

Abstract
Neuronal and non-neuronal cells express the huntingtin (HTT) protein, yet neurodegeneration in Huntington’s disease (HD) is largely selective, affecting most prominently striatal medium spiny neurons and cortical pyramidal neurons. Selective toxicity of full-length human mutant HTT (fl-mHTT) may be due in part to its expression in non-neuronal cells. While studies suggest neuronal–glial interactions are important in HD and fl-mHTT is expressed in astrocytes, it has not been determined whether the expression of fl-mHTT in astrocytes is necessary for HD pathogenesis. To directly assess the necessity of fl-mHTT in astrocytes for HD pathogenesis, we used a mouse genetic approach and bred the conditional mHTT-expressing BACHD mouse model with GFAP-CreERT2 mice. We show that GFAP-CreERT2 expression in these mice is highly selective for astrocytes, and we are able to significantly reduce the expression of fl-mHTT protein in the striatum and cortex of BACHD/GFAP-CreERT2-tam mice. We performed behavioral, electrophysiological and neuropathological analyses of BACHD and BACHD/GFAP-CreERT2-tam mice. Behavioral analyses of BACHD/GFAP-CreERT2-tam mice demonstrate significant improvements in motor and psychiatric-like phenotypes. We observe improvements in neuropathological and electrophysiological phenotypes in BACHD/GFAP-CreERT2-tam mice compared to BACHD mice. We observed a restoration of the normal level αB-crystallin in the striatum of the BACHD/GFAP-CreERT2 mice, indicating a cell autonomous effect of mHTT on its expression. Taken together, this work indicates that astrocytes are important contributors to the progression of the behavioral and neuropathological phenotypes observed in HD.
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder characterized clinically by cognitive, psychiatric and motor deficits. HD is caused by a CAG repeat expansion in the gene encoding the widely expressed protein huntingtin (HTT) (1,2). The HTT protein is found in both neuronal and non-neuronal cells throughout the brain; however, neurodegeneration is most prominent in striatal medium spiny neurons (MSNs) and cortical pyramidal neurons (3–6).
Astrocytes are thought to have a key role in HD pathogenesis. They are important for cellular homeostasis, which includes providing the neuron with energy and substrates for neurotransmission (7,8). Astrocyte processes are associated with both the pre-synaptic and post-synaptic components of the synapse (9,10), and astrocytes help to remove synaptic glutamate through glutamate transporters (11,12).
In HD brains, reactive astrocytes are prominent, and the severity of reactive astrocyte morphology increases with disease progression (13). There is a decrease in the level of mRNA of the astroglial-specific glutamate transporter EAAT2 in HD (14) and a decrease in mouse Glt-1 mRNA, protein or glutamate uptake in mutant HTT (mHTT)-expressing mouse models (13,15–17). This transporter has a key role in removing synaptic glutamate (11,12), and its loss may contribute to downstream excitotoxicity in HD (16,18,19). Another study using R6/2 mice demonstrated decreased astrocyte Kir4.1 K+ channel activity that could lead to altered MSN excitability (20,21). We have found a marked dysregulation of glutamate release from cultured astrocytes from full-length human mutant HTT (fl-mHTT)-expressing BACHD mice (22). Additional astrocytic mechanisms, including decreased BDNF release and decreased expression of the small heat shock protein αB-crystallin, have been implicated (23–25). Finally, others have reported that when truncated mHTT is expressed in cultured primary astrocytes in the presence of wild-type neurons, these cells are capable of inducing neuronal cell death through a variety of mechanisms including excitotoxicity (23,26–28). Together, these studies implicate astrocytic expression of mHTT in HD pathogenesis.
We have taken an in vivo mouse genetic approach using the conditional BACHD mouse model to examine the contribution of astrocytic fl-mHTT to disease pathogenesis in vivo. We used a GFAP-CreERT2 transgene to decrease fl-mHTT to determine whether the presence of mHTT in astrocytes is necessary for HD pathogenesis. We found that reduction of fl-mHTT in astrocytes of BACHD/GFAP-CreERT2 mice after tamoxifen injection significantly decreases the total mHTT burden in the brain. In addition, reducing fl-mHTT expression in astrocytes slows the progression of HD-relevant phenotypes displayed by these mice. Furthermore, our data demonstrates that reducing fl-mHTT in astrocytes leads to a redistribution of mHTT aggregates in neurons in the BACHD mice, restores the level of synaptic marker proteins and reverses the electrophysiological abnormalities in excitatory transmission at corticostriatal synapses. These data point to the potential of modulating astrocytic mHTT to achieve enhanced neuronal function and neuroprotection.
Results
fl-mHTT reduction in BACHD/GFAP-CreERT2 mice
In order to address the hypothesis that fl-mHTT within astrocytes contributes to HD phenotypes, we used the conditional BACHD mouse model (29,30). In these studies, we combined the BACHD model with an inducible GFAP-CreERT2 mouse model where Cre recombinase is active after tamoxifen injection to perform selective deletion of the fl-mHTT transgene. This inducible Cre strategy was necessary because the GFAP promotor is active in glial progenitors that give rise to astrocytes, neurons and oligodendrocytes (31–33); thus early deletion with a non-regulatable GFAP-Cre might lead to non-selective deletion.
To evaluate the effectiveness of the GFAP-CreERT2 recombination (34), we first bred these mice to the Ai14 Rosa26 tdTomato reporter mouse line (35). The mice were weaned at 21 days, and then tamoxifen was administered for 10 days. Histological studies were used to determine recombination efficiency and specificity in these mice (Fig. 1A–C). Cells where GFAP-CreERT2 was active in the Ai14 mice were red based on the removal of a floxed (LoxP-flanked) stop codon (35). Nearly 85% of the tdTomato positive astrocytes (n = 300) were also stained with an antibody to Aldh1l1, a marker of mature astrocytes (36). We also stained neurons on sections from Ai14/GFAP-CreERT2-tam mice using the NeuN antibody at 1.5 months of age. We counted tdTomato positive cells (n = 200) and observed no overlapping signal with NeuN in the striatum of these mice (Fig. 1C). In the cortex, we found that less than 0.4% of the tdTomato positive cells were also NeuN positive (n = 300). Together, these data show that the GFAP-CreERT2 mouse and the tamoxifen treatment protocol used for these studies provided efficient and highly selective recombination in astrocytes and thus were suitable for our studies.
Figure 1.
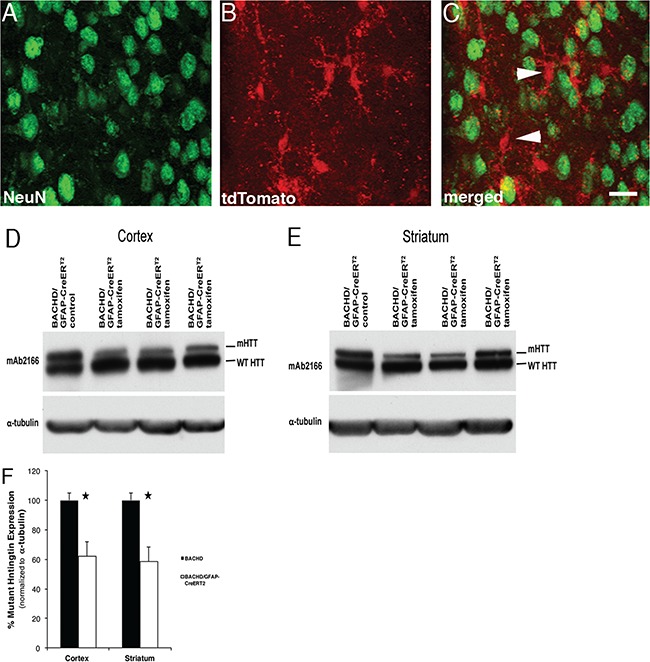
Full-length mHTT reduction in astrocytes. GFAP-CreERT2 mice crossed to Ai14 Rosa tdTomato mice and injected with tamoxifen reveal recombination in astrocytes. Ai14/GFAP-CreERT2-tam mice. (A) NeuN positive staining (green) of neurons in the striatum. (B) tdTomato positive astrocytes (arrow) and merged in (C). Western blot demonstrating fl-mHTT reduction in BACHD/GFAP-CreERT2-tam mouse (D) cortex and (E) striatum. (F) Analyses of fl-mHTT levels in BACHD/GFAP-CreERT2-tam mice reveals a 40% (±8.42) reduction in fl-mHTT levels in cortex (P = 0.027) and 42% (±10.05) reduction in striatum (P = 0.023) when normalized to α-tubulin. Data are mean ± SEM, n = 4 BACHD and n = 3 BACHD/GFAP-CreERT2.
We then bred BACHD mice to GFAP-CreERT2 mice and administered tamoxifen for 10 days after weaning. To evaluate the effect of transgene deletion on brain HTT levels, we extracted protein from the cortex and striatum of BACHD/GFAP-CreERT2-vehicle–injected and BACHD/GFAP-CreERT2-tamoxifen–injected mice at 2 months of age. We performed western blots and probed for fl-mHTT and endogenous mouse HTT with mAb2166 (Fig. 1D and E). When fl-mHTT levels in the BACHD and BACHD/GFAP-CreERT2 tam were compared after normalizing to α-tubulin, we found an average reduction of fl-mHTT levels by 40% (±8.42%, P < 0.027) in the cortex and about 42% (±10.05%, P < 0.023) in the striatum of BACHD/GFAP-CreERT2-tam–injected mice when compared to BACHD (Fig. 1F; n = 4 BACHD and n = 3 BACHD/GFAP-CreERT2 mice; Student’s t-test). There were no differences in endogenous mouse HTT protein expression levels in the striatum or cortex in wild-type and GFAP-CreERT2 mice (data not shown). Thus, we concluded that elimination of astrocyte fl-mHTT reduces the overall brain load of fl-mHTT.
