/mol, when compared to the here investigated structures and it had a variation of ±0.3 kcal/mol in comparison to indomethacin (IMN), whereas the other ones had a variation ranging from ±1.3 to ±0.4 kcal/mol. All these values are shown in Figure 7.
Figure 7.

Binding affinity provided by AutoDock/Vina software of the phenylbutazone derivatives (structures 1, 7–15) and standard ligands. Ligand control for COX-2 organism Mus musculus and Homo sapiens were indomethacin and rofecoxib.
Furthermore, we observed that ligand RCX showed a higher binding affinity values (−10.4 kcal/mol) to COX-2 (Homo sapiens), when compared to phenylbutazone (1) and phenylbutazone derivatives (structures 7–15) as well. Indomethacin showed the lowest affinity (−7.7 kcal/mol). However, compound 7 had a higher binding affinity (−9.7 kcal/mol) regarding to the studied structures and had a variation ±0.7 kcal/mol in comparison to RCX and the other one structures had a variation ranging from ±1.5 to ±0.8 kcal/mol.
With these data, we propose that the phenylbutazone (1) and phenylbutazone derivatives (7–15) are able to make binding interaction on both organisms (Mus musculus and Homo sapiens). However, 1, 13–15 derivatives have greater binding affinity to enzyme from Mus musculus but 1, 7, 8, 14 and 15 derivatives showed greater binding affinity to the human enzyme.
According to the studies of Beretta et al. (2005) [23], the examination of IC80 values of the four non-steroidal anti-inflammatory drugs among them phenylbutazone (+134.4%) and flunixin (+29.7%). Phenylbutazone show greater selectivity for COX-2 in accordance to 50% inhibitory concentration. In fact, we suggested that phenylbutazone derivatives has both anti-inflammatory and peripheral analgesic effects and Beretta et al. (2005) [23] has demonstrated that phenylbutazone and flunixin in horses when compared to meloxicam, retains a best selectivity at higher concentrations. We can also to observe higher interaction numbers of indomethacin which is a potent non-selective inhibitor of the enzyme cyclooxygenase (COX), being a fundamental element of the cascade of arachidonic acid, the metabolic pathway that allows the synthesis of prostaglandins and thromboxanes.
Figure 8 shows all interactions for individually docked structure 14 that had similar interactions to the active sites of COX-2 (Mus musculus) for indomethacin to around the alpha helix between the amino acids residues Ser353, Val523 and Gly526, in β-leaf the amino acids residue Ala527. There is also a hydrophobic interaction with residues Val349, Ser353, Tyr385, Val523, Met522 and Ala527.
Figure 8.

Interactions of structure between 14 and COX-2 of Mus musculus. Hydrophobic effect of alkyl or Pi-alkyl, Pi-sulfur, hydrogen bonds and Pi-Pi interactions are shown.
All amino-acids residues between ligands with therapeutic targets COX-2 for the Mus musculus organism are shown in Tables S1 and S2 (see supplementary material). Similarly, analyzing the interaction sites for structure 7, as shown in the Figure 9 and comparing with the interaction sites rofecoxib, we observed similar results between Homo sapiens and Mus musculus organism. Both active sites of COX-2 have some amino-acid residues around the α-helix identified by the residue Arg120 and Val116, for β-leaf the amino acids residues Tyr 355 and Leu384, Ala 516 and Met522. In addition, the residues Val523 and Ala527 were identified close to the active site.
Figure 9.

Interactions of structure 7 (COX-2 organism Homo sapiens). Hydrophobic interactions alkyl, Pi-alky, Pi-sulfur, hydrogen bonds and Pi-Pi interactions can be seen.
Quantitative data of distances and binding free energies (ΔG) between the phenylbutazone derivatives and COX-2 for both receptors of Mus musculus and Homo sapiens organisms can be seen in Tables S1–S4 (see supplementary material). It is possible to verify that, among the template structures, such as indomethacin (IMN) and rofecoxib (RCX) inhibitors, the increase in the number of interactions resulted in diminution of the binding free energy, which indicates a higher degree of spontaneity of the interactions. This type of analysis provides additional information of features of the compounds to get a better understanding of the chemical behavior for new drugs.
This effect is noticeable for phenylbutazone derivatives with binding free energy similar to the observed for the template structures. In fact, common interactions with the amino-acids occurred in all the analyzed structures and they can indicate that they play a relevant play in the anti-inflammatory activity for the structures here investigated—Tables S1–S4 (see supplementary material).
Comparing phenylbutazone derivatives with the template molecules indomethacin and rofecoxib, we can observe that diminution of the binding free energy is similar because they have two different interactions, one with Leu352 and another with Ser353. Our results show that the interactions are a favorable factor for lowest values of ΔG here obtained.
