

Abstract
The retinal disease gene peripherin 2 (PRPH2) is essential for the formation of photoreceptor outer segments (OSs), where it functions in oligomers with and without its homologue ROM1. However, the precise role of these proteins in OS morphogenesis is not understood. By utilizing a knock-in mouse expressing a chimeric protein comprised of the body of Rom1 and the C-terminus of Prph2 (termed RRCT), we find that the Prph2 C-terminus is necessary and sufficient for the initiation of OSs, while OS maturation requires the body of Prph2 and associated large oligomers. Importantly, dominant-negative physiological and biochemical defects in RRCT heterozygous rods are rescued by removing Rom1, suggesting Rom1 is a regulator for OS formation. Our experiments evaluating Prph2 trafficking show that Rom1 is a key determinant of whether Prph2 complexes utilize conventional versus unconventional (Golgi bypass) secretory pathways to reach the OS. These findings significantly advance our understanding of the molecular underpinnings of OS morphogenesis and particularly the role of Rom1.
Introduction
Peripherin-2/retinal degeneration slow (PRPH2/RDS) is a photoreceptor-specific gene required for the formation of outer segments (OSs) (1–3). Mutations in PRPH2 cause widely varying forms of incurable inherited retinal degeneration including autosomal dominant retinitis pigmentosa (RP), digenic RP and various forms of macular degeneration (4). Because some PRPH2 mutations lead to rod-dominant diseases while others lead to more cone-dominant conditions, much effort has been dedicated to understanding the differential behavior of Prph2 in rod and cone photoreceptors and the specific functions of the different domains of Prph2 (5–8).
Prph2 is a tetraspanin glycoprotein confined to the disc/lamellae rim where it co-localizes with its non-glycosylated homologue, rod outer segment membrane protein 1 (Rom1) (9,10). Prph2 and Rom1 have cytoplasmic N- and C-termini, four transmembrane domains and a large extracellular/intradiscal loop (D2). The D2 loops in Prph2 and Rom1 are essential for their oligomerization (7,11,12) into non-covalently linked homo- and hetero-tetramers (13,14). The tetramers further assemble into heteromeric intermediate-sized complexes and large Prph2 homo-oligomers (15). These larger complexes are held together by intermolecular disulfide bonds at cysteine residue 150 (C150) (12,16). Covalently linked Prph2 complexes are required for the proper growth and maturation of OSs and are thought to be specifically required for rim stability. For example, in animal models in which Prph2 cannot form covalently linked complexes (C150S), OS and disc formation are initiated, but OSs do not elongate and rapidly degrade (16–18). In contrast, animals lacking Prph2 (in the Prph2−/−, also known as rds−/− or rd2) do not initiate OS formation (19).
While the D2 loop of Prph2 is thought to be critical for rim stability and elongation/maturation of OSs due to its role in oligomerization, the C-terminus is a likely candidate for initiation of OS structures. The C-terminus of Prph2 is intrinsically disordered (20) and has been shown to be important for OS targeting (8), capable of promoting membrane fusion (21,22), and involved in the generation or regulation of membrane curvature (23–25). Recent evidence showed that the Prph2 C-terminus suppresses ciliary ectosome release (26), a process thought to be critical for the initiation of OSs. Support for this model of the complementary functions of the Prph2 D2 loop and C-terminus comes from experiments performed on transgenic mice overexpressing a chimeric protein in which the Rom1 D2 loop was replaced with the Prph2 D2 loop (27). The Rom1-Prph2-D2 chimera did not initiate OS formation in the Prph2−/− model but rescued the Prph2+/− phenotype, in which disc formation is initiated by the presence of one allele of wild-type (WT) Prph2.
In contrast to Prph2, no pathogenic mutations in ROM1 have been reported so far. Rom1−/− retinas are largely normal, exhibiting minor defects in photoreceptor disc size and late-onset structural and functional degeneration (28). Rom1 alone (e.g. in Prph2−/−) cannot initiate OS formation. However, recent evidence suggests that Rom1 may be more important than once thought. We found that while Prph2 overexpression is well tolerated by photoreceptors (29), overexpression of Rom1 can be toxic to cones (30). In addition, Rom1 may act as a disease modifier contributing to phenotypic heterogeneity in patients and animal models with Prph2-associated RP or pattern dystrophy (31,32). Likewise, digenic RP in patients with PRPH2 and ROM1 mutations has been reported (33,34). Rom1 lacks the OS-targeting sequence identified in the Prph2 C-terminus and past hypotheses suggested that the inability of Rom1 to support OS development was due to an inability to traffic to the OS in the absence of Prph2 (8). However, we recently showed that Rom1 can target to the OS without Prph2 (35), so the inability of Rom1 to support OS formation clearly has another cause. Two differences between Prph2 and Rom1 stand out at the molecular level. First, the proteins exhibit very little homology in their C-termini, and second, Prph2 forms very large covalently linked homo-oligomers while Rom1 is found only in tetramers and more intermediate-sized complexes. Though covalently linked complexes are required for proper OS formation (16–18), it is not clear what distinct role is played by the large homo-oligomers compared to the intermediate-sized heteromeric complexes.
To help further explore the divergent roles of Prph2 and Rom1, we generated a chimeric knock-in mouse model in which the body of Prph2 (through the end of the fourth transmembrane domain) was replaced by the body of Rom1 (termed RRCT for Rom1 body + Rds C-terminus). We find that the resulting chimeric RRCT protein initiates OS formation, and some OS function, but does not support full elaboration of OS structures. In addition, when expressed in the presence of WT Prph2, the RRCT protein exhibits dominant-negative structural and functional defects. Many of these defects are alleviated in rods by the absence of endogenous Rom1. These structure–function studies significantly advance our understanding of the divergent roles of Prph2 and Rom1 and their differential roles in rod versus cone photoreceptors.
Results
RRCT protein is expressed at very low levels in the retina
We generated an in vitro expression vector carrying the RRCT complementary deoxyribonucleic acid (cDNA), comprising the cytomegalovirus (CMV) promoter, an N-terminus FLAG tag, the Rom1 coding region up through the end of the fourth transmembrane domain, followed by the Prph2 C-terminus sequence (Supplementary Material, Figs S1 and S2). COS cells have been widely used for evaluating oligomerization and various other properties of WT and mutant forms of Prph2 (12,13,36). COS-7 cells were transfected with plasmids carrying WT Prph2 with a Myc tag, RRCT with a FLAG tag, Rom1 or C214S-Prph2 [a control mutant that is known to be retained in the endoplasmic reticulum (ER) that exhibits trafficking defects (36)]. Similar to WT Prph2 and Rom1 and in contrast to C214S-Prph2, RRCT protein did not accumulate in the ER and exhibited a WT distribution on post-ER membranes and stable RRCT protein was detected on western blots at the predicted size (∼35 kDa) (Supplementary Material, Fig. S3).
We thus proceeded with in vivo experiments. The RRCT knock-in mouse model was generated by knocking Rom1 exon 1 (from the translation start site), intron 1, exon 2, intron 2 and the first part of exon 3 into the Prph2 locus, replacing the corresponding region of the Prph2 gene (Supplementary Material, Figs S1 and S2) and resulting in a chimeric protein comprising the body of Rom1 and the C-terminus of Prph2. For clarity’s sake we refer to mice homozygous/heterozygous for the RRCT allele as Prph2R/R and Prph2+/R, respectively. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) evaluation at postnatal day (P) 30 showed that the RRCT transcript was expressed at levels similar to WT Prph2, suggesting that the message was expressed and regulated normally in the retina (Fig. 1A).
Figure 1.
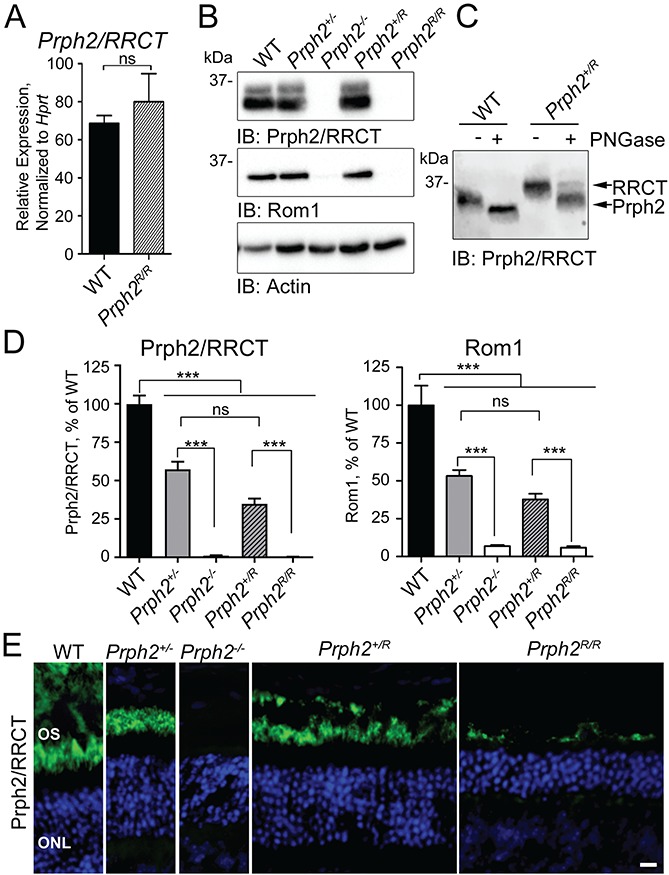
The RRCT knock-in is expressed in the retina. (A) qRT-PCR was performed on P30 retinal cDNAs using primers that recognize both WT Prph2 and RRCT knock-in transcripts. Values were normalized to Hprt as a housekeeping gene. N = 3 retinas/genotype (mean ± SEM). ns = not significant (P > 0.05) by two-sided Student’s t-test. (B) P30 retinal extracts were separated on 10% reducing SDS-PAGE. Blots were probed with antibodies for Prph2/RRCT (RDS-CT), Rom1 (ROM1-CT) or actin (as a loading control). Shown is a representative blot; blots were repeated with retinal extracts from different animals a minimum of five times. (C) Western blot of untreated and PNGase F treated retinal extracts from WT and Prph2R/R probed for Prph2/RRCT (RDS-CT). (Repeated on extracts from five different animals.) (D) Levels of Prph2/RRCT as percent of WT were measured from the density of the bands in (B) and plotted as means ± SEM from N = 5–10 retinas/genotype. ***P < 0.001, ns: not significant by one-way ANOVA with Tukey’s post-hoc comparison. (E) P30 retinal sections were immunofluorescently labeled for Prph2/RRCT (RDS-CT, green) and nuclei were counterstained with DAPI (blue), images were captured at 40×. OS: outer segment, ONL: outer nuclear layer. Scale bar: 10 μm. Immunofluorescence experiments were repeated at least three times using sections from 3–5 different animals.