Improvement in behavioral phenotypes in BACHD/GFAP-CreERT2 mice: motor performance
In order to assess the effect of fl-mHTT reduction in astrocytes on behavioral phenotypes observed in BACHD mice, we studied BACHD, BACHD/GFAP-CreERT2-tam and wild-type mice. The BACHD/GFAP-CreERT2 mice were injected with tamoxifen after weaning (beginning at 22–23 days old) one time/day for 10 days. Motor impairment has been observed in BACHD mice beginning at 2 months of age and is more prominent at 6 months of age (29,30,37). Due to the robustness of the rotarod test to identify motor coordination deficits in the BACHD model and other mice expressing mHTT, we focused on this test and used an accelerating rotarod paradigm to assess motor performance in our cohort of mice at 2, 6 and 12 months of age in the present study. The mice are weighed at each age when tested (Table 1). The mice are trained first at 2 months for three trials/day over 2 days and then tested for three trials/day over 3 days.
Table 1.
Body weight of animals used for behavior experiments
| Light–Dark | ||||||
|---|---|---|---|---|---|---|
| 2 months | 6 months | 12 months | ||||
| Mean (g) | SEM | Mean (g) | SEM | Mean (g) | SEM | |
| Wild type | 26.53 | 0.78 | 32.64 | 1.47 | 35.25 | 0.92 |
| BACHD/GFAP/CreERT2 | 28.76 | 0.76 | 38.13 | 1.22 | 38.53 | 2.59 |
| BACHD | 31.21 | 0.74 | 38.92 | 0.96 | 41.39 | 1.52 |
| Rotarod/forced swim | ||||||
|---|---|---|---|---|---|---|
| 2 months | 6 months | 12 months | ||||
| Mean (g) | SEM | Mean (g) | SEM | Mean (g) | SEM | |
| Wild type | 23.19 | 0.55 | 23.23 | 0.54 | 31.06 | 0.95 |
| BACHD/GFAP-CreERT2 | 26.95 | 0.59 | 28.72 | 0.51 | 40.94 | 1.19 |
| BACHD | 27.92 | 0.62 | 29.22 | 0.79 | 43.75 | 1.14 |
We found with rotarod testing, BACHD mice showed a marked impairment in rotarod performance beginning at 6 months of age when compared to wild-type control, and all mice demonstrated a progressive decline over the subsequent 6 months [BACHD n = 13, wild type n = 11, BACHD/GFAP-CreERT2-tam n = 17 (age X genotype: F (4, 556) = 16.9, P < 0.0001; Fisher’s least significant difference (LSD): P = 0.0084)]. The most severe decline in BACHD mice was observed at 12 months, as has been observed in previous studies (29,30,37). The BACHD/GFAP-CreERT2-tam mice, while demonstrating an initial decline similar to BACHD mice at 6 months, subsequently showed a reduced rate of decline thereafter (Table 2). At the 12-month time point, their performance was better than BACHD mice [29.4% decrease in BACHD/GFAP-CreERT2-tam versus 42.6% decrease in BACHD mice (genotype: F (3, 101) = 11.5, P < 0.0001)]. Importantly, the improvements observed in the rotarod phenotype were present in both females and males of BACHD/GFAP-CreERT2-tam mice.
Table 2.
Statistical results of behavior experiments
| N | 2 months | 6 months | 12 months | |
|---|---|---|---|---|
| Rotarod, cohort 1 | ||||
| BACHD | 13 | -- (4.389 +/− 0.06) | -- (4.079 +/− 0.06) | -- (3.303 +/− 0.10) |
| BACHD/GFAP-CreERT2 | 17 | NS (4.354 +/− 0.05) | NS (4.033 +/− 0.05) | ****(3.956 +/− 0.06) |
| Wild type | 11 | NS (4.321 +/− 0.06) | NS (4.029 +/− 0.06) | ****(4.266 +/− 0.06) |
| Rotarod, cohort 2 | ||||
| BACHD | 33 | -- (3.327 +/− 0.05) | -- (2.181 +/− 0.05) | -- (1.791 +/− 0.05) |
| BACHD/GFAP-CreERT2 | 40 | NS (3.412 +/− 0.04) | ****(2.618 +/− 0.04) | ****(2.321 +/− 0.04) |
| Wild type | 32 | ****(4.538 +/− 0.05) | ****(4.144 +/− 0.05) | ****(3.890 +/− 0.06) |
| Rotarod, N-GFAP-CreERT2 | ||||
| BACHD | 12 | -- (4.740 +/− 0.05) | -- (4.228 +/− 0.05) | -- (3.527 +/− 0.05) |
| BACHD/N-GFAP-CreERT2 | 10 | NS (4.715 +/− 0.05) | *(4.386 +/− 0.05) | **(3.778 +/− 0.05) |
| Wild type | 11 | ****(5.263 +/− 0.05) | ****(5.070 +/− 0.05) | ****(4.700 +/− 0.05) |
| Light–dark box choice | ||||
| BACHD | 21 | -- (31.21 +/− 3.6) | -- (37.28 +/− 3.2) | -- (32.02 +/− 4.6) |
| BACHD/GFAP-CreERT2 | 24 | NS (32.15 +/− 3.2) | NS (27.83 +/− 3.2) | NS (25.46 +/− 5.7) |
| Wild type | 32 | *(40.8 +/− 1.9) | NS (43.13 +/− 3.6) | *(54.64 +/− 7.2) |
| Forced swimming | ||||
| BACHD | 32 | -- (27.69 +/− 4.6) | -- (35.13 +/− 6.2) | -- (38.29 +/− 5.9) |
| BACHD/GFAP-CreERT2 | 40 | NS (28.43 +/− 3.9) | NS (28.25 +/− 3.8) | *(27.23 +/− 4.2) |
| Wild type | 27 | *(1.22 +/− 0.5) | *(1.91 +/− 0.7) | *(1.78 +/− 0.9) |
NS = not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. All P-values compared to BACHD (Fisher’s LSD).
To further validate these observations on rotarod performance of BACHD and BACHD/GFAP-CreERT2-tam mice, a second larger independent cohort of mice was studied (Fig. 2A–C; BACHD n = 33, wild type n = 32, BACHD/GFAP-CreERT2-tam n = 40; Table 2—Rotarod 2nd cohort). Cohort 2 mice were trained and tested the same as cohort 1 mice. This replicate experiment yielded a consistent and similar result [age X genotype: F (6, 2402) = 24.2, P < 0.0001; Fisher’s LSD: P < 0.0001]. Although motor learning on the rotarod was likely not significantly different within this cohort based on previous work (29,30), training data for a subset of the cohort was recorded during the 2-month rotarod test and analyzed to assess whether a learning deficit was present in the BACHD (n = 20) or BACHD/GFAP-CreERT2 mice (n = 25) when compared to wild type (n = 20). Analysis of variance (ANOVA) revealed no significant difference among genotypes for average performance on either Day 1 [F (2,38) = 1.8095, P = 0.1766] or Day 2 [F (2,39) = 1.7101, P = 0.1938] (Supplementary Material, Fig. S1); therefore, we concluded that mice of all genotypes were able to learn the behavior appropriately at this age. Since it is known that BACHD mice have increased weight when compared to wild-type mice (Table 1) (29,30,40), in this second cohort, we obtained body weight and used this as a covariate in our analyses. We find that body weight was correlated to some degree with the observed rotarod fall latency at 6 months [R2 = 0.39, F (1, 129) = 81.4, P < 0.0001] and at 12 months [R2 = 0.35,F (1, 118) = 64.3, P < 0.0001]. However, when body weight and its interaction with genotype, sex and age were assessed in a full repeated measures mixed model, weight was not found to be a significant predictor of performance (F (1, 187) = 1.8, P = 0.1821).
Figure 2.

