

Abstract
E-selectin mediates the rolling of circulating leukocytes during inflammatory processes. Previous genome-wide association studies in European and Asian individuals have identified the ABO locus associated with E-selectin levels. Using Trans-Omics for Precision Medicine whole genome sequencing data in 2249 African Americans (AAs) from the Jackson Heart Study, we examined genome-wide associations with soluble E-selectin levels. In addition to replicating known signals at ABO, we identified a novel association of a common loss-of-function, missense variant in Fucosyltransferase 6 (FUT6; rs17855739,p.Glu274Lys, P = 9.02 × 10−24) with higher soluble E-selectin levels. This variant is considerably more common in populations of African ancestry compared to non-African ancestry populations. We replicated the association of FUT6 p.Glu274Lys with higher soluble E-selectin in an independent population of 748 AAs from the Women’s Health Initiative and identified an additional pleiotropic association with vitamin B12 levels. Despite the broad role of both selectins and fucosyltransferases in various inflammatory, immune and cancer-related processes, we were unable to identify any additional disease associations of the FUT6 p.Glu274Lys variant in an electronic medical record-based phenome-wide association scan of over 9000 AAs.
Introduction
E-selectin is a C-type lectin, expressed on endothelial cells, activated by cytokines, and mediates the rolling of circulating leukocytes during inflammatory processes. E-selectin recognizes and binds to sialylated carbohydrates (Lewis x antigen groups) present on surface proteins of leukocytes. Endothelial cell dysfunction is an early step of atherosclerotic plaque formation and leads to the activation of endothelial cells (1). Activated endothelial cells have increased expression of monocyte interaction, chemoattractants and adhesion molecules, such as selectins (2). When E-selectin binds to leukocytes’ ligands on the cell surface, leukocyte rolling is slowed, which facilitates transmigration into the intimal space of the arterial wall (3). Entry of the leukocyte beyond the wall leads to monocytes differentiation into foam cells and initiation of fatty streak formation (4). Furthermore, through intimal thickening caused by foamy macrophages, a necrotic core develops into a fibroathermoa whose rupture can lead to myocardial infarction or sudden cardiac death (5). Mutations associated with E-selectin may disrupt the binding to ligands expressed by neutrophils, monocytes, eosinophils, memory-effector T-like lymphocytes or natural killer cells, leading to increased leukocyte recruitment (6,7). E-selectin has potential implications for both risk stratification and therapeutic development for a variety of autoimmune and cardiovascular diseases (CVDs) (8–12) characterized by excessive inflammatory response (13,14).
A soluble form of E-selectin, shed from damaged or activated endothelial cells and released into the circulation, has been associated with various cardiometabolic disorders (15). Soluble E-selectin levels differ by sex and ethnicity (15,16) and demonstrate significant heritability (17). In several genome-wide association studies (GWAS) of soluble E-selectin conducted among individuals of European ancestry, the ABO locus (chromosomal region 9q24), which encodes a glycosyltransferase that determines the ABO blood group, has been the only genome-wide significant locus, explaining 9–19% of the phenotypic variance of E-selectin levels (18–20). In particular, SNPs that tag the A1 blood group (rs651007, rs579459) are associated with lower soluble E-selectin levels compared to individuals who are genetically inferred type O (18,19,21,22).
To further elucidate the genetic determinants of soluble E-selectin levels, we analyzed whole genome sequence data from a large African American cohort [the Jackson Heart Study (JHS)] with soluble E-selectin measured. Given the lower effect sizes expected for non-coding variation and the difficulty of replication given the limited whole genome sequencing (WGS) sample sizes available in African Americans (AAs), we performed aggregate tests only for rare coding variants, and single-variant tests for only low frequency and common variation [minor allele frequency (MAF) > =0.1%]. However, the availability of sequencing data in JHS still provided a number of advantages for our analysis, notably better assessment of low frequency variants and improved haplotype construction in the ABO region (23). Past GWAS tended to exclude AAs, despite the population being disproportionately affected by many severe diseases (24). Misclassification of risk variants (e.g. as false genetic positive by ethnic group) may also contribute to health disparities, especially in instances of ambiguous diagnoses (25). In addition, individuals of African ancestry have, on average, greater genetic diversity than individuals of European ancestry. This facilitated our discovery of significant genetic variant associations that were missed by previous GWAS. We followed up newly discovered loci in a sample of AAs from the Women’s Health Initiative (WHI) and compared association results between two different soluble E-selectin assay methods, enzyme-linked immunosorbent assay (ELISA) and an aptamer-based proteomic technology. Finally, we performed a phenome-wide association study (PheWAS) of newly identified soluble E-selectin genetic loci using electronic medical record (EMR)-linked biobanks to investigate the potential clinical impact of our findings.
Results
WGS (at 30 × depth) following by multi-sample genotype calling was performed on a sample of 3128 AAs from JHS (see Materials and Methods for details). Of the 3128, 2249 had available E-selection measures (see Supplementary Material, Table S1 for baseline characteristics). Genome-wide discovery was performed for 30 095 928 variants [single nucleotide variants (SNVs) and small indels] with MAF > = 0.001. There were three genome-wide significant loci at 9q24 (85 variants), 1q24 (70 variants) and 19p13 (31 variants), with sentinel variants ABO rs635634 (P = 7.61 × 10−76), SELL-SELE rs3917422 (P = 1.64 × 10−45) and FUT6 rs17855739 (P = 3.49 × 10−22), respectively. (Fig. 1; Table 1). As the FUT6 rs17855739 association represented the only new locus for E-selectin levels, we only sought replication for rs17855739. There was no evidence of genomic inflation within different allele frequency bins (Fig. 1). Among related individuals from JHS, we estimated the age and sex adjusted heritability of E-selectin at 0.741.
Figure 1.