Evaluating Prph2, Rom1 and RRCT protein levels in RRCT knock-in retinas was tricky since many antibody epitopes (black bars in Supplementary Material, Fig. S1A) were present in multiple proteins (i.e. Prph2 and RRCT or Rom1 and RRCT). Rom1 protein could be assessed separately from RRCT using ROM1-CT antibody. However, RRCT and Prph2 protein could not be distinguished from one another. To help simplify interpretations, immunoblots and images are labeled for the detected proteins, e.g. Prph2/RRCT or Rom1/RRCT. Reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE/western blots of P30 retinal extracts from WT, Prph2+/−, Prph2−/− Prph2+/R and Prph2R/R were probed with antibodies against Prph2/RRCT (Fig. 1B). RRCT protein was undetectable in Prph2R/R retinas by immunoblot (Fig. 1B) while, in Prph2+/R retinas, total Prph2/RRCT protein levels were reduced to those seen in the Prph2+/− (Fig. 1B and D). Rom1 levels in Prph2+/R were also reduced to those seen in Prph2+/−, and levels in the Prph2R/R were similar to the Prph2−/− (Fig. 1B and D). To distinguish between Prph2 and RRCT in Prph2+/R animals, we took advantage of the fact that because RRCT contains the body of Rom1, it is non-glycosylated and therefore does not change size when treated with Peptide:N-glycosidase F (PNGase F). In WT retinas treated with PNGase F, Prph2 is fully deglycosylated, while two bands are present in the Prph2+/R, one corresponding to WT Prph2, the other to non-glycosylated RRCT. Densitometric quantification of these bands showed that in the Prph2+/R, ∼28 ± 3% of total Prph2/RRCT was RRCT while the remainder was WT Prph2 (Fig. 1C).
The low levels of RRCT in Fig. 1C suggest that the chimeric protein is either inherently unstable or degraded because no OSs are formed in the absence of WT Prph2. This latter situation is often the case with OS proteins; for example, in the Prph2−/− retina (which lacks OSs) both rhodopsin and Rom1 levels are severely reduced since there is no OS in which to reside. To help evaluate these possibilities, retinal sections were labeled for Prph2/RRCT (green, Fig. 1E). Small amounts of RRCT protein are detected in the Prph2R/R retina, but only in a very thin layer, quite different from the thicker OS layer in the WT, Prph2+/− or the Prph2+/R (Fig. 1E). To further evaluate the localization of the RRCT protein, we co-labeled retinal sections for Prph2/RRCT (green) and other photoreceptor markers. Co-localization between RRCT (Prph2R/R) and rhodopsin, S-opsin and M-opsin (Fig. 2A–C, arrows) coupled with a lack of co-labeling between RRCT and the inner segment (IS)/axoneme marker acetylated alpha-tubulin (Fig. 2D) confirms that RRCT protein traffics out of the IS in the Prph2R/R in rods and cones. However, though the RRCT protein also localizes with Rom1 in Prph2R/R retina, it does not appear to elaborate lengthy OSs (Fig. 2E). Prph2/RRCT distribution in the Prph2+/R was similar to that of WT, with no accumulation outside the OS.
Figure 2.
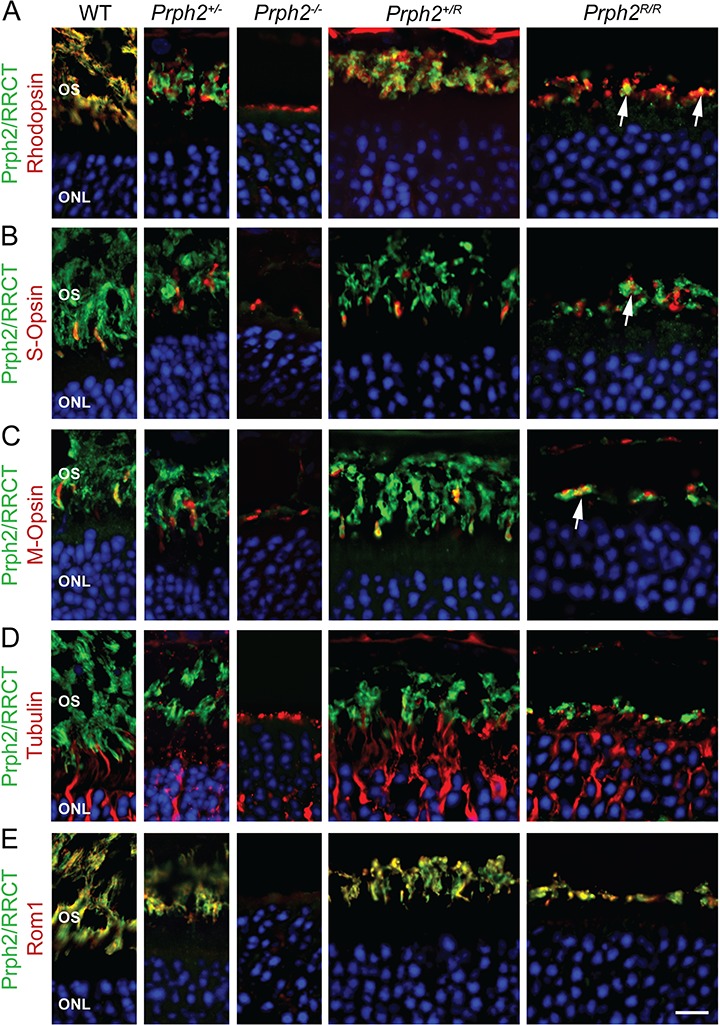
RRCT protein is targeted to the OS. (A–E) P30 retinal sections were immunofluorescently labeled for Prph2/RRCT (green, RDS-CT or mAB 2B7) and rhodopsin (red, mAB 1D4, (A)), S-opsin (red, goat polyclonal, (B)), M-opsin (red, Craft polyclonal, (C)), acetylated α-tubulin (red, (D)) and Rom1 (red, ROM1-CT, (E)). Nuclei are counterstained with DAPI (blue). Scale bar: 10 μm (captured at 100×) OS: outer segment, ONL: outer nuclear layer. Immunofluorescence experiments were repeated at least three times using sections from 3–5 different animals.
RRCT protein alone does not support full OS formation
No OS layer was detected by light microscopy in the Prph2R/R at P30 similar to the Prph2−/− (Fig. 3A). Both genotypes exhibited thinning of the outer nuclear layer (ONL) indicative of degeneration; 5 and 6 rows of nuclei were observed in the Prph2R/R versus 6 and 7 in the Prph2−/− while 9 and 10 were seen in the WT retina. However, on the electron microscopy (EM) level, very small OSs were observed on some cells in the Prph2R/R (arrows, Fig. 3B), in contrast to the Prph2−/− where no OSs were detected. These small OSs likely corresponded to the RRCT-expressing structures seen by immunofluorescence. When examined at higher magnification (Fig. 3C and Supplementary Material, Fig. S4A), these structures exhibited a whorl-like morphology, characterized by the formation of lengthy disc membranes. To understand whether these structures were rods or cones (or both), sections were immunogold (IG) labeled (Fig. 3D). The whorl-like OSs largely labeled with rhodopsin, while S-opsin containing OS membrane exhibited a more open, less tightly packed morphology (additional examples are in Supplementary Material, Fig. S4B).
Figure 3.
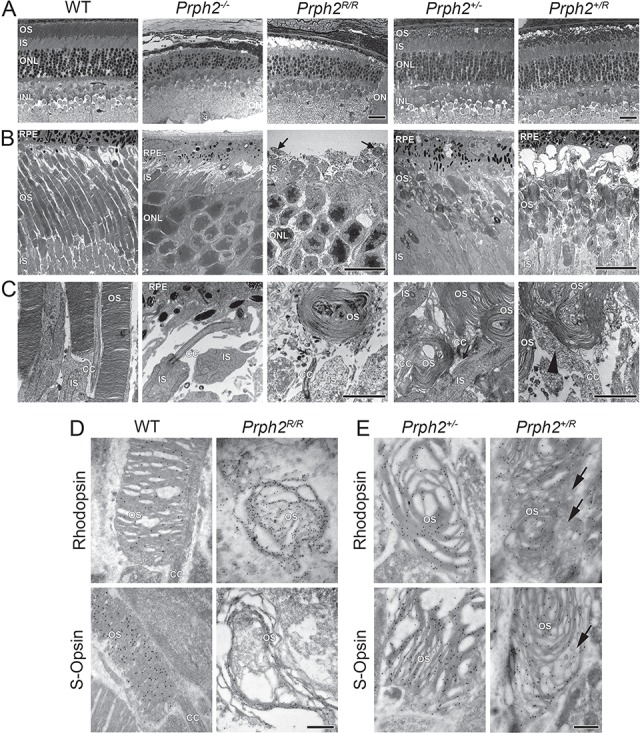
RRCT alone cannot support OS formation while expression of RRCT and WT Prph2 results in accumulation of abnormal vesicular structures. (A) Shown are representative light microscopy images collected at P30. Scale bar: 25 μm. (B and C) Shown are representative P30 EM images collected at 3000× (B) and 15 000× (C). Scale bars: 10 μm (B), 2 μm (C). Arrows indicate tiny nubs of OS in the Prph2R/R, an example of which is shown at higher magnification in (C). Light microscopy and EM were repeated on eyes from at least three different animals. (D and E) Rod OSs were identified by IG labeling with rhodopsin (Fliesler polyclonal) while cone OSs were identified by labeling with S-opsin (Craft polyclonal) antibodies (50 000×). Scale bar: 500 nm. IG labeling was repeated on eyes from at least three different animals. (A–D). RPE: retinal pigment epithelium, OS: outer segment, IS: inner segment, ONL: outer nuclear layer, INL: inner nuclear layer, ON: optic nerve and CC: connecting cilium.
To evaluate the structural effects of RRCT protein when expressed in the presence of the WT protein, Prph2+/R was compared to the Prph2+/− at P30. Overall retinal structure in these genotypes was similar (Fig. 3A), showing proper lamination and slight degeneration: 8 and 9 rows of nuclei in the central retina in the Prph2+/R and 7 and 9 in the Prph2+/− versus 9 and 10 in the WT. Prph2+/R and Prph2+/− retinas have OSs with a globular, whorl-like morphology rather than the columnar OSs of the WT (Fig. 3B). However, evaluation at higher magnification demonstrated different morphology in the Prph2+/R retina compared to the Prph2+/−. In addition to the large whorl-like OS structures, Prph2+/R OSs showed significant accumulation of large vesicular structures in the connecting cilium and base of the OS (Fig. 3C, arrowhead and Supplementary Material, Fig. S4C). These vesicular structures are largely absent in the Prph2+/− but occur in rods and cones in the Prph2+/R (Fig. 3E, and Supplementary Material, Fig. S4D). Rhodopsin and S-opsin were concentrated in the whorl-like discs in Prph2+/− and the Prph2+/R and were largely absent from the abnormal vesicular structures in the Prph2+/R (Fig. 3E, arrows and Supplementary Material, Fig. S4D, arrows). In contrast, IG immunoreactivity for Prph2/RRCT and Rom1/RRCT was concentrated in the large vesicular structures in the OS adjacent to the discs (Fig. 4A–D, arrowheads and Supplementary Fig. S4E, arrowheads). In the WT and Prph2+/−, Prph2 and Rom1 were largely restricted to the disc rims as expected (Fig. 4A and C, arrows). These data suggest that the presence of RRCT leads Prph2/Rom1/RRCT complexes to accumulate outside the discs and that these complexes cannot support normal rim formation.