The alkyl or aryl interactions have strong hydrophobic characteristic and they are related to favorable entropic factors can promote the lowering of the free energy of ligand-receptor binding and all the tested ligands were capable of forming hydrogen interactions with residues at their respective binding sites [24,25,26,27,28]. This result confirms that proposed phenylbutazone derivatives have in silico anti-inflammatory activity via COX inhibition, as well as, in silico study is a valuable tool as a support for in vivo assays and from the in silico analysis, mechanisms can be clarified at the molecular level.
In the rational drug design process, availability of 3D (three-dimensional) protein structures is crucially important. The X-ray crystallography, considered an efficient tool in determining protein 3D poses, is a time-consuming and expensive method. In addition, certain proteins cannot be successfully crystallized or are difficult to crystallize, for example the membrane proteins. Therefore, a very low number of membrane proteins structures has been determined. The recent breakthroughs indicate that NMR is indeed a very powerful tool in determining the 3D structures of membrane proteins [29,30,31,32] but it is also time-consuming and costly. To acquire the structural information in a timely manner, a series of 3D protein structures were developed by means of homology (or comparative) technique and were found very useful for drug development [33,34,35].
2.6. Toxicological Properties
In this present study, only structures 1, 7, 8, 14 and 15 showed good results of molecular docking as well as predictions of toxicological properties, in comparison to the results obtained for the template molecules (Indomethacin, Refocoxib and Phenylbutazone). The toxicological properties of the structures containing toxicity alarms are shown in Table 3. It is noted that indomethacin in the toxicity analysis did not show any alert, a fact that could be due to the low concentration in which the compound acts inside the active site. Analysis of the toxicological properties allowed us to observe that all the structures point out toxicity alerts characterized as “plausible” or “acceptable”—see Table 3.
Table 3.
Predictions of the toxicological properties of structures investigated.
| Structures | Toxicity Prediction Alert (Lhasa Prediction) | Toxicophoric Group | Toxicity Alert | Toxicity Prediction (Custom Prediction) |
|---|---|---|---|---|
| Indometacin | - | - | - | Nothing to declare |
| Refecoxib | Hepatotoxicity in human, mouse and rat | Furan | PLAUSIBLE | Nothing to declare |
| Phenylbutazone (1) | Hepatotoxicity in human and mouse | 1,2-Diphenyl-3,5-pyrazolidinedione derivative | CERTAIN | Nothing to declare |
| Hepatotoxicity in rat | 1,2-Diphenyl-3,5-pyrazolidinedione derivative | PROBABLE | ||
| Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | ||
| 7 | Hepatotoxicity in human, mouse and rat | 1,2-Diphenyl-3,5-pyrazolidinedione derivative | PLAUSIBLE | Nothing to declare |
| Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | ||
| 8 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| 13 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| 14 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| 15 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
Refecoxib showed an alert of hepatotoxicity respective to furan derivatives. Many of these compounds have been reported to cause a dose-related hepatic necrosis in rodents including furosemide to produce midzonal to centrilobular necrosis in mice and rats [36,37,38,39]. The mechanism of toxicity is proposed to involve oxidative ring opening of the furan ring leading to formation of reactive intermediates, which can alkylate proteins or form adducts with DNA [40]. Evidence of liver toxicity for furan-containing compounds in humans is limited. For example furosemide has not been associated with liver effects in humans but it has caused jaundice and attention has been drawn to its potential hepatotoxicity in case of high dose administration to patients with renal failure [38,41,42]. Furan is a liver toxicant and hepatocarcinogen in rodents [43,44].
The hepatotoxicity of furan and related compounds is thought to result from the formation of chemically reactive metabolites [37,40]. The oxidative opening of the furan ring is primarily catalyzed by cytochrome P450s and is thought to proceed via the initial formation of a furan-epoxide. The scission of the ring leads to the formation of an electrophilic alpha, β-unsaturated dicarbonyl moiety, which can covalently bind to macromolecules via a Michael-type addition [40].
The scope of this alert has been defined by the common structural features of the active compounds in this class, namely the furan ring moiety. The monosubstituted 2- and 3-methylfurans are excluded on the basis they can be readily oxidized to carboxylic acids and excreted without causing toxic effects as supported by experimental studies in rodents [38,39]. Based on this data, carboxylic acids and their derivatives are also excluded from the furan ring. Nitro and alkyl amine compounds are also omitted as they are thought to induce toxicity via a different mechanism [45].
In Table 3, structures 1 and 7 are shown containing alerts of hepatotoxicity for 1,2-diphenyl-3,5-pyrazolidinedione derivatives. During their use as non-steroidal anti-inflammatory drugs (NSAIDs), this class has been reported to cause necrosis, steatosis and granulomas in the liver [26]. The mechanism of hepatotoxicity is thought to be an immune-mediated reaction but a dose related intrinsic effect has also been reported for phenylbutazone [46].