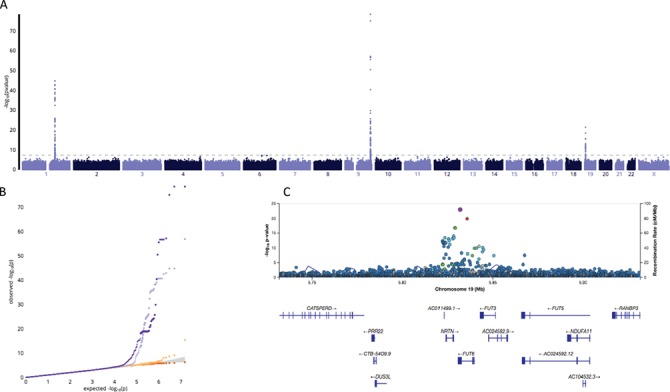
Manhattan plot of genome-wide associations with soluble E-selectin levels (A); quantile-quantile plots (QQplots) of P-values from the genome-wide association tests with soluble E-selectin levels (B) at four different MAF bins (red: 0.001 < = MAF < 0.003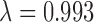 , orange: 0.003 < = MAF < 0.011
, orange: 0.003 < = MAF < 0.011 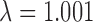 , violet: 0.011 < = MAF < 0.066
, violet: 0.011 < = MAF < 0.066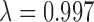 , purple: 0.066 < = MAF < = 0.5
, purple: 0.066 < = MAF < = 0.5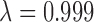 ); Regional plot of E-selectin associations at the FUT6 locus (C), with the rs17855739 variant highlighted in purple and pairwise LD denoted by color from red (strong) to green (moderate) to blue (weak).
); Regional plot of E-selectin associations at the FUT6 locus (C), with the rs17855739 variant highlighted in purple and pairwise LD denoted by color from red (strong) to green (moderate) to blue (weak).
Table 1.
Summary statistics of top associations with E-selectin levels
| rsID | Chr:pos | Ref/Alt | MAF | Gene region | Beta | SE | P |
|---|---|---|---|---|---|---|---|
| rs635634 | 9:133279427 | T/C | 0.095 | ABO | 0.925 | 0.0483 | 7.61 × 10−76 |
| rs3917422 | 1:169729619 | T/G | 0.021 | SELE | −1.472 | 0.1017 | 1.64 × 10−45 |
| rs17855739 | 19:5831829 | C/T | 0.294 | FUT6 | 0.3211 | 0.0328 | 3.49 × 10−22 |
SE: standard error
Using the gene-based Sequence Kernel Association Test (SKAT)-O test for rare-variant associations within coding regions, we observed a single strong genome-wide significant association at SELE (P = 1.96 × 10−37). The association signals at each of the three loci are described further below.
ABO
The E-selectin association signal on chromosome 9q24 locus encompassed a ∼25 kb region. The sentinel variant rs635634 (MAF = 0.095) is an upstream SNV near ABO, which encodes glycosyltransferases responsible for the ABO blood group system (Table 1). In JHS, the sentinel SNV rs635634 is in moderately strong linkage disequilibrium (LD) with two previously reported ABO genetic variants associated with lower E-selectin levels, rs651007 and rs579459 (Rsq = 0.76), both of which tag the A1 blood group allele. The rs635634 signal was consistent across the ELISA and SOMAScan platforms (Supplementary Material, Table S2) in a subset of 210 JHS participants, suggesting this signal is robust to E-selectin assay. After conditioning on rs635634, neither rs651007 nor rs579459 remained associated with soluble E-selectin (P = 0.688 and 0.760, respectively); however, a region-wide significant signal was additionally observed in conditional analysis at rs8176722 (MAF = 0.13; P = 1.35 × 10−7), which is in moderately strong LD (Rsq∼0.9) with several ABO missense variants (rs8176741, rs8176746, rs8176747) that define the B blood group allele. We therefore sought to characterize the associations between each of the genetically determined blood groups and soluble E-selectin levels. Consistent with previous reports, we present evidence that the O, A1 and B diplotype blood groups, with OO as referent blood group, are independently associated with lower soluble E-selectin levels, whereas the A2 blood group (both A2O and A2A2) does not appear to have an effect (Supplementary Material, Table S3).
Fucosyltransferase 6
We identified a novel soluble E-selectin genome-wide significant association signal at chromosome 19p13 encompassing Fucosyltransferase 6 (FUT6)-FUT3, with the sentinel signal at a missense variant in FUT6 (rs17855739, Glu274Lys, MAF = 0.294) associated with higher E-selectin levels (Table 1) and explaining ∼5% of the phenotypic variance. Replication in an independent sample (N = 765) of AAs from WHI confirmed the E-selectin - rs17855739 association [beta coefficient (beta) =0.2398, P = 4.03 × 10−5, MAF = 0.28]. Previous GWAS of soluble E-selectin included only individuals of European and Asian ancestry and did not identify the FUT6 rs17855739, which is more frequent among individuals of African ancestry. In the 1000 Genomes Phase 1 (HaploReg v4.1), the frequency of the FUT6 rs17855739 variant allele in Africans is 0.39 (compared to 0.294 in JHS AAs), while the frequency in European and East Asian ancestral groups ranged from 0.03–0.06. Sex-stratified analyses of the rs17855739 association yielded similar effect sizes for males and females (beta = 0.152 and beta = 0.158, respectively) and a non-significant (P = 0.66) interaction term for sex.
After conditioning on the sentinel E-selectin-associated FUT6 rs17855739 variant in JHS, there were no further genome-wide significant signals in the genomic region, though there were some residual association signals in FUT6 (Supplementary Material, Table S4). Therefore, we cannot completely rule out a modest secondary signal. In a subset of 210 JHS individuals who had soluble E-selectin measured using both antibody-based ELISA and aptamer-based SOMAScan platforms, the association with FUT6 rs17855739 was consistent across platforms (ELISA beta = 0.308, P = 3.3 × 10−3, SOMAScan beta = 0.303, P = 3.9 × 10−3) (Supplementary Material, Table S2).