Motor deficits are decreased in BACHD/GFAP-CreERT2-tam mice. Rotarod analysis of a cohort of BACHD (B), BACHD/GFAP-CreERT2-tam (BG) and wild-type (WT) mice at 2, 6 and 12 months of age. There is a decreased latency to fall from the rotarod beginning at 2 months of age in BACHD mice. (A) Latency to fall in BACHD/GFAP-CreERT2-tam mice is increased at 6 and 12 months when all mice are grouped together, BACHD (n = 33), wild type (n = 32), BACHD/GFAP-CreERT2-tam (n = 40). This change is observed in male (B) and female (C) BACHD/GFAP-CreERT2-tam mice. An additional GFAP-CreERT2 (N-GFAP-CreERT2) mouse was bred to confirm that the original findings were not due to the specific Cre mouse chosen (D–F). The latency to fall of the new BACHD/N-GFAP-CreERT2-tam (BG-N) mice is reduced for the mice as a group at 6 and 12 months, BACHD (n = 12), wild type (n = 11), BACHD/N-GFAP-CreERT2 -tam (n = 10); the robust observed difference in male mice likely drives this result. All data are log-transformed and presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Given the concern that some behavioral phenotypes may be observed when using only one conditional or inducible Cre mouse model, we also studied mice produced by a cross with an additional GFAP-CreERT2 (called N-GFAP-CreERT2 for simplicity here)-expressing mouse model (32) to the BACHD mice. We performed repeated measure rotarod studies using BACHD (n = 12), BACHD/N-GFAP-CreERT2-tam (BG-N) (n = 10) and wild-type (n = 11) mice at 2, 6 and 12 months (Fig. 2D—F, Table 2). Although we used fewer mice in this cohort, we nonetheless observe a slowing of the decline in rotarod performance by 6 months of age in the BACHD/N-GFAP-CreERT2-tam mice [age X genotype: F (8, 1028) = 10.9, P < 0.0001; Fisher’s LSD: P = 0.0013]. This was similar to the change observed in the BACHD/GFAP-CreERT2-tam mice. The appearance of a similar phenotype in these studies using two different GFAP-CreERT2 mice to perform selective deletion of the BACHD transgene supports the argument that the reduced rate of decline in rotarod performance is due to the decrease of fl-mHTT in GFAP-CreERT2–expressing astrocytes.
We also performed the open field test to analyze general motor activity of BACHD, wild-type and BACHD/GFAP-CreERT2-tam mice at 2 and 6 months of age (Supplementary Material, Fig. S2). We did not observe any significant difference between the mice at 2 months of age. However, we observed a significant difference in performance of BACHD mice as compared to wild-type mice at 6 months of age where they show less total distance moved. In addition, the velocity of the movement in the BACHD mice is less than the wild type. When we examined the open field behavior of the BACHD/GFAP-CreERT2 mice, we found no difference in their performance at 2 months when compared to wild type or BACHD. There is a statistically significant difference in the performance of the BACHD/GFAP-CreERT2 mice at 6 months when compared to wild-type (P < 0.0001) as well as the BACHD mice (P < 0.0001) for total distance moved, as well as the velocity. This result reveals that while there is an improvement in both the total distance moved and velocity in the BACHD/GFAP-CreERT2 mice, there is only a partial contribution of fl-mHTT-expressing astrocytes to this motor activity. Unfortunately, due to technical challenges, we were unable to assess the performance of this group of mice at 12 months of age. Nonetheless, taking both the rotarod and open field data together, we identify fl-mHTT-expressing astrocytes as contributors to motor deficits in HD.
Improvement in behavioral phenotypes in BACHD/GFAP-CreERT2-tam mice: neuropsychiatric features
HD patients display psychiatric changes (39,41), and non-motor phenotypes can be identified in mouse models. These changes can manifest in HD patients prior to overt motor dysfunction (38). We have previously shown that BACHD mice display anxiety-like deficits in performance using the light–dark box paradigm (29). We tested the BACHD/GFAP-CreERT2 mice to determine if the expression of fl-mHTT in astrocytes contributed to the observed deficit in the BACHD mice. A difference between BACHD and BACHD/GFAP-CreERT2-tam mice was present at 2 months (Fig. 3A–C), with sex [F (1, 76) = 42.37, P < 0.0001] and genotype [F (2, 76.3) = 16.27, P < 0.0001] as significant predictors of performance. No interactions were significant in the experiment. Follow-up with Tukey’s honest significant difference (HSD) multiple comparison indicated that performance of BACHD and wild type (WT) mice was significantly different. The performance of BACHD/GFAP-CreERT2-tam mice, while intermediate to both BACHD and WT performance, was different from neither.
Figure 3.
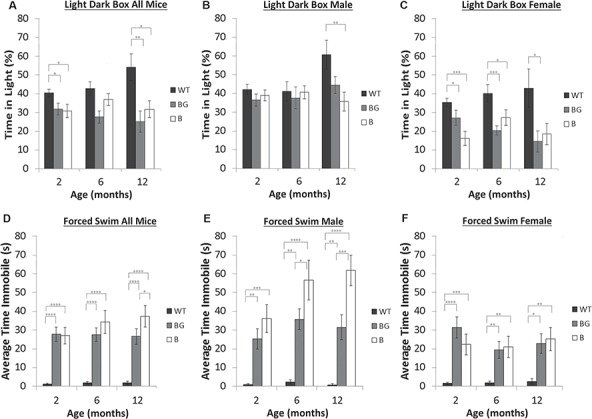
Psychiatric-like deficits in BACHD/GFAP-CreERT2-tam mice. The light–dark box test was used to assess anxiety-like behaviors at 2, 6 and 12 months of age. As a group, there is a significant difference in performance at 2 and 12 months of age between BACHD and wild type. BACHD (n = 21), BACHD/GFAP-CreERT2-tam (n = 24) and wild type (n = 32). As a group, BACHD/GFAP-CreERT2-tam mice also have reduced performance when compared to wild type at 2 and 12 months. The difference in performance is observed primarily in females. (D–F) The forced swim test was used to analyze depressive-like phenotypes in the mice; BACHD (n = 32), BACHD-GFAP-CreERT2-tam (n = 40) and wild type (n = 27). As a group, BACHD and BACHD/GFAP-CreERT2 mice have increased immobility at 2 months as compared to wild-type mice. (D) By 12 months of age, the BACHD/GFAP-CreERT2 mice are more mobile than BACHD mice. The decrease in immobility is due to changes in BACHD-GFAP-CreERT2-tam male mice at 6 and 12 months (E) and not female BACHD-GFAP-CreERT2-tam mice (F). Data are mean ± SEM. *P < 0.05, **P < 0.01.
Because the impact of sex was immediately apparent, mice were stratified by this variable and considered in separate models. Age, genotype and their interaction were included in the model. For female mice, genotype remained a significant predictor of performance [F (2,42) = 16.08, P < 0.0001]; age and the age X genotype interaction remained insignificant. Follow-up with Tukey’s HSD multiple comparison in female mice indicated that performance in BACHD and WT mice was different. For male mice, age, genotype and their interactions were not significant predictors of performance, and there were no differences between BACHD, WT and BACHD/GFAP-CreERT2-tam mice.
We used the forced swim test to analyze depression-like phenotypes in the BACHD/GFAP-CreERT2-tam mice (Fig. 3D–F). We have previously shown that BACHD mice spend more time immobile using this test, which suggests a depression-like phenotype. We tested male and female mice at 2, 6 and 12 months. Beginning at 2 months, BACHD and BACHD/GFAP-CreERT2-tam mice spent a significantly greater amount of time immobile in the water when compared to their WT counterparts, indicating an increase in depressive-like behavior [age X genotype: F (6, 206) = 2.4, P = 0.0338]. Interestingly, BACHD mice showed a continued and progressive decline in performance over the 12-month period of study, significantly increasing their average time spent immobile from 2 months to 6 months (P = 0.0102). The BACHD/GFAP-CreERT2-tam mice in contrast showed no further worsening after 2 months (P = 0.3677) or between 6 and 12 months (P = 0.3157). Although we observed a trend towards increased immobility in male BACHD/GFAP-CreERT2-tam mice (Fig. 3E), this effect was small, and neither sex nor its interaction with genotype was significant in multivariate analysis [age X sex: F (2, 206) = 3.96, P = 0.0205], (Fig. 3E and F).
The effect of weight was also assessed for this cohort, and while body weight was positively correlated with time spent immobile at both 6 months (r = 0.4642, P < 0.0001) and 12 months (r = 0.3808, P < 0.0001), neither were significant predictors in the mixed effect model (6 months body weight (BW): P = 0.4989; 12 months BW: P = 0.2269), and body weight accounted for only 21.5% of the variance at 6 months and 14.5% of the variance at 12 months.
Neuropathological changes are reduced in BACHD/GFAP-CreERT2-tam mice
Significant atrophy and cell death are observed in the brains of HD patients. We have previously shown that BACHD mouse brains show neuropathological changes including decreased brain weight and brain volume as compared to wild-type mice (29,30). Therefore, we compared brain weight in BACHD/GFAP-CreERT2-tam, BACHD and wild-type mice (n = 24 BACHD, n = 25 WT and n = 27 BACHD/GFAP-CreERT2-tam), [ANOVA, F (2, 77) = 8.7563, P = 0.0004] (Fig. 4A). BACHD brain weight (average (avg.) = 0.414 g) is reduced compared to wild-type brain weight (avg. = 0.445 g) at 13–14 months, (P < 0.0001—Fisher’s LSD post hoc test). We found an increase in brain weight of the BACHD/GFAP-CreERT2-tam mice at 13–14 months (avg. = 0.4295) when compared to BACHD mice (0.414 g), although this increase was not statistically significant (P = 0.094). While the BACHD/GFAP-CreERT2-tam mouse brain weight is increased, it is still significantly different from wild type (P = 0.011) (Fig. 4A). This demonstrates that decreasing fl-mHTT expression in astrocytes may be beneficial, despite not being statistically significant at the age analyzed. Additionally, BACHD cortical and striatal volume were analyzed in these mice at 13–14 months (n = 20 wild-type, n = 20 BACHD/GFAP-CreERT2, n = 24 BACHD mice) (Fig. 4B and C). ANOVA revealed that there was a significant difference in cortical volume among these mice [F (2, 43) = 8.093, P = 0.0007]. Post hoc analysis revealed a significant decrease (P = 0.001) in cortical volume in BACHD mice as compared to wild type. No statistically significant differences were observed between BACHD and BACHD/GFAP-CreERT2 mice (P = 0.158) or wild-type and BACHD/GFAP-CreERT2 mice (P = 0.104). The striatal volume of BACHD mice is significantly reduced as compared to wild-type (P = 0.001) and BACHD/GFAP-CreERT2-tam mice (P = 0.017). The BACHD/GFAP-CreERT2 striatal volume was also significantly different from wild type (P = 0.048) (Fig. 4C). Thus, there is a partial rescue of striatal volume in these mice. Overall, this data demonstrates that decreasing fl-mHTT expression in astrocytes positively affected these neuropathological measures in the BACHD/GFAP-CreERT2 mice, although not all of the parameters reached statistical significance.