Figure 4.
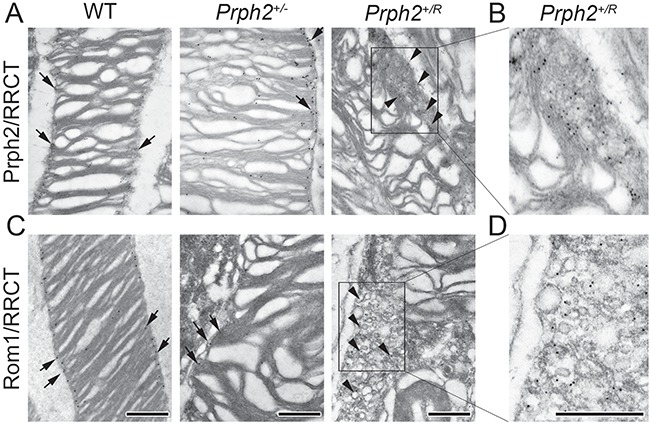
Prph2, ROM-1 and RRCT localize to the abnormal vesicles in the Prph2+/R. IG coupled with TEM was performed on retinas at P30. IG labeling was repeated on eyes from at least three different animals. (A) Prph2/RRCT were labeled using RDS-MPCT. (B) Shown is enlarged section of (A). (C) Rom1/RRCT were labeled using mAB 2H5. (D) Shown is enlarged section of (C). (A–D) All images were captured at 50 000× except bottom left which was captured at 40 000×. Arrowheads indicate accumulation of Prph2/Rom1/RRCT on vesicles in Prph2+/R while arrows show Prph2/Rom1 labeling largely restricted to the rims region in the other genotypes. Scale bar: 500 nm.
In keeping with this idea, we observed that many disc rims in the Prph2+/R lacked the hairpin-like pinched morphology characteristic of WT and Prph2+/− discs (Fig. 5A, arrowhead), while other rims appeared normal (Fig. 5A, arrow). To better characterize this phenotype, we measured the diameter of the outer and inner portions of the disc rim and calculated mean ‘% pinched’ for each disc where % pinched = (Di-Do)/Do*100 and Di is the inner diameter of the rim and Do is the outer diameter of the rim (Fig. 5B, see diagram, Supplementary Material, Fig. S4F, and methods section for more details). Thus, for a disc with a normal hairpin structure, values are negative since the inner diameter is smaller than the outer diameter. In contrast, positive values represent discs with no hairpin structure where the inner diameter is larger than the outer diameter. Even though the Prph2+/− exhibited extremely elongated discs that adopted a distinctive abnormal whorl-like morphology, rims were only slightly less pinched when compared to WT (Prph2+/− mean % pinched is −26.6% ± 0.877% versus −31.74% ± 0.876% for WT, Fig. 5B). In contrast, discs in the Prph2+/R were much less pinched than either WT or Prph2+/−, with a mean % pinching of −7.2% ± 1.33%. In addition there was a much wider range of rim morphologies as can be appreciated by the broadened Prph2+/R peak in the frequency distribution in Fig. 5C. When outer and inner rim diameter were plotted separately, we observed no significant differences in mean outer rim diameter among the three genotypes; however, consistent with decreased overall disc pinching in the Prph2+/R, inner rim diameter was significantly larger in the Prph2+/R than either the WT or Prph2+/− (Fig. 5D). These data show that rim morphology is significantly altered in the presence of RRCT: many discs lack the typical pinched hairpin structure and exhibit a high degree of variability in rim shape.
Figure 5.
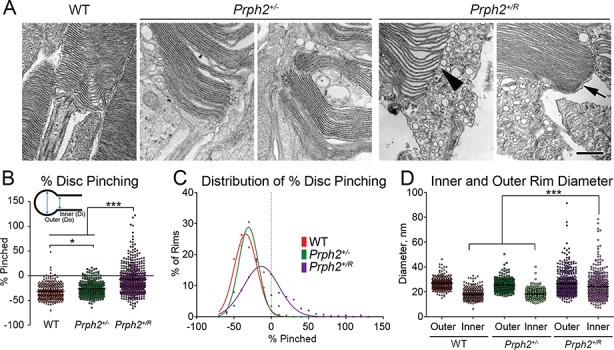
Expression of the RRCT knock-in allele alters the structure of the disc rim region. (A) Shown are representative high magnification EMs captured at 40 000× at P30. Arrowhead shows abnormally dilated rim in Prph2+/R while arrow shows normally structured rim. Inner and outer rim diameters were measured from 250 to 450 individual rims from 3 to 4 eyes/genotype. Scale bar 500 nm. (B and C) Percent pinching for each rim measured was calculated, black line indicates mean rim diameter and results were plotted on a histogram. (C) *P < 0.05 ***P < 0.001 by one-way ANOVA with Tukey’s post-hoc comparison. (D) Inner and outer rim diameters are plotted. ***P < 0.001 by one-way ANOVA with Tukey’s post-hoc comparison.
RRCT exerts dominant negative effects on rod and cone function
To determine whether the structural changes observed in animals carrying the RRCT allele resulted in functional changes, electroretinography (ERG) was performed at P30 and P180. Haploinsufficiency in the Prph2+/− led to the well-established reduction in scotopic ERG values by P30 (mean scotopic a-wave amplitudes in Prph2+/− are 53% of WT, Fig. 6A and B). Scotopic ERGs were further reduced in the Prph2+/R (28% of WT). This difference persisted at P180 when scotopic a-wave values fell to 38% of WT in the Prph2+/− and 14% of WT in the Prph2+/R. Mean scotopic b-wave values mirrored these patterns, with maximum amplitudes in the Prph2+/R significantly reduced compared to both Prph2+/− and WT at P30 and P180 (Fig. 6C). Consistent with the presence of small nubs of OSs in the Prph2R/R, ERG values were quite low. Scotopic a-wave values in the Prph2R/R were reduced to 7.8% of WT at P30 and 6.9% of WT at P180 (Fig. 6B). However, although scotopic a-wave and scotopic b-wave amplitudes were not statistically significantly different between Prph2−/− and Prph2R/R, the shape of the scotopic waveform was better in the Prph2R/R than in the Prph2−/− (Fig. 6A), suggesting that some small amount of true signal was preserved in the Prph2R/R at P30.
Figure 6.
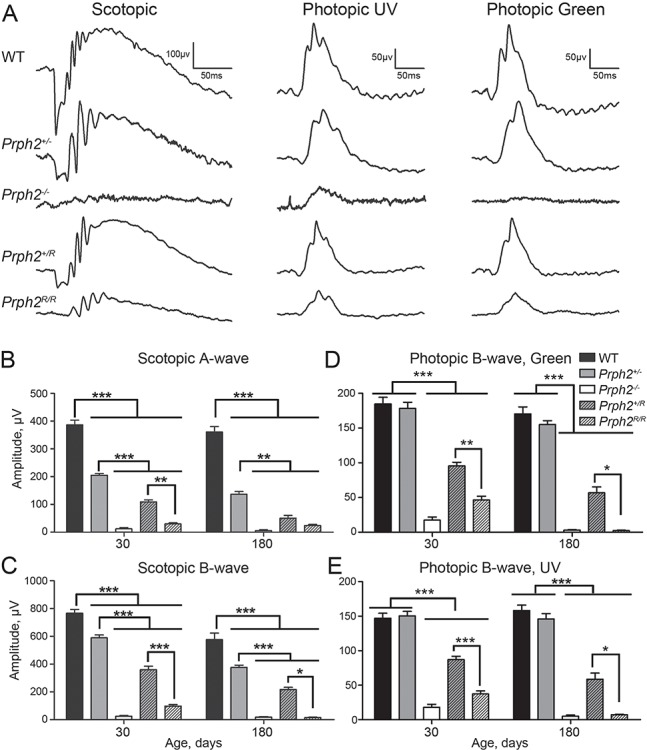
Heterozygous RRCT animals exhibit dominant negative defects. (A) ERGs were recorded under scotopic or photopic conditions at P30 and P180. Shown are representative waveforms at P30. (B–E) Plotted are mean (±SEM) scotopic a- and b-wave amplitudes (B and C), and mean (±SEM) photopic b-wave amplitudes in response to green light and UV light (D and E). N = 5–15 animals/genotype/age. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA with Tukey’s post-hoc comparison.
S- and M- cone function was measured by photopic ERGs recorded in response to ultraviolet (UV) and green light, respectively (Fig. 6D and E). The Prph2+/− retina does not exhibit early-onset cone defects, but age-matched Prph2+/R exhibited significant reductions in both S- and M- cone photopic b-wave amplitudes when compared to WT and Prph2+/− (UV responses in the Prph2+/R were 59% of WT and green responses were 51% of WT at P30). By P180, UV responses in the Prph2+/R attained only 37.1% of WT and green responses were 33% of WT. At P30, Prph2R/R had better preservation of cone function than rod function (S- and M-cone responses were reduced to 25% of WT while rod responses were 7.8% of WT). However, S- and M- cone functions in the Prph2R/R was drastically reduced by P180 to less than 5% of WT, similar to the Prph2−/−. Combined, these data show that retinas containing only RRCT protein exhibit better function than those lacking Prph2, but RRCT alone does not support proper retinal function. Importantly, the Prph2+/R shows a dominant-negative effect of RRCT (in the presence of WT Prph2) on both rod and cone function.
RRCT leads to biochemical abnormalities which are partially corrected by elimination of Rom1
Prph2 and Rom1 interact via non-covalent bonds and covalent disulfide bonds mediated by the D2 loop (Supplementary Material, Fig. S1A). Co-immunoprecipitation (IP) from COS-7 cells double transfected with Prph2 and Rom1, RRCT and Rom1 or RRCT and Prph2 (Supplementary Material, Fig. S5) showed that RRCT retained the ability to interact with WT Prph2 and Rom1. To determine whether Rom1 and RRCT interact in vivo, we performed similar experiments on retinal extracts (Fig. 7A–C). Immunoprecipitation (IP) for RRCT from Prph2R/R retinal extracts pulled down Rom1 (Fig. 7B), even though this model has very low levels of RRCT and Rom1. Because of limitations of antibody epitopes, evaluating interactions between Prph2 and RRCT in vivo is not possible in the presence of endogenous Rom1, so we crossed the RRCT allele onto the Rom1−/− background, enabling detection of RRCT (using antibodies against the Rom1 D2 loop) independently of Prph2. IP with antibodies against Prph2/RRCT pulled down RRCT (Fig. 7C) in retinal extracts from the Prph2+/R/Rom1−/− confirming RRCT interacts with Prph2 and Rom1.