Phenylbutazone and its pharmacologically active metabolites oxyphenbutazone and kebuzone are used as NSAIDs. They may cause various side effects including several forms of hepatic injury occurring during treatment. In many reports of human liver toxicity by phenylbutazone (daily doses varying from 300 mg to 800 mg) the hepatocellular injury was described as being acute and the severity ranged from mild to moderate with or without cholestasis [36,46].
Phenylbutazone has been reported to cause steatosis, cholestasis and centrilobular necrosis, often with ballooning degeneration and hypertrophy of Kupffer cells. Noncaseating granulomas in the portal and periportal areas are observed in cases of hypersensitivity to this drug [46]. In addition, it has been reported that its active metabolites cause similar toxic effects [47,48]. Idiosyncratic liver injury was reported after administration of feprazone, (daily dose of 600 mg) when biopsies of individuals showed inflammation of the portal tract and parenchyma [49]. Sulfinpyrazone, an uricosuric agent, has been described as an inducer of microsomal enzymes and shown to cause hepatic injury in isolated cases [50].
Mice exposed to phenylbutazone by gavage (150–300 mg/kg) during a 2 years study were found to have hepatomegaly, steatosis, cellular degeneration and coagulative necrosis [51]. The mechanism of hepatotoxicity is not clear for this class but is reported to be mostly idiosyncratic, although the toxicity of phenylbutazone has been reported to have two components. The intrinsic toxicity of phenylbutazone is thought to cause cholestasis and necrosis, while cases of hypersensitivity indicated by the presence of granulomas, are thought to be due to idiosyncratic reactions [46].
Results obtained for compounds 1, 7, 8, 13–15 indicating toxicity alerts also include coverage for hydrazone precursors of hydrazines. The presence of a skin sensitization structural alert within a molecule indicates the molecule has the potential to cause skin sensitization. Whether or not the molecule will be a skin sensitizer will also depend upon its percutaneous absorption. Generally, small lipophilic molecules are more readily absorbed into the skin and are therefore more likely to cause sensitization [52,53,54]—see Table 3.
3. Materials and Methods
3.1. Theoretical
Initially, the calculations of Density Functional Theory (DFT) have also been employed in the determination of geometry and physic-chemical properties such as partition oil-water (Log P) and volume (V) for the phenylbutazone and its related derivatives [55]. It was done using computational program Gaussian 09 molecular package [56]. In order to understand the interaction way at the molecular level on phenylbutazone derivatives, their structural and electronic properties were obtained by parameters of reactivity and theoretical properties such as energies occupied molecular orbital of highest energy (HOMO) and the energy unoccupied molecular orbital of lowest energy molecular orbital (LUMO) and ionization potential (IP).
To examine the reactivity of the calculated compounds (HOMO-LUMO) GapL-H obtained by the difference in energy between the HOMO and LUMO [57] (Equation (1)).
| GapL-H = ELUMO − EHOMO | (1) |
The DFT method was employed, using the B3LYP functional hybrid [58,59] and the 6-31+G(d,p) basis set to phenylbutazone derivatives (PD) which act as electron donating group. The ionization potential (IP) was calculated as the energy difference between the neutral molecule (EPD) and the free radical cation (EPD•+) (Equation (2)). In addition, the stabilization energy (ΔEiso) was calculated by means of the energy’s difference between the phenylbutazone cation free radical (EPBT•+) plus neutral form of derivatives (EPD) and the respective phenylbutazone neutral molecule (EPBT) plus cation free radical of their derivatives (EPD•+) (Equation (3)).
| IP = EPD•+ − EPD | (2) |
| ΔEiso = (EPBT•+ + EPD) − (EPBT + EPD•+) | (3) |
3.2. Molecular Docking Simulations Study
The cyclooxygenase-2 (COX-2) structures resolved by X-ray diffraction was downloaded from the Protein Data Bank (PDB) with PDB codes 4COX (Mus musculus) and 5KIR (Homo sapiens) with resolution of 2.9 and 2.7 Å, respectively.
The control standard ligands used in docking molecular study were indomethacin (IMN) and rofecoxib (RCX) downloaded at PDB server in sdf format ensuring the bioactive conformation. All phenylbutazone derivatives structures were optimized using the density functional theory method (DFT) in theory level B3LYP/6-31+G(d,p). Also, the frequencies were also calculated using B3LYP/6-31+G(d,p) and observed that there were no negative frequencies, thereby ensuring that local minimum energy structure [60]. The phenylbutazone derivatives and therapeutic targets structures for molecular docking study were prepared using the software Discovery Studio 5.0 (San Diego, CA, USA) [61], removing the water molecules and for docking validations were downloaded with the therapeutic targets crystal structures and crystallographic inhibitors were used in molecular docking study, as well as, in validation through of the root-mean-square deviation (RMSD) value between the experimental data with computational data, according to studies carried out by Macêdo et al. (2015) [62] and Federico et al. (2017) [63].
