

Abstract
Human-induced Pluripotent Stem Cell (hiPSC)-derived models have advanced the study of neurodegenerative diseases, including Parkinson’s disease (PD). While age is the strongest risk factor for these disorders, hiPSC-derived models represent rejuvenated neurons. We developed hiPSC-derived Aged dopaminergic and cholinergic neurons to model PD and related synucleinopathies. Our new method induces aging through a `semi-natural’ process, by passaging multiple times at the Neural Precursor Cell stage, prior to final differentiation. Characterization of isogenic hiPSC-derived neurons using heterochromatin and nuclear envelope markers, as well as DNA damage and global DNA methylation, validated our age-inducing method. Next, we compared neurons derived from a patient with SNCA-triplication (SNCA-Tri) and a Control. The SNCA-Tri neurons displayed exacerbated nuclear aging, showing advanced aging signatures already at the Juvenile stage. Noteworthy, the Aged SNCA-Tri neurons showed more α-synuclein aggregates per cell versus the Juvenile. We suggest a link between the effects of aging and SNCA overexpression on neuronal nuclear architecture.
Introduction
During the past decade, the use of human-induced Pluripotent Stem Cells (hiPSCs) has revolutionized the research of neurodegenerative diseases and leveraged the in vitro model systems available for studying these diseases (1,2). Reprogramming somatic cells into hiPSCs resets their donor’s age, resulting in rejuvenated cells that lack the aging signatures of the cells from which they are derived (3–6), introducing a shortcoming in utilizing hiPSC-derived models for the study of age-related neurodegenerative diseases, such as dementia conditions and Lewy Bodies spectrum disorders (7,8). Therefore, it is imperative to establish hiPSC-based systems that recapitulate molecular and cellular phenotypes related to advanced aging (9) and are fully suitable for the research of age-related neurodegenerative diseases (10,11).
Several strategies have been developed for inducing aging-like features in hiPSC-derived neurons (12) including exposure to toxic compounds that induce mitochondrial stress or reactive oxygen species (ROS) (13), ectopic expression of progerin gene (11) and telomerase manipulation (14). An alternative strategy to maintain the aging signature in derived neurons applies directed conversion of fibroblasts into induced Neurons (iNs) that display an age-related transcriptional profile (5). These are appealing strategies to induce or retain aging; however, each of the existing approaches has inherent limitations (12) that may introduce extraneous variables that could potentially pose other challenges for the research of age-related brain diseases, in particular for the deep exploration of the genetic determinants and molecular mechanisms of these diseases. Therefore, it is important to develop additional strategies for inducing aging in hiPSC-based models of late-onset neurodegenerative diseases such as synucleinopathies.
Synucleinopathies are a group of neurodegenerative diseases that share a common pathological lesion of intracellular protein inclusions largely composed of α-synuclein aggregates (15). The SNCA gene, encoding the α-synuclein protein, has been implicated in the etiology of synucleinopathies, and it has been suggested that SNCA expression levels are critical for the development of these diseases (16). Among synucleinopathies, Parkinson’s disease (PD) has been extensively studied using various model systems including hiPSC-derived midbrain dopaminergic neurons (mDA) (17), while hiPSC-derived models have been underutilized in the study of other synucleinopathies such as Dementia with Lewy body (DLB) and Lewy body variants of Alzheimer’s disease (18). To further understand the genetic etiologies and molecular mechanisms that are commonly perturbed in synucleinopathies, and those that may underlie the heterogeneity among the different diseases in this group, it is important to characterize in-depth isogenic hiPSC-derived models of different pathology-relevant neurons derived from patients and healthy subjects in the context of aging.
Here, we developed and optimized a new alternative approach to induce aging in hiPSC-derived neurons using a natural-like process that does not involve treatment with toxins or ectopic gene expression. To validate our approach, we characterized age-related phenotypes associated with nuclear architecture (11). Specifically, we measured markers for heterochromatin organization and changes in the nuclear envelope structure that indicate aged nuclei (11,19). In addition, we evaluated DNA damage (20) and changes in global DNA methylation (21), DNA features associated with aging. We applied our method to induce aging in hiPSC-derived dopaminergic and cholinergic neurons to model PD and DLB, respectively. hiPSC lines were derived from an apparently healthy control and a patient with triplication of the SNCA (SNCA-Tri) locus. The levels of SNCA-mRNA in patients with SNCA-Tri are constitutively 2-fold higher compared with subjects with diploid copy of SNCA, and these patients manifest early-onset PD and DLB (13,22). Therefore, cell lines obtained from patients with SNCA-Tri represent an adequate model to study the mechanisms that mediate the pathogenic effect of SNCA overexpression on the development of synucleinopathies. In this study, we established a link between overexpression of SNCA, neuronal aging and nuclear abnormalities.
Results
Generation and characterization of Juvenile and Aged hiPSC-derived neurons
Multiple passaging prior to final differentiation induces aging features
In order to establish a hiPSC-based system to model common synucleinopathies, we differentiated two hiPSCs lines, derived from a patient with SNCA-Tri and an apparently healthy control (Control), into mDA and basal forebrain cholinergic neurons (BFCNs), which are primarily affected in PD and DLB, respectively. To generate mDA and BFCN from the parental hiPSC lines we applied two different differentiation protocols optimized specifically for each neuronal type, following our previous publication (18). Specifically, hiPSCs were first differentiated into ventral midbrain (MD) and Medial Ganglionic Eminence (MGE) progenitors following by terminally differentiation into mDA and BFCN, respectively (Fig. 1A). Cells were characterized throughout the differentiation process at the hiPSC, Neural Progenitor Cell (NPC) and mature neuron stages and according to their respective neuronal type. The successful differentiation of both hiPSC-derived lines for each neuronal type was characterized and validated by immunocytochemistry and real-time Reverse-Transcription PCR using specific markers (Fig. 1; Supplementary Material, Figs S1–S5) (18). In prior work, we successfully differentiated 83.1% of the hiPSC into MD progenitors while 28% were fully matured to give mDAs, similarly, 84.4% of the hiPSC were differentiated into MGE progenitors, while 36.4% went through final maturation into BFCN (18).
Figure 1.
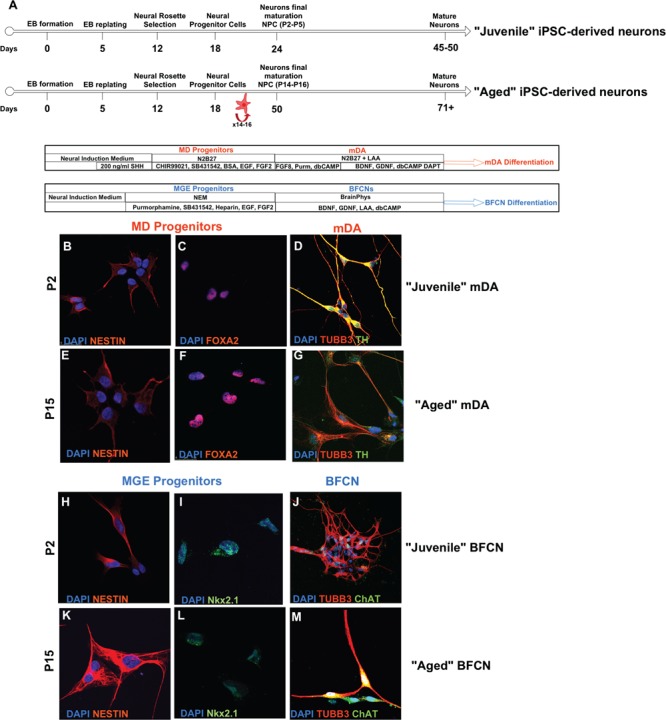
Differentiation of Juvenile and Aged hiPSC-derived neurons. (A) Timeline of the differentiation steps of Juvenile and Aged hiPSC-derived mDA and BFCN. (B–M) Representative immunocytochemistry images throughout the differentiation protocols from Control hiPSCs. (B and C) MD progenitors and (H and I) MGE progenitors co-expressed Nestin and FoxA2, and Nestin and Nkx2.1 at P2. (D) Juvenile mDA neurons and (J) Juvenile BFCN, differentiated from P2–P5 NPCs, were characterized for the co-expression of the mature neuronal marker TUBB3 and TH, and TUBB3 and ChAT, respectively. After prolonged passaging (E and F) MD progenitors and (K and L) MGE progenitors retain the co-expression of Nestin and FoxA2 and Nestin and Nkx2.1. (G) Aged mDA neurons and (M) Juvenile BFCN were successfully differentiated from P14–P16 NPCs, and they showed the co-expression of the mature neuronal marker TUBB3 and TH, and TUBB3 and ChAT, respectively. See also Supplementary Material, Figures S1–S5.