Figure 7.
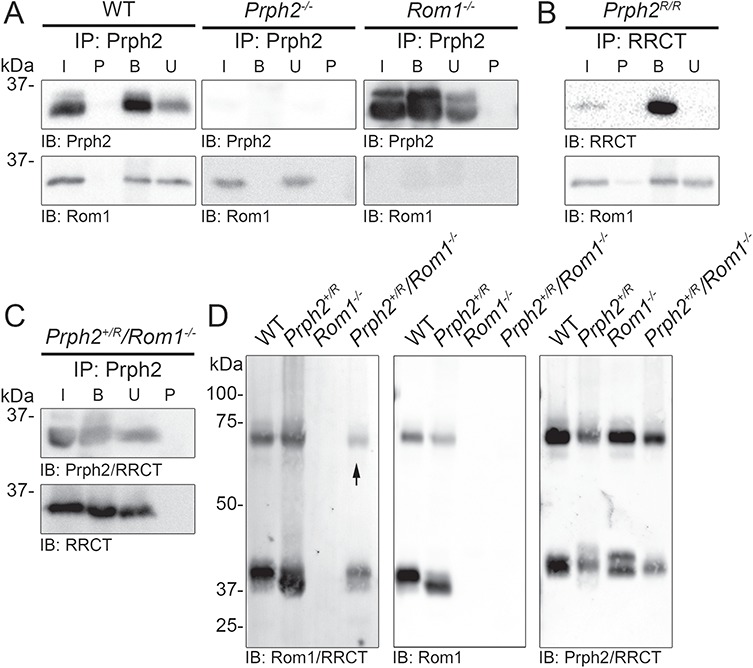
RRCT protein retains the ability to bind ROM-1. (A) IP was performed on P30 retinal extracts for Prph2 (RDS-CT), and blots were probed for Prph2 (mAB 2B7) and Rom1 (ROM1-CT). (B) IP was performed for RRCT (RDS-CT), and blots were probed for RRCT (mAB 2B7) and Rom1 (ROM1-CT). (C) IP was performed for Prph2/RRCT (RDS-CT) and blots were probed for Prph2/RRCT (RDS-CT) or RRCT (mAB 2H5). (A–C) IPs were repeated three times each (with retinas from different animals). (D) Retinal extracts were separated on 10% non-reducing SDS-PAGE. Blots were probed for ROM-1/RRCT (mAB 2H5), Rom1 (ROM1-CT) or Prph2/RRCT (RDS-CT). Arrow indicates RRCT dimer. Blots were repeated 3–5 times on retinal extracts from different animals.
In the retina, non-covalently linked Prph2/Rom1 homo- and hetero-tetramers assemble into covalently linked intermediate-sized Prph2/Rom1 hetero-oligomers and covalently linked Prph2 homo-oligomers (11,15,16). On a non-reducing gel, non-covalently linked Prph2/Rom1 complexes run as monomers while covalently linked complexes run as dimers. As expected, under non-reducing conditions, Prph2 and Rom1 were found in both monomeric and dimeric forms in control retinas. In addition, both monomers and dimers were detected in the Prph2+/R/Rom1−/− using antibodies that recognize RRCT, indicating that RRCT protein participates in covalently linked complexes (Fig. 7D, arrow).
Previous work has shown that higher-order Prph2 homo-oligomers are found in gradient fractions 1–3, intermediate complexes are in fractions 4 and 5 and tetramers are in fractions 6–9 after non-reducing velocity sedimentation using 5–20% sucrose gradients. WT Prph2 was distributed in all these fractions (black line, Fig. 8A, plots show the percent of total Prph2 or Rom1 present in each fraction) while WT Rom1 was not found in the fractions corresponding to higher order complexes (black line, Fig. 8B). In the Prph2+/R, the total pool of Prph2/RRCT was shifted to the right (green line, Fig. 8A), reflecting a decrease in higher-order complexes and a concomitant increase in tetramers. We also saw increased immunoreactivity in fraction 10, which usually has very little Prph2, suggesting that in the presence of RRCT, some Prph2 or RRCT may not have even assembled into tetramers, but rather was present as dimers or monomers. When blots of Prph2+/R gradient fractions were probed for Rom1/RRCT, a similar right shift was noted (green line, Fig. 8B); the percent of Rom1/RRCT found in fractions associated with intermediate-sized complexes (4 and 5) was decreased, with an increase in Rom1/RRCT in tetrameric fractions (8 and 9) as well as in the lighter fractions (10 and 11). A small amount of Rom1/RRCT was also found in fraction 1, which usually contained no detectable Rom1. When blots from the Prph2+/R were probed with antibodies for Rom1 that do not recognize RRCT, the pattern was similar to that seen for Rom1/RRCT: specifically, a right shift in the major peak and a small increase in fraction 1, indicating that native Rom1 is found abnormally in fractions 1, 10 and 11. These findings suggest that RRCT affects overall complex formation rather than merely skewing the distribution because the RRCT alone assembles into abnormal complexes.
Figure 8.
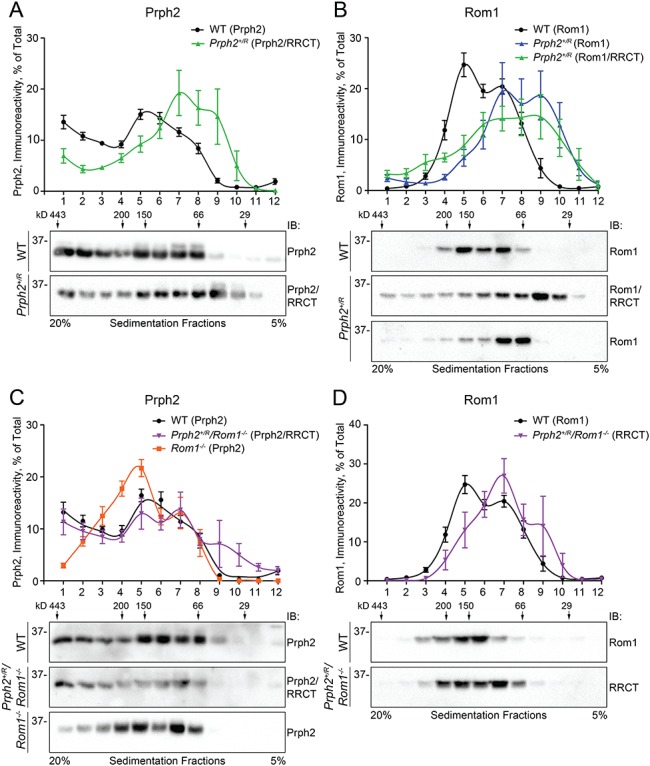
RRCT protein alters the formation of Prph2/Rom1 complexes. (A and B) Retinal extracts from P30 WT (circles) and Prph2+/R (triangles) were separated on continuous 5–20% non-reducing sucrose gradients. (A and B) Gradient fractions were separated on reducing SDS-PAGE and blots were probed for Prph2/RRCT (mAB 2B7, (A)), Rom1 (ROM1-CT, black and blue lines, (B)) or Rom1/RRCT (mAB 2H5, green line (B)). (C and D) Retinal extracts from P30 WT (circles), Prph2+/R/Rom1−/− (triangles) and Rom1−/− (squares) were separated on continuous 5–20% non-reducing sucrose gradients. Gradient fractions were separated on reducing SDS-PAGE and blots were probed for Prph2/RRCT (RDS-CT) (C) or Rom1/RRCT (mAB 2H5) (D). (A–D) Graphs plot mean (±SEM) percent of the total RDS/ROM-1 in each fraction. N = 5–7 gradients/genotype using retinas from different animals for each gradient. Numbers on top of the blots indicate the location of control molecular weight markers in the gradient as in (15).
We wondered whether the shifts in Prph2/Rom1 complex formation in the Prph2+/R retina could be due to RRCT acting as ‘extra’ Rom1. Therefore, we evaluated Prph2/Rom1 complex assembly in the Prph2+/R/Rom1−/− with Rom1−/− serving as a control. In the Rom1−/− (orange line, Fig. 8C), Prph2 complexes were shifted away from higher-order fractions (1–3) toward intermediate fractions (4 and 5), likely to compensate for the missing Rom1 in fractions 4 and 5. In contrast, the distribution of Prph2/RRCT complexes in the Prph2+/R/Rom1−/− (purple line, Fig. 8C) was similar to WT (black lines in Fig. 8C and D were replotted from Fig. 8A and B for comparison) and was not dramatically right-shifted as it was in the Prph2+/R (green line, Fig. 8A). These data suggest that RRCT may harbor some functions of Rom1, a hypothesis largely supported by gradient blots probed for Rom1/RRCT (Fig. 8D). In the Prph2+/R/Rom1−/−, RRCT complex distribution was partially rescued to that of WT Rom1 and did not exhibit the dramatic right shift and simultaneous increase in fraction 1 seen in the Prph2+/R.
Eliminating Rom1 also increased levels of Prph2/RRCT: mean Prph2/RRCT levels in P30 Prph2+/R retinas were 34% of WT, while levels in Prph2+/R/Rom1−/− retinas were 59% of WT (Fig. 9A). Evaluation of Prph2+/R/Rom1−/− retinas with antibodies against Rom1/RRCT showed that the RRCT allele generates protein levels ∼44% of WT Rom1 levels (Fig. 9B), but it is not possible to tell whether this much RRCT protein is present when WT Rom1 is expressed, (i.e. in the Prph2+/R). Immunofluorescent evaluation of Prph2+/R/Rom1−/− retinas also enabled us to evaluate whether RRCT protein was properly targeted to the OS in the presence of WT Prph2. We found no evidence of co-localization between RRCT (green, Fig. 9C) and the IS marker Syntaxin 3B (Fig. 9C and D, red) in the Prph2+/R/Rom1−/− retina; all RRCT protein was properly localized to the OS. Similarly, when sections were co-labeled for RRCT (Fig. 9E, green) and cone arrestin that labels cone OS and IS (Fig. 9E, red), RRCT was detected in cones in the OS layer (Fig. 9E, arrows) but not cone IS (Fig. 9E, arrowheads).
Figure 9.