The FUT6 gene encodes plasma alpha (1,3) fucosyltransferase, a Golgi stack membrane protein involved in the creation of sialyl-Lewis X, an E-selectin ligand. Mollicone et al. (30) described the identical FUT6 Glu274Lys mutation in the homozygous state as the apparent cause of plasma alpha (1,3) fucosyltransferase deficiency, suggesting that it may be a loss of function (LoF), recessively acting mutation. Indeed, the SIFT score for rs17855739 was reported at 0.015 (predicted deleterious) and the PolyPhen HVAR score at 0.832 (possibly damaging). In screening the FUT6 rs17855739 LoF variant for association with other plasma biomarkers measured in JHS, there was a nominal association with higher vitamin B12 levels (P = 0.012). Interestingly, we found a better model fit for a quadratic model of the T allele for rs17855739 compared to the additive model (P-value for the F-test = 2.2 × 10−5) for E-selectin levels but that the T allele acted recessively for vitamin B12 levels (Table 2). In addition, we found there to be strong evidence of interaction between ABO blood groups and rs17855739 on E-selectin levels, with differential effects of rs17855739 in the presence of the A1-O and B-O groups in comparison to O-O (Supplementary Material, Table S5). To investigate whether rs17855739 is associated with any other clinical phenotypes, we conducted a PheWAS in a total of 9066 AAs from the Biobank of Vanderbilt University (BioVU) and BioMe EMR-based biobanks (see Materials and Methods) under both additive and recessive models. No results reached significance after Bonferroni correction for testing 339 diagnostic codes.
Table 2.
Akaike information criterion (AIC) and Bayesian information criterion (BIC) (in parentheses) of three different models for rs17855739
| Analyte | Additive | Recessive | Quadratic |
|---|---|---|---|
| E-selectin | 3010 (3027) | 3013 (3030) | 2994 (3017) |
| Vitamin B12 | 1418 (1433) | 1413 (1428) | 1415 (1435) |
The three different models were additive, recessive and quadratic.
SELE
The association signal on chromosome 1q24 spanned a broad 500 kb region including the E-selectin (SELE) and L-selectin (SELL) structural genes (Fig. 1). The most strongly associated marker (rs3917422, MAF = 0.021) is a missense variant (p.Gln257Pro), located in the extracellular domain of the E-selectin protein. The SIFT score for rs3917422 was reported at 0.26928 (not deleterious) and the PolyPhen HVAR score at 0.001 (benign). Previous GWAS of soluble E-selectin did not identify the SELE variant as only individuals of European and Asian ancestry were included, and the variant was more frequent among individuals of African ancestry. The frequency of the SELE variant allele in Africans is 2%, while the frequency in European and Chinese ancestral groups is less than 0.1%.
In gene burden analysis, we also observed a strong association of rare variants in SELE with soluble E-selectin levels (SKAT-O P = 1.96 × 10−37). Following adjustment for the lead single-variant association rs3917422 variant, the SKAT-O association signal was markedly diminished (P = 0.06), suggesting the gene burden signal is largely driven by rs3917422 (see Supplementary Material, Table S6). Notably, in the subset of 210 JHS individuals who had soluble E-selectin measured using both ELISA and aptamer-based platforms, the SELE rs3917422 sentinel variant was only associated with soluble E-selectin measured by ELISA (beta = −1.785, P = 8.80 × 10−9), but not by SOMAScan (beta = 0.019, P = 0.954) (Supplementary Material, Table S2), suggesting that the underlying variant[s] being tagged through linkage disequilibrium likely affect ELISA antibody binding.
Discussion
E-selectin is highly expressed on endothelial cells when activated by cytokines or chemokines (26,27) and is specifically involved in the tether and rolling phases of the leukocyte–endothelial adhesion during various inflammatory and immune responses (28). The amount of soluble E-selectin shed from damaged or activated endothelial cells correlates with the amount of cell surface E-selectin expression (29). We report the first genome-wide analysis of soluble E-selectin in a large AA cohort. By using WGS, we were able to perform a comprehensive investigation of common and lower frequency variants and directly assess the greater genetic diversity of African ancestry individuals that may be missed with commercial genome-wide genotyping arrays. In addition to generalizing the previously known association with the ABO locus to AAs, we report two novel association signals for soluble E-selectin, a missense LoF variant at the FUT6 locus and a signal of extended LD at the cognate SELE locus.
The native ligand for E-selectin is the fucosylated tetrasaccharide sialyl Lewis X, which is structurally related to blood group antigens and is present on specific leukocyte glycoproteins containing O/N-linked glycans or glycosphingolipids, such as PSGL-1, CD44 and ESL-1 (26–28,30). Plasma alpha(1,3)-fucosyltransferase, encoded by FUT6, is involved in the formation of sialyl-Lewis X (Le(x)) (31). The African FUT6 rs17855739 variant encodes a non-conservative amino acid substitution (p. Glu274Lys) located in the enzyme’s catalytic region. This variant co-segregates with plasma alpha(1,3)-fucosyltransferase deficiency and leads to inactivation of fucosyltransferase activity and absence of cell-surface sialyl Le(x) expression in vitro (30). Reduced expression of the sialyl Le(x) on circulating or tissue leukocytes in individuals carrying the FUT6 LoF variant may lead to lower E-selectin ligand binding affinity and/or decreased E-selectin clearance from the circulation, which may be responsible for the higher soluble E-selectin concentration observed in FUT6 allele carriers. Alternatively, immunoassays are potentially susceptible to differential glycosylation of their target protein, and it is possible that decreased fucosylation of soluble E-selectin in carriers of the FUT6 LoF variant might enhance ELISA antibody-binding affinity and possibly lead to spuriously higher levels of soluble E-selectin. However, the consistency of FUT6 LoF variant association results as measured by both ELISA and aptamer-based E-selectin detection methods makes this explanation less likely, unless the aptamer binds to the same antigenic region.
