

Abstract
The extracellular signal-related kinase 1 and 2 (ERK1/2) pathway is a highly conserved signaling cascade with numerous essential functions in development. The scaffold protein Shoc2 amplifies the activity of the ERK1/2 pathway and is an essential modulator of a variety of signaling inputs. Germline mutations in Shoc2 are associated with the human developmental disease known as the Noonan-like syndrome with loose anagen hair. Clinical manifestations of this disease include congenital heart defects, developmental delays, distinctive facial abnormalities, reduced growth and cognitive deficits along with hair anomalies. The many molecular details of pathogenesis of the Noonan-like syndrome and related developmental disorders, cumulatively called RASopathies, remain poorly understood. Mouse knockouts for Shoc2 are embryonic lethal, emphasizing the need for additional animal models to study the role of Shoc2 in embryonic development. Here, we characterize a zebrafish shoc2 mutant, and show that Shoc2 is essential for development, and that its loss is detrimental for the development of the neural crest and for hematopoiesis. The zebrafish model of the Noonan-like syndrome described here provides a novel system for the study of structure–function analyses and for genetic screens in a tractable vertebrate system.
Introduction
The evolutionarily conserved extracellular signal-regulated kinase 1 and 2 (ERK1 and ERK2) signaling pathway is implicated in the regulation of a wide range of processes in developing and adult tissues (1,2). To facilitate the accuracy of the distribution, range and quality of the transduced cellular signals, the ERK1/2 pathway is modulated at many levels. Thus, to understand how ERK1/2 signals are controlled, it is necessary to define the specific roles and the contributions to the final response of each architectural component of the pathway. Scaffold proteins are central to the mechanisms that define cellular localization, coordinate positive and negative feedback signals and protect activated signaling molecules (3,4). Yet, contributions of scaffolds to the ERK1/2 pathway hierarchy are not fully understood, and thus require further careful analysis.
The non-enzymatic scaffold protein Shoc2 (sur-8, Soc-2) plays a key role in the activation of the ERK1/2 signaling pathway by epidermal growth factor (EGF) and platelet-derived growth factor receptor tyrosine kinases (5,6). This ubiquitously expressed protein with a large leucine-rich repeat (LRR) domain and an unstructured N-terminus, recruits the signaling proteins M-Ras and Raf-1, and is regulated via an elegant mechanism that couples post-translational modifications and intracellular localization to the complex mechanism of protein module remodeling (7). In the absence of cell stimulation, Shoc2 is distributed throughout the cytosol. Upon receptor activation and transmission of ERK1/2 signals, the E3 ligase HUWE1 ubiquitinates Shoc2 thereby prompting an inhibitory feedback loop that controls Shoc2’s ability to accelerate ERK1/2 signals. We further demonstrated that in response to ERK1/2 pathway activation, Shoc2 complexes undergo remodeling on the surface of endocytic vesicles to where complexes are recruited via their interaction with the small (AAA+) ATPase PSMC5 (8–10).
Improper localization of Shoc2 complexes or deficiencies in their assembly leads to a Noonan-like syndrome with loose anagen hair (NSLH; OMIM: 607721). NSLH is an autosomal dominant condition that belongs to the group of developmental diseases cumulatively called RASopathies (11). Patients carrying a missense mutation in Shoc2 are diagnosed by displaying cardiac (most often valvuloseptal) abnormalities, proportional short stature, facial dysmorphia (e.g. ocular hypertelorism) and a variety of less penetrant defects (e.g. cognitive, genitourinary, auditory abnormalities and unique loss of anagen hair) (12–16). Importantly, recently published studies indicate that Shoc2 RASopathy is much broader phenotypically and often presents with symptoms more severe than Noonan patients (12,17–19). The precise molecular pathology underlying Shoc2 mutations is an area of active investigation. While previous studies suggested that the disease-causing S2G allele results in a gain-of-function, we have found in cell culture assays that the mutant protein behaves as a loss-of-function (13,20). Thus, more work is needed to determine the genotype–phenotype relationships in NSLH. Outstanding questions also include how mutations in Shoc2 contribute to a syndrome with a wide-spectrum phenotype, and what are the biological activities regulated by Shoc2 transmitted ERK1/2 signals. As a knockout of Shoc2 in the mouse results in early embryonic lethality, additional animal models are needed to dissect the role of this important scaffolding protein during embryogenesis.
To investigate the role of Shoc2 in tissue development and morphogenesis, we have exploited the powerful zebrafish model (Danio rerio). Molecular pathways of zebrafish are highly conserved and the external development and transparency of zebrafish embryos enables monitoring and high-resolution imaging of very early developmental events. Previous studies using zebrafish have played a key role in defining the core Ras-ERK1/2 pathway and have provided fundamental insights into the understanding of developmental abnormalities of other RASopathies (21).