Figure 4.
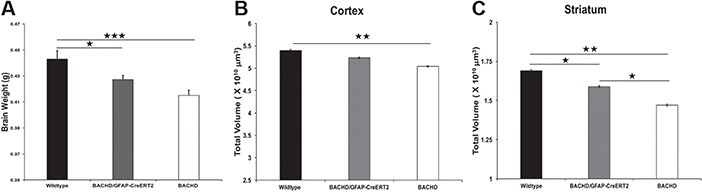
Neuropathology in BACHD/GFAP-CreERT2-tam mice. (A) Brain weight is decreased in BACHD mice at 13–14 months. BACHD/GFAP-CreERT2-tam mice have increased brain weight as compared to BACHD mice, although it is not statistically significant. BACHD/GFAP-CreERT2-tam mice brain weight is statistically significant when compared to wild type; BACHD (n = 25), BACHD/GFAP-CreERT2-tam (n = 28) and wild type (n = 26). Striatal (B) and cortical (C) volume was assessed at 13–14 months. Striatal and cortical volume is decreased in BACHD mice as compared to wild type (B, C). BACHD/GFAP-CreERT2-tam mice have decreased cortical and striatal volume when compared to wild-type mice (n = 10 mice/genotype). Data are mean ± SEM. *P < 0.05, **P < 0.01. One-way ANOVA and Tukey’s HSD post hoc test were used for analysis.
To further explore whether decreasing fl-mHTT in astrocytes of the BACHD/GFP-CreERT2 mice affects other neuropathological deficits observed in the BACHD mice, we assessed synaptic marker proteins in the striatum. There is a decrease in synaptic marker expression in both HD patients and mHTT-expressing mouse models (29,44,45). It is known that astrocytes are important players in neurotransmission; therefore, a reduction in the fl-mHTT protein in these cells may affect the post-synaptic marker protein change observed in HD. We assessed two post-synaptic markers that are affected in the BACHD mice—post-synaptic density 95 (PSD-95) and α-actinin2 (α-actn2). These two proteins are primarily localized to the PSD and are known to interact with and contribute to dysfunction of other proteins implicated in HD pathogenesis, including NMDA receptors. We performed immunofluorescence staining of brain sections taken from wild-type, BACHD and BACHD/GFP-CreERT2 mice at 13–14 months of age (wild-type n = 4 mice/11 sections, BACHD n = 5 mice/14 sections, BACHD/GFP-CreERT2n = 5/15 sections). We quantified the staining intensity of PSD-95 and α-actn2 in the striatum of these mice and identified a significant difference in the expression of α-actn2 [F (2,37) = 141.92, P < 0.0001] and PSD-95 [F (2, 78) = 38.42, P < 0.0001] among the genotypes. We identified a significant decrease in immunofluorescence staining intensity of α-actn2 (Fig. 5A–C) and PSD-95 (Fig. 5D–F) in the striatum of BACHD mice as compared to wild type, which is consistent with previous work. The α-actn2 expression is increased in the BACHD/GFAP-CreERT2 mice and is significantly higher than in BACHD mice (P < 0.0001), although still less than what is observed in wild-type mice (P < 0.0001, Fig. 5G); the PSD-95 expression in BACHD/GFAP-CreERT2 striatum is restored to wild-type levels (P = 0.428, Fig. 5H). Taken together, these changes suggest that decreasing mHTT expression in astrocytes affects the expression of these post-synaptic proteins and thus may affect neurotransmission in these mice.
Figure 5.
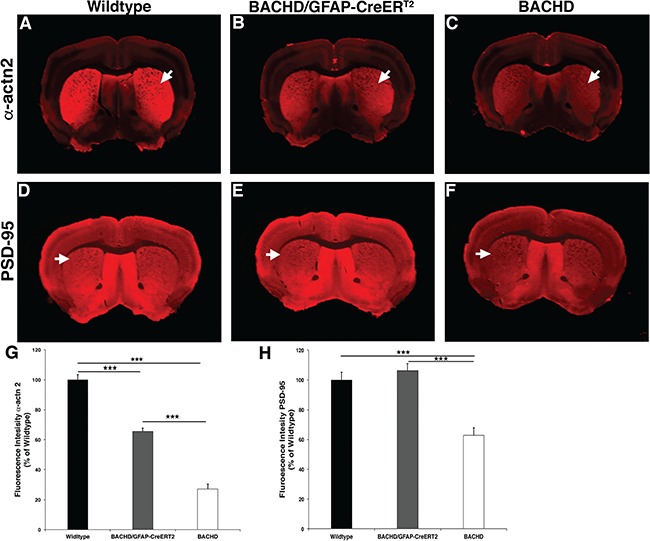
Striatal post-synaptic protein expression in BACHD/GFAP-CreERT2-tam mice. Post-synaptic protein expression level of α-actn2 is decreased in the striatum of BACHD mice at 13- to 14-month-old mice. Immunofluorescence staining of α-actn2 (A) wild-type (n = 4), (B) BACHD/GFAP-CreERT2 (n = 5) and (C) BACHD (n = 5) mice stained with α-actn2 antibody reveals a decrease in BACHD mice, arrows indicate striatal staining in all panels. Immunofluorescence staining with PSD-95 in (D) wild-type, (E) BACHD/GFAP-CreERT2 and (F) BACHD mice also reveals a decrease in BACHD striatum. (G, H) Charts detailing the reduction of α-actn2 and PSD-95 in BACHD mice and increased expression in BACHD/GFAP-CreERT2 mice striata. ***P < 0.0001. Data are mean ± SEM. One-way ANOVA and Tukey’s HSD post hoc test were used for analysis.
Expression of astrocyte-enriched proteins implicated in HD pathogenesis
There have been reports of EAAT2 RNA expression changes in patient tissue and Glt-1 protein expression changes or abnormal glutamate transport in some transgenic mice expressing mHTT (16). Additionally, levels of the astrocyte-enriched potassium channel Kir4.1 were reduced in the striatum of R6/2 mic. Given this and other data, we evaluated the levels of expression of Kir4.1 and Glt-1 in protein homogenate on western blot from the striatum of 13- to 14-month-old wild-type, BACHD and BACHD/GFAP-CreERT2 mice. There was no significant difference in expression of Glt-1 [F (2,8) = 0.399] or Kir4.1 [F (2,8) = 3.4] among the genotypes (Fig. 6A and B). These data show that astrocytic expression of mHTT in the striatum of BACHD mice does not lead to altered expression of these highly enriched astrocyte proteins at this age.
Figure 6.
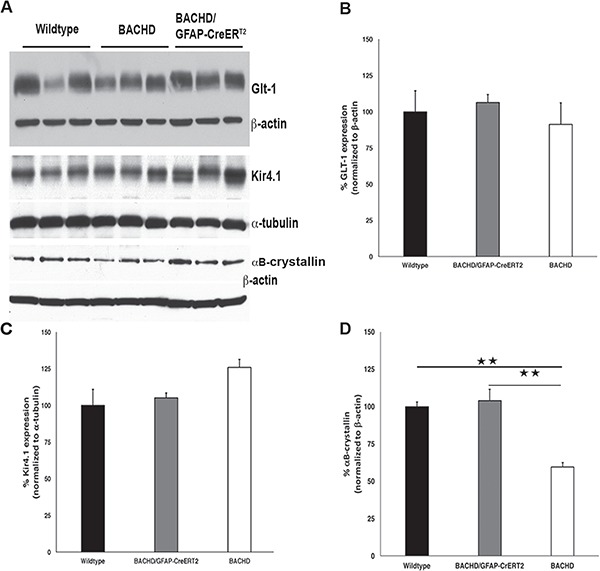
Expression of astrocyte-enriched proteins. Western blot analyses of astrocyte-enriched proteins in striatal protein homogenate from 13- to 14-month-old mice. (A) Western blots using Glt-1 and βactin as loading control (top panel), Kir4.1 and α-tubulin as loading control (middle panel) and αB-crystallin with βactin as loading control (bottom panel). Charts demonstrating no significant differences in the expression level of (B) Glt-1 [F (2,8) = 0.69, P = 0.54] or (C) Kir4.1, [F (2,8) = 3.4, P = 0.101] in the striatum of BACHD mice at this age. (D) Expression of αB-crystallin is changed in the group of samples [F (2,8) = 24.45, P = 0.0013]. It is decreased in BACHD mice (P = 0029) and normalized in BACHD/GFAP-CreERT2 mice (P = 0.7994) as compared to wild-type levels. BACHD/GFAP-CreERT2 levels are significantly increased as compared to BACHD levels. n = 3 BACHD, n = 3 BACHD/GFAP-CreERT2, n = 3 wild type. Data are mean ± SEM. **P < 0.004. One-way ANOVA and Tukey’s HSD post hoc test were used for analysis.