Soluble E-selectin levels have been correlated with risk of various cardiovascular, autoimmune and metabolic disorders including diabetes, hypertension, coronary heart disease and Graves’ disease (32). Moreover, interaction of E-selectin on endothelial cells and sialyl Lewis X expressed on leukocytes has been implicated as a crucial step in cancer metastasis, particularly, breast, prostate and colon cancer (33). FUT6 deficiency due to homozygosity for the rs17855739 LoF variant is relatively common among certain populations (frequency ∼10% in Africans, Indonesians), but is not known to be associated with any particular adverse health effects (34,35). We were unable to detect any significant association between FUT6 rs17855739 (either additively or recessively) and various disease phenotypes, except a nominally significant relationship with vitamin B12 levels. Fucosyltransferases may influence vitamin B12 absorption through intestinal host-microbial interaction or through glycosylation of vitamin B12-binding proteins and such as TCN1 and gastric intrinsic factor (36–38).
The absence of any detectable relationship between the FUT6 rs17855739 LoF variant and chronic inflammatory, CVDs or cancer in our AA PheWAS may suggest no direct causal relationship between genetically determined higher soluble E-selectin levels with these common disorders. Nonetheless, larger AA samples with well-defined disease outcomes may be necessary to more fully characterize the biologic impact and clinical consequences of FUT6 deficiency and genetically determined high soluble E-selectin. In particular, it should be noted that GWAS have identified various other non-coding genetic variants of FUT6 to be associated not only with other N-glycosylation phenotypes (39) and vitamin B12 levels (40–42), but also with serum carcinoembryonic antigen levels (a cancer biomarker) (39), basophil count (43) and advanced age-related macular degeneration (44).
Consistent with the results of GWAS and genetically determined ABO blood group analyses in whites and other ethnicities, the strongest signal we observed at the ABO locus was the association of lower soluble E-selectin with variants tagging the A1 blood group allele (rs635634, rs579459 and rs651007) (19,22,45,46). In conditional analyses, we also observed evidence for at least two, and possible three, independent variants associated with soluble E-selectin at the ABO locus (led by rs8176722, an LD proxy of the B blood group). The conditional regression results were confirmed in JHS by an analysis of ABO diplotypes using genetically defined ABO alleles; individuals carrying one or more A1 or B alleles had significantly lower soluble E-selectin compared to O/O individuals. Together, ABO alleles explained about 20% of the phenotypic variation in JHS AAs. Our results are largely consistent with prior studies showing significant differences in soluble E-selectin according to genetically inferred ABO genotypes in whites. However, an independent effect of the B allele on lower soluble E-selectin was reported in some studies (19,22) but not by others (21,47). In the Multi-Ethnic Study of Atherosclerosis, the A2 allele was associated with higher levels in Hispanics compared to Type O participants but lower levels in European Americans (47); the interpretation of those findings is likely limited by the relatively small number of A2 carriers. Thus, further studies are warranted.
Variants that tag the A1 glycosytransferase allele (such as rs507666) have been associated with lower levels of other circulating adhesion molecules (P-selectin, soluble Intercellular Adhesion Molecule 1) (21,45,48), as well as higher total and low density lipoprotein (LDL)-cholesterol, and higher risk of coronary artery disease (49–53). The rs8176722 B group tagging variant was associated with severe malaria in African individuals (54). In most instances, the mechanisms underlying these pleotropic phenotypic associations are unknown, including the seemingly paradoxical relationship between lower soluble adhesion molecule concentration and increased CVD risk associated with the A (or non-O) blood group allele. It has been hypothesized that the A antigen or the A glycosyltransferase may lower soluble E-selectin levels by either interfering with cleavage of E-selectin from the endothelial cell surface or altering clearance from the circulation (55). It is also possible that soluble ABO antigens may interfere with soluble E-selectin capture or detection in antibody-based assays. In one study, this possibility was excluded by performing analytic interference or mixing studies with A blood group antigen, which showed no differential effects on E-selectin measurement (18). The lack of analytical interference is also supported by the detection of a strong A1 allele association in both ELISA and aptamer-based platforms, both in JHS and in a recent aptamer-based GWAS (56).
In contrast to FUT6 and ABO, the association signal at the SELE locus detected by ELISA was not reproducible using an aptamer-based assay method. It is likely that one or more SELE coding variants (e.g. rs3917422 or p.Gln257Pro) contained within the extended block of LD tagged by our lead variant causes a structural change in the E-selectin protein that interferes with ELISA antibody binding, as opposed to true changes on circulating E-selectin levels. There are other examples of common but population-specific missense polymorphisms located within the structural gene encoding a particular plasma protein that interfere with antibody-based protein detection and induce falsely measured low values, such as with intracellular ICAM-1 (45) and vitamin D-binding protein (57). Soluble adhesion molecules and other inflammatory mediators involved in host pathogen defense are often subject to evolutionary selective pressures (e.g. malaria), and therefore may be particularly prone to such ethnicity-specific biomarker assay interference issues. These findings have potential implications for investigations into the relationships between soluble adhesion molecules and cardiovascular and other disease outcomes, particularly when comparing results across different ethnic groups.
In summary, we report two novel association signals for soluble E-selectin in AAs: a missense LoF variant at the FUT6 locus and a signal of extended LD at the cognate SELE locus. The latter likely represents an artifact due to African-specific protein polymorphism that specifically interferes with E-selectin detection by commercial immunoassay. The African FUT6 rs17855739 LoF variant, which is associated with higher soluble E-selectin and plasma alpha(1,3)-fucosyltransferase deficiency, should be further investigated for association with chronic disease outcomes in AAs, particularly given the potential importance of E-selectin interactions as a therapeutic target for various chronic inflammatory conditions (58).