The rejuvenation of hiPSC lines poses a challenge for studying late-onset age-related synucleinopathies. In order to address the loss of age during reprograming, we developed a new method to generate Aged hiPSC-derived neurons. The principle of our approach is built on prolonging cultures to imitate `natural’ physiological aging. Prolonging the maturation of the final BFCN and mDA resulted in cell death after 4 weeks (data not shown). Therefore, inducing aging at the mature neuron stage is limited. Thus, to induce aging features, we extended the time at the NPC stage prior to final maturation (Fig. 1A), such that the generation of mature neurons occurs after several weeks of culturing during which the NPCs were passaged every 2 days. To optimize the method, we prolonged the culturing of the NPCs until passage 24 (P24). After P24, we observed extensive cell death for both cell lines and differentiation protocols; furthermore, the cells lost their ability to proliferate, indicating that the NPCs can be cultured and passaged up to P24.
To further optimize the induced aging method, we tested the ability of the NPCs to undergo final maturation post-multiple passaging. We differentiated NPCs at P20 by applying the final maturation medium and observed that these cells failed to complete the maturation. Therefore, we reduced the prolonged culture of the NPCs to passage 15 (P15). MGE and MD progenitors at P15 were differentiated into final mDA and BFCN, respectively, and showed the expression of TUBB3 and TH, or TUBB3 and ChAT, respectively (Fig. 1E–G, K–M; Supplementary Material, Figs S2–S5), indicating the successful differentiation into mature neurons following prolonged passaging of NPCs to P15.
Next, we evaluated whether prolonging NPCs cultures induced age-like phenotypes. For this analysis, we used age-associated cellular markers that were reported to reliably measure age in vitro. Specifically, we measured the heterochromatin marker, Heterochromatin protein 1 γ (HP1γ) and the nuclear envelope marker, Lamin A/C (11). To determine the passage at which the NPCs on one hand feature age-related phenotype and on the other hand are viable and able to undergo successful final differentiation, we compared these markers between the NPCs at three passage groups, P2–P4, their isogenic partners NPCs at P15 and P20–P24 (viable but unable to mature) (Fig. 2). We observed a progressive loss of HP1γ signal (Fig. 2M and N) and higher frequency of nuclear folding for both MGE and MD lines (Fig. 2A–F) with increasing passages (Supplementary Material, Tables S2–S5). NPCs at late passages, P15 and P20–P24, were characterized by significant lower intensities of the HP1γ signal compared with the early passage NPCs P2–P4 (Fig. 2G–N). Consistently, P15 and P20–P24 showed a 4–8-fold increase in nuclear folding compared with the NPCs at P2–P5 (Fig. 2A–F). Mitochondrial dysfunction, measured by higher mitochondrial ROS production, has been associated in aging (11). Thus, in addition, we also evaluated mitochondrial superoxide production. NPCs at P15 showed an increase in mitochondrial superoxide production compared with NPCs at P2–P5 (Fig. 2O). Together we demonstrated that prolonged passaging induces aging-related features. Furthermore, the age-related markers for P15 were significantly different from their matched NPCs at earlier passages and exhibited the same trends as the latest passages, indicating that passaging to P15 is sufficient to induce aging for both MGE and MD progenitors. Therefore, we optimized the protocol to extend the culture of the NPCs to P15 at which they showed aging-related features, were viable, retained the ability to proliferate and were able to undergo successful differentiation to final mature neurons.
Figure 2.
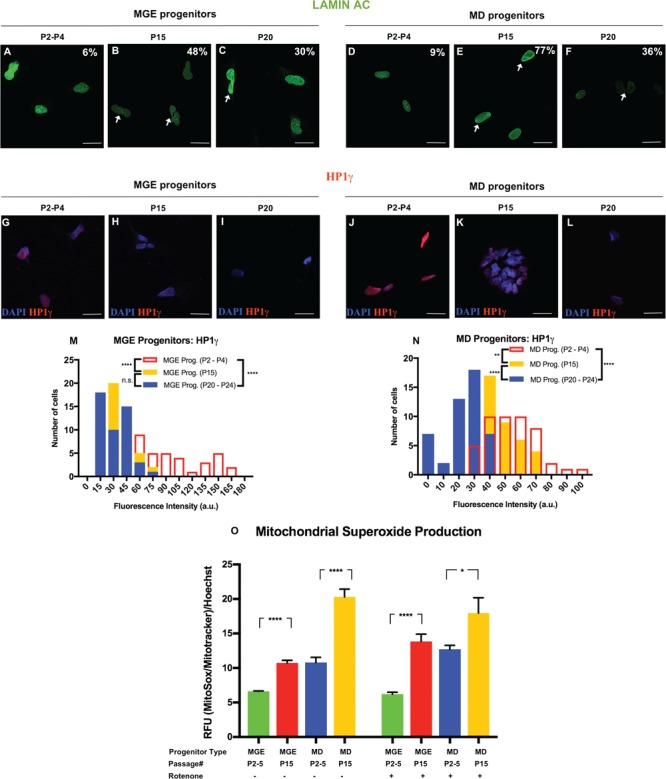
Characterization MGE and MD Progenitors after prolonged passaging. (A–F) Immunocytochemistry for Lamin A/C in (A–C) MGE progenitors and (D–F) MD progenitors. Percentages indicate the proportion of cells with folded/blebbed nuclear morphology, white arrows denote abnormal nuclei. (G–L) Immunocytochemistry for HP1γ in (G–I) MGE progenitors and (J–L) MD progenitors. (M) Quantification of the HP1γ marker in MGE progenitors demonstrated the reduction in the signal intensity at different passages. (N) Quantification of the HP1γ marker in MD progenitors demonstrated the reduction in the signal intensity at different passages. The data are plotted as frequency distributions of relative fluorescence intensity for 200 cells. a.u., arbitrary units. n = 2 independent differentiation protocols. n.s. not significant **P < 0.01, ****P < 0.0001 according to Kolmogorov–Smirnov test (M and N). Scale bars: 25 μm. See also Supplementary Material, Tables S2–S5 for all statistical comparisons. (O) Mitochondrial superoxide production was measured in MGE and MD Progenitors at P2–P5 and P15. Cells were treated with 20 μm Rotenone for 18 h (right panel). Bars represent mean ± standard error of the mean (SEM). n = 6 for two independent experiments ****P < 0.0001, *P < 0.05; two-tailed Student’s t-test. See Supplementary Material, Table S6 for all statistical comparisons.
Mature neurons derived normal and disease hiPSCs maintain nuclear-aging markers
In order to validate our method of inducing aging in hiPSC-derived neurons we next evaluated the ability of the mature neurons to maintain the aging-associated phenotype upon final differentiation. Aging has been associated with changes in chromatin organization (11,23,24). We utilized two markers of peripheral heterochromatin: (1) HP1γ and (2) the Trimethylation Marker on Histone 3 Lysine 9 (H3K9me3) (11). The HP1γ and H3K9me3 markers were previously defined and showed to correlate with the donor’s age, whereas a global loss of both markers was associated with aging in vitro (11). Juvenile and Aged BFCNs and mDA neurons were differentiated from P2–P5 NPCs and P14–P16 NPCs, respectively, for both the Control and SNCA-Tri lines. The levels of the HP1γ and H3K9me3 were measured in all eight lines, and the signal intensities were compared for each set of neuronal type, i.e. BFCN and mDA. For the purposes of method validation, we analyzed the differences between the Aged and the Juvenile neurons within each isogenic hiPSC line, i.e. Control or SNCA-Tri. The Control hiPSC-derived Aged BFCNs and mDA neurons showed global loss in the signal intensity of the heterochromatin markers HP1γ and H3K9me3 compared with their counter partner Juvenile BFCN (Fig. 3A, B, I, K, L and S; Supplementary Material, Tables S7 and S12) and mDA neurons (Fig. 3E, F, J, O, P and T; Supplementary Material, Tables S8 and S13), respectively. The same trends were observed for the SNCA-Tri lines, that is, comparison of the Aged relative to the Juvenile in each neuronal type showed global reduction in the heterochromatin markers (Fig. 3C, D, G–J, M, N, Q–T; Supplementary Material, Tables S7, S8, S12, S13). These results demonstrated that the isogenic hiPSC-BFCN and mDA-Aged neurons acquire and maintain heterochromatin age-related features.
Figure 3.