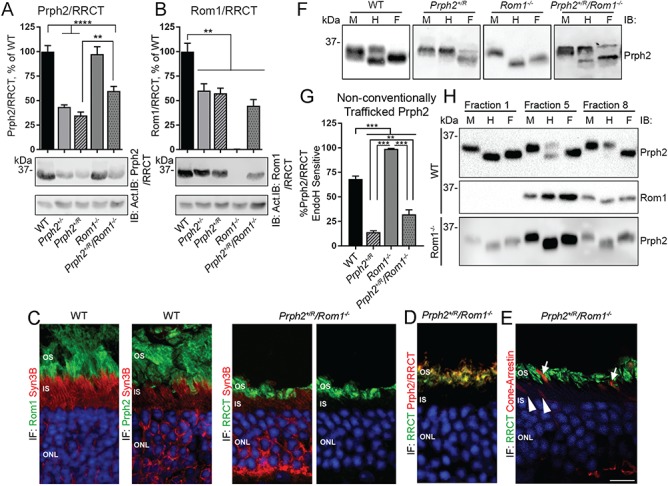
Eliminating Rom1 corrects biochemical defects in the Prph2+/R. (A and B) P30 retinal extracts were separated on reducing SDS-PAGE. Blots were probed for Prph2/RRCT (RDS-CT), Rom1/RRCT (ROM1-2H5) or actin (as a loading control). Bands were measured densitometrically and plotted as means ± SEM from N = 5–10 retinas/group. (C–E) Retinal sections were immunofluorescently labeled, and shown are single planes from confocal image stacks captured at 100×. Images in (C) were labeled with Syntaxin 3B (IS marker) in red and either Rom1/RRCT (ROM1-2H5) or Prph2 (mAB 2B7) in green. Image in (D) was labeled for Prph2/RRCT (red) and RRCT (green). Image in (E) was labeled for cone arrestin (red) and RRCT (ROM1-2H5, green). Arrowheads show cone IS lacking RRCT and arrows show cone OS positive for RRCT. Scale bar: 10 μm. OS: outer segments, IS: inner segments, ONL: outer nuclear layer, IF: immunofluorescence. Repeated at least three times using sections from 3 to 5 different animals (F). Retinal extracts were treated with EndoH (H), PNGase F (F) or mock treated (M). Blots were probed for Prph2/RRCT (RDS-CT). (G) Percent of Prph2/RRCT that is sensitive to EndoH (bottom band from ‘H’ lane of panel F) digestion calculated from the blots in (F) and plotted as means ± SEM, N = 5–15 retinas/group. **P < 0.01, ***P < 0.001 by one-way ANOVA with Tukey’s post-hoc comparison. (H) WT and Rom1−/− retinas underwent sucrose gradient velocity sedimentation under non-reducing conditions. Fractions 1, 5 and 8 subsequently underwent digestion with EndoH (H), PNGase F (F) or were mock treated (M). Repeated five times using gradients from different eyes.
Prph2 traffics to the OS via the conventional secretory pathway and an unconventional secretory pathway that bypasses the trans-Golgi (37,38). N-linked glycans on proteins that bypass the trans-Golgi do not fully mature and therefore retain sensitivity to Endoglycosidase H (EndoH). Unfortunately, we cannot directly assess which trafficking pathway is utilized by Rom1 or RRCT since neither is glycosylated; however, to assess the extent to which Prph2 is trafficked via the unconventional secretory pathway in the presence of RRCT, we treated retinal extracts with EndoH and plotted the fraction of total Prph2/RRCT, which was sensitive to EndoH (Fig. 9F, bottom bands, H column). PNGase F treatment (which removes all N-linked glycans, ‘F’) and mock digested (‘M’) were included as controls. Approximately 68% of Prph2 is EndoH sensitive in the WT (Fig. 9G) and EndoH sensitivity varies dramatically with complex size. When fractions from sucrose gradients of WT retinal extracts are treated with EndoH, all the Prph2 in the fraction associated with large Prph2 homo-oligomers (fraction 1, Fig. 9H) is EndoH sensitive, while Prph2 in fractions associated with Prph2/Rom1 hetero-tetramers (fraction 8, Fig. 9H) is almost entirely EndoH resistant and the fraction associated with intermediate-sized oligomers (fraction 5, Fig 9H) exhibits both EndoH sensitive and resistant populations. Strikingly, we observe that in the Rom1−/− retina, virtually all of the Prph2 is EndoH sensitive (Fig. 9F), regardless of complex size (Fig. 9H). These findings suggest that large Prph2 homo-oligomers largely bypass the trans-Golgi and that Rom1 is important for trafficking its associated Prph2 via conventional pathways. Based on the idea that the RRCT protein acts like excess Rom1, we predicted that unconventionally secreted (i.e. EndoH sensitive) Prph2/RRCT in the Prph2+/R retina would be reduced. This was the case; in the Prph2+/R retina, <10% of Prph2/RRCT was EndoH sensitive, in contrast to ~68% in WT retinas (Fig. 9F–G). In common with other biochemical defects, this decrease in sensitivity in the Prph2+/R was partially corrected in the Prph2+/R/Rom1−/− (Fig. 9F and G).
Eliminating Rom1 ameliorates structural and functional defects associated with RRCT in rods
To see if the biochemical abnormalities that are corrected in the Prph2+/R/Rom1−/− versus the Prph2+/R are associated with structural and functional changes, EM and ERG were performed on Prph2+/R/Rom1−/− and controls. OSs in the Rom1−/− are only mildly abnormal [(28) and Fig. 10A]. OS structure in the Prph2+/R/Rom1−/− was rescued to that seen in the Prph2+/− and was improved compared to that seen in the Prph2+/R (Fig. 10B–D). Specifically, while the Prph2+/− and Prph2+/R/Rom1−/− OSs were shorter than WT and had large whorls, they exhibited fairly normal connecting cilia (Fig. 10B–C, arrows) and had much less of the abnormal vesicle accumulation seen in the Prph2+/R at the base of the OS and in the connecting cilium (Fig. 10D, arrowheads).
Figure 10.
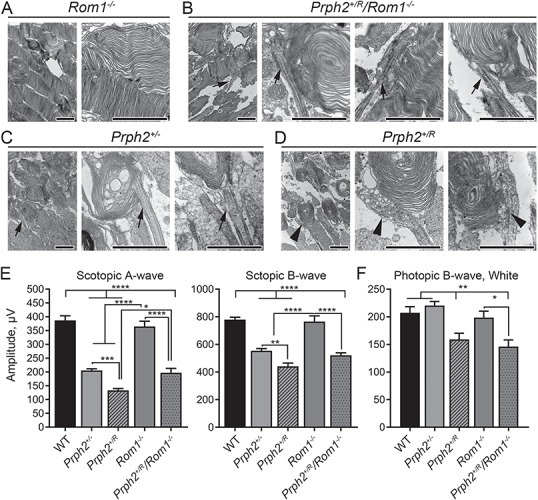
Elimination of Rom1 improves structural and functional phenotypes seen in the Prph2+/R. (A–D) Shown are representative medium magnification (left image in each panel 10 000×) and high magnification (all other images, 40 000×) EMs from Rom1−/− (A), Prph2+/R/Rom1−/− (B), Prph2+/− (C) and Prph2+/R (D). Arrows indicate similar cilia/OS bases in the Prph2+/− and Prph2+/R/Rom1−/−, while arrowheads show the abnormally dilated and vesicles filled cilia/OS bases in the Prph2+/R. Scale bars 2 μm. EM was performed on three different animals/genotype. (E) Full-field ERGs were recorded under scotopic or photopic conditions at P30. Plotted are mean (±SEM) maximum scotopic a-, scotopic b- and photopic b-wave amplitudes. N = 5–15 animals/genotype/age. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA with Tukey’s post-hoc comparison.
This structural improvement in the Prph2+/R/Rom1−/− compared to the Prph2+/R was reflected in improved rod ERG function. Scotopic a-wave amplitudes in Prph2+/R/Rom1−/− eyes were significantly improved compared to the Prph2+/R and were rescued to levels seen in the Prph2+/− (Fig. 10E). In contrast, photopic cone responses were not improved in the Prph2+/R/Rom1−/− compared to the Prph2+/R, and remained significantly lower than the WT, Prph2+/− and Rom1−/− (Fig. 10F).
Discussion
Here, we report that a chimeric protein comprising the body of Rom1 and the C-terminus of Prph2 initiates OS formation and supports some ERG function, but does not support OS elongation or maturation. In addition, the RRCT protein exerts dominant-negative structural and functional defects when expressed in the presence of one copy of WT Prph2. These phenotypic abnormalities are alleviated in rods by removing endogenous Rom1. Removal of endogenous Rom1 (i.e. in Prph2+/R/Rom1−/−) also corrected biochemical defects seen in the Prph2+/R and the Rom1−/−, specifically rescuing complex formation and imbalances in the distribution of Prph2 between conventional and unconventional secretory pathways. In contrast, cone defects associated with RRCT are not alleviated by elimination of Rom1, suggesting that the two cell types may utilize Prph2 and Rom1 differently.
Why should excess Rom1 body (here in the form of the RRCT protein in the Prph2+/R) cause severe defects when its absence is fairly well tolerated (28)? Overexpression of Prph2 is well tolerated in rods and cones and does not lead to degeneration (29). However, overexpression of other photoreceptor proteins such as rhodopsin is toxic (39,40), and we have reported cone toxicity when Rom1 is overexpressed (30). The biochemical mechanism for this Rom1 toxicity likely lies in altered distribution and trafficking of Prph2 complexes as in the Prph2+/R. The Prph2+/R has fewer large covalently linked complexes than WT, abnormal inclusion of Rom1 in remaining large covalent complexes and an increased fraction of Prph2 trafficking via conventional secretory pathways. These changes are likely due to excess Rom1 body (i.e. RRCT) in the Prph2+/R sequestering Prph2 in intermediate and tetrameric complexes at the expense of large Prph2 homo-oligomers. Interestingly, even when the gain-of-function defects in complex formation are corrected in the Prph2+/R by removing Rom1 (i.e. in the Prph2+/R/Rom1−/−), rod structure and function improved only to levels seen in the haploinsufficient Prph2+/− rather than to WT levels, suggesting that the RRCT allele also has loss-of-function effects. Although Prph2/RRCT levels are higher in the Prph2+/R/Rom1−/− than in the Prph2+/R, the total Prph2/RRCT levels in the Prph2+/R/Rom1−/− are ~60% of WT, insufficient to correct Prph2-associated haploinsufficiency (29).
One of our long-term goals is to understand how Prph2 and Rom1 function differently in rod versus cone cells in order to elucidate mechanism underlying PRPH2-associated disease. Here, dominant-negative defects in Prph2+/R rods but not cones were corrected by the removal of endogenous Rom1, suggesting that the Rom1 body has a different role in the two cell types. Approximately 95% of mouse photoreceptors are rods, so our studies showing biochemical improvement in Prph2 oligomerization in the Prph2+/R/Rom1−/− largely represent the case in rods and provide a logical explanation for improved rod structure and function. It is unclear, however, why cones are not improved in this case. Though excess Rom1 leads to structural and functional defects in cones (30), clearly excess Rom1 body does not entirely account for the dominant-negative effects of the RRCT allele in cones, as cones were not improved by reducing Rom1 levels. We have observed differences in Prph2 OS targeting as a function of differences in complex formation in rods versus cones (16,17), but we find no such differences here; rods and cones target RRCT to the OS (both with and without endogenous Prph2). Thus, overt differences in OS targeting are unlikely to underlie the rod versus cone differences. We have previously found that rods and cones use Prph2 differently during the initiation of OS formation, namely that rods require Prph2 to begin forming OSs/discs, while cones can elaborate partly functional OSs without Prph2 (5,6), and this difference in morphogenesis may be tied to the differences in rod versus cone response to removal of Rom1 in the Prph2+/R/Rom1−/−. Future studies may explore the biochemical defects that occur in cones with excess Rom1 (or RRCT), specifically with regard to Prph2/Rom1 complex formation and use of conventional versus non-conventional secretory pathways.