Materials and Methods
JHS discovery sample and analysis
JHS is a population-based longitudinal study based in Jackson, Mississippi, designed to unravel the genetic and environmental determinants of CVD in AAs (Supplemental methods) (59). The design and sampling of JHS have been previously described (60). A total of 5306 male and female participants, ranging from 21–94 years of age, were recruited between 2000 and 2004. At visit 3 (years 2009–2013), E-selectin was measured in plasma in a random subset of 2249 JHS participants by the R&D systems ELISA. The Beckman Coulter Biomek NXp was used for vitamin B12 measurements at baseline (61,62). JHS participants (N = 3128 post quality control) underwent 30 × WGS through the Trans-Omics for Precision Medicine (TOPMed) project at the Northwest Genome Center at University of Washington; genotype calling was performed by the Informatics Resource Center at the University of Michigan, as previously described (63). After quality control, a total of 30 095 928 variants (MAF > = 0.001) and 2249 JHS participants with WGS and E-selectin measurements were included in the discovery analysis. To test for rare variant associations, we ran gene-based tests of non-synonymous variants with MAF < 5% using the SKAT-O test (64). Log-E-selectin levels were regressed on age, sex and the first ten ancestry principal components (PCs), and the residuals were rank-based inverse normally transformed. These transformed values were analyzed for association using a linear mixed model approach to account for familial relationships, as implemented in EPACTS 3.2.6 on the University of Michigan ENCORE server (https://encore.sph.umich.edu). Genome-wide significance was pre-defined as P < 1.66 × 10−9 or 0.05/30 095 928 variants tested. The same approach was used to analyze log-Vitamin B12 levels as well.
To identify ABO diplotypes, SNVs and indels were haplotype phased (Beagle v4.1) (65) and cross-referenced with ABO alleles listed by the International Society of Blood Transfusion (ISBT) (66). For cross-referencing, cDNA coordinates associated with >200 ABO ISBT alleles were converted to genomic coordinates. Final diplotype assessment was based on the `best’ matched ABO allele and based on prioritization of LoF O alleles over non-LoF of function alleles (e.g. A or B alleles).
Linear regression was used to assess the relationship of soluble E-selection using an indicator variable for each ABO diplotype (compared to the referent diplotype of O/O), and adjusting for covariates age, sex and 10 PCs. Individuals carrying rare ABO alleles [cisAB, B(A) or weak A] were excluded from analysis. To assess interaction between ABO type and FUT6 on E-selectin levels, an interaction term between ABO diplotype and FUT6 rs17855739 genotype was added to the linear regression model.
Heritability was assessed using variance components methods implemented in the Sequential Oligogenic Linkage Analysis Routines program version 8.1.1 from a subset of 1077 related JHS individuals (67).
WHI replication sample and analysis
WHI is one of the largest US cohort studies (n = 161 808). A diverse population including 26 045 (17%) women from minority groups were recruited from 1993–1998 across the USA. Of the minority participants enrolled in WHI 8515 were self-identified AAs who had consented to genetic research and were thus eligible for the WHI-SNP Health Association Research project. The Affymetrix 6.0 platform was used for genome-wide genotyping in all participants. Genotyping and quality control procedures have been previously described in detail (68). Imputation into the 1000 Genomes Phase 3 reference panel was conducted on the Michigan Imputation Server. The genotypes for rs17855739 were imputed with high confidence (Rsq = 0.936).
Of the ∼8000 WHI AAs with 1000 Genomes imputed data, 765 were measured for E-selectin levels on the R&D ELISA assay. As in the discovery analysis, the log of E-selectin levels was regressed on age and the first 10 PCs, after which the residuals were rank-based inverse-normal transformed. The association with rs17855739 was tested using a simple linear regression of these transformed values against the dosage of the imputed genotypes.
PheWAS
We performed a PheWAS of rs17855739 using two AA EMR-linked biobanks, the BioVU and BioMe of Mount Sinai. In BioVU, rs12019136 (r2 = 0.94 with rs17855739) was used as an LD proxy for the PheWAS. Phenotype-codes with at least five cases were considered, with a total of 339 codes tested under both and additive and recessive model. Logistic regressions adjusted for age, sex and the first 10 PCs were run within BioVU and BioME. The results were combined using a fixed-effect inverse-variance weighted meta-analysis.
Supplementary Material
Acknowledgements
Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for `NHLBI TOPMed: The Jackson Heart Study’ (phs000964.v1.p1) was performed at the University of Washington Northwest Genomics Center (HHSN268201100037C). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1). Phenotype harmonization, data management, sample-identity QC and general study coordination were provided by the TOPMed Data Coordination Center (3R01HL-120393-02S1). We gratefully acknowledge the studies and Participants who provided biological samples and data for TOpMed. The Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201300049C and HHSN268201300050C), Tougaloo College (HHSN268201300048C) and the University of Mississippi Medical Center (HHSN268201300046C and HHSN268201300047C) contracts from NHLBI and the National Institute for Minority Health and Health Disparities (NIMHD). The authors also wish to thank the staffs and participants of JHS. The Women’s Health Initiative (WHI) program is funded by NHLBI, the National Institutes of Health, the US Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C and HHSN268201600004C. The authors thank the WHI investigators and staff for their dedication and the study participants for making the program possible. The Mount Sinai BioMe Biobank is supported by the Andrea and Charles Bronfman Philanthropies. P.L.A., A.P.R. and L.A.L. were supported by NHLBI R01HL132947. L.M.R. was supported by NHLBI T32 HL129982. R.J.F.L. was supported by National Institute of Health (R01DK110113, U01HG007417, R01DK107786, R01HL142302).
Conflict of Interest statement. None declared.