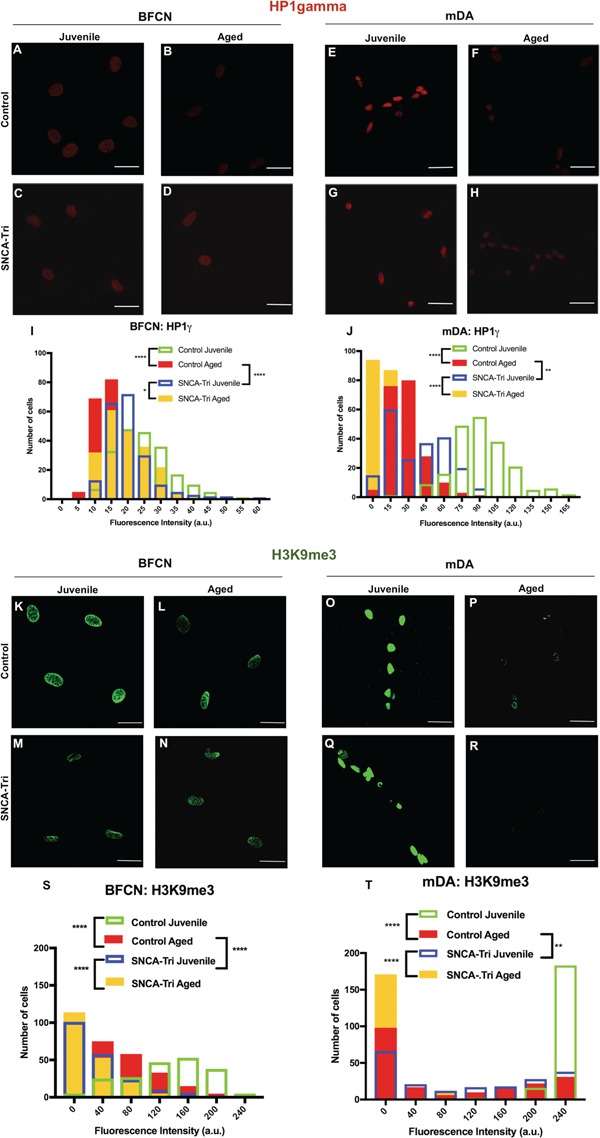
The effects of inducing aging and SNCA multiplication on heterochromatin markers. (A–H) Immunocytochemistry for HP1γ in (A–D) BFCN and (E–H) mDA Control and SNCA-Tri. Aged hiPSC-derived BFCN and mDA neurons showed global loss in the signal of HP11γ. (I) Quantification of the HP1γ marker in BFCN demonstrated the reduction in the signal intensity in the Aged versus Juvenile in the Control and SNCA-Tri lines. (J) Quantification of the HP1γ marker in mDA demonstrated the reduction in the signal intensity in the Aged versus Juvenile in the Control and SNCA-Tri lines. SNCA-Tri neurons demonstrated reduction in the signal intensities compared with the respective Control neurons. The data are plotted as frequency distributions of relative fluorescence intensity for 200 cells. a.u., arbitrary units. n = 2 independent differentiation protocols. *P < 0.05, **P < 0.01, ****P < 0.0001 according to Kolmogorov–Smirnov test (I and J). Scale bars: 25 μm. See also Supplementary Material, Tables S7, S8, S11 for all statistical comparisons. (A–H) Immunocytochemistry for H3K9me3 in (K–N) BFCN and (O–R) mDA Control and SNCA-Tri. Aged hiPSC-derived BFCN and mDA neurons showed global loss in the signal of H3K9me3. (S) Quantification of the H3K9me3 marker in BFCN demonstrated the reduction in the signal intensity for the Aged versus Juvenile neurons in the Control and SNCA-Tri lines. (T) Quantification of the H3K9me3 marker in mDA demonstrated the reduction in the signal intensity in the Aged versus Juvenile neurons in the Control and SNCA-Tri lines. SNCA-Tri neurons demonstrated reduction in the signal intensities compared with the respective Control neurons. The data are plotted as frequency distributions of relative fluorescence intensity for 200 cells. a.u., arbitrary units. n = 2 independent differentiation protocols. **P < 0.01, ****P < 0.0001 according to Kolmogorov–Smirnov test (I and J). Scale bars: 25 μm. See also Supplementary Material, Tables S12, S13, S16 for all statistical comparisons.
The loss of the integrity of the nuclear envelope has been associated with aging and disease (11,19). To further validate our method for inducing aging in hiPSC-derived BFCN and mDA neurons, we analyzed two nuclear envelope markers: Lamin A/C (11) and Lamin B1 (19). Lamins are involved in the structural integrity of the nuclear envelope, whereas Lamins A, C and B1 contribute to the nuclear shape (25–27). To analyze the nuclear envelope, Juvenile and Aged hiPSC-derived BFCN and mDA neurons from both Control and SNCA-Tri lines were stained for Lamin A/C (Fig. 4A–H) and Lamin B1 (Fig. 4I–P). First, we analyzed the percentage of abnormal nuclei using Lamin A/C marker by counting folded and `blebbed’ nuclei as abnormal (11). The Aged Control BFCNs and mDA neurons showed an increase in the percentage of the folded nuclei compared with the Juvenile Control BFCNs (13.5 versus 47.9%, Fig. 4A and B) and mDA neurons (14.5 versus 44.8%, Fig. 4E and F), respectively. Similar results were obtained in the analysis of the SNCA-Tri line, for both types of neurons the Aged exhibited higher percentage of folded nuclei in comparison to their matched Juvenile neurons (56.1 versus 65.1% and 44.7 versus 56.5%, Fig. 4C, D, G, H). Another marker for aging, related to the nuclear envelope, is the decrease in the nuclear circularity (19). We next measured the nuclear circularity by analyzing the Lamin B1 marker (Fig. 4I–P). A significant decrease in the nuclear circularity was observed for each isogenic Aged compared with the Juvenile neurons, for each of the two neuronal types (Fig. 4Q and R; Supplementary Material, Tables S17 and S18). These results demonstrated that the isogenic hiPSC-BFCN and mDA Aged neurons acquire and maintain age-related abnormalities of the nuclear envelope.
Figure 4.
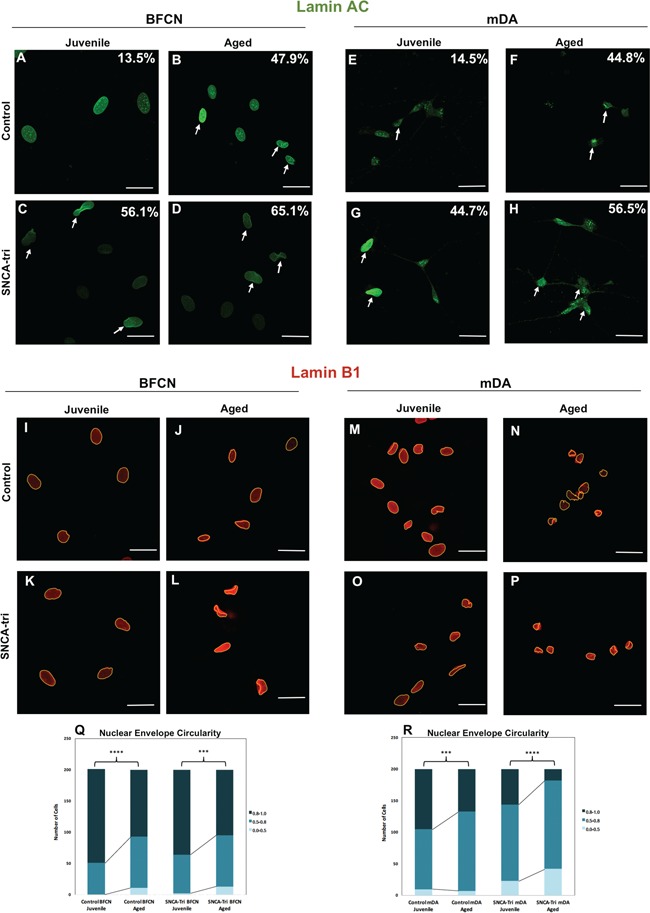
The effects of inducing aging and SNCA multiplication on abnormal nuclear envelope morphology. (A–H) Immunocytochemistry for Lamin A/C in (A–D) BFCN and (E–H) mDA Control and SNCA-Tri lines. Percentages indicate the proportion of cells with folded/blebbed nuclear morphology, white arrows denote abnormal nuclei. Aged hiPSC-derived BFCN and mDA neurons showed higher percentage of abnormal nuclei. SNCA-Tri neurons demonstrated higher percentage of folded/blebbed nuclei compared with the respective Control neurons. (I–P) Immunocytochemistry for Lamin B1 in (I–L) BFCN and (M–P) mDA Control and SNCA-Tri lines. Aged hiPSC-derived BFCN and mDA neurons showed a loss in the nuclear envelope circularity. (Q) Quantification of the nuclear envelope circularity in BFCN demonstrated the loss of nuclear envelope circularity in the Aged versus Juvenile neurons in the Control and the SNCA-Tri lines. (R) Quantification of the nuclear envelope circularity in mDA demonstrated the reduction in the nuclear envelope circularity in the Aged versus Juvenile neurons in the Control and SNCA-Tri lines. A loss in nuclear envelope circularity was observed for the SNCA-Tri lines compared with the respective Control neurons. The data are plotted as frequency distributions of for 200 cells. n = 2 independent differentiation protocols. ***P < 0.001, ****P < 0.0001 according to Kolmogorov–Smirnov test (Q and R). Scale bars: 25 μm. See also Supplementary Material, Tables S17–S19 for all statistical comparisons.
To further validate our inducing-aging method in hiPSC-derived neurons, we measured DNA damage and loss in global DNA methylation, age-related features of the DNA (20,21). DNA damage was analyzed using the comet assay, specifically measures of the Olive Tail Moment (OTM) (28). The OTM is a comprehensive measure of DNA damage that includes the smallest detectable parts of migrating DNA as well as the number of broken DNA in the tail. The Aged Control hiPSC-derived BFCNs and mDA neurons showed an increase in OTM compared with the Juvenile Control BFCN and mDA (11.81 versus 18.90% and 21.96 versus 25.94%, Fig. 5A, B, E, F, I and J; Supplementary Material, Table S20), respectively. Similarly, the analysis of the SNCA-Tri neuronal lines also demonstrated higher OTM values for the Aged compared with the Juvenile BFCN and mDA neurons (29.3 versus 34.88% and 26.35 versus 32.41%, Fig. 5C, D, G–J; Supplementary Material, Table S20). These results indicated an increase in DNA damage in the Aged neurons compared with the counter partner Juvenile.