In this study, we show that both morpholino (MO)-mediated knockdown and CRISPR/Cas9 mutagenesis of Shoc2 in zebrafish result in multiple deficiencies during embryogenesis, including gross defects in blood cell differentiation and abnormal craniofacial development. Our observations that shoc2 morphants and Shoc2 CRISPR/Cas9 mutants (crispants) have systemic defects in neural crest specification and hematopoiesis underscore the central role of Shoc2 in embryogenesis. Our finding that the loss of Shoc2 induces an array of developmental defects emphasizes its essential role in coordinating activities of the linear components of the ERK1/2 pathway. Moreover, we have developed and characterized a heritable model of Shoc2 that exhibits many of the clinical hallmarks associated with the RASopathies, which can now be used to decipher the molecular mechanisms underlying these diseases.
Results
Shoc2 in zebrafish
Bioinformatics searches identified one Shoc2 zebrafish orthologue that is highly homologous to human Shoc2 at the amino acid level, sharing 88% identity (10). The zebrafish shoc2 gene contains nine exons, is located to chromosome 22 and predicts a 62.7 kDa protein of 561 amino acids. Comparison to the human Shoc2 protein sequences reveals significant conservation in the fold of the LRR domains, suggesting that functional motifs have been conserved across evolution (Fig. 1A). To determine the temporal expression pattern of shoc2 we performed reverse transcription polymerase chain reaction (RT-PCR) using RNA templates obtained from embryos at 3, 6, 12, 24, 48 and 72 h post fertilization (hpf, Fig. 1B). RT-PCR confirmed that shoc2 is maternally inherited and is expressed through all the analyzed time points. Shoc2 protein expression at early stages was confirmed using specific antibodies (Fig. 1C). We also examined the expression of shoc2 by whole-mount in situ hybridization between the stages of 6 hpf and 2 days post fertilization (dpf) and found that shoc2 is expressed ubiquitously during early development. Notably at 24 hpf, shoc2 was expressed in the dorsal aorta (DA) and was detectable in the somite boundaries (Supplementary Material, Fig. S1).
Figure 1.
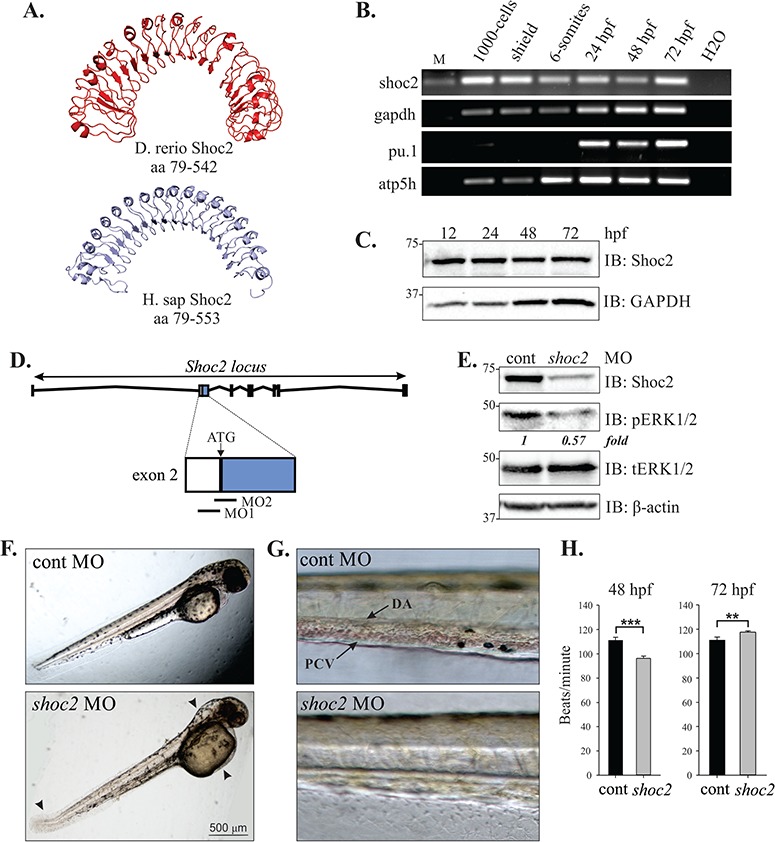
Knockdown of Shoc2 disrupts morphogenesis and causes severe anemia. (A) Homology models of zebrafish and human Shoc2, residues 79-561 and 79-582, respectively, were constructed using the published structure of Arabidopsis Flg22-FLS2-BAK1 immune complex (PDB id: 4MN8) and Leptospira interrogans LRR protein LIC11098 (PDB id: 4U08) as templates for zebrafish and human Shoc2 models correspondingly. The modeling was done using the I-TASSER server and figures were prepared using PyMol software. For modeling detail see (52,53). (B) shoc2 mRNA was detected using reverse transcription polymerase chain reaction (RT-PCR) at the indicated times. pu.1, atp5h and gapdh were used as reference genes. pu.1 was previously shown to express after 16 hpf (54). (C) Western blot analysis of zebrafish embryos. Embryos were harvested for immunoblotting at indicated time points. The expression of Shoc2 and GAPDH was analyzed using specific antibodies. GAPDH was used as a loading control. (D) Schematic representation of Shoc2 loci and MO-targeting sites. (E) Embryos injected with shoc2 and control MO were harvested for immunoblotting at 72 hpf. The expression of indicated proteins was analyzed using specific antibodies. (F) Representative body images of control and shoc2 morphants at 48 hpf (taken from the set of embryos analyzed in E, G and H). One-cell stage embryos were injected with the standard control MO and shoc2 ATG MO1. The optimal dose for microinjection of MO was determined as that which resulted in specific defects but did not cause gross lethality or global defects. As shown, at 48 hpf, 70.4% (57 out of 81) of shoc2 morphants displayed an enlarged hindbrain (arrowhead), enlarged yolk, a posterior kink in the tail (arrow). (G) Representative body images of control and shoc2 morphants at 72 hpf. shoc2 MO-injected embryos showed reduced numbers of cells in circulation when compared to control embryos. (H) The cardiac heart rate of control and shoc2 morphants was measured at 48 and 72 hpf. Significant difference in heart rate was observed in shoc2 morphants when compared to WT. Error bars represent means with SEM. ***P < 0.001 and **P < 0.05 (Student’s t-test). MO: morpholino, hpf: hours post fertilization, M: DNA marker, PVC: posterior cardinal vein, DA: dorsal aorta.