We also used strital homogenates from BACHD and BACHD/GFAP-CreERT2 mice to examine the protein expression level of αB-crystallin, small heat shock protein that is capable of binding to unfolded proteins to inhibit aggregation (42), which is expressed primarily by astrocytes and oligodendrocytes in the nervous system (46). It has been shown that expression of αB-crystallin is reduced in whole-brain homogenates from BACHD mice, and astrocytic overexpression of αB-crystallin is capable of rescuing behavioral and neuropathological deficits observed in BACHD mice (24). However, it is not clear if the decrease observed is due to mHTT expression in astrotyes. Since protein expression can be affected non-cell autonomously, it is important to determine if reducing mHTT expression in astrocytes is capable of restoring the levels of expression of this highly enriched astrocyte protein in the brains of BACHD/GFAP-CreERT2 mice. We observed a significant change in the αB-crystallin protein levels among the genotypes [F (2,8) = 24.45, P = 0.0013]. We observed the previously reported significant decrease in αB-crystallin in BACHD mice on western blots using striatal protein samples from mice 13–14 months of age (Fig. 6A, bottom panel; P = 0029). We also observed a significant increase in the levels of αB-crystallin in striatal protein samples taken from BACHD/GFAP-CreERT2 mice at 13–14 months of age, with no significant difference from wild type found (P = 0.7994) (Fig. 6A, bottom panel). The increase in αB-crystallin expression in the BACHD/GFAP-CreERT2 mice was significantly different from BACHD levels (P = 0017). These data suggest that mHTT expression within the astrocyte acts cell autonomously to control αB-crystallin expression and does not significantly affect the expression of Kir4.1 or Glt-1 in the brains of the BACHD mice at the age examined.
NMDA receptor function in BACHD/GFAP-CreERT2-tam mice
Notably, we have shown that decreasing fl-mHTT levels in astrocytes of BACHD/GFAP-CreERT2 mice leads to an increase in post-synaptic proteins that may have an effect on neurotransmission. Furthermore, astrocytes are known to modulate neuronal activity by releasing gliotransmitters in response to neurotransmitter release from neurons (47–51). It is known that BACHD mice have clear deficits in the cortical–striatal circuit, which has been demonstrated by the observation of decreases in evoked NMDAR-mediated synaptic currents (29,30). As astrocytes play a critical role in the modulation of synaptic activity, we examined the electrophysiological phenotype of striatal MSNs in the BACHD and BACHD/GFAP-CreERT2-tam mice compared to their WT littermates at 15–16 months of age. Specifically, we examined basic membrane properties and evoked synaptic NMDAR currents, both of which are significantly altered in BACHD mice. The BACHD mice showed a significant decrease in cell membrane capacitance compared to WT littermates [46.1 ± 2.5 pF BACHD (n = 6 mice, n = 11 cells) versus 58.8 ± 3.8 pF WT (n = 10 mice, n = 16 cells, P = 0.01)], but membrane resistance, decay time constant and holding current were not significantly altered. In the BACHD/GFAP-CreERT2-tam mice, the capacitance was restored compared to BACHD mice [46.1 ± 2.5 pF BACHD (n = 11 cells) versus 61.1 ± 2.9 pF BACHD/GFAP-CreERT2-tam (n = 6 mice, n = 13 cells, P = 0.001)].
After blocking GABAA and AMPA receptors with BIC (10 μM) and CNQX (20 μM), respectively, electrical stimulation via a monopolar electrode placed on the corpus callosum evoked an NMDAR-mediated response in MSNs. A significant decrease in peak amplitude of the NMDAR-mediated current occurred in BACHD compared to WT mice [107.0 ± 15.4 pA BACHD (n = 11) versus 216.2 ± 42.3 pA WT (n = 16, P = 0.05)] (Fig. 7A and BA), while there was no significant change in area, half-width, rise time or decay time. This decrease in amplitude was restored in BACHD/GFAP-CreERT2-tam mice [107.0 ± 15.4 pA BACHD (n = 11) versus 200.3 ± 33.1 pA BACHD/GFAP-CreERT2-tam (n = 13, P = 0.03)]. These data demonstrate that the expression of fl-mHTT in astrocytes also contributes to the abnormal neuronal function observed in BACHD mice and that deletion of fl-mHTT can restore normal striatal synaptic physiology.
Figure 7.
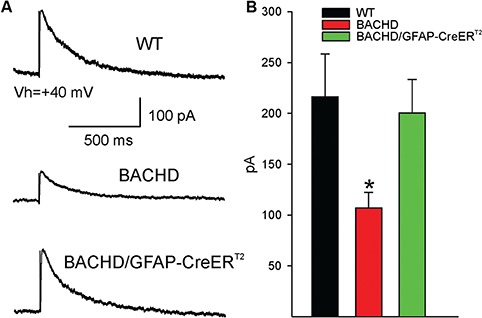
NMDA-mediated currents are restored in BACHD/GFAP-CreERT2-tam mice. (A) Example traces of NMDA currents evoked in MSNs by electrical stimulation of the corpus callosum (average of three responses) in WT (n = 16), BACHD (n = 11) and BACHD/GFAP-CreERT2-tam (n = 13) mice. (B) The bar graph represents the average NMDA current peak amplitude in MSNs from the three groups. BACHD current peak amplitude is significantly different from wild type, while BACHD-GFAPCreERT2-tam MSN peak amplitude is increased back to wild-type levels. *P < 0.05, one-way ANOVA.
Discussion
This study illustrates the important contribution of human fl-mHTT-expressing astrocytes to HD pathogenesis. We were able to significantly reduce the overall level of fl-mHTT in the brains of BACHD mice using GFAP-CreERT2-tam mice. When fl-mHTT is reduced in astrocytes, there is a reduction in the rate of disease progression as measured by a decreased rate of decline of motor dysfunction and a reduction of neuropsychiatric deficits. There is a normalization of synaptic marker expression and a restoration of normal physiological properties of MSNs with normalization of NMDA receptor–mediated neurotransmission.
Using a conditional mouse transgenic approach (22), we broadly and temporally assessed the contribution of human fl-mHTT-expressing mature astrocytes to the various HD-relevant phenotypes observed in the BACHD mice. This approach decreased the likelihood of affecting neuronal precursors expressing GFAP and allowed us to target mature astrocytes rather than neurons (34,52). In BACHD/GFAP-CreERT2-tam mice, we observed rotarod performance that is initially the same as BACHD mice at 2 months of age. However, as a group, the performance of BACHD/GFAP-CreERT2-tam mice is better than BACHD mice at subsequent time points. Furthermore, the performance in the open field test is also improved at 6 months. Our results with BACHD/GFAP-CreERT2 mice are similar to what has been observed in amyotrophic lateral sclerosis (ALS) models, where there is a slowing of disease progression when mutant SOD1 expression is reduced in astrocytes (53,54). Therefore, while fl-mHTT is expressed in astrocytes, based on this result, these cells are unlikely to be initiators of the HD motor phenotypes but are contributing to the continued progression and worsening of the motor phenotypes.
There is considerable data to support astrocyte involvement in the development of depression through multiple mechanisms, including inhibition of vesicular release (55) and glutamate transporters (56,57). Using the forced swim test, we observed a significant increase in immobility time of BACHD mice as a group as early as 2 months that persisted until 12 months of age. However, as a group, we did not observe differences between BACHD and BACHD/GFAP-CreERT2-tam mice until 12 months that was more robust in male BACHD/GFAP-CreERT2-tam mice. The change that we observe in this behavior in the BACHD/GFAP-CreERT2-tam male mice is early and sustained, which therefore suggests a long-term and early change to the circuits mediating this behavior. Interestingly, the depressive-like phenotype seemed to worsen with age only in male BACHD mice.
While there is an increase in cortical volume in the BACHD/GFAP-CreERT2-tam mice at 12 months of age, it did not reach statistical significance. However, striatal volume is significantly increased in the BACHD/GFAP-CreERT2-tam mice. This difference might represent the differences in deficits in astrocytes present in these two brain regions caused by the presence of mHTT. It is becoming clearer that astrocytes from different brain regions are diverse with different physiological and morphological properties (58). Nonetheless, this change, coupled with the change in electrophysiological measures, clearly indicates that these cells are involved in improving neurological function in these mice.
We did not observe a significant decrease in the level of expression Kir4.1 or Glt-1 protein in the striatum of these mice, although total tissue expression of Kir4.1 and Glt-1 in the BACHD mice may not be truly indicative of the expression or localization on a cell-by-cell basis. It is possible that the effects of decreases in both Glt-1 and Kir4.1 levels affect microcircuits and are not appreciated in a whole protein homogenate. In BACHD mice, αB-crystallin decreases progressively, and it is noteworthy that this decrease is not observed until after 6 months of age. This is coincident with the most robust behavioral changes in BACHD mice and the age at which some of the behaviors are partially rescued in the BACHD/GFAP-CreERT2 mice. This result further suggests that astrocytes likely contribute to HD progression and not onset. This is also interesting given the recent finding that mHTT decreases the expression of αB-crystallin and impairs exosome secretion from astrocytes further demonstrating the importance of these cells in HD pathogenesis (25).