The indomethacin structure is a non-selective inhibitor (Mus musculus, PDB 4COX) and rofecoxib structure bound to human COX-2 (Homo sapiens, PDB 5KIR) were used via AutoDock 4.2/Vina 1.1.2 (Scripps Research Ins., San Diego, CA, USA) by graphical interface Pyrx (version 0.8.30) with default parameters via the genetic algorithm, following the protocol described previously by Padilha et al. (2016), Pereira et al. (2018) and Costa et al. (2018) [64,65,66]. The population size was 100, selection-pressure was 1.1, the number of operations was 10,000, the number of islands was 1, the niche size was 2, operator weights for migrate was 0, mutate was 100 and crossover was 100. Spatial coordinates used for the center of the grid is described in Table 4.
Table 4.
Data from protocols used for molecular docking validation purposes.
| Receptor | Ligand | Ligand Coordinates of the Grid Center | Grid Size (Points) |
|---|---|---|---|
| COX-2 (PDB code: 4COX) Mus musculus |
Indomethacin | X = 11.5685 Y = 12.3903 Z = 7.2430 |
26 x 30 y 28 z |
| COX-2 (PDB code: 5KIR) Homo sapiens |
Rofecoxib | X = 26.6293 Y = 40.1834 Z = 3.8484 |
26 x 30 y 28 z |
The coordinates of the grid box located at the pocket of interest sere selected based on interactions observed between inhibitor and enzyme, inside a 10 Å radius sphere then defined. Ten solutions were calculated for each phenylbutazone derivatives and minimum binding energy conformations were analyzed.
Docking studies both COX-2 and their complexed specific ligands were carried out using the AutoDock 4.2/Vina 1.1.2 and PyRx 0.8.30 software. Spatial coordinates for PDB ID 4COX and PDB ID 5KIR were chosen according to the interaction observed between the two COX enzymes and their respective original ligands. The visualizations and distance of ligands interactions with COX were in Discovery Studio 5.0.
The energy score function was used to assess the binding free energy (ΔG) of interactions between COX and phenylbutazone derivatives in PyRx 0.8.30. Analysis of each pose (conformation + orientation) of the ligands were also taken into account for selection of the best ΔG values as well as binding affinity, using AutoDock 4.2/Vina 1.1.2, in order to evaluate the selectivity (and binding affinity) of the phenylbutazone derivatives regarding the targeted COX-2 structures here used (from Mus musculus and Homo sapiens). Both the indomethacin and rofecoxib were used as templates or positive control controls for inhibiting COX-2 because they are known potent COX-2 inhibitors.
3.3. Toxicological Predictions
Toxicity profile of the structures was evaluated using the Deductive Estimation of Risk from Existing Knowledge (DEREK) 10.0.2 software (Leeds, UK) [67]. We have considered DEREK alerts of toxicity involving the human, mouse and rat species. DEREK makes the prediction of toxicity of the structures in a qualitative way, since it is an expert system that focuses attention on the toxic action of chemical compounds. The system performs this analysis based on implemented rules and depicts the relationship between a structural feature and a toxicophoric group present in the compounds as possible inducers of certain types of toxicity. It is considered that, in addition to toxicity, DEREK can identify specific aspects related to carcinogenicity, mutagenicity, skin sensitization, irritation, teratogenicity and neurotoxicity [68,69].
4. Conclusions
Our results using DFT/B3LYP/6-31+G(d,p) calculations showed that the molecular orbitals, ionization potential and spin density contributions of phenylbutazone can be used to explain their oxidation prediction and chemical reactivity. Both phenyl rings have more influence under electron donation capacity than alkyl moiety. Aromatic hydroxylation ring has more impact on electron donation capacity than alkyl hydroxylation. The spin densities contribution showed that a regioselective hydroxylation is more favored at the para-positions of phenyl ring than other positions. Docking results here obtained indicate that compounds 1, 7, 8 and 13–15 have potential capacity of COX-2 inhibition in human and mice enzyme as well, due to contain similar interactions to the observed for the templates or control compounds (indomethacin and refecoxib). The toxicity could be here related to an aromatic hydroxylation. In accordance to our calculations, the proposed derivatives are potentially more active and safer than phenylbutazone and only compounds 8 and 13–15 were the most promising. These results could explain the biological properties observed for phenylbutazone and they support the design of novel and safer derivative candidates.