References
- 1. Gimbrone M.A. Jr. and Garcia-Cardeña G. (2016) Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res., 118, 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Osborn L., Hession C., Tizard R., Vassallo C., Luhowskyj S., Chi-Rosso G. and Lobb R. (1989) Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell, 59, 1203–1211. [DOI] [PubMed] [Google Scholar]
- 3. Ramos C.L., Huo Y., Jung U., Ghosh S., Manka D.R., Sarembock I.J. and Ley K. (1999) Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ. Res., 84, 1237–1244. [DOI] [PubMed] [Google Scholar]
- 4. Tabas I., Garcia-Cardeña G. and Owens G.K. (2015) Recent insights into the cellular biology of atherosclerosis. J. Cell Biol., 209, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Otsuka F., Yasuda S., Noguchi T. and Ishibashi-Ueda H. (2016) Pathology of coronary atherosclerosis and thrombosis. Cardiovasc. Diagn. Ther., 6, 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Revelle B.M., Scott D., Kogan T.P., Zheng J. and Beck P.J. (1996) Structure–function analysis of P-selectin-sialyl LewisX binding interactions. Mutagenic alteration of ligand binding specificity. J. Biol. Chem., 271, 4289–4297. [DOI] [PubMed] [Google Scholar]
- 7. Snapp K.R., Wagers A.J., Craig R., Stoolman L.M. and Kansas G.S. (1997) P-selectin glycoprotein ligand-1 is essential for adhesion to P-selectin but not E-selectin in stably transfected hematopoietic cell lines. Blood, 89, 896–901. [PubMed] [Google Scholar]
- 8. Jilma B., Kovar F.M., Hron G., Endler G., Marsik C.L., Eichinger S. and Kyrle P.A. (2006) Homozygosity in the single nucleotide polymorphism Ser128Arg in the E-selectin gene associated with recurrent venous thromboembolism. Arch. Intern. Med., 166, 1655–1659. [DOI] [PubMed] [Google Scholar]
- 9. Jilma B., Marsik C., Kovar F., Wagner O.F., Jilma-Stohlawetz P. and Endler G. (2005) The single nucleotide polymorphism Ser128Arg in the E-selectin gene is associated with enhanced coagulation during human endotoxemia. Blood, 105, 2380–2383. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida M., Takano Y., Sasaoka T., Izumi T. and Kimura A. (2003) E-selectin polymorphism associated with myocardial infarction causes enhanced leukocyte-endothelial interactions under flow conditions. Arterioscler. Thromb. Vasc. Biol., 23, 783–788. [DOI] [PubMed] [Google Scholar]
- 11. Alessandro R., Seidita G., Flugy A.M., Damiani F., Russo A., Corrado C., Colomba P., Gullotti L., Buettner R., Bruno L. and De Leo G. (2007) Role of S128R polymorphism of E-selectin in colon metastasis formation. Int. J. Cancer, 121, 528–535. [DOI] [PubMed] [Google Scholar]
- 12. Zemlin A.E., Matsha T.E., Kengne A.P., Hon G.M. and Erasmus R.T. (2017) Correlation of E-selectin concentrations with carotid intima-media thickness and cardio-metabolic profile of mixed ancestry South Africans: a cross-sectional study. Ann. Clin. Biochem., 54, 92–100. [DOI] [PubMed] [Google Scholar]
- 13. Preston R.C., Rabbani S., Binder F.P., Moes S., Magnani J.L. and Ernst B. (2014) Implications of the E-selectin S128R mutation for drug discovery. Glycobiology, 24, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ernst B. and Magnani J.L. (2009) From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov., 8, 661–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roldan V., Marin F., Lip G.Y. and Blann A.D. (2003) Soluble E-selectin in cardiovascular disease and its risk factors. A review of the literature. Thromb. Haemost., 90, 1007–1020. [DOI] [PubMed] [Google Scholar]
- 16. Lutsey P.L., Cushman M., Steffen L.M., Green D., Barr R.G., Herrington D., Ouyang P. and Folsom A.R. (2006) Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: the MESA study. J. Thromb. Haemost., 4, 2629–2635. [DOI] [PubMed] [Google Scholar]
- 17. Lee M., Czerwinski S.A., Choh A.C., Demerath E.W., Sun S.S., Chumlea W.C., Towne B. and Siervogel R.M. (2006) Quantitative genetic analysis of cellular adhesion molecules: the Fels Longitudinal Study. Atherosclerosis, 185, 150–158. [DOI] [PubMed] [Google Scholar]
- 18. Karakas M., Baumert J., Kleber M.E., Thorand B., Dallmeier D., Silbernagel G., Grammer T.B., Rottbauer W., Meisinger C., Illig T., März W. and Koenig W. (2012) A variant in the ABO gene explains the variation in soluble E-selectin levels-results from dense genotyping in two independent populations. PLoS One, 7, e51441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paterson A.D., Lopes-Virella M.F., Waggott D., Boright A.P., Hosseini S.M., Carter R.E., Shen E., Mirea L., Bharaj B., Sun L. et al. (2009) Genome-wide association identifies the ABO blood group as a major locus associated with serum levels of soluble E-selectin. Arterioscler. Thromb. Vasc. Biol., 29, 1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun W., Kechris K., Jacobson S., Drummond M.B., Hawkins G.A., Yang J., Chen T.H., Quibrera P.M., Anderson W., Barr R.G. et al. (2016) Common genetic polymorphisms influence blood biomarker measurements in COPD. PLoS Genet., 12, e1006011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kiechl S., Paré G., Barbalic M., Qi L., Dupuis J., Dehghan A., Bis J.C., Laxton R.C., Xiao Q., Bonora E. et al. (2011) Association of variation at the ABO locus with circulating levels of soluble intercellular adhesion molecule-1, soluble P-selectin, and soluble E-selectin: a meta-analysis. Circ. Cardiovasc. Genet., 4, 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Voight B.F., Scott L.J., Steinthorsdottir V., Morris A.P., Dina C., Welch R.P., Zeggini E., Huth C., Aulchenko Y.S., Thorleifsson G. et al. (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet., 42, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Auer P.L. and Leal S.M. (2017) From exomes to genomes: challenges and solutions in population-based genetic association studies. Eur. J. Hum. Genet., 25, 395–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bentley A.R., Callier S. and Rotimi C.N. (2017) Diversity and inclusion in genomic research: why the uneven progress? J. Community Genet., 8, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manrai A.K., Funke B.H., Rehm H.L., Olesen M.S., Maron B.A., Szolovits P., Margulies D.M., Loscalzo J. and Kohane I.S. (2016) Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med., 375, 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Somers W.S., Tang J., Shaw G.D. and Camphausen R.T. (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell, 103, 467–479. [DOI] [PubMed] [Google Scholar]