In this study, we used 15- to 16-month-old BACHD mice and confirmed previous results demonstrating a decrease in peak amplitude of evoked NMDAR-mediated response in MSNs. This reduction was restored in BACHD/GFAP-CreERT2-tam mice. While the decreased input at this age in BACHD mice could be due to either pre- or post-synaptic neuronal mechanisms, astrocytes are also known contributors to synaptic activity. The increase in post-synaptic proteins PSD-95 and α-actn2 at the late time points suggests that fl-mHTT in astrocytes contributes to this change in HD in a non-cell autonomous manner. The mechanism for this non-cell autonomous contribution to the expression of these two important post-synaptic proteins was not explored here. We also did not explore whether an increase in the expression of these proteins occurs at 6 months of age, which is the age at which some of the phenotypes are improved in the BACHD/GFAP-CreERT2 mice. Taken together, a decrease of fl-mHTT expression in astrocytes could decrease an aberrant activity caused by the expression of fl-mHTT in astrocytes, thus restoring normal synaptic activity.
The electrophysiological data reported here are from BACHD and BACHD/GFAP-CreERT2-tam mice at 15–16 months of age. There is considerable data from HD models showing biphasic changes in MSN synaptic activity. In BACHD MSNs at early stages, there is a significant increase in frequency of sEPSCs; at late stages, there are significant decreases in sEPSCs (30,59). Thus, it will be important to analyze MSN changes observed in BACHD/GFAP-CreERT2-tam mice at earlier stages to ascertain the temporal contribution of fl-mHTT-expressing astrocytes to the observed changes in synaptic activity. Furthermore, it will be important to determine if those changes are different in the dopamine receptor D2– and dopamine receptor D1–expressing MSNs (59–61).
We have shown that our approach using conditional BACHD mice with GFAP-CreERT2 mice will provide a tractable and reproducible genetic system for identifying and studying disease mechanisms at play in astrocytes in HD. It is intriguing that targeting mHTT in astrocytes could decrease disease pathogenesis. There has been considerable work performed recently concerning targeting of mHTT in the brains of HD mice (62,63). The methodologies target HTT broadly without giving consideration to specific cell types. Given our data, it may be reasonable to specifically target mHTT in astrocytes. Additionally, therapeutic approaches that take advantage of CRISPR/Cas technology may be used to ‘correct’ the DNA in human astrocytes and those cells used for transplantation (43) to reduce the total burden of mHTT and reduce toxicity that may be contributed by mHTT-expressing astrocytes.
Materials and Methods
Animals
All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Alabama at Birmingham (UAB) Institutional Care and Use Committee and the University of California Los Angeles Institutional Care and Use Committee. BACHD mice were maintained by breeding with FvB/NJ (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Non-transgene-positive mice were used as controls. GFAP-CreERT2 mice on the C57BL6 background (32,34) were backcrossed to FvB/NJ for three generations, and the percentage of FvB/NJ was determined using the Jackson Laboratories genome scanning service for development of congenic lines (Supplementary Material, Table S1). The subsequent positive GFAP-CreERT2 mouse covering at least 94% of FvB/NJ genome was backcrossed one additional time. The resulting GFAP-CreERT2 mice (now 4th generation from speed congenics) were used to expand the GFAP-CreERT2 FvB/NJ colony and breed to BACHD mice.
Tamoxifen treatment of mice
At 24–28 days after weaning, the BACHD/GFAP-CreERT2 and GFAP-CreERT2/Ai14 mice were weighed to determine volume of tamoxifen required for injection. These mice were injected intraperitoneally with tamoxifen (Sigma-Aldrich, St. Louis, MO, T5648) one time/day with 4 mg/ml for 10 days. Five to ten days after the last tamoxifen injection, some BACHD/GFAP-CreERT2 mice were euthanized, and the cortex and striatum were dissected in ice-cold 0.01 M phosphate buffered saline solution (PBS). GFAP-CreERT2/Ai14 mice exposed to the same tamoxifen injection paradigm as described above were perfused with ice-cold PBS, followed by ice-cold 4% paraformaldehyde (PFA). The brains of the mice were dissected and post-fixed overnight at 4°C in 4% PFA, followed by cryoprotection in 30% sucrose at 4°C. Brains were then frozen on dry ice; 35-μm sections were cut on a cryostat or microtome and stored in cryoprotectant.
Preparation of protein lysates
Mouse brains were dissected in ice-cold 100-mm PBS supplemented with Complete Protease Inhibitor Cocktail tablets (Thermo Scientific, Waltham, MA, 88665). Lysates were prepared as previously described. Briefly, protein was extracted from brain tissue homogenized in a modified RIPA buffer using 10–12 strokes with a Potter–Elvehjem homogenizer on ice, followed by centrifugation at 4°C for 15 min at 16 100 g. The resulting soluble supernatant was removed, and protein quantity was determined using Pierce BCA Protein Assay kit (Thermo Scientific, Waltham, MA).
Western blotting
Protein samples were prepared for loading by heating in NuPAGE LDS buffer (Invitrogen, Carlsbad, CA) for 10 min at 70°C and resolved on 3–8% Tris-acetate NuPAGE gels (Invitrogen) using Tris-acetate running buffer then wet-transferred onto PVDF membranes using NuPAGE transfer buffer (Invitrogen). Gels were loaded with 25–30 μg of protein and immunoblots were probed with antibodies against HTT, (1:3000, MAB2166, mouse, EMD Millipore Burlington, MA) and either α-tubulin (1:3000, T9026, mouse, Sigma-Aldrich, St. Louis, MO) or βactin (1:5000, A7811, mouse, Sigma-Aldrich, St. Louis, MO) in 5% non-fat dry milk shaking overnight at 4°C; 5–10 □g of protein was used for Kir4.1 (1:1000, APC-035, rabbit, Alomone Labs Ltd., Jerusalem, Israel), GLT-1 (1:10,000, AB1783, guinea pig, EMD Millipore Burlington, MA) and αB-crystallin (1:2000, ADI-SPA-223, Enzo Biochem, New York, NY) in 5% non-fat dry milk. The HRP-labeled secondary antibodies from Jackson Immunoresearch—anti-rabbit (1:10,000), anti-guinea pig (1:10,000) and anti-mouse (1:10,000)—were all incubated for 1–2 h at room temperature. Chemiluminescent detection was accomplished using SuperSignal West Pico Chemiluminescent Substrate reagents (Thermo Scientific, Waltham, MA).
Immunohistochemistry
Mice were perfused with ice-cold PBS, followed by ice-cold 4% PFA. The brains of the mice were dissected and post-fixed overnight at 4°C in 4% PFA, followed by cryoprotection in 30% sucrose at 4°C. Brains were then frozen on dry ice, and 35-μm sections were cut on a cryostat or microtome and stored in cryopreserve. Sections were rinsed 3× in 0.01-M PBS and blocked with 2% normal donkey serum and 3% bovine serum albumin in 0.01-M PBS. Sections were incubated with α-actn2 (1:3000, mouse, Sigma-Aldrich, A7811) and PSD-95 (1:5000, UC Davis/NIH NeuroMab mouse mAb 75–028) (29,30,64). After washing three times in 0.01-M PBS, (Alexa Fluor®, Invitrogen, Carlsbad, CA) 647, A21241, goat anti-mouse (1:2000) was used for α-actn2 and PSD-95. Tissue sections labeled with PSD-95 and α-actn2 were imaged on a LiCor Odyssey (Licor Biosciences, Lincoln, NE) and quantified using Licor ImageStudioLite software package. For each section, the striatum was defined with the oval selection tool, and the signal intensity within the region was recorded. Signal intensity for BACHD, BACHD/GFAP-CreERT2 and wild-type sections was compared.
Stereology
BACHD, BACHD/GFAP-CreERT2-tam and wild-type animals were perfused with 4% PFA, and 35-μm coronal brain sections were cut using a Leica SM 2010 K microtome. The first section was chosen at random from the first 10 sections containing striatum, and every 10th section thereafter was immunostained with NeuN (1:500) antibody to label neurons. Sections were rinsed 3× in 0.01-M PBS and blocked with 2% normal donkey serum and 3% bovine serum albumin in 0.01-M PBS. Sections were incubated with the mouse NeuN in 0.01-M PBS with 2% normal donkey serum, 3% bovine serum albumin and 0.2% Triton X-100 according to an established protocol (30,64). A biotinylated donkey anti-mouse secondary (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was incubated for 2 h at RT. Sections were mounted and coverslipped with Permount. Stereo Investigator software (Microbrightfield, MBF Bioscience, Williston, VT) and the Cavalieri method were used to determine the total volume of the striatum and cortex at 13–14 months of age.
Behavior
Accelerating rotarod
All behavioral studies were performed in the UAB Behavior Assessment Core (NIH/NINDS P30 NS47466). Wild-type, BACHD and BACHD/GFAP-CreERT2-tam male and female littermates were trained on a San Diego Instruments Rotor-Rod for three trials per day for 2 days at 2 months of age. Animals were tested using an accelerating rotator paradigm of 4–40 rpm over 5 min for three trials per day for 3 days. Animals rested 30 min between each trial. At 6 and 12 months, mice were given an additional 5 min of training on the first day of the rotarod test. Animals from the same cohort of mice were measured at 2–3 months, 6–7 months and 12–13 months. All rotarod tests were performed during the light phase of the light cycle.