Rom1 appears to be controlling how much Prph2 is processed via conventional versus unconventional trans-Golgi bypass plasma membrane targeting pathways. Tetramers and intermediate complexes that contain Rom1 traffic largely through conventional pathways, while large Prph2 homo-oligomers bypass the Golgi. Previous experiments in Xenopus showed that the C-terminus of Prph2 was sufficient to target green fluorescent protein (GFP) to the OS, leading to the designation of this region (residues 317–336) as an OS targeting signal (8), and a single C-terminal valine residue (V332) is essential (41). In contrast, the Rom1 C-terminus did not promote OS localization, though full-length Rom1 in mouse retina can target to the OS in the absence of Prph2 (35). The Prph2 C-terminus-targeting sequence may promote trafficking through the unconventional pathway, while this signal is overridden in complexes containing Rom1, specifically Rom1 body (as in RRCT), which then traffic more traditionally. Multiple unconventional secretory pathways for membrane resident proteins exist (42,43), and Prph2 is thought to utilize a pathway dependent on COPII-mediated exit from the ER and independent of GRASP55 in the medial Golgi (38). This pathway may be similar to that utilized by polycystin 2 (44), another protein targeted (like Prph2) to the primary cilium. When the polycystin 2 targeting is mutated, unconventional protein trafficking is abolished and the protein traffics through the canonical Golgi-mediated pathway, but without specific ciliary localization. Similarly, when valine 332 is mutated, Prph2 no longer targets to the cilia (OS), rather appearing elsewhere in the photoreceptor. Thus, the hypothesis that the Prph2 C-terminus-targeting sequence/valine 332 is involved in unconventionally targeted Prph2 is a logical, if untested, one. Future experiments will explore the links between the Prph2-targeting sequence, unconventional trafficking pathways, the role of Rom1 in promoting the trafficking of a subset of Prph2 and the function of conventionally versus unconventionally trafficked Prph2.
One of the key findings here is that the Prph2 C-terminus (i.e. in the Prph2R/R) is capable of initiating OS/disc morphogenesis without any Prph2 body present. This ability is due to the Prph2 C-terminus (rather than the body of Rom1) because in the presence of Rom1 without Prph2 [in the Prph2−/− or in the Prph2−/-Rho−/− in which Rom1 levels are stabilized (35)], no OS/disc formation occurs. The ability of RRCT and the Prph2 C-terminus to initiate disc formation is consistent with recent work demonstrating that Prph2−/− OSs that have been electroporated with a construct comprising the body of rhodopsin and the C-terminus of Prph2 exhibit membranous material inside the nascent OS (26). These enclosed membranes do not fully flatten, expand or stack as normal discs do, but they are attached to the OS and represent elaborated membranes that share some morphological similarities to nascent discs during photoreceptor development. The OS morphology in Prph2R/R is significantly better than that observed in the rhodopsin Prph2 C-terminus chimera. Though small and whorl-shaped, OSs in the Prph2R/R exhibit stacked membranous discs with rims, while the membranous structures in the Rho-Prph2 C-terminus chimera are more open, smaller and randomly arranged (26). This is likely because the presence of Rom1 body in the RRCT permits RRCT protein to oligomerize (in contrast to the rhodopsin chimera), suggesting that the role of the Prph2 C-terminus in initiating OS/disc morphogenesis partly relies on oligomerization.
Other previous work on the Prph2 C-terminus has focused on two additional functions of the Prph2 C-terminus: its ability to mediate membrane fusion (21,22) and its ability to regulate membrane curvature (23,24). Recent in vitro experiments found that Prph2 C-terminal deletion mutants promoted enhanced membrane curvature compared to full-length Prph2 (23). Though our results do not directly analyze the ability of the Prph2 C-terminus to induce membrane curvature, we do find that the Prph2+/R photoreceptors exhibit less curved rims than WT or Prph2+/− counterparts, consistent with the in vitro findings using the deletion mutants. These data suggest that one role of the Prph2 C-terminus may be in regulating the size/shape of the rim region and that the ratio of C-terminus to Prph2 body is essential for proper rim shape. In vitro Prph2 has also been found to promote membrane fusion (45), and while Prph2 complex formation with Rom1 is thought to be important for this activity (i.e. mutations in the Prph2 D2 loop impair it) (22,46), the Prph2 C-terminus alone is capable of promoting membrane fusion (47). This fusogenic role has been hypothesized to be important in both in disc shedding and in disc morphogenesis, where newly evaginated discs must fuse away from the plasma membrane as they mature into separate structures. While we do not assess membrane fusion here, the finding that the Prph2+/R photoreceptors accumulate abnormal small to medium vesicular structures may suggest that an increase in the ratio of Prph2 C-terminus to Prph2 body leads to increased fusion and the formation of these vesicular structures. Further exploration of these ideas will require additional studies.
In conclusion, these structure function studies are a striking example of the divergent yet complementary functions for two homologous proteins and provide additional evidence that Prph2 and Rom1 function differently in rod and cone photoreceptors. We show that the C-terminus of Prph2 is capable of initiating OS morphogenesis and disc formation but that the Prph2 body and large Prph2 oligomers are required for proper rim shape and the structural stability needed for OS maturation/elongation. In addition, we show that excess Rom1 is toxic, possibly because of its ability to regulate the unconventional trafficking of Prph2. These findings advance our understanding of the process of OS biogenesis, a cell biological phenomenon that remains incompletely understood.
Materials and Methods
Animal care and use
All experimental animal procedures, maintenance and handling were approved by the Institutional Animal Care and Use Committees (IACUCs) at the University of Oklahoma Health Sciences Center and at the University of Houston, as well as the guidelines set by the Association for Research in Vision and Ophthalmology. RRCT knock-in mice were generated by inGenious Targeting Laboratory, Inc. (Ronkonkoma, New York, USA). In the knock-in allele, the Prph2 genomic sequence beginning from the translation start site (ATG) and ending after the CTC corresponding to Leu 282 of Prph2 was replaced with the Rom1 genomic sequence beginning at the Rom1 translation start site (ATG) and ending with the TTG corresponding to Leu 286 of Rom1 (Supplementary Material, Figs S1 and S2). This substitution resulted in a protein corresponding to Rom1 through the fourth transmembrane domain with the C-terminus of Prph2, expressed from the Prph2 locus and with the Prph2 5′ and 3′ UTRs. The targeting construct included a long homology arm extending ∼6.0 kb upstream of the Prph2 ATG start site in exon 1, 1307 bp of Rom1 genomic sequence including exon 1 beginning at the ATG, Rom1 intron 1, exon 2, intron 2 and the first 21 base pairs of exon 3. This was followed by Prph2 exon 3 beginning at position 22 from the beginning of exon 3. The LoxP/FRT-Neo cassette was inserted 170 bp downstream of Prph2 exon 3. The short homology arm extended 2.0 kb past the end of the Neo cassette. The final targeting vector was 16.7 kb, and restriction analysis and sequencing confirmed that no other mutations were introduced into the coding sequences of Prph2 or Rom1. Following electroporation of linearized targeting construct, embryonic stem cells were screened for the presence of the desired allele and positive clones were injected into C57BL/6 blastocysts and implanted. Chimeric founders were bred to identify mice with germline transmission and then bred to FLPeR expressing mice (Stock#003946, The Jackson Laboratory, Bar Harbor, ME) to remove the Neo cassette. These mice as well as all other strains we use (e.g. Rom1−/− and Prph2−/−) were backcrossed onto our in-house ‘WT’ strain. This strain was created by breeding FVB mice to C57BL/6, eliminating the rd1 and rd8 mutations and then inbreeding for 10 generations. Polymerae chain reaction (PCR) genotyping confirmed that none of the mice used in this study carry the rd8 mutation. Other mouse lines used in this study included Prph2−/− (also known as rds and rd2) bred from founders provided by Dr Neeraj Agarwal (currently at the National Institutes of Health, Bethesda, MD) and Rom1−/− bred from founders provided by Dr Roderick McInnes (McGill University, Montreal, Canada). Animals of both genders were used and were reared under cyclic lighting conditions (12 h L/D, ~30 lux). Animal groups were not randomized, but all groups included both male and female mice. No animals were excluded from the study unless they were euthanized for meeting a humane endpoint as described in the IACUC protocol, or if eyes were grossly injured/abnormal. These criteria are common in all our studies. For each experiment, sample sizes were estimated based on our prior work utilizing similar models. For clarity’s sake we refer to mice homozygous/heterozygous for the RRCT allele as Prph2R/R and Prph2+/R, respectively.
Immunofluorescence
Eyes were enucleated, fixed, dissected and cryoprotected as described previously (17,48). Frozen retinal cross sections (10 μm) were collected at the optic nerve. Sections were incubated in 1% NaBH4, phosphate buffered saline (PBS) and blocking buffer [PBS with 5% BSA (Sigma-Aldrich, St. Louis, MO), 1% fish gelatin (Sigma-Aldrich), 2% donkey serum (Jackson ImmunoResearch, West Grove, PA) and 1% Triton X-100 (VWR, Radnor, PA)]. Sections were incubated overnight with primary antibodies (Table 1) diluted in blocking buffer, then rinsed, incubated with appropriate AlexaFluor-conjugated secondary antibodies (ThermoFisher, Waltham MA, USA), rinsed again and (DAPI; ThermoFisher). Images were captured on a BX-62 spinning disc confocal microscope equipped with an ORCA-ER camera (Olympus, Japan) and analyzed with Slidebook™ 4.2 software (Intelligent Imaging Innovations, Denver, CO) or on a Fluoview 1000 laser scanning confocal microscope (Olympus) and analyzed with ImageJ. All images are presented as single planes from a confocal stack unless otherwise specified. Images from the spinning disc confocal microscope were deconvolved using the nearest neighbors paradigm; images from the laser scanning confocal were not deconvolved. Images were captured with 60×/1.42 oil or 100×/1.40 oil objectives, and exposure times and display settings (brightness and contrast) for all images were normalized to a control section where primary antibody was omitted during processing. No gamma adjustments were made to immunofluorescent images.
Table 1.