- 27. Beauharnois M.E., Lindquist K.C., Marathe D., Vanderslice P., Xia J., Matta K.L. and Neelamegham S. (2005) Affinity and kinetics of sialyl Lewis-X and core-2 based oligosaccharides binding to L- and P-selectin. Biochemistry, 44, 9507–9519. [DOI] [PubMed] [Google Scholar]
- 28. Scott D.W. and Patel R.P. (2013) Endothelial heterogeneity and adhesion molecules N-glycosylation: implications in leukocyte trafficking in inflammation. Glycobiology, 23, 622–633. [DOI] [PubMed] [Google Scholar]
- 29. Kjaergaard A.G., Dige A., Krog J., Tonnesen E. and Wogensen L. (2013) Soluble adhesion molecules correlate with surface expression in an in vitro model of endothelial activation. Basic Clin. Pharmacol. Toxicol., 113, 273–279. [DOI] [PubMed] [Google Scholar]
- 30. Mollicone R., Reguigne I., Fletcher A., Aziz A., Rustam M., Weston B.W., Kelly R.J., Lowe J.B. and Oriol R. (1994) Molecular basis for plasma alpha(1,3)-fucosyltransferase gene deficiency (FUT6). J. Biol. Chem., 269, 12662–12671. [PubMed] [Google Scholar]
- 31. Zarbock A., Ley K., McEver R.P. and Hidalgo A. (2011) Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood, 118, 6743–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao F., Mamatyusupu D., Wang Y., Fang H., Wang H., Gao Q., Dong H., Ge S., Yu X., Zhang J. et al. (2016) The Uyghur population and genetic susceptibility to type 2 diabetes: potential role for variants in CAPN10, APM1 and FUT6 genes. J. Cell. Mol. Med., 20, 2138–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trinchera M., Aronica A. and Dall'Olio F. (2017) Selectin ligands sialyl-Lewis a and sialyl-Lewis x in gastrointestinal cancers. Biology (Basel), 6, 16–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Elmgren A., Borjeson C., Mollicone R., Oriol R., Fletcher A. and Larson G. (2000) Identification of two functionally deficient plasma alpha 3-fucosyltransferase (FUT6) alleles. Hum. Mutat., 16, 473–481. [DOI] [PubMed] [Google Scholar]
- 35. Caillard T., Le Pendu J., Ventura M., Mada M., Rault G., Mannoni P. and Oriol R. (1988) Failure of expression of alpha-3-L-fucosyltransferase in human serum is coincident with the absence of the X (or Le(x)) antigen in the kidney but not on leucocytes. Exp. Clin. Immunogenet., 5, 15–23. [PubMed] [Google Scholar]
- 36. Lee H.S., Choe G., Kim W.H., Kim H.H., Song J. and Park K.U. (2006) Expression of Lewis antigens and their precursors in gastric mucosa: relationship with Helicobacter pylori infection and gastric carcinogenesis. J. Pathol., 209, 88–94. [DOI] [PubMed] [Google Scholar]
- 37. Sheu B.S., Sheu S.M., Yang H.B., Huang A.H. and Wu J.J. (2003) Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut, 52, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Degnan P.H., Taga M.E. and Goodman A.L. (2014) Vitamin B12 as a modulator of gut microbial ecology. Cell Metab., 20, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang Y., Tang W., Huang T., Gao Y., Tan A., Yang X., Zhang H., Hu Y., Qin X., Li S. et al. (2014) Genetic variations affecting serum carcinoembryonic antigen levels and status of regional lymph nodes in patients with sporadic colorectal cancer from Southern China. PLoS One, 9, e97923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nongmaithem S.S., Joglekar C.V., Krishnaveni G.V., Sahariah S.A., Ahmad M., Ramachandran S., Gandhi M., Chopra H., Pandit A., Potdar R.D. et al. (2017) GWAS identifies population-specific new regulatory variants in FUT6 associated with plasma B12 concentrations in Indians. Hum. Mol. Genet., 26, 2551–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin X., Lu D., Gao Y., Tao S., Yang X., Feng J., Tan A., Zhang H., Hu Y., Qin X. et al. (2012) Genome-wide association study identifies novel loci associated with serum level of vitamin B12 in Chinese men. Hum. Mol. Genet., 21, 2610–2617. [DOI] [PubMed] [Google Scholar]
- 42. Grarup N., Sulem P., Sandholt C.H., Thorleifsson G., Ahluwalia T.S., Steinthorsdottir V., Bjarnason H., Gudbjartsson D.F., Magnusson O.T., Sparsø T. et al. (2013) Genetic architecture of vitamin B12 and folate levels uncovered applying deeply sequenced large datasets. PLoS Genet., 9, e1003530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Astle W.J., Elding H., Jiang T., Allen D., Ruklisa D., Mann A.L., Mead D., Bouman H., Riveros-Mckay F., Kostadima M.A. et al. (2016) The allelic landscape of human blood cell trait variation and links to common complex disease. Cell, 167, 1415–1429e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fritsche L.G., Igl W., Bailey J.N., Grassmann F., Sengupta S., Bragg-Gresham J.L., Burdon K.P., Hebbring S.J., Wen C., Gorski M. et al. (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet., 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paré G., Chasman D.I., Kellogg M., Zee R.Y., Rifai N., Badola S., Miletich J.P. and Ridker P.M. (2008) Novel association of ABO histo-blood group antigen with soluble ICAM-1: results of a genome-wide association study of 6,578 women. PLoS Genet., 4, e1000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teng M.S., Hsu L.A., Wu S., Chou H.H., Chang C.J., Sun Y.Z., Juan S.H. and Ko Y.L. (2013) Mediation analysis reveals a sex-dependent association between ABO gene variants and TG/HDL-C ratio that is suppressed by sE-selectin level. Atherosclerosis, 228, 406–412. [DOI] [PubMed] [Google Scholar]