Figure 5.
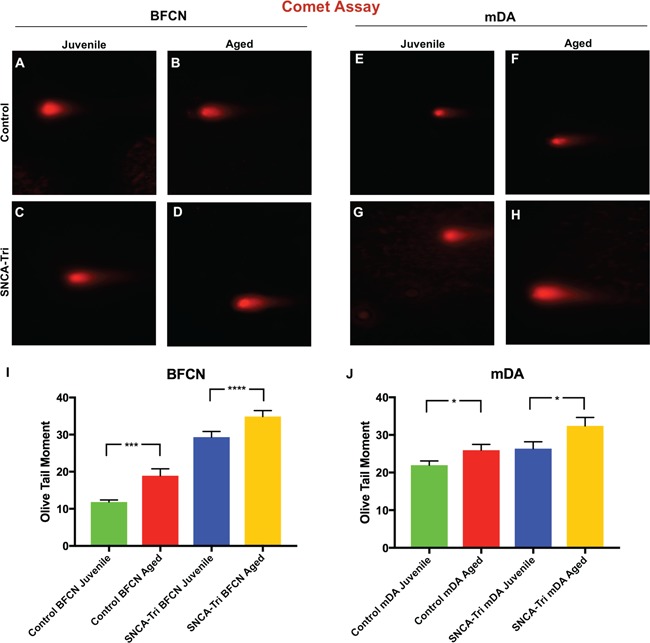
The effects of inducing aging and SNCA multiplication on DNA damage. (A–H) Comet assay photographs of hiPSC-derived (A–D) BFCN and (E–H) mDA neurons for the Control and SNCA-Tri lines. Aged hiPSC-derived BFCN and mDA neurons showed increased DNA damage. (I and J) Comet assay analysis of the OTM demonstrated an increase in the OTM for the Aged (I) BFCN and (J) mDA compared with the isogenic (Control, SNCA-Tri) Juvenile neurons. SNCA-Tri neurons demonstrated increased DNA damage compared with the respective Control neurons. Each column represents the mean of 50 cells for each of the two biological replicates. The error bars represent the SEM. ****P < 0.0001, ***P < 0.001, *P < 0.05; Student’s t-test. See Supplementary Material, Tables S20 and S21 for all statistical comparison.
Next, to evaluate global DNA methylation we measured the percentage of the 5 methylcitosine (5 mC%) (21). The Aged Control hiPSC-derived BFCNs and mDA neurons showed lower 5 mC% compared with their counter partner Juvenile BFCNs (2.02 versus 1.15%, Fig. 6A) and mDA neurons (1.23 versus 0.65%, Fig. 6B), respectively, indicating loss in global DNA methylation. The same trends were observed for the SNCA-Tri neuronal lines (1.98 versus 0.75% and 3.42 versus 2.48%, Fig. 6A and B) showing a decrease in the 5 mC% in the Aged versus the Juvenile neurons.
Figure 6.
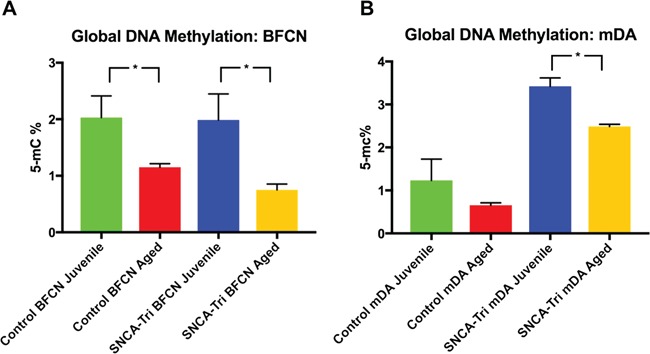
Aged hiPSC-derived BFCN and mDA neurons show a loss in global DNA methylation. Global 5 mC% analysis of the hiPSC-derived (A) BFCN and (B) mDA neurons of the Control and SNCA-Tri lines. Comparisons of the 5 mC% found a decrease in the global DNA methylation in the Aged neurons versus the Juvenile isogenic (Control and SNCA-Tri) and corresponding neuronal types (BFCN and mDA). Each column represents the mean of two biological and technical replicates. The error bars represent the SEM. *P < 0.05; Student’s t-test. See Supplementary Material, Tables S22 and S23 for all statistical comparisons.
Collectively, these results validated our method to induce aging in hiPSC-derived BFCN and mDA neurons demonstrating that passaging multiple times prior to final differentiation induces and maintains previously defined and well-established nuclear aging-related markers.
The effect of SNCA-Tri on neuronal aging phenotypes
It has been suggested that overexpression of SNCA plays a role in the development of PD and other synucleinopathies (16). We showed that SNCA-Tri hiPSC-derived neurons exhibit in ∼2-fold higher levels of SNCA-mRNA and protein compared with the Control neurons (18,29) (Supplementary Material, Fig. S5H). To evaluate the effect of SNCA overexpression on neuronal aging, we analyzed the differences between the Control and the SNCA-Tri neurons for each neuronal type and within each age, i.e. Juvenile or Aged. Using the heterochromatin and nuclear envelope markers we showed that the SNCA-Tri neurons were characterized by exacerbated nuclear aging features compared with the age-matched Controls. Specifically, in the analysis of the heterochromatin markers, HP1γ and H3K9me3, the Juvenile SNCA-Tri showed an overall lower signal intensity compared with the Juvenile Control for both hiPSC-derived BFCN and mDA neurons (Fig. 3I, J, S, T; Supplementary Material, Tables S7,S8,S12,S13). Similar trends for both types of neurons were observed in the comparisons of the Aged SNCA-Tri to the Aged Control hiPSC-derived neurons, whereas the SNCA-Tri lines were characterized by lower signal intensities (Fig. 3I, J, S, T; Supplementary Material, Tables S7,S8,S12S13). Noteworthy, we observed that the Juvenile SNCA-Tri neurons displayed signal intensity distributions that were comparable, to some extent, to those of the Aged Control hiPSC-derived neurons (Fig. 3I, J, S, T; Supplementary Material, Tables S7,S8,S12,S13). Moreover, the Juvenile SNCA-Tri hiPSC-derived BFCN exhibited an overall reduction for the H3K9me3 marker compared with the Aged Control hiPSC-derived BFCN (Fig. 3S), indicating the possibility of further exacerbation of the aging-related features in the SNCA-Tri BFCNs. We also observed that the global loss in the signal intensity of the heterochromatin markers between Juvenile and Aged neurons was more pronounced for the Control than the SNCA-Tri hiPSCs-derived neurons, possibly because the SNCA-Tri neurons undergo aging-related nuclear processes already at the Juvenile stage (Supplementary Material, Fig. S6 and Tables S9,S10,S14,S15). Altogether, these results suggested that overexpression (∼2-fold) of SNCA gene is associated with an exacerbation of the heterochromatin aging-related phenotypes.
Figure 7.
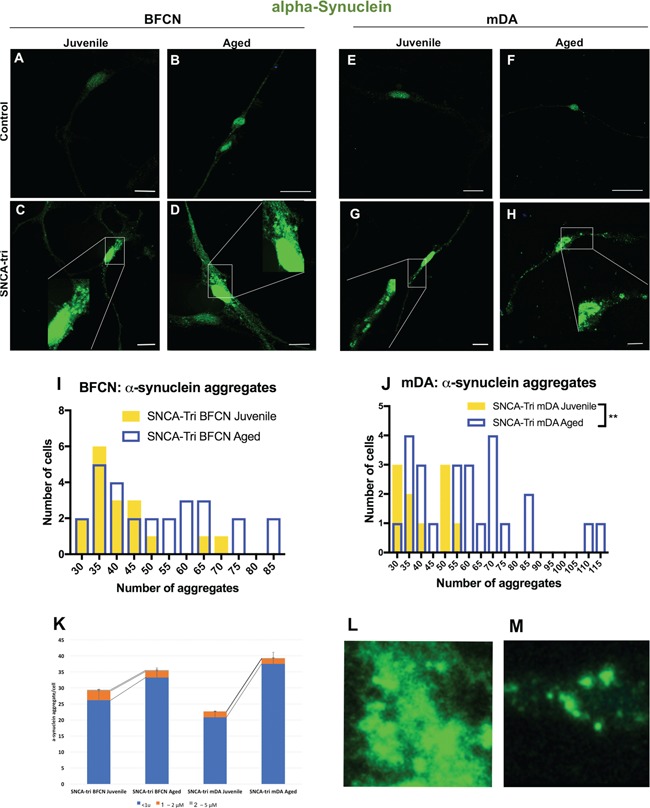
Characterization of α-synuclein aggregates in SNCA-Tri hiPSC-derived BFCN and mDA neurons. (A–H) Immunocytochemistry for α-synuclein protein in (A–D) BFCN and (E–H) mDA, Control and SNCA-Tri lines. Small and medium aggregates can be observed in SNCA-Tri (C and D) BFCN and (G and H) mDA neurons. (I and J) Quantification of the number of aggregates in (I) SNCA-Tri BFCN and (J) SNCA-Tri mDA. (K) Analysis of aggregates size per cell in Juvenile versus Aged SNCA-Tri BFCN and mDA. (L and M)α-synuclein aggregates in SNCA-Tri derived neurons showed distinct morphologies, diffused versus punctuated, in (L) BFCN (M) SNCA-Tri mDA, respectively. (I–K) The data are plotted as frequency distributions of number of α-synuclein aggregates for 50 cells. **P < 0.01, Student’s t-test. Scale bars: (B, F) 25 μm (A, C–E, G and H) 10 μm. See also Supplementary Material, Tables S24–S29 for all statistical comparisons.