Supplementary Materials
The following are available online.
Author Contributions
Conceptualization, R.S.B.; tautomerism study, I.C.P.; molecular simplification, S.S.B.O., electronic study, R.B.M.; molecular graphics V.A.B.; writing—review & editing, C.H.T.P.S., J.M.C., R.S.R., R.C.S., J.d.S.C. and C.B.R.S.
Funding
We acknowledge the support provided by the PROPESP/UFPA and CNPq for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Lees P., Toutain P. Pharmacokinetics, pharmacodynamics, metabolism, toxicology and residues of phenylbutazone in humans and horses. Vet. J. 2013;196:294–303. doi: 10.1016/j.tvjl.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 2.MacAllister C.G., Morgan S.J., Borne A.T., Pollet R.A.J. Comparison of adverse effects of phenylbutazone, flunixin meglumine. and ketoprofen in horses. J. Am. Vet. Med. Assoc. 1993;202:71–77. [PubMed] [Google Scholar]
- 3.Lees P., Higgins A.J. Clinical pharmacology and therapeutic uses of non-steroidal anti-inflammatory drugs in the horse. Equine Vet. J. 1985;17:83–96. doi: 10.1111/j.2042-3306.1985.tb02056.x. [DOI] [PubMed] [Google Scholar]
- 4.Mathews K.A. Non-steroidal anti-inflammatory analgesics: A review of current practice. J. Vet. Emerg. Cris. Care. 2002;12:89–97. doi: 10.1046/j.1435-6935.2002.00007.x. [DOI] [Google Scholar]
- 5.Fitzpatrick J.L., Nolan A.M., Lees P., May S.A. Inflammation and pain. In: Andrews A.H., Blowey R.W., Boyd H., Eddy R.G., editors. Bovine Medicine Diseases Husandry Cattle. 2nd ed. Blackwell Science Ltd.; Oxford, UK: 2004. pp. 1050–1056. [Google Scholar]
- 6.Collins L.G., Tyler D.E. Phenylbutazone toxicosis in the horse: A clinical study. J. Am. Vet. Med. Assoc. 1984;184:699–703. [PubMed] [Google Scholar]
- 7.Triggs E.J., Whyatt P.L., Eckert G. Bioavailability evaluation of an enteric-coated phenylbutazone formulation. and cross-over comparison with a sugar-coated product. Med. J. Aust. 1977;2:830–833. doi: 10.5694/j.1326-5377.1977.tb99335.x. [DOI] [PubMed] [Google Scholar]
- 8.Silva B.H.S., Barros T.G., Barros C.A.L., Vieira J.L.F., Borges R.S. An electronic study of biphenyl derivatives chlorinated. J. Comp. Theor. Nanosc. 2011;8:216–219. doi: 10.1166/jctn.2011.1680. [DOI] [Google Scholar]
- 9.Levy G.N. Prostaglandin H synthases. nonsteroidal anti-inflammatory drugs and colon cancer. FASEB J. 1997;11:234–247. doi: 10.1096/fasebj.11.4.9068612. [DOI] [PubMed] [Google Scholar]
- 10.Kidd B.L., Urban L.A. Mechanisms of inflammatory pain. Br. J. Anaesth. 2001;87:3–11. doi: 10.1093/bja/87.1.3. [DOI] [PubMed] [Google Scholar]
- 11.Queiroz A.N., Mendes A.P.S., Leal M.R.S., Chaves Neto A.M.J., Borges R.S. Tautomerism and radical-scavenging activity of edaravone by DFT methods. J. Comp. Threoret. Nanosc. 2010;7:153–156. doi: 10.1166/jctn.2010.1339. [DOI] [Google Scholar]
- 12.Antonczak S. Electronic description of four flavonoids revisited by DFT method. J. Mol. Struct. TheoChem. 2008;856:38–45. doi: 10.1016/j.theochem.2008.01.014. [DOI] [Google Scholar]
- 13.Queiroz A.N., Gomes B.A.Q., Moraes W.M.J., Borges R.S. A theoretical antioxidant pharmacophore for resveratrol. Eur. J. Med. Chem. 2009;44:1644–1649. doi: 10.1016/j.ejmech.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 14.Lobo V., Patil A., Phatak A., Chandra N. Free radicals antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010;4:118–126. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wood C.M. Toxic responses of the gill. In: Schlenk D., Benson W.H., editors. Target Organ Toxicity in Marine and Freshwater Teleosts. 2nd ed. CRC Press; London, UK: 2001. pp. 1–26. [Google Scholar]
- 16.Borges R.S., Queiroz A.N., Mendes A.P.S., Araújo S.C., França L.C.S., Franco E.C.S., Gomes-Leal W., Silva A.B.F. Density functional theory (DFT) study of edaravone derivatives as antioxidants. Int. J. Mol. Sci. 2012;13:7594–7606. doi: 10.3390/ijms13067594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvalho E.S., Santa-Brígida S.A., Queiroz A.N., Silva J.R., Silva O.P.P., Barros C.A.L., Borges R.S. Edaravone toxicity can be related to redox properties of their oxidized derivatives. Chem. Data Collect. 2017;7–8:51–57. doi: 10.1016/j.cdc.2017.02.001. [DOI] [Google Scholar]