Primary antibodies
| Antigen | Antibody | Species | Source | Concentration |
|---|---|---|---|---|
| Prph2/RRCT | RDS-CT | Rbt-PC | In house (15,16) | WB/IF 1:1000 |
| Prph2/RRCT | mAB 2B7, mAB 2E7 | Ms-MC | In house (49) | WB/IF 1:1000 |
| Prph2/RRCT | RDS-MPCT | Rbt-PC | Dr Andrew Goldberg (51,52) | IG 1:10 |
| Rom1 | ROM1-CT | Rbt-PC | In-house (15,16) | WB/IF 1:1000 |
| Rom1/RRCT | mAB 2H5 | Ms-MC | In-house (49,51) | WB/IF/IG 1:5 |
| Rhodopsin | Rhodopsin | Rbt-PC | Dr Steven Fliesler (35,40) | IG 1:10 |
| Rhodopsin | mAB 1D4 | Ms-MC | Dr Robert Molday (35,53) | IF 1:1000 |
| S-opsin | OPN1SW (N-20) | Gt-PC | Santa Cruz Biotechnology, cat# sc-14363 (6,49) | IF 1:500 |
| S-opsin | S-opsin | Rbt-PC | Dr Cheryl Craft (36,54) | IG 1:10 |
| M-opsin | Opsin 1 (Medium Wave) | Rbt-PC | Novus Biologicals cat# 110-74730 (55) | IF 1:1000 |
| Cone Arrestin | C-Arr | Rbt-PC | Dr Cheryl Craft (6,54) | IF 1:5000 |
| Syntaxin 3B | Syn3B | Rbt-PC | Synaptic Systems (cat# 110–032) (17,37) | IF 1:1000 |
| Acetylated Alpha-Tubulin | Ac-A-Tub, Clone 611B-1 | Ms-MC | Sigma (cat# T7451) (6) | IF 1:500 |
| Calreticulin | Calreticulin | Ckn-PC | Abcam (Ab #2908) (31,36) | IF 1:500 |
PC: polyclonal, MC: monoclonal, WB: western blot, IF: immunofluorescence, IG: immunogold. Epitope locations for Prph2/Rom1/RRCT antibodies are shown in Supplementary Material, Fig. 1.
Protein chemistry
For protein chemistry, retinas were added to solubilization buffer [PBS, pH 7.0, containing 1% (v/v) TX-100, 5 mM EDTA, 5 mg/ml N-ethyl maleimide (NEM), and protease inhibitors]; briefly sonicated and then incubated at 4°C for 1 h. After pelleting insoluble material in a microfuge, soluble protein content in extracts was quantified using Bradford reagent (Bio-Rad) per the manufacturer’s directions. Immunoprecipitation (100 μg retinal protein extract per sample) and reducing/non-reducing SDS-PAGE/western blot analyses (15 μg retinal protein extract per lane) were performed as described previously (16,49). Non-reducing velocity sedimentation was performed on whole retinal extracts using continuous 5–20% (w/v) sucrose gradients as described previously (15,49). For gradients, 100 μg protein was loaded per gradient for WT and Rom1−/− while 200 μg was loaded per gradient for Prph2+/R and Prph2+/R/Rom1−/− (due to the lower Prph2 levels). Retinas were not pooled, in each experiment individual retinas from different animals were used (i.e. N = 5 for any given experiment corresponds to five retinas examined individually from five different animals). Primary antibodies used for protein chemistry are listed in Table 1, secondary antibodies were anti-mouse and anti-rabbit HRP (SeraCare, Milford, MA), or anti-mouse 800IR/anti-rabbit 680IR (Licor, Lincoln, NE). All blots analyzed had unsaturated bands (contrast and brightness have been increased to improve visualization) and bands were analyzed densitometrically using Image Studio (Version 5.2, Licor). For glycosidase treatments, retinal extracts (5 μg per sample for WT and Rom1−/− and 24 μg per sample for Prph2+/R and Prph2+/R/Rom1−/−) or gradient fractions (9 μl per fraction) were denatured for 10 min at 100°C using the denaturing buffer provided with the EndoH/PNGase F (New England Biolabs, Ipswich, MA) in a total reaction volume of 30 μl. This was then separated into three aliquots of 10 μl each and an additional 10 μl of enzyme master mix was added to each sample for a final reaction volume of 20 μl. The EndoH sample received 0.1 μl of EndoH plus manufacturer indicated buffer and water; the PNGase sample received 1 μl PNGase F plus manufacturer indicated buffer, detergent and water; and the third sample received EndoH buffer and water without enzyme. All samples were incubated at 37°C for 2 h and then analyzed by SDS-PAGE/western blot. Optimization experiments demonstrated that 2 h was sufficient for complete EndoH/PNGase digestion and incubation for longer periods did not change the outcomes. Experiments were repeated on 3–10 independent retinas/gradient fractions per group.
qRT-PCR
For qRT-PCR, total RNA was isolated, cDNA was prepared and real-time PCR was performed as described previously (50). Briefly, total RNA was isolated from retinas as described previously (50) using TRIzol reagent (ThermoFisher, Waltham, MA) and treated with RNase-free DNase I (Promega, Madison, WI) according to the manufacturer’s recommendations. Reverse transcription was performed using an oligo-dT primer and superscript III reverse transcriptase (ThermoFisher). qRT-PCR was done using a real-time PCR detection system (C1000 Thermal Cycler, Bio-rad, Temecula, CA) and was performed as previously described (5). Values were normalized to the housekeeping gene Hprt. Primer sequences were as follows: Prph2 and RRCT F- 5′ AGGGTCCTTAGGGCAGGTTA-3′, R- 5′-GGTTGGGATGCAGAACTTGT-3′, Hprt. F-5′-GCAAACTTTGCTTTCCCTCGGTT-3′, R-5′- CAAGGGCATATCCAACAACA-3′.
Histology and electron microscopy
Tissue collection, dissection, fixation and plastic embedment for light microscopy (histology), conventional transmission EM and EM IG labeling were as described previously (5). Antibodies used for IG labeling are listed in Table 1. For quantitative analysis of disc rim morphology, an observer blinded to genotype measured the mean outer disc rim diameter and the mean inner disc rim diameter (Fig. 5B and Supplementary Material, Fig. S2) in 250–450 rims/genotype. Rims were measured in multiple images per eye (N = 10−20) and multiple eyes per genotype (N = 3−5) by an individual blinded to genoytpe. These values were used to calculate mean ‘% pinched’ for each disc where % pinched = (Di-Do)/Do*100 and Di is the inner diameter of the rim and Do is the outer diameter of the rim. Image analysis was done using Adobe Photoshop CS6 (Adobe, San Jose, CA).
Electroretinography
Full-field scotopic and spectral photopic ERGs were performed at P30 or P180 using a BigShot® Ganzfeld and UTAS system (LKC, Gaithersburg, MD) as described previously (16,49). Briefly, the eyes of dark-adapted, anesthetized animals were dilated (1% Cyclogyl®; Pharmaceutical Systems, Inc., Tulsa, OK) and scotopic measurements were recorded in response to a single strobe flash at 157 cd.s/m2. For photopic recordings, animals were light-adapted for 5 min (29.03 cd/m2) followed by exposure to 25 flashes at 530 nm (M-cones, 12.5 cd.s/m2). Finally, after a 1 min recovery period, they were exposed to 25 flashes at 365 nm (S-cones, 0.79 cd.s/m2). In some experiments, photopic recordings were done under white light, where measurements were an average of 25 flashes at 77 cd.s/m2. Body temperature was maintained throughout by the use of a warming pad.
Transfection and vectors
Constructs carrying WT Prph2, C214S Prph2 and Rom1 cDNA under the control of the CMV promoter in the pcDNA3.1 vector have been described previously (36). A vector carrying the RRCT cDNA in the same background was generated and all vectors were sequenced prior to use. For immunocytochemistry, COS-7 cells were seeded onto poly-L-lysine (P4832, Sigma) coated 22 mm cover slips in six well plates and transfected with 2 μg of each vector. For protein chemistry, COS-7 cells were seeded into 10 cm dishes and transfected with 5 μg of WT/Rom1 vector and either 5 or 7.5 μg RRCT vector. Calcium phosphate transfection was used as described previously (37); in brief, 2X BBS buffer (50 mM BES, 280 mM NaCl, 1.4 mM Na2HPO4 and pH 6.96) was added to 250 mM CaCl2 containing the appropriate amount of dissolved vector drop by drop then allowed to sit for 30 min prior to being gently added to the DMEM media of cultures (~70% confluent). This was allowed to sit overnight, then the media was replaced with fresh and allowed to sit for an additional 24 h prior to harvesting and downstream processing. Immunoprecipitation, western blotting and immunofluorescence were performed as described in the body of the paper.
Statistical analysis
Differences between groups were analyzed by one-way analysis of variance (ANOVA) with Tukey’s post-hoc comparisons or two-sided Student’s t-test (where only two groups were analyzed). To confirm that data came from a Gaussian distribution prior to ANOVA, they were analyzed by the D’Agostino–Pearson omnibus normality test. Analysis was done using GraphPad Prism version 7.4 (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgements
The authors thank Chen Chen, Barb Nagel, Layne Rodden and Marc Banworth for technical assistance and Muayyad Al-Ubaidi for assistance with the manuscript. The authors also thank Roderick McInnes, Robert Molday, Steven Fliesler, Cheryl Craft and Andrew Goldberg for reagents as indicated in the text.
Conflict of Interest statement. None declared.
Funding
National Eye Institute (R01EY10609 to M.I.N.); Oklahoma Center for the Advancement of Science and Technology (OCAST HR14-150 to S.M.C.); Presbyterian Health Foundation (to S.M.C.); OUHSC Live Animal Imaging and Analysis Core (P30EY027125); Mentoring Diabetes Research in Oklahoma Core (7P20GM104934-10).
Author contributions
S.M.C. and M.I.N. designed the experiments. All authors participated in conducting and analyzing experiments. S.M.C. wrote the manuscript. All authors reviewed and approved the manuscript.