Similar trends were found in the analysis of the nuclear envelope markers. Consistently, we demonstrated exacerbated nuclear aging characteristics in the SNCA-Tri neurons compared with the age-matched Controls. We observed an increased percentage of abnormal nuclear envelope in the Juvenile SNCA-Tri BFCN and mDA in comparison to the Juvenile Control neurons (Fig. 4A, C, E, G). The Aged neurons showed the same effect, but the SNCA-Tri BFCN and mDA showed a relative subtle increase in abnormal nuclei compared with their counter partners Control (Fig. 4B, D, F, H). Also, the SNCA-Tri Juvenile neurons had similar percentages of abnormal nuclei to those of the Aged Control hiPSC-derived neurons (Fig. 4B, C, F, G). These results were in line with the observations for the heterochromatin markers, suggesting that SNCA overexpression results in earlier initiation of aging-related nuclear processes. Next, we compared the nuclear circularity and found a greater loss in the SNCA-Tri mDA versus the age-matched Control mDA neurons (Fig. 4Q and R; Supplementary Material, Table S18). The Juvenile SNCA-Tri mDA presented a decrease in nuclear circularity even compared with the Aged Control mDA. These results suggested that overexpression (∼2-fold) of SNCA gene correlates with an exacerbation of aging-related phenotypes of the nuclear envelope. The extent of the SNCA overexpression effect was different for the BFCN. The Juvenile SNCA-Tri BFCN showed subtle decrease in nuclear circularity in comparison to the Juvenile Control BFCN; however, the Aged SNCA-Tri BFCN showed no differences in circularity compared with the Aged Control BFCN (Fig. 4Q and R; Supplementary Material, Table S17).
In addition, we compared the DNA damage and the global DNA methylation between the Control and SNCA-Tri neurons. An increase in DNA damage was detected for all the SNCA-Tri neuronal lines compared with the respective neuronal type and age-matched Control lines (Fig. 5; Supplementary Material, Table S20), suggesting an impact of SNCA overexpression on DNA damage. The magnitude of the SNCA overexpression effect on DNA damage was more substantial for the BFCN neurons (∼2–2.5 increase in OTM values), while the effects were subtle for the mDA lines. Remarkably, the extent of DNA damage in the Juvenile SNCA-Tri mDA neurons was similar to that of the Aged Control hiPSC-derived neurons (Fig. 5E–H, J; Supplementary Material, Table S20), and the Juvenile SNCA-Tri hiPSC-derived BFCN showed even a greater DNA damage compared with the Aged Control hiPSC-derived BFCN (Fig. 5A–D and I; Supplementary Material, Table S20), indicating that SNCA overexpression has a comparable or greater effect on DNA damage than that of aging. On the other hand, analysis of the global DNA methylation showed no consistent trends in the comparisons between SNCA-Tri neuronal lines and their respective Control neurons (Fig. 6). The results were inconclusive, implying that the perturbations to the nuclear phenotypes caused by SNCA overexpression do not involve loss in global DNA methylation normally occurring in aging.
Collectively, our results showed that SNCA-Tri neurons exhibited enhanced aging-related nuclear architecture and DNA integrity features, suggesting that SNCA overexpression affects the aging of neuronal nuclei. This effect is cell-type specific, indicating differential exacerbation of some nuclear architecture phenotypes between the isogenic lines. These findings suggested neuronal type-specific effects of SNCA overexpression on nuclear aging, strengthening the power of our system to model the heterogeneity in synucleinopathies. In conclusion, the results of the nuclear aging phenotypes suggested a link between SNCA overexpression, exacerbation of nuclear aging and neuronal type differential susceptibility to aging.
The effect of induced aging on α-synuclein aggregates
Aging was associated with increased protein aggregation (30–32). To examine the presence of α-synuclein aggregates in our model, Juvenile and Aged hiPSC-derived neurons from both Controls and SNCA-Tri cells were stained for α-synuclein. In the Control hiPSC-derived BFCN and mDA neurons, we detected a predominant signal in the nuclei (Fig. 7A, B, E, F), whereas in the SNCA-Tri-derived neurons we observed an intense signal in the nuclei but also throughout the cell body (Fig. 7C, D, G, H). Next, we characterized the α-synuclein signal within the cell body of 50 randomly selected neurons. We detected α-synuclein aggregates in the SNCA-Tri BFCN and mDA neurons and defined the aggregates based on their size into small (<1 μm), medium (1–2 μm) and large (2–5 μm) (33). The majority (>85%) of the total aggregates were classified as small aggregates (Fig. 7K), while few aggregates were in the ranges of medium and large sizes. A significant increase in the number of small aggregates was found in the Aged SNCA-Tri mDA neurons compared with the Juvenile mDA, while no significant differences were observed between the Aged and Juvenile SNCA-Tri BFCN (P < 0.0001 and P = 0.1257, respectively; Supplementary Material, Table S24). This observation indicated differential phenotypic consequences upon inducing aging in the context of SNCA overexpression related to the neuronal type, which may contribute to the heterogeneity in synucleinopathies (Fig. 7K).
Next, we analyzed the number of aggregates per cell. To determine the `noise’ we evaluated the Control iPSC-derived BFCN and mDA and found that the maximum number of α-synuclein aggregates (all small, <1 μm) in these neurons was 30 aggregates/cell, of note only 2% of the cells exhibited 30 aggregates (Supplementary Material, Fig. S7). Accordingly, we set the threshold and considered in our analysis only cells that displayed more than 30 aggregates. To evaluate the effect of aging, we analyzed the number of aggregates in the Juvenile compared with the Aged SNCA-Tri BFCN and mDA neurons. We observed a significant increase in the number of aggregates in the Aged mDA compared with the Juvenile (Fig. 7J), while no significant difference was detected in Juvenile versus Aged BFCN (Fig. 7I).
We further characterized the aggregates and identified differences in the conformation of the aggregates between SNCA-Tri BFCN and mDA neurons. Specifically, the aggregates in the BFCN had a more diffuse conformation (Fig. 7L), while aggregates in the mDA appear more defined with a punctuate structure (Fig. 7M).
Taken together, these analyses demonstrated different characteristics of the α-synuclein aggregation between the isogenic BFCN and mDA neurons. The overall results suggest that the interplay between aging and SNCA overexpression have a differential effect on different neuronal types primarily affected in synucleinopathies, providing new insights into the heterogeneity in synucleinopathies and an adequate model system to investigate the common and distinct mechanisms underlying synucleinopathies.
Discussion
In this study, we suggested a link between the overexpression of SNCA, neuronal nuclear structure and aging. We developed a new in vitro system, hiPSC-derived Aged neurons. Using our neuronal models, we found that the effects of SNCA overexpression on neuronal nuclear signatures, including nuclear architecture, chromatin organization and DNA damage, are comparable to the effects of inducing aging, suggesting that SNCA overexpression exacerbates nuclear aging.
There is an unmet need for in vitro systems to model age-related brain diseases. We developed a new method to generate Aged hiPSC-derived neurons to address the current challenges related to recapitulating aging features in hiPSC-derived models so that they are fully suitable for studying late-onset neurodegenerative diseases. We described an attractive strategy for inducing aging by multiple passaging at the NPCs stage. We next demonstrated increased defects in the nuclear architecture and DNA integrity and loss in global DNA methylation in the Aged hiPSC-derived neurons compared with the isogenic Juvenile neurons, validating that our new method is fully suitable to induce aging features in hiPSC-derived neurons. Previously described strategies to induce aging applied external interventions (11,13,14,19), and while suitable to induce aging phenotypes, they present some limitations. For example, it is not clear whether the exposure to toxins will force the cells to show an artificial disease phenotype rather than a physiological change correlated to the aging process and how the ectopic expression of genes, i.e. progerin, might induce molecular changes that are preferably associated with progerin overexpression rather than molecular mechanisms of the normal aging. An alternative approach is the directed conversion of fibroblasts into iNs. This strategy presents several strengths, primarily the ability to retain the aging signature of the donor’s cells as demonstrated by age-related transcriptional profile (5). iNs are generated by overexpression of specific developmental transcription factors (34–37) to trigger the differentiation of fibroblasts to a specific neuronal fate, a method that requires an intense optimization for the efficient delivery of the reprogramming factors essential to the conversion. While iNs represent an alternative strategy in the study of age-related phenotypes and diseases, the method inherent variability across repeated experiments of generating the iNs and may also present other confounding factors related to the ectopic expression of transcription factors. Other strategies apply small molecules for the conversion of fibroblasts into iNs. While these protocols overcome constrains related to ectopic expression of transcription factors, the major caveat is that the majority ∼80% of the generated neurons are glutamatergic neurons that do not represent the primary neuronal types involved in PD–DLB spectrum disorders (38,39). Our protocol applies a compelling approach by mimicking, to a certain extent, `natural aging’ process, without applying extraneous factors that may introduce confounding effects and allows reproducibility across experiments. Our novel method generates a system that is applicable and fully suitable for modeling late-onset neurodegenerative diseases and highly valuable for the research of a broad range of diseases of the aging brain including movement disorders and the spectrum of dementias.