- 47. Larson N.B., Bell E.J., Decker P.A., Pike M., Wassel C.L., Tsai M.Y., Pankow J.S., Tang W., Hanson N.Q., Alexander K. et al. (2016) ABO blood group associations with markers of endothelial dysfunction in the multi-ethnic study of atherosclerosis. Atherosclerosis, 251, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paré G., Ridker P.M., Rose L., Barbalic M., Dupuis J., Dehghan A., Bis J.C., Benjamin E.J., Shiffman D., Parker A.N. and Chasman D.I. (2011) Genome-wide association analysis of soluble ICAM-1 concentration reveals novel associations at the NFKBIK, PNPLA3, RELA, and SH2B3 loci. PLoS Genet., 7, e1001374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harst P. and Verweij N. (2018) Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res., 122, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nelson C.P., Goel A., Butterworth A.S., Kanoni S., Webb T.R., Marouli E., Zeng L., Ntalla I., Lai F.Y., Hopewell J.C. et al. (2017) Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet., 49, 1385–1391. [DOI] [PubMed] [Google Scholar]
- 51. Surakka I., Horikoshi M., Magi R., Sarin A.P., Mahajan A., Lagou V., Marullo L., Ferreira T., Miraglio B., Timonen S. et al. (2015) The impact of low-frequency and rare variants on lipid levels. Nat. Genet., 47, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suhre K., Arnold M., Bhagwat A.M., Cotton R.J., Engelke R., Raffler J., Sarwath H., Thareja G., Wahl A., DeLisle R.K. et al. (2017) Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun., 8, 14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou L., He M., Mo Z., Wu C., Yang H., Yu D., Yang X., Zhang X., Wang Y., Sun J. et al. (2013) A genome wide association study identifies common variants associated with lipid levels in the Chinese population. PLoS One, 8, e82420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Band G., Le Q.S., Jostins L., Pirinen M., Kivinen K., Jallow M., Sisay-Joof F., Bojang K., Pinder M., Sirugo G. et al. (2013) Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet., 9, e1003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Blann A.D., Daly R.J. and Amiral J. (1996) The influence of age, gender and ABO blood group on soluble endothelial cell markers and adhesion molecules. Br. J. Haematol., 92, 498–500. [DOI] [PubMed] [Google Scholar]
- 56. Benson M.D., Yang Q., Ngo D., Zhu Y., Shen D., Farrell L.A., Sinha S., Keyes M.J., Vasan R.S., Larson M.G. et al. (2018) Genetic architecture of the cardiovascular risk proteome. Circulation, 137, 1158–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Uitterlinden A.G., Fang Y., Van Meurs J.B., Pols H.A. and Van Leeuwen J.P. (2004) Genetics and biology of vitamin D receptor polymorphisms. Gene, 338, 143–156. [DOI] [PubMed] [Google Scholar]
- 58. Silva M., Videira P.A. and Sackstein R. (2017) E-selectin ligands in the human mononuclear phagocyte system: implications for infection, inflammation, and immunotherapy. Front. Immunol., 8, 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sempos C.T., Bild D.E. and Manolio T.A. (1999) Overview of the Jackson Heart Study: a study of cardiovascular diseases in African American men and women. Am. J. Med. Sci., 317, 142–146. [DOI] [PubMed] [Google Scholar]
- 60. Taylor H.A.,.J., Wilson J.G., Jones D.W., Sarpong D.F., Srinivasan A., Garrison R.J., Nelson C. and Wyatt S.B. (2005) Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn. Dis., 15, S6-4-17. [PubMed] [Google Scholar]
- 61. Henry O.R., Benghuzzi H., Taylor H.A. Jr., Tucci M., Butler K. and Jones L. (2012) Suppression of homocysteine levels by vitamin B12 and folates: age and gender dependency in the Jackson Heart Study. Am. J. Med. Sci., 344, 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hwang S.J., Ballantyne C.M., Sharrett A.R., Smith L.C., Davis C.E., Gotto A.M. Jr. and Boerwinkle E. (1997) Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk in Communities (ARIC) study. Circulation, 96, 4219–4225. [DOI] [PubMed] [Google Scholar]
- 63. Raffield L.M., Zakai N.A., Duan Q., Laurie C., Smith J.D., Irvin M.R., Doyle M.F., Naik R.P., Song C., Manichaikul A.W. et al. (2017) D-dimer in African Americans: whole genome sequence analysis and relationship to cardiovascular disease risk in the Jackson Heart Study. Arterioscler. Thromb. Vasc. Biol., 37, 2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., NHLBI Go Exome Sequencing—ESP Lung Project Team, Christiani D.C., Wurfel M.M. and Lin X. (2012) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet., 91, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Browning S.R. and Browning B.L. (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet., 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Storry J.R., Castilho L., Chen Q., Daniels G., Denomme G., Flegel W.A., Gassner C., Haas M., Hyland C., Keller M. et al. (2016) International society of blood transfusion working party on red cell immunogenetics and terminology: report of the Seoul and London meetings. ISBT Sci. Ser., 11, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Almasy L. and Blangero J. (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet., 62, 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reiner A.P., Lettre G., Nalls M.A., Ganesh S.K., Mathias R., Austin M.A., Dean E., Arepalli S., Britton A., Chen Z. et al. (2011) Genome-wide association study of white blood cell count in 16,388 African Americans: the continental origins and genetic epidemiology network (COGENT). PLoS Genet., 7, e1002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.