References
- 1. Sanyal S. and Jansen H.G. (1981) Absence of receptor outer segments in the retina of rds mutant mice. Neurosci. Lett., 21, 23–26. [DOI] [PubMed] [Google Scholar]
- 2. Reuter J.H. and Sanyal S. (1984) Development and degeneration of retina in rds mutant mice: the electroretinogram. Neurosci. Lett., 48, 231–237. [DOI] [PubMed] [Google Scholar]
- 3. Connell G., Bascom R., Molday L., Reid D., McInnes R.R. and Molday R.S. (1991) Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc. Natl. Acad. Sci. U.S.A., 88, 723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boon C.J., Hollander A.I., Hoyng C.B., Cremers F.P., Klevering B.J. and Keunen J.E. (2008) The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res., 27, 213–235. [DOI] [PubMed] [Google Scholar]
- 5. Farjo R., Skaggs J.S., Nagel B.A., Quiambao A.B., Nash Z.A., Fliesler S.J. and Naash M.I. (2006) Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J. Cell Biol., 173, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conley S.M., Al-Ubaidi M.R., Han Z. and Naash M.I. (2014) Rim formation is not a prerequisite for distribution of cone photoreceptor outer segment proteins. FASEB J., 28, 3468–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding X.Q., Stricker H.M. and Naash M.I. (2005) Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry (Mosc), 44, 4897–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tam B.M., Moritz O.L. and Papermaster D.S. (2004) The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Mol. Biol. Cell, 15, 2027–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moritz O.L. and Molday R.S. (1996) Molecular cloning, membrane topology, and localization of bovine rom-1 in rod and cone photoreceptor cells. Invest. Ophthalmol. Vis. Sci., 37, 352–362. [PubMed] [Google Scholar]
- 10. Arikawa K., Molday L.L., Molday R.S. and Williams D.S. (1992) Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J. Cell Biol., 116, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goldberg A.F. and Molday R.S. (1996) Subunit composition of the peripherin/rds-rom-1 disk rim complex from rod photoreceptors: hydrodynamic evidence for a tetrameric quaternary structure. Biochemistry (Mosc), 35, 6144–6149. [DOI] [PubMed] [Google Scholar]
- 12. Goldberg A.F., Loewen C.J. and Molday R.S. (1998) Cysteine residues of photoreceptor peripherin/rds: role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry (Mosc), 37, 680–685. [DOI] [PubMed] [Google Scholar]
- 13. Loewen C.J. and Molday R.S. (2000) Disulfide-mediated oligomerization of Peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J. Biol. Chem., 275, 5370–5378. [DOI] [PubMed] [Google Scholar]
- 14. Goldberg A.F., Fales L.M., Hurley J.B. and Khattree N. (2001) Folding and subunit assembly of photoreceptor peripherin/rds is mediated by determinants within the extracellular/intradiskal EC2 domain: implications for heterogeneous molecular pathologies. J. Biol. Chem., 276, 42700–42706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chakraborty D., Ding X.Q., Fliesler S.J. and Naash M.I. (2008) Outer segment oligomerization of Rds: evidence from mouse models and subcellular fractionation. Biochemistry (Mosc), 47, 1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chakraborty D., Ding X.Q., Conley S.M., Fliesler S.J. and Naash M.I. (2009) Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum. Mol. Genet., 18, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chakraborty D., Conley S.M., Stuck M.W. and Naash M.I. (2010) Differences in RDS trafficking, assembly and function in cones versus rods: insights from studies of C150S-RDS. Hum. Mol. Genet., 19, 4799–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zulliger R., Conley S.M., Mwoyosvi M.L., Al-Ubaidi M.R. and Naash M.I. (2018) Oligomerization of Prph2 and Rom1 is essential for photoreceptor outer segment formation. Hum. Mol. Genet., epub ahead of print. 10.1093/hmg/ddy2402018/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jansen H.G. and Sanyal S. (1984) Development and degeneration of retina in rds mutant mice: electron microscopy. J. Comp. Neurol., 224, 71–84. [DOI] [PubMed] [Google Scholar]
- 20. Ritter L.M., Arakawa T. and Goldberg A.F. (2005) Predicted and measured disorder in peripherin/rds, a retinal tetraspanin. Protein Pept. Lett., 12, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boesze-Battaglia K., Lamba O.P., Napoli A.A. Jr., Sinha S. and Guo Y. (1998) Fusion between retinal rod outer segment membranes and model membranes: a role for photoreceptor peripherin/rds. Biochemistry (Mosc), 37, 9477–9487. [DOI] [PubMed] [Google Scholar]
- 22. Boesze-Battagliaa K. and Stefano F.P. (2002) Peripherin/rds fusogenic function correlates with subunit assembly. Exp. Eye Res., 75, 227–231. [DOI] [PubMed] [Google Scholar]
- 23. Milstein M.L., Kimler V.A., Ghatak C., Ladokhin A.S. and Goldberg A.F.X. (2017) An inducible amphipathic helix within the intrinsically disordered C terminus can participate in membrane curvature generation by peripherin-2/rds. J. Biol. Chem., 292, 7850–7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khattree N., Ritter L.M. and Goldberg A.F. (2013) Membrane curvature generation by a C-terminal amphipathic helix in peripherin-2/rds, a tetraspanin required for photoreceptor sensory cilium morphogenesis. J. Cell. Sci., 126, 4659–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kevany B.M., Tsybovsky Y., Campuzano I.D., Schnier P.D., Engel A. and Palczewski K. (2013) Structural and functional analysis of the native peripherin/ROM1 complex isolated from photoreceptor cells. J. Biol. Chem., 288, 36272--36284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salinas R.Y., Pearring J.N., Ding J.D., Spencer W.J., Hao Y. and Arshavsky V.Y. (2017) Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J. Cell Biol., 216, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kedzierski W., Weng J. and Travis G.H. (1999) Analysis of the rds/peripherin.rom1 complex in transgenic photoreceptors that express a chimeric protein. J. Biol. Chem., 274, 29181–29187. [DOI] [PubMed] [Google Scholar]
- 28. Clarke G., Goldberg A.F., Vidgen D., Collins L., Ploder L., Schwarz L., Molday L.L., Rossant J., Szel A., Molday R.S. et al. (2000) Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat. Genet., 25, 67–73. [DOI] [PubMed] [Google Scholar]
- 29. Nour M., Ding X.Q., Stricker H., Fliesler S.J. and Naash M.I. (2004) Modulating expression of peripherin/rds in transgenic mice: critical levels and the effect of overexpression. Invest. Ophthalmol. Vis. Sci., 45, 2514–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chakraborty D., Conley S.M., Nash Z., Ding X.Q. and Naash M.I. (2012) Overexpression of ROM-1 in the cone-dominant retina. Adv. Exp. Med. Biol., 723, 633–639. [DOI] [PubMed] [Google Scholar]
- 31. Conley S.M., Stuck M.W., Watson J.N. and Naash M.I. (2017) Rom1 converts Y141C-Prph2-associated pattern dystrophy to retinitis pigmentosa. Hum. Mol. Genet., 26, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poloschek C.M., Bach M., Lagreze W.A., Glaus E., Lemke J.R., Berger W. and Neidhardt J. (2010) ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest. Ophthalmol. Vis. Sci., 51, 4253–4265. [DOI] [PubMed] [Google Scholar]
- 33. Kajiwara K., Berson E.L. and Dryja T.P. (1994) Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 264, 1604–1608. [DOI] [PubMed] [Google Scholar]
- 34. Dryja T.P., Hahn L.B., Kajiwara K. and Berson E.L. (1997) Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci., 38, 1972–1982. [PubMed] [Google Scholar]
- 35. Chakraborty D., Conley S.M., Al-Ubaidi M.R. and Naash M.I. (2014) Initiation of rod outer segment disc formation requires RDS. PLoS One, 9, e98939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Conley S.M., Stricker H.M. and Naash M.I. (2010) Biochemical analysis of phenotypic diversity associated with mutations in codon 244 of the retinal degeneration slow gene. Biochemistry (Mosc), 49, 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zulliger R., Conley S.M., Mwoyosvi M.L., Stuck M.W., Azadi S. and Naash M.I. (2015) SNAREs interact with retinal degeneration slow and rod outer segment membrane protein-1 during conventional and unconventional outer segment targeting. PLoS One, 10, e0138508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian G., Ropelewski P., Nemet I., Lee R., Lodowski K.H. and Imanishi Y. (2014) An unconventional secretory pathway mediates the cilia targeting of peripherin/rds. J. Neurosci., 34, 992–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wen X.H., Shen L., Brush R.S., Michaud N., Al-Ubaidi M.R., Gurevich V.V., Hamm H.E., Lem J., Dibenedetto E., Anderson R.E. et al. (2009) Overexpression of rhodopsin alters the structure and photoresponse of rod photoreceptors. Biophys. J., 96, 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tan E., Wang Q., Quiambao A.B., Xu X., Qtaishat N.M., Peachey N.S., Lem J., Fliesler S.J., Pepperberg D.R., Naash M.I. et al. (2001) The relationship between opsin overexpression and photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci., 42, 589–600. [PubMed] [Google Scholar]
- 41. Salinas R.Y., Baker S.A., Gospe S.M. 3rd and Arshavsky V.Y. (2013) A single valine residue plays an essential role in peripherin/rds targeting to photoreceptor outer segments. PLoS One, 8, e54292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Malhotra V. (2013) Unconventional protein secretion: an evolving mechanism. EMBO J., 32, 1660–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rabouille C., Malhotra V. and Nickel W. (2012) Diversity in unconventional protein secretion. J. Cell. Sci., 125, 5251–5255. [DOI] [PubMed] [Google Scholar]
- 44. Hoffmeister H., Babinger K., Gurster S., Cedzich A., Meese C., Schadendorf K., Osten L., Vries U., Rascle A. and Witzgall R. (2011) Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol., 192, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boesze-Battaglia K., Stefano F.P., Fenner M. and Napoli A.A. Jr. (2000) A peptide analogue to a fusion domain within photoreceptor peripherin/rds promotes membrane adhesion and depolarization. Biochim. Biophys. Acta, 1463, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boesze-Battaglia K., Stefano F.P., Fitzgerald C. and Muller-Weeks S. (2007) ROM-1 potentiates photoreceptor specific membrane fusion processes. Exp. Eye Res., 84, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boesze-Battaglia K., Goldberg A.F., Dispoto J., Katragadda M., Cesarone G. and Albert A.D. (2003) A soluble peripherin/Rds C-terminal polypeptide promotes membrane fusion and changes conformation upon membrane association. Exp. Eye Res., 77, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stricker H.M., Ding X.Q., Quiambao A., Fliesler S.J. and Naash M.I. (2005) The Cys214–>Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem. J., 388, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Conley S.M., Stuck M.W., Burnett J.L., Chakraborty D., Azadi S., Fliesler S.J. and Naash M.I. (2014) Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum. Mol. Genet., 23, 3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cai X., Nash Z., Conley S.M., Fliesler S.J., Cooper M.J. and Naash M.I. (2009) A partial structural and functional rescue of a retinitis pigmentosa model with compacted DNA nanoparticles. PLoS One, 4, e5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stuck M.W., Conley S.M. and Naash M.I. (2014) The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum. Mol. Genet., 23, 6260–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goldberg A.F., Ritter L.M., Khattree N., Peachey N.S., Fariss R.N., Dang L., Yu M. and Bottrell A.R. (2007) An intramembrane glutamic acid governs peripherin/rds function for photoreceptor disk morphogenesis. Invest. Ophthalmol. Vis. Sci., 48, 2975–2986. [DOI] [PubMed] [Google Scholar]
- 53. MacKenzie D., Arendt A., Hargrave P., McDowell J.H. and Molday R.S. (1984) Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry (Mosc), 23, 6544–6549. [DOI] [PubMed] [Google Scholar]
- 54. Zhu X., Li A., Brown B., Weiss E.R., Osawa S. and Craft C.M. (2002) Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol. Vis., 8, 462–471. [PubMed] [Google Scholar]
- 55. Chakraborty D., Conley S.M., Zulliger R. and Naash M.I. (2016) The K153Del PRPH2 mutation differentially impacts photoreceptor structure and function. Hum. Mol. Genet., 25, 3500–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.