Here, we suggest a new pathogenic effect of the overexpression of SNCA. We described dysmorphic nuclear envelope shape, loss of heterochromatin markers and substantial DNA damage in hiPSC-derived neurons from a patient with SNCA-Tri, indicating a link between SNCA overexpression and nuclear architecture. The perturbations to the nuclear envelope, chromatin organization and nuclear DNA integrity in relation to SNCA overexpression were not described before. Over two decades ago, SNCA was identified as a genetic determinant for familial PD (40); following the original discovery, Genome Wide Association Studies found that SNCA was also associated with sporadic PD (41) and other synucleinopathies (42). However, the normal function of the protein encoded by the SNCA gene and the pathway that mediates its pathogenic effect in disease state are still unknown (15). It has been proposed that α-synuclein is involved in membrane interactions, protein degradation, maintaining supply of synaptic vesicles, regulation of dopamine release and transport, mitochondrial dysfunction and autophagy (43). Overexpression of SNCA was implicated in the pathogenesis of synucleinopathies (16), and its perturbing effects have been widely studied. The effect of elevated levels of α-synuclein has been evaluated in various cell organelle in context to multiple cellular pathways, such as in the mitochondria in relation to oxidative stress, the lysosomes in relation to autophagy–lysosome pathway, in the endoplasmic reticulum-to-Golgi trafficking (43,44). However, its role within the nucleus has been understudied. Although, α-synuclein was historically identified as a neuron-specific protein, localized to a region of the nuclear envelope (45), to date, only few reports examined α-synuclein in the context of the nucleus (46–49), in particular the interaction of α-synuclein with histones and its effects on the post-transcriptional modification of histones (50–52). However, these few studies were conducted in vitro or performed in Drosophila models. Here, for the first time, we utilized hiPSC-derived neurons that represent the relevant human cell types for synucleinopathies to investigate the role of SNCA in the nucleus and in relation to the chromatin remodeling. We found that overexpression of SNCA exacerbated age-related nuclear abnormalities and proposed that SNCA contributes directly or indirectly to the maintenance of the nuclear envelope integrity, heterochromatin structure and DNA integrity. Based on our results and the previous publication above, we speculate a role for SNCA in chromatin remodeling, such that elevated levels of SNCA lead to extensive loss in heterochromatin markers. As a consequence, the DNA is more accessible and exposed to potential damage. These changes in chromatin organization may also result in abnormal shape of the nuclear envelope.
The involvement of the nucleus in the pathogenesis of PD and more generally in synucleinopathies has not been explored. Consistent with the effect of SNCA overexpression on nuclear abnormalities we identified in this study, only one paper reported an effect of another PD associated gene, Leucine-rich repeat kinase 2 (LRKK2), on nuclear phenotypes (19). The authors characterized hiPSC-derived NPCs with the LRKK2 pathogenic mutation G2019S and found aberrant nuclear envelope organization that was rescued by correcting the mutation to the wild-type allele (19). These data, along with our findings, strengthen the hypothesis that the nucleus plays a role in the pathogenesis of PD, and exacerbation of neuronal nuclear abnormalities contributes to the development of PD. Our study extended on these observations, suggesting a possible role of the nucleus not only in PD but also in DLB, and that changes in the nuclear architecture and chromatin organization might play a mechanistic role in the pathogenesis of synucleinopathies in general.
The model system we described in this study is suitable for deep exploration of common and distinctive cell-specific mechanisms underlying synucleinopathies. Each disease in this group shows distinct characteristics regarding the neuronal cell types and the brain regions containing the Lewy bodies (53). Specifically, in early stages of diseases, mDA neurons are primarily affected in PD, while the BFCNs show primarily hallmarks of the disease in DLB (53). In this study, the isogenic hiPSC-derived dopaminergic and cholinergic neurons showed neuronal type-specific differences related to the extent of abnormalities in the nuclear architecture, the effect of inducing aging and the characteristics of α-synuclein aggregation. We observed that BFCN and mDA are differentially susceptible to the aging protocol, whereas the mDAs exhibited stronger effects upon inducing aging for most characterized phenotypes (Supplementary Material, Tables S11, S16, S19, S21, S23–S27). The exacerbation of some nuclear-related phenotypes as a consequence of SNCA overexpression was also more pronounced in the mDA (Supplementary Material, Tables S11, S16, S19, S21, S23–S27). We also detected α-synuclein aggregates within SNCA-Tri cells that were morphologically different between the cholinergic and mDA, recapitulating the differences between the defined versus diffused Lewy bodies in PD versus DLB, respectively. Recently, we also showed using a similar strategy a neuron-type-specific expression profiles of miRNAs in isogenic hiPSC-derived cholinergic and mDA, suggesting a microRNA-mediated differential regulation of SNCA-mRNA (18). Collectively, these results suggest the importance of investigating physiological- and disease-related mechanisms in pathology-relevant cells to achieve a deeper understanding of the mechanisms underlying the etiology and heterogeneity in synucleinopathies.
Here, to the best of our knowledge, for the first time we described an interplay between the overexpression of SNCA, neuronal nuclear phenotypes and aging. We suggest that wild-type SNCA functions in pathways related to neuronal nuclear architecture and chromatin organization. This hypothesis warrants further investigations to gain a deeper understanding of the link between SNCA and nuclear architecture in health and disease. Establishing the link between synucleinopathies and an exacerbated nuclear aging opens new avenues for therapeutic interventions.
Materials and Methods
Cell culture and neuronal differentiation
hiPSCs from an apparently healthy individual (GM23280, RRID: CVCL_F179) and a patient with SNCA-Tri (ND34391, RRID:CVCL_F202) were purchased from Coriell Cell Repositories, NJ, USA (http://ccr.coriell.org/) and from the NINDS Human Cell and Data Repository, NJ, USA (https://nindsgenetics.org), respectively. GM23280 and ND34391 lines have a normal karyotype. hiPSCs were cultured under feeder-independent conditions in mTeSR™1 medium (StemCell Technologies, Vancouver, Canada) onto hESC-qualified Matrigel-coated plates. Cells were passaged using Gentle Cell Dissociation Reagent (StemCell Technologies) according to the manufacturer’s manual.
The mDA derive from the MD, while the BFCN derive from the MGE (54). We therefore used specific protocols to differentiate hiPSCs to mDA and BFCN (55–57). hiPSCs were differentiated as we previously described (18). Briefly, differentiation into mDA was performed using an embryoid body (EB)-based protocol. hiPSCs were dissociated with Accutase (StemCell Technologies) and seeded into Aggrewell 800 plates (10 000 cells per microwell; Stem Cell Technologies) in Neural Induction Medium (NIM; Stem Cell Technologies) supplemented with Y27632 (10 μm) to form EBs. On day 5, EBs were replated onto Matrigel-coated plates in NIM. On day 6, NIM was supplemented with 200 ng/mL SHH (Peprotech, NJ, USA) leading to the formation of neural rosettes. On day 12, neural rosettes were selected with Neural Rosette Selection reagent (used per the manufacturer’s instructions, StemCell Technologies) and replated in Matrigel-coated plates in N2B27 medium supplemented with 3 μm CHIR99021, 2 μm SB431542, 5 μg/ml BSA, 20 ng/ml bFGF and 20 ng/ml EGF, leading to the formation of neural precursor cells (NPCs). Differentiation of NPCs into mDA was initiated 1 day after passaging the NPCs on poly-L-ornithine (PLO)/laminin-coated plates. NPC maintenance medium was substituted by final differentiation medium consisting of N2B27 medium supplemented with 100 ng/ml FGF8 (Peprotech), 2 μm Purmorphamine, 300 ng/ml Dibutyryl cAMP (db-cAMP) and 200 μm L-ascorbic acid (L-AA) for 14 days. From day 14, cells were fed with maturation medium consisting of 20 ng/ml GDNF, 20 ng/ml BDNF, 10 μm DAPT, 0.5 mm db-cAMP and 200 μm L-AA. Medium was changed every other day.