- 18.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 19.Gowtham U., Jayakanthan M., Sundar D. Molecular docking studies of dithionitrobenzoic acid and its related compounds to protein disulfide isomerase: Computational screening of inhibitors to HIV-1 entry. BMC Bioinform. 2008;9:1–10. doi: 10.1186/1471-2105-9-S12-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hevener K.E., Zhao W., Ball D.M., Babaoglu K., Qi J., White S.W., Lee R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inform. Model. 2009;49:444–460. doi: 10.1021/ci800293n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurumbail R.G., Stevens A.M., Gierse J.K., Mcdonald J.J., Stegeman R.A., Pak J.Y., Gildehaus D., Iyashiro J.M., Penning T.D., Seibert K., et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 22.Orlando B.J., Malkowski M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. 2016;72:772–776. doi: 10.1107/S2053230X16014230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beretta C., Garavaglia G., Cavalli M. COX-1 and COX-2 inhibition in horse blood by phenylbutazone. flunixin. carprofen and meloxicam: An in vitro analysis. Pharm. Res. 2005;52:302–306. doi: 10.1016/j.phrs.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Costa J.S., Costa K.S.L., Cruzb J.V., Ramos R.S., Silva L.B., Brasil D.S.B., Tomich C.S., Rodrigues C.B., Macêdo W.J.C. Virtual screening and statistical analysis in the design of new caffeine analogues molecules with potential epithelial anticancer activity. Curr. Pharm. Des. 2018;24:576–594. doi: 10.2174/1381612823666170711112510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruz J.V., Neto M.F.A., Silva L.B., Ramos R., Costa J., Brasil D.S.B., Lobato C.C., Costa G.V., Bittencourt J.A.H.M., Silva C.H.T.P., et al. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules. 2018;23:453. doi: 10.3390/molecules23020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos C.B.R., Ramos R.S., Ortiza B.L.S., Silva G.M., Giuliatti S., Navarrete J.L.A., Carvalho J.C.T. Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol. 2018;222:107–120. doi: 10.1016/j.jep.2018.04.041. [DOI] [PubMed] [Google Scholar]
- 27.Teles Fujishima M.A., Silva N.S.R., Ramos R.S., Batista Ferreira E.F., Santos K.L.B., Silva C.H.T.P., Silva J.O., Campos Rosa J.M., Santos C.B.R. An antioxidant potential, quantum-chemical and molecular docking study of the major chemical constituents present in the leaves of Curatella americana Linn. Pharmaceuticals. 2018;11:72. doi: 10.3390/ph11030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz J.V., Serafim R.B., Silva G.M., Giuliatti S., Rosa J.M.C., Araújo Neto M.F., Leite F.H.A., Taft C.A., Silva C.H.T.P., Santos C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening and molecular dynamics. J. Mol. Model. 2018;24:225. doi: 10.1007/s00894-018-3756-y. [DOI] [PubMed] [Google Scholar]
- 29.Oxenoid K., Dong Y.S., Cao C., Cui T., Sancak Y., Markhard A.L., Grabarek Z., Kong L., Liu Z., Ouyang B., et al. Architecture of the mitochondrial calcium uniporter. Nature. 2016;533:269–273. doi: 10.1038/nature17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dev J., Park D., Fu Q., Chen J., Ha H.J., Ghantous F., Herrmann T., Chang W., Liu Z., Frey G., et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science. 2016;353:172–175. doi: 10.1126/science.aaf7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.OuYang B., Xie S., Berardi M.J., Zhao X.M., Dev J., Yu W., Sun B., Chou J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature. 2013;498:521–525. doi: 10.1038/nature12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Q., Fu T.M., Cruz A.C., Sengupta P., Thomas S.K., Wang S., Siegel R.M., Wu H., Chou J.J. Structural basis and functional role of intramembrane trimerization of the Fas/CD95 death receptor. Mol. Cell. 2016;61:602–613. doi: 10.1016/j.molcel.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X.B., Wang S.Q., Xu W.R., Wang R.L. Novel Inhibitor Design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS ONE. 2011;6:e28111. doi: 10.1371/journal.pone.0028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Y., Wang S.Q., Xu W.R., Wang R.L. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS ONE. 2012;7:e38546. doi: 10.1371/journal.pone.0038546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou K.C. Review: Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem. 2004;11:2105–2134. doi: 10.2174/0929867043364667. [DOI] [PubMed] [Google Scholar]