Light–dark box exploration
The light/dark exploration box is made of Plexiglas with a clear (light) side and a smaller, fully opaque (dark) side, separated by a partition with a small opening for the mouse to travel through. The dark side is completely closed to light. The lighted side is illuminated with a 60-watt light bulb or light with an illuminance of about 400 lux. Light/dark exploration was performed during the early phase of the light/dark cycle. Mice were placed in the testing room at least 1 h prior to testing. The mice were then tested for 10 min in the light/dark box and assessed for movements from one side to the other. All tests were video recorded and scored by researchers blind to the genotypes of the mice.
Forced swimming
Mice were allowed to acclimate to the experimental room at least 1 h before testing period. Mice were placed in transparent plastic cylinders 13 cm in diameter × 24 cm high containing water that is 25°C. Animals were placed in the water and recorded with a video camera for 6 min. Analysis of swimming behavior was performed for the last 4 min of the test. Immobility duration was scored as the time spent immobile, excluding the movements necessary to keep the animal’s head above water. The researchers independently analyzed the video and were blind to the genotypes of the mice.
Open field
Mice were placed in the testing room at least 1 h prior to the start of the dark cycle to acclimate to the experimental room. The animals were tested 1–2 h after the room lights have shut off during the early phase of the light–dark cycle. The mice were tested in an open field for 15 min each during the dark phase of the light–dark cycle using EthoVision (Noldus, Wageningen, The Netherlands) software to track the movement and general activity of the mouse.
Electrophysiology
The mice were deeply anaesthetized with isoflurane and decapitated. The brains were quickly removed and placed in oxygenated ice-cold slice solution containing (in mm) 208 sucrose, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1.3 MgCl2, 8 MgSO4 and 10 glucose. Coronal slices (350 μm) were cut and transferred to an incubating chamber containing artificial cerebro-spinal fluid (containing in mm: 130 NaCl, 2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 MgCl2, 2 CaCl2 and 10 glucose), oxygenated with 95% O2–5% CO2 (pH 7.2–7.4, 290–310 mOsm, 25 ± 2°C). After 1-h incubation, MSNs were selected for recordings. Cells were visualized using a 40× water-immersion lens on an Olympus microscope (BX50WI, Tokyo, Japan) equipped with differential interference contrast optics. MSNs were identified with infrared videomicroscopy (QICAM-IR Fast 1394, Qimaging, Surrey, BC, Canada). All experiments were performed in voltage clamp mode, with patch electrodes (3–5 MΩ) filled with the following internal solution (in mm): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 5 MgATP, 5 EGTA, 10 HEPES, 0.5 GTP, 10 phosphocreatine, (pH 7.25–7.3, osmolality, 280–290 mOsm).
Synaptic stimulation: to assess input–output functions, a monopolar glass electrode (impedance 1 MΩ) was placed in the corpus callosum 200–300 μm from the MSN. To examine NMDAR-mediated currents, stimuli were applied every 20 s. NMDAR-mediated currents were recorded at a holding potential of +40 mV in the presence of BIC (10 μM) and 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM). For this experiment, the current intensity that evoked 75% of the peak amplitude response was used. Peak amplitude, area, rise and decay time were determined using ClampFit software (Molecular Devices, San Jose, CA). Decay times were assessed using the 90–10% portion of the decay phase, where 100% was the peak amplitude and 0% was at baseline.
Statistical analysis
Rotarod
Male and female mice were tested at 2–3 months, 6–7 months and 12–13 months of age. Statistical analysis was performed using the PROC MIXED procedure in SAS using an alpha of 0.05. Significant interactions were followed up using the Fisher’s LSD multiple comparison procedure. Age, genotype, sex, weight and their interactions were considered in the model. The observed data conformed to a logarithmic distribution, as is consistent with time-to-failure tests, and were therefore log transformed for analysis.
Open field
Mice were tested at 2 and 6 months of age. Statistical analysis was performed using the PROC MIXED procedure in SAS using an alpha of 0.05. Significant interactions were followed up using the Fisher’s LSD multiple comparison procedure or Tukey’s HSD multiple comparison.
Light–dark box and forced swim
Mice were tested at 2, 6 and 12 months of age. Statistical analysis was performed using the PROC MIXED procedure in SAS using an alpha of 0.05. Significant interactions were followed up using the Fisher’s LSD multiple comparison procedure or Tukey’s HSD multiple comparison. Age, genotype, sex and their interactions were considered in the models.
Brain weight and brain volume
After perfusion and cryoprotection, total brain weight (in grams) was measured and forebrain weight was recorded after removal of the cerebellum. The data are expressed as mean +/- the standard error of the mean (SEM). Differences among the groups were assessed by one-way ANOVA followed by Fisher’s LSD multiple comparison procedure or Tukey’s HSD multiple comparison.
Supplementary Material
Acknowledgements
We thank David Standaert for discussions and critical reading of the manuscript. We thank Christopher Evans and Bhindyaa Kaur for help with animal colony maintenance.
Conflict of Interest statement. None declared.
Funding
This work was supported by Hereditary Disease Foundation and the National Institutes of Health/National Institute of Neurological Disorders and Stroke [NS069614, NS089750-01A1 to M.G. and NS41574, NS96994, U54HD087101 to M.S.L.].
References
- 1. Group, T.H.s.D.C.R (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Sharp A.H., Loev S.J., Schilling G., Li S.H., Li X.J., Bao J., Wagster M.V., Kotzuk J.A., Steiner J.P., Lo A. et al. (1995) Widespread expression of Huntington's disease gene (IT15) protein product. Neuron, 14, 1065–1074. [DOI] [PubMed] [Google Scholar]
- 3. Cudkowicz M. and Kowall N.W. (1990) Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann. Neurol., 27, 200–204. [DOI] [PubMed] [Google Scholar]
- 4. Nana A.L., Kim E.H., Thu D.C., Oorschot D.E., Tippett L.J., Hogg V.M., Synek B.J., Roxburgh R., Waldvogel H.J. and Faull R.L. (2014) Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington's disease. J. Huntingtons Dis., 3, 45–64. [DOI] [PubMed] [Google Scholar]
- 5. Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D. and Richardson E.P. Jr. (1985) Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol., 44, 559–577. [DOI] [PubMed] [Google Scholar]
- 6. Vonsattel J.P. and DiFiglia M. (1998) Huntington disease. J. Neuropathol. Exp. Neurol., 57, 369–384. [DOI] [PubMed] [Google Scholar]
- 7. Haydon P.G. and Carmignoto G. (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev., 86, 1009–1031. [DOI] [PubMed] [Google Scholar]
- 8. Anderson C.M. and Nedergaard M. (2003) Astrocyte-mediated control of cerebral microcirculation. Trends Neurosci., 26, 340–344author reply 344–345. [DOI] [PubMed] [Google Scholar]
- 9. Ventura R. and Harris K.M. (1999) Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci., 19, 6897–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Halassa M.M., Fellin T. and Haydon P.G. (2007) The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med., 13, 54–63. [DOI] [PubMed] [Google Scholar]
- 11. Maragakis N.J. and Rothstein J.D. (2001) Glutamate transporters in neurologic disease. Arch. Neurol., 58, 365–370. [DOI] [PubMed] [Google Scholar]
- 12. Bergles D.E., Diamond J.S. and Jahr C.E. (1999) Clearance of glutamate inside the synapse and beyond. Curr. Opin. Neurobiol., 9, 293–298. [DOI] [PubMed] [Google Scholar]
- 13. Faideau M., Kim J., Cormier K., Gilmore R., Welch M., Auregan G., Dufour N., Guillermier M., Brouillet E., Hantraye P. et al. (2010) In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum. Mol. Genet., 19, 3053–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arzberger T., Krampfl K., Leimgruber S. and Weindl A. (1997) Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease—an in situ hybridization study. J. Neuropathol. Exp. Neurol., 56, 440–454. [DOI] [PubMed] [Google Scholar]
- 15. Lievens J.C., Woodman B., Mahal A., Spasic-Boscovic O., Samuel D., Kerkerian-Le Goff L. and Bates G.P. (2001) Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol. Dis., 8, 807–821. [DOI] [PubMed] [Google Scholar]
- 16. Behrens P.F., Franz P., Woodman B., Lindenberg K.S. and Landwehrmeyer G.B. (2002) Impaired glutamate transport and glutamate–glutamine cycling: downstream effects of the Huntington mutation. Brain, 125, 1908–1922. [DOI] [PubMed] [Google Scholar]
- 17. Huang K., Kang M.H., Askew C., Kang R., Sanders S.S., Wan J., Davis N.G. and Hayden M.R. (2010) Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol. Dis., 40, 207–215. [DOI] [PubMed] [Google Scholar]
- 18. Hassel B., Tessler S., Faull R.L. and Emson P.C. (2008) Glutamate uptake is reduced in prefrontal cortex in Huntington's disease. Neurochem. Res., 33, 232–237. [DOI] [PubMed] [Google Scholar]
- 19. Cross A.J., Slater P. and Reynolds G.P. (1986) Reduced high-affinity glutamate uptake sites in the brains of patients with Huntington's disease. Neurosci. Lett., 67, 198–202. [DOI] [PubMed] [Google Scholar]