The differentiation into BFCN was performed using our previously described protocol (18,55,56). Briefly, EBs were formed into Aggrewell 800 plates in NIM. On day 5, EBs were replated, and the medium was changed daily. From day 8, neural rosettes were grown into NEM (7 parts KO-DMEM to 3 parts F12, 2 mm Glutamax, 1% penicillin and streptomycin, supplemented with 2% B27 (all Life Technologies), plus 20 ng/ml FGF, 20 ng/ml EGF, 5 μg/ml heparin, 20 μm SB431542 and 10 μm Y27632, 1.5 μm Purmorphamine (55). On day 12, neural rosettes were selected with Neural Rosette Selection Reagent and replated in NEM onto Matrigel-coated plates. On day 23, Y27632 was withdrawn and final differentiation was performed onto PLO/laminin-coated plates in the presence of BrainPhys Medium (Stemcell Technologies) supplemented with N2, B27, BDNF, GDNF, L-AA and db-cAMP until day 45–50. Medium was changed every other day.
Generation of Juvenile and Aged neurons
NPCs were passaged every 2 days in their respective medium. NPCs were passaged with Accutase (StemCell Technologies) and plated on Matrigel-coated plates (2.5*104 cells/cm2). To generate the Juvenile neurons, final differentiation procedures were applied to the NPCs at P2–P5 following the protocol outlined above. For the generation of the Aged neurons, NPCs underwent multiple passaging and at P14–P16 were differentiated to final neurons.
Overall, eight cell lines were included in the analyses: Juvenile Control BFCN, Aged Control BFCN, Juvenile Control mDA, Aged Control mDA, Juvenile SNCA-Tri BFCN, Aged SNCA-Tri BFCN, Juvenile SNCA-Tri mDA and Aged SNCA-Tri mDA.
Immunocytochemistry and imaging
Prior to immunostaining, cells were plated onto PLO/Laminin Coated Cells Imaging Coverglasses (Eppendorf, 0030742060). Cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton X-100. Immunocytochemistry was performed as follows: cells were blocked in 5% goat serum for 1 h before incubating with primary antibodies overnight at 4°C (Supplementary Material, Table S1). Secondary antibodies (Alexafluor, Life Technologies, MA, USA) were incubated for 1 h at room temperature. Nuclei were stained with NucBlue® Fixed Cell ReadyProbes® Reagent (Thermofisher, MA, USA), according to the manufacturers’ instructions. Images were acquired on the Leica SP5 confocal microscope using a 40× objective. All images were collected by two blinded observers.
Immunocytochemistry quantification of nuclear markers
Immunofluorescent images of HP1γ and H3K9me3 markers were analyzed using ImageJ software (58). For each cell line a total of 200 cells were analyzed, 100 cells per staining from two independent experiments. Frequency distribution plots represent two experimental replicates binned by arbitrary unit increments based on the natural groupings of the data.
Nuclear folding was analyzed using the Lamin A/C marker, and folded nuclear envelope shape was considered as abnormal. Nuclear circularity was quantified using the built-in ImageJ, WI, USA circularity plug-in and assessed based on the Lamin B1 marker. A circularity value of 1.0 indicates a perfect circle. A value approaching 0 indicates an increasingly elongated polygon (58). One hundred cells per staining were analyzed for two independent experiments. Data analyses were performed by observers that were blinded to the type of neuron and condition they were evaluating.
Mitochondrial superoxide assay
Control MGE and MD progenitors at P2–P5 and P14–P16 were seeded at 3.5 × 104 cells/mm2 and cultured in high glucose NEM and N2B27 medium without phenol red, respectively, in black 96 well plates (Grenier, NC, USA). High-throughput screening plate reader analysis (FLUOstar Omega, BMG, NC, USA) was conducted following a previously described protocol (59). Briefly, 24 h after plating, NPCs were treated with 20 μm rotenone for 18 h or with DMSO only. The MitoSox assay was used for the detection of mitochondria-associated superoxide levels. Adherent NPCs in 96 well plates were incubated with 2 μm MitoSOX™ (Ex./Em. 510/580 nm) and 2 μm MitoTracker® Green (485/520 nm) (both Life Technologies) in high glucose medium without phenol red for 15 min at 37°C in the dark. Cells were washed twice with medium containing 1 μm Hoechst 33342. Fluorescence was detected by sequential readings, and MitoSOX™ signals were normalized to mitochondrial content (Mitrotracker®) and cell number (Hoechst). Each experiment was performed in six replicates per cell lines and treatment, and each experiment was repeated three times independently.
Comet assay
Comet assay was used to measure DNA damage in hiPSC-derived neurons applying a protocol based on a previously published method (60). Briefly, mature neurons were lysed in alkaline conditions by placing the slides in A1 solution [1.2 M NaCl, 100 mm Na2EDTA, 0.1% sodium lauryl sarcosinate, 0.26 M NaOH (pH > 13)] at 4°C in the dark for 18–20 h. Slides were washed three times using A2 solution [0.03 M NaOH, 2 mm Na2EDTA (pH 12.3)], and electrophoresis was conducted for 25 min at a voltage of 0.6 V/cm in fresh A2 solution. Slides were then washed twice in distilled H2O for 5 min, subsequently immerged in 70% ethanol, dried for 15 min at room temperature and stained with SYBR Green for 30 min. After washing the excess of staining, cells were imaged using a Zeiss Axio Observer Widefield Fluorescence Microscope, Germany. Comets were analyzed using the OpenComet Software, MA, USA to determine the OTM (28), the parameter selected as the quantitative measure for each comet. The OTM was determined in 100 cells, 50 cells per each of two independent Comet experiments.
Global DNA methylation
DNA from each cell line was extracted using DNeasy Blood and Tissue Kit (Qiagen, Germany). Global DNA methylation was assessed using a commercially available 5 Methylcytosine (5 mC)-based immunoassay platform [MethylFlash™ Global DNA Methylation (5 mC) ELISA Kit, Epigentek, NY, USA], according to the manufacturer’s instructions. Briefly, purified DNA (50 ng) and unmethylated (negative) control DNA (10 ng) were incubated in strip wells with a solution to promote DNA binding and adherence to the well. Samples in the strip wells were treated with solutions containing the diluted 5 mC capture and the detection antibodies. The methylated fraction of DNA was quantified colorimetrically by absorbance readings using a FLUOstar Optima, BMG. The percentage of methylated DNA was calculated as a proportion of the optical density (OD), according to manufacturers’ instructions using the formula
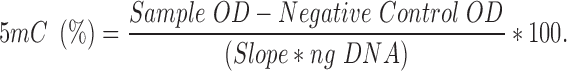 |
The percentage of 5 mC was determined using two replicates in each of the two independent experiments.
Immunocytochemistry quantification of α-synuclein aggregates
Immunofluorescent images of α;-synuclein aggregates were analyzed using Leica Application Suite X Software, Germany. The number and size of aggregates were analyzed for 50 cells per cell line. The baseline for number of aggregates per cells included in the analysis was determined in reference to the number of aggregates observed in the Control cell lines. Size of aggregates was defined in three groups according to Poehler et al. (33): small (<1 μm), medium (1–2 μm) and large (2–5 μm). Frequency distribution plots represent aggregates number and size binned by arbitrary unit increments based on the natural groupings of the data.
Statistical analyses
The differences between the Juvenile and Aged neurons and across the different cell lines were analyzed statistically using the following data comparisons tests (GraphPad Prism7): (i) analysis of corresponding cumulative distributions using the Kolmogorov–Smirnov test; (ii) multiple comparisons of the median using the Kruskal–Wallis test; (iii) two-group comparisons using Student’s t-tests; and (iv) multiple comparisons using the Tukey–Kramer Honestly Significant Difference test.
Supplementary Material
Acknowledgements
We thank Mr Thomas Ribar, the director of the iPSC-core facility at Duke University for his assistance with the hiPSCs work. We thank Dr Yasheng Gao, the microscopy specialist of the Light Microscopy Core Facility at Duke University for technical assistance.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health/National Institute of Neurological Disorders and Stroke (R01 NS085011 to O.C.); National Center for Advancing Translational Sciences of the National Institutes of Health (UL1TR001117).