- 36.Zimmerman H.J. Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver. 2nd ed. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 1999. p. 789. [Google Scholar]
- 37.McMurtry R.J., Mitchell J.R. Renal and hepatic necrosis after metabolic activation of 2-substituted furans and thiophenes, including furosemide and cephaloridine. Toxicol. Appl. Pharmacol. 1977;42:285–300. doi: 10.1016/0041-008X(77)90005-9. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell J.R., Potter W.Z., Hinson J.A., Jollow D.J. Hepatic necrosis caused by furosemide. Nature. 1974;251:508–511. doi: 10.1038/251508a0. [DOI] [PubMed] [Google Scholar]
- 39.Wiley R.A., Traiger G.J., Baraban S., Gammal L.M. Toxicity-distribution relationships among 3-alkylfurans in mouse liver and kidney. Toxicol. Appl. Pharmacol. 1984;74:1–9. doi: 10.1016/0041-008X(84)90263-1. [DOI] [PubMed] [Google Scholar]
- 40.Dalvie D.K., Kalgutkar A.S., Khojasteh-Bakht S.C., Obach R.S., O’Donnell J.P. Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol. 2002;15:269–299. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 41.Alvarez-Diez T.M., Zheng J. Mechanism-based inactivation of cytochrome P450 3A4 by 4-ipomeanol. Chem. Res. Toxicol. 2004;17:150–157. doi: 10.1021/tx034143l. [DOI] [PubMed] [Google Scholar]
- 42.Gordon W.P., Forte A.J., McMurtry R.J., Gal J., Nelson S.D. Hepatotoxicity and pulmonary toxicity of pennyroyal oil and its constituent terpenes in the mouse. Toxicol. Appl. Pharmacol. 1982;65:413–424. doi: 10.1016/0041-008X(82)90387-8. [DOI] [PubMed] [Google Scholar]
- 43.Peterson L.A., Naruko K.C., Predecki D.P. A reactive metabolite of furan, cis-2-butene-1,4-dial, is mutagenic in the Ames assay. Chem. Res. Toxicol. 2000;13:531–534. doi: 10.1021/tx000065f. [DOI] [PubMed] [Google Scholar]
- 44.Hamadeh H.K., Jayadev S., Gaillard E.T., Huang Q., Stoll R., Blanchard K., Chou J., Tucker C.J., Collins J., Maronpot R., et al. Integration of clinical and gene expression endpoints to explore furan-mediated hepatotoxicity. Mutat. Res. 2004;549:169–183. doi: 10.1016/j.mrfmmm.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 45.Nelson S.D. Structure toxicity relationships—How useful are they in predicting toxicities of new drugs? Adv. Exp. Med. Biol. 2001;500:33–43. doi: 10.1007/978-1-4615-0667-6_4. [DOI] [PubMed] [Google Scholar]
- 46.Benjamin S.B., Ishak K.G., Zimmerman H.J., Grushka A. Phenylbutazone liver injury: A clinical-pathologic survey of 23 cases and review of the literature. Hepatology. 1981;1:255–263. doi: 10.1002/hep.1840010311. [DOI] [PubMed] [Google Scholar]
- 47.Gaisford W. Fatality after oxyphenbutazone in Still’s disease. Br. Med. J. 1962;2:1517. doi: 10.1136/bmj.2.5318.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kunze K.D., Porst H., Tschopel L. Morphology and pathogenesis of liver injury produced by dihydralazine, propranolol and ketophenylbutazone. Zentralbl. Allg. Pathol. Pathol. 1985;130:509–518. (In German) [PubMed] [Google Scholar]
- 49.Wiggins J., Scott D.L. Hepatic injury following feprazone therapy. Rheumatol. Rehabil. 1981;20:44–45. doi: 10.1093/rheumatology/20.1.44. [DOI] [PubMed] [Google Scholar]
- 50.Muller H.G. Jaundice following sulfinpyrazone administration. Dtsch. Med. Wochenschr. 1981;106:1117. (In German) [PubMed] [Google Scholar]
- 51.Kari F., Bucher J., Haseman J., Eustis S., Huff J. Long-term exposure to the anti-inflammatory agent phenylbutazone induces kidney tumors in rats and liver tumors in mice. Jpn. J. Cancer Res. 1995;86:252–263. doi: 10.1111/j.1349-7006.1995.tb03048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kayser D., Schlede E. Chemikalien und Kontaktallergie—Eine bewertende Zusammenstellung. Verlag Urban & Vogel; Munique, Germany: 2001. (In German) [Google Scholar]