- 20. Jiang R., Diaz-Castro B., Looger L.L. and Khakh B.S. (2016) Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington's disease model mice.J. Neurosci., 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tong X., Ao Y., Faas G.C., Nwaobi S.E., Xu J., Haustein M.D., Anderson M.A., Mody I., Olsen M.L., Sofroniew M.V. et al. (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat. Neurosci., 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee W., Reyes R.C., Gottipati M.K., Lewis K., Lesort M., Parpura V. and Gray M. (2013) Enhanced Ca-dependent glutamate release from astrocytes of the BACHD Huntington's disease mouse model. Neurobiol. Dis., 58C, 192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hong Y., Zhao T., Li X.J. and Li S. (2016) Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci., 36,8790–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oliveira A.O., Osmand A., Outeiro T.F., Muchowski P.J. and Finkbeiner S. (2016) alphaB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington's disease. Hum. Mol. Genet., 25,1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hong Y., Zhao T., Li X.J. and Li S. (2017) Mutant huntingtin inhibits alphaB-crystallin expression and impairs exosome secretion from astrocytes. J. Neurosci., 37, 9550–9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shin J.Y., Fang Z.H., Yu Z.X., Wang C.E., Li S.H. and Li X.J. (2005) Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol., 171, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bradford J., Shin J.Y., Roberts M., Wang C.E., Li X.J. and Li S. (2009) Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. U.S.A., 106, 22480–22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chou S.Y., Weng J.Y., Lai H.L., Liao F., Sun S.H., Tu P.H., Dickson D.W. and Chern Y. (2008) Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J. Neurosci., 28, 3277–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang N., Gray M., Lu X.H., Cantle J.P., Holley S.M., Greiner E., Gu X., Shirasaki D., Cepeda C., Li Y. et al. (2014) Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat. Med., 20, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gray M., Shirasaki D.I., Cepeda C., Andre V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E. et al. (2008) Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci., 28, 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casper K.B. and McCarthy K.D. (2006) GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol. Cell Neurosci., 31, 676–684. [DOI] [PubMed] [Google Scholar]
- 32. Ganat Y.M., Silbereis J., Cave C., Ngu H., Anderson G.M., Ohkubo Y., Ment L.R. and Vaccarino F.M. (2006) Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J. Neurosci., 26, 8609–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Namba T., Mochizuki H., Onodera M., Mizuno Y., Namiki H. and Seki T. (2005) The fate of neural progenitor cells expressing astrocytic and radial glial markers in the postnatal rat dentate gyrus. Eur. J. Neurosci., 22, 1928–1941. [DOI] [PubMed] [Google Scholar]
- 34. Casper K.B., Jones K. and McCarthy K.D. (2007) Characterization of astrocyte-specific conditional knockouts. Genesis, 45, 292–299. [DOI] [PubMed] [Google Scholar]
- 35. Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. et al. (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci., 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A. et al. (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci., 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menalled L., El-Khodor B.F., Patry M., Suarez-Farinas M., Orenstein S.J., Zahasky B., Leahy C., Wheeler V., Yang X.W., MacDonald M. et al. (2009) Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol. Dis., 35, 319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duff K., Paulsen J.S., Beglinger L.J., Langbehn D.R., Stout J.C. and Predict H.D.I.O.T.H.S.G (2007) Psychiatric symptoms in Huntington's disease before diagnosis: the predict-HD study. Biol. Psychiatry, 62, 1341–1346. [DOI] [PubMed] [Google Scholar]
- 39. Craufurd, J. D.S. (2002) In Bates G.P., Tabrizi S., & Jones L. (eds), Huntington’s Disease. Oxford University Press, Oxford, In press, pp. 62–94. [Google Scholar]
- 40. Kudwa A.E., Menalled L.B., Oakeshott S., Murphy C., Mushlin R., Fitzpatrick J., Miller S.F., McConnell K., Port R., Torello J. et al. (2013) Increased body weight of the BAC HD transgenic mouse model of Huntington's disease accounts for some but not all of the observed HD-like motor deficits. PLoS Curr., 5 doi: 10.1371/currents.hd.0ab4f3645aff523c56ecc8ccbe41a198 10.1371/currents.hd.0ab4f3645aff523c56ecc8ccbe41a198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stout J.C., Paulsen J.S., Queller S., Solomon A.C., Whitlock K.B., Campbell J.C., Carlozzi N., Duff K., Beglinger L.J., Langbehn D.R. et al. (2011) Neurocognitive signs in prodromal Huntington disease. Neuropsychology, 25, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Horwitz J. (2003) Alpha-crystallin. Exp. Eye Res., 76, 145–153. [DOI] [PubMed] [Google Scholar]
- 43. Benraiss A., Wang S., Herrlinger S., Li X., Chandler-Militello D., Mauceri J., Burm H.B., Toner M., Osipovitch M., Jim Xu Q. et al. (2016) Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun., 7, 11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raymond L.A., Andre V.M., Cepeda C., Gladding C.M., Milnerwood A.J. and Levine M.S. (2011) Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience, 198, 252–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goto S. and Hirano A. (1990) Synaptophysin expression in the striatum in Huntington's disease. Acta Neuropathol., 80, 88–91. [DOI] [PubMed] [Google Scholar]
- 46. Iwaki T., Kume-Iwaki A. and Goldman J.E. (1990) Cellular distribution of alpha B-crystallin in non-lenticular tissues. J. Histochem. Cytochem., 38, 31–39. [DOI] [PubMed] [Google Scholar]
- 47. Parpura V., Basarsky T.A., Liu F., Jeftinija K., Jeftinija S. and Haydon P.G. (1994) Glutamate-mediated astrocyte-neuron signalling. Nature, 369, 744–747. [DOI] [PubMed] [Google Scholar]
- 48. Araque A., Li N., Doyle R.T. and Haydon P.G. (2000) SNARE protein-dependent glutamate release from astrocytes. J. Neurosci., 20, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Araque A., Sanzgiri R.P., Parpura V. and Haydon P.G. (1999) Astrocyte-induced modulation of synaptic transmission. Can. J. Physiol. Pharmacol., 77, 699–706. [PubMed] [Google Scholar]
- 50. Bezzi P., Vesce S., Panzarasa P. and Volterra A. (1999) Astrocytes as active participants of glutamatergic function and regulators of its homeostasis. Adv. Exp. Med. Biol., 468, 69–80. [DOI] [PubMed] [Google Scholar]
- 51. Mothet J.P., Parent A.T., Wolosker H., Brady R.O. Jr., Linden D.J., Ferris C.D., Rogawski M.A. and Snyder S.H. (2000) D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A., 97, 4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brenner M., Kisseberth W.C., Su Y., Besnard F. and Messing A. (1994) GFAP promoter directs astrocyte-specific expression in transgenic mice. J. Neurosci., 14, 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yamanaka K., Chun S.J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D.H., Takahashi R., Misawa H. and Cleveland D.W. (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci., 11, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun S., Sun Y., Ling S.C., Ferraiuolo L., McAlonis-Downes M., Zou Y., Drenner K., Wang Y., Ditsworth D., Tokunaga S. et al. (2015) Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. U.S.A., 112, E6993–E7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cao X., Li L.P., Wang Q., Wu Q., Hu H.H., Zhang M., Fang Y.Y., Zhang J., Li S.J., Xiong W.C. et al. (2013) Astrocyte-derived ATP modulates depressive-like behaviors. Nat. Med., 19, 773–777. [DOI] [PubMed] [Google Scholar]
- 56. Bechtholt-Gompf A.J., Walther H.V., Adams M.A., Carlezon W.A. Jr., Ongur D. and Cohen B.M. (2010) Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology, 35, 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. John C.S., Smith K.L., Van't Veer A., Gompf H.S., Carlezon W.A. Jr., Cohen B.M., Ongur D. and Bechtholt-Gompf A.J. (2012) Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology, 37, 2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chai H., Diaz-Castro B., Shigetomi E., Monte E., Octeau J.C., Yu X., Cohn W., Rajendran P.S., Vondriska T.M., Whitelegge J.P. et al. (2017) Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron, 95, 531–549e539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andre V.M., Cepeda C., Fisher Y.E., Huynh M., Bardakjian N., Singh S., Yang X.W. and Levine M.S. (2011) Differential electrophysiological changes in striatal output neurons in Huntington's disease. J. Neurosci., 31, 1170–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Botelho E.P., Wang E., Chen J.Y., Holley S., Andre V., Cepeda C. and Levine M.S. (2014) Differential synaptic and extrasynaptic glutamate-receptor alterations in striatal medium-sized spiny neurons of aged YAC128 Huntington's disease mice. PLoS Curr., 6 doi: 10.1371/currents.hd.34957c4f8bd7cb1f5ec47381dfc811c3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Andre V.M., Fisher Y.E. and Levine M.S. (2011) Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington's disease. Front. Syst. Neurosci., 5, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kordasiewicz H.B., Stanek L.M., Wancewicz E.V., Mazur C., McAlonis M.M., Pytel K.A., Artates J.W., Weiss A., Cheng S.H., Shihabuddin L.S. et al. (2012) Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron, 74, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DiFiglia M., Sena-Esteves M., Chase K., Sapp E., Pfister E., Sass M., Yoder J., Reeves P., Pandey R.K., Rajeev K.G. et al. (2007) Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. U.S.A., 104, 17204–17209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gu X., Li C., Wei W., Lo V., Gong S., Li S.H., Iwasato T., Itohara S., Li X.J., Mody I. et al. (2005) Pathological cell–cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron, 46, 433–444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.