References
- 1. Qiang L., Fujita R. and Abeliovich A. (2013) Remodeling neurodegeneration: somatic cell reprogramming-based models of adult neurological disorders. Neuron, 78, 957–969. [DOI] [PubMed] [Google Scholar]
- 2. Doege C.A. and Abeliovich A. (2014) Dementia in a dish. Biol. Psychiatry, 75, 558–564. [DOI] [PubMed] [Google Scholar]
- 3. Rohani L., Johnson A.A., Arnold A. and Stolzing A. (2014) The aging signature: a hallmark of induced pluripotent stem cells? Aging Cell, 13, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lapasset L., Milhavet O., Prieur A., Besnard E., Babled A., Ait-Hamou N., Leschik J., Pellestor F., Ramirez J.M., De Vos J. et al. (2011) Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev., 25, 2248–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mertens J., Paquola A.C., Ku M., Hatch E., Bohnke L., Ladjevardi S., McGrath S., Campbell B., Lee H., Herdy J.R. et al. (2015) Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell, 17, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K. and Yamanaka S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 7. Wyss-Coray T. (2016) Ageing, neurodegeneration and brain rejuvenation. Nature, 539, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Niccoli T. and Partridge L. (2012) Ageing as a risk factor for disease. Curr. Biol., 22, R741–R752. [DOI] [PubMed] [Google Scholar]
- 9. Gitler A.D., Dhillon P. and Shorter J. (2017) Neurodegenerative disease: models, mechanisms, and a new hope. Dis. Model. Mech., 10, 499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cornacchia D. and Studer L. (2017) Back and forth in time: directing age in iPSC-derived lineages. Brain Res., 1656, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller J.D., Ganat Y.M., Kishinevsky S., Bowman R.L., Liu B., Tu E.Y., Mandal P.K., Vera E., Shim J.W., Kriks S. et al. (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell, 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Studer L., Vera E. and Cornacchia D. (2015) Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell, 16, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Byers B., Cord B., Nguyen H.N., Schule B., Fenno L., Lee P.C., Deisseroth K., Langston J.W., Pera R.R. and Palmer T.D. (2011) SNCA triplication Parkinson's patient's iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One, 6, e26159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vera E., Bosco N. and Studer L. (2016) Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep., 17, 1184–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goedert M., Jakes R. and Spillantini M.G. (2017) The synucleinopathies: twenty years on. J. Parkinsons Dis., 7, S53–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tagliafierro L. and Chiba-Falek O. (2016) Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics, 17, 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao B., Ng H.H., Takahashi R. and Tan E.K. (2016) Induced pluripotent stem cells in Parkinson's disease: scientific and clinical challenges. J. Neurol. Neurosurg. Psychiatry, 87, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tagliafierro L., Glenn O.C., Zamora M.E., Beach T.G., Woltjer R.L., Lutz M.W. and Chiba-Falek O. (2017) Genetic analysis of alpha-synuclein 3′ untranslated region and its corresponding microRNAs in relation to Parkinson's disease compared to dementia with Lewy bodies. Alzheimers Dement., 13, 1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu G.H., Qu J., Suzuki K., Nivet E., Li M., Montserrat N., Yi F., Xu X., Ruiz S., Zhang W. et al. (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature, 491, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoeijmakers J.H. (2009) DNA damage, aging, and cancer. N. Engl. J. Med., 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- 21. Jones M.J., Goodman S.J. and Kobor M.S. (2015) DNA methylation and healthy human aging. Aging Cell, 14, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R. et al. (2003) Alpha-synuclein locus triplication causes Parkinson's disease. Science, 302, 841. [DOI] [PubMed] [Google Scholar]
- 23. Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A. et al. (2015) Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science, 348, 1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Larson K., Yan S.J., Tsurumi A., Liu J., Zhou J., Gaur K., Guo D., Eickbush T.H. and Li W.X. (2012) Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet., 8, e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robin J.D. and Magdinier F. (2016) Physiological and pathological aging affects chromatin dynamics, structure and function at the nuclear edge. Front. Genet., 7, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burke B. and Stewart C.L. (2006) The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu. Rev. Genomics Hum. Genet., 7, 369–405. [DOI] [PubMed] [Google Scholar]
- 27. Lammerding J., Schulze P.C., Takahashi T., Kozlov S., Sullivan T., Kamm R.D., Stewart C.L. and Lee R.T. (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest., 113, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olive P.L., Banath J.P. and Durand R.E. (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the `comet’ assay. Radiat. Res., 122, 86–94. [PubMed] [Google Scholar]
- 29. Chiba-Falek O. (2017) Structural variants in SNCA gene and the implication to synucleinopathies. Curr. Opin. Genet. Dev., 44, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. David D.C. (2012) Aging and the aggregating proteome. Front. Genet., 3, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones R. (2010) Protein aggregation increases with age. PLoS Biol., 8, e1000449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindner A.B. and Demarez A. (2009) Protein aggregation as a paradigm of aging. Biochim. Biophys. Acta, 1790, 980–996. [DOI] [PubMed] [Google Scholar]
- 33. Poehler A.M., Xiang W., Spitzer P., May V.E., Meixner H., Rockenstein E., Chutna O., Outeiro T.F., Winkler J., Masliah E. et al. (2014) Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy, 10, 2171–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Graf T. and Enver T. (2009) Forcing cells to change lineages. Nature, 462, 587–594. [DOI] [PubMed] [Google Scholar]
- 35. Masserdotti G., Gascon S. and Gotz M. (2016) Direct neuronal reprogramming: learning from and for development. Development, 143, 2494–2510. [DOI] [PubMed] [Google Scholar]
- 36. Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Sudhof T.C. and Wernig M. (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature, 463, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Broccoli V. (2017) Reprogramming of somatic cells: iPS and iN cells. Prog. Brain Res., 230, 53–68. [DOI] [PubMed] [Google Scholar]
- 38. Hu W., Qiu B., Guan W., Wang Q., Wang M., Li W., Gao L., Shen L., Huang Y., Xie G. et al. (2015) Direct conversion of normal and Alzheimer's disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell, 17, 204–212. [DOI] [PubMed] [Google Scholar]
- 39. Li X., Zuo X., Jing J., Ma Y., Wang J., Liu D., Zhu J., Du X., Xiong L., Du Y. et al. (2015) Small-molecule-driven direct reprogramming of mouse fibroblasts into functional neurons. Cell Stem Cell, 17, 195–203. [DOI] [PubMed] [Google Scholar]
- 40. Nussbaum R.L. (2017) The identification of alpha-synuclein as the first Parkinson disease gene. J. Parkinsons Dis., 7, S45–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simon-Sanchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G. et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet., 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bras J., Guerreiro R., Darwent L., Parkkinen L., Ansorge O., Escott-Price V., Hernandez D.G., Nalls M.A., Clark L.N., Honig L.S. et al. (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet., 23, 6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stefanis L. (2012) Alpha-synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med., 2, a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Villar-Pique A., Lopes da Fonseca T. and Outeiro T.F. (2016) Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J. Neurochem., 139 (Suppl. 1), 240–255. [DOI] [PubMed] [Google Scholar]
- 45. Maroteaux L., Campanelli J.T. and Scheller R.H. (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci., 8, 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goncalves S. and Outeiro T.F. (2013) Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol. Neurobiol., 47, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McLean P.J., Ribich S. and Hyman B.T. (2000) Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J. Neural Transm. Suppl., 58, 53–63. [DOI] [PubMed] [Google Scholar]
- 48. Mori F., Tanji K., Yoshimoto M., Takahashi H. and Wakabayashi K. (2002) Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res., 941, 118–126. [DOI] [PubMed] [Google Scholar]
- 49. Iwata A., Miura S., Kanazawa I., Sawada M. and Nukina N. (2001) Alpha-synuclein forms a complex with transcription factor Elk-1. J. Neurochem., 77, 239–252. [DOI] [PubMed] [Google Scholar]
- 50. Goers J., Manning-Bog A.B., McCormack A.L., Millett I.S., Doniach S., Di Monte D.A., Uversky V.N. and Fink A.L. (2003) Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry, 42, 8465–8471. [DOI] [PubMed] [Google Scholar]
- 51. Kontopoulos E., Parvin J.D. and Feany M.B. (2006) Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet., 15, 3012–3023. [DOI] [PubMed] [Google Scholar]
- 52. Sugeno N., Jackel S., Voigt A., Wassouf Z., Schulze-Hentrich J. and Kahle P.J. (2016) Alpha-synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep., 6, 36328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Seidel K., Mahlke J., Siswanto S., Kruger R., Heinsen H., Auburger G., Bouzrou M., Grinberg L.T., Wicht H., Korf H.W. et al. (2015) The brainstem pathologies of Parkinson's disease and dementia with Lewy bodies. Brain Pathol., 25, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu H. and Zhang S.C. (2011) Specification of neuronal and glial subtypes from human pluripotent stem cells. Cell. Mol. Life Sci., 68, 3995–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hu Y., Qu Z.Y., Cao S.Y., Li Q., Ma L., Krencik R., Xu M. and Liu Y. (2016) Directed differentiation of basal forebrain cholinergic neurons from human pluripotent stem cells. J. Neurosci. Methods, 266, 42–49. [DOI] [PubMed] [Google Scholar]
- 56. Crompton L.A., Byrne M.L., Taylor H., Kerrigan T.L., Bru-Mercier G., Badger J.L., Barbuti P.A., Jo J., Tyler S.J., Allen S.J. et al. (2013) Stepwise, non-adherent differentiation of human pluripotent stem cells to generate basal forebrain cholinergic neurons via hedgehog signaling. Stem Cell Res., 11, 1206–1221. [DOI] [PubMed] [Google Scholar]
- 57. Lin L., Goke J., Cukuroglu E., Dranias M.R., VanDongen A.M. and Stanton L.W. (2016) Molecular features underlying neurodegeneration identified through in vitro modeling of genetically diverse Parkinson's disease patients. Cell Rep., 15, 2411–2426. [DOI] [PubMed] [Google Scholar]
- 58. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mittal S., Bjornevik K., Im D.S., Flierl A., Dong X., Locascio J.J., Abo K.M., Long E., Jin M., Xu, B.et al. (2017) β2-adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science, 357, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Olive P.L. and Banath J.P. (2006) The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc., 1, 23–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.