- 53.Wahlberg J.E., Boman A. Guinea pig maximization test. Curr. Problems Dermatol. 1985;14:59–106. doi: 10.1159/000411607. [DOI] [PubMed] [Google Scholar]
- 54.Rycroft R.J.G., Wilkinson J.D. Irritants and sensitisers. In: Champion R.H., Burton J.L., Ebling F.J.G., editors. Textbook of Dermatology. 5th ed. Volume 1. Blackwell Science Ltd.; Oxford, UK: 1991. pp. 717–754. [Google Scholar]
- 55.Parr R.G., Yang W. Density Functional Theory of Atoms and Molecules. Oxford University Press; Oxford, UK: 1989. [Google Scholar]
- 56.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian 09 Revision A.02. Gaussian Inc.; Wallingford, CT, USA: 2009. [Google Scholar]
- 57.Horton W., Peerannawar S., Török B., Török M. Theoretical and experimental analysis of the antioxidant features of substituted phenol and aniline model compounds. Struct. Chem. 2018 doi: 10.1007/s11224-018-1183-4. [DOI] [Google Scholar]
- 58.Becke A.D.J. Density-funtional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993;98:1372–1377. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 59.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1998;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 60.Santos C.B.R., Lobato C.C., Braga F.S., Costa J.S., Favacho H.A.S., Carvalho J.C.T., Macedo W.J.C., Brasil D.S.B., Silva C.H.T.P., Hage-Melim L.I.S. Rational design of antimalarial drugs using molecular modeling and statistical analysis. Curr. Pharm. Desig. 2015;21:4112–4127. doi: 10.2174/1381612821666150528121423. [DOI] [PubMed] [Google Scholar]
- 61.Discovery Studio Modeling Environment. Dassault Systemes BIOVIA; San Diego, CA, USA: 2015. version 5.0. [Google Scholar]
- 62.Macêdo W.J.C., Braga F.S., Santos C.F., Costa J.S., Melo G.S., Mello M.N., Sousa D.S., Carvalho J.C.T., Brasil D.S.B., Santos C.B.R. Antimalarial artemisinins derivatives study: Molecular modeling and multivariate analysis: PCA. HCA. KNN. SIMCA and SDA. J. Comp. Threoret. Nanosc. 2015;12:3443–3458. doi: 10.1166/jctn.2015.4138. [DOI] [Google Scholar]
- 63.Federico L., Santos C.B.R., Lobato C., Gomes J., Rosa J.M.C., Silva C.H.T.P. Ligand- and structure-based drug design of novel calcium channel blockers. J. Comp. Threoret. Nanosc. 2017;14:3489–3502. doi: 10.1166/jctn.2017.6519. [DOI] [Google Scholar]
- 64.Padilha E.C., Serafim R.B., Sarmiento D.Y.R., Santos C.F., Santos C.B.R., Silva C.H.T.P. New PPARα/γ/δ optimal activator rationally designed by computational methods. Braz. Chem. Soc. 2016;27:1636–1647. doi: 10.5935/0103-5053.20160043. [DOI] [Google Scholar]
- 65.Pereira A.L.E., Santos G.B., Franco M.S.F., Federico L.B., Silva C.H.T.P., Santos C.B.R. Molecular modeling and statistical analysis in the design of derivatives of human dipeptidyl peptidase IV. J. Biomol. Struct. Dyn. 2018;36:318–334. doi: 10.1080/07391102.2016.1277163. [DOI] [PubMed] [Google Scholar]
- 66.Costa J.S., Ramos R.S., Costa K.S.L., Brasil D.S.B., Silva C.H.T.P., Ferreira E.F.B., Borges R.S., Campos J.M., Macêdo W.J.C., Santos C.B.R. An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules. 2018;23:2801. doi: 10.3390/molecules23112801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Derek for Windows. Department of Chemistry, University of Leeds; Leeds, UK: 2007. version 10.0.2; User Guide. Lhasa Limited. [Google Scholar]
- 68.Rindings J.E., Barratt M.D., Cary R. Computer prediction of possible toxic action from chemical structure, an update of the DEREK system. Toxicology. 1996;106:267–279. doi: 10.1016/0300-483X(95)03190-Q. [DOI] [PubMed] [Google Scholar]
- 69.Barcellos M.P., Santos C.B.R., Federico L.B., Almeida P.F., Silva C.H.T.P., Taft C.A. Pharmacophore and structure-based drug design, molecular dynamics and admet/tox studies to design novel potential pad4 inhibitors. J. Biomol. Struct. Dyn. 2018:1–33. doi: 10.1080/07391102.2018.1444511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.