MO knockdown of shoc2
To explore the function of Shoc2 during embryogenesis, we first used two different translation-blocking morpholino antisense (MO) oligonucleotides to inhibit expression of shoc2 at early stages of development (Fig. 1D). The effectiveness of gene knockdown by these translation-blocking MOs was confirmed by western blot analysis, and showed that the shoc2 MO was highly efficient and its translation-blocking activity lasted through 3 dpf (Fig. 1E). Western blots of 3 dpf whole-embryo extracts derived from shoc2 morphants also revealed a clear reduction of phospho-ERK1/2 in knockdown embryonic tissue, confirming that depletion of Shoc2 leads to decreased levels of phosphorylated ERK1/2 (pERK1/2) in zebrafish (Fig. 1E). Both MOs produced similar phenotypes when injected into one-cell stage embryos (data not shown), and MO1 was used for all subsequent analyses. A standard control MO was used to control for non-specific phenotypes resulting from the microinjection procedure. Survival of MO-injected and uninjected embryos between 8 and 24 hpf was not significantly different across shoc2 morphants (76.7 ± 10.7%), control morphants (84.6 ± 17.5%) and uninjected embryos (81.9 ± 21.3%).
Control and shoc2 MO-injected embryos were categorized based upon their morphology and developmental stage at 24 and 48 hpf using standard staging criteria (22). At 24 and 48 hpf, morphological markers (including the presence or absence of a beating heart) were scored to determine the developmental stage. Nearly all embryos injected with the standard control MO were at the correct developmental stage at 24 hpf (98%) and showed no overt morphological changes. Among the shoc2 morphants without developmental delay (90%), the body shape, head shape and presence/absence of pericardial effusion were scored at 48 hpf, and the embryos were categorized as mild, moderate or severe. Shoc2 morphants in the `mild’ category displayed no developmental delay or overt morphological changes when compared to control morphants. This category represented a very small number of shoc2 morphants (2%). shoc2 morphants in the `moderate’ category were also at the correct developmental stage, but displayed mild body torqueing, with spinal curvature. Moderate category shoc2 morphants also displayed mild brain malformation, large yolks and kinked tails. The majority of shoc2 morphants were categorized as `severe’ and displayed a body torque, with malformations of the tail and brain, and showed reduced circulation of blood cells within the DA and posterior veins. We did not observe profound alterations in shoc2 morphant heart structure; however, severe morphants did display a slightly faster, statistically significant, heartbeat rate at 72 hpf (Fig. 1F, G and H). All embryos in this category displayed enlarged yolks. This phenotype was observed in 94% of shoc2 MO1-injected embryos (n > 200, from four independent experiments). No pooling of red blood cells was found at any site away from circulation in the shoc2 morphants.
Since hematopoietic stem cells and angioblasts originate from shared pluripotent precursors, blood and blood vessel formation are closely related in early developmental stages (23). Thus, we examined whether vascular angiogenesis was affected by Shoc2 knockdown. Shoc2 MOs were injected into zebrafish embryos in which endothelial cells were marked by the expression of green fluorescent protein (fli1a:EGFP) allowing for a more detailed analysis of vascular development (24). At 22–48 hpf, the major vessels, such as DA, caudal artery, posterior cardinal vein and caudal vein appeared normal indicating normal vasculogenesis (not shown). However, at 72 hpf defective vasculature branching was visible with the most significant changes in the subintestinal vein (SIV; Supplementary Material, Fig. S2). The SIV of 90% (n = 61, from three independent experiments) of shoc2 MO-injected embryos exhibited reduced size/number of vessel branches and/or ectopic blood vessels when compared to the basal defect level of control embryos. Vascular defects did not appear to correlate directly with cardiac defects, as we observed embryos with vascular abnormalities that did not display major pericardial effusion or valvular insufficiency as assessed by a to-and-fro movement of blood within the heart.
To validate hematopoietic defects further, we utilized o-dianisidine staining to detect hemoglobin of erythropoietic cells (25,26). In control embryos, hemoglobin-positive cells were found soon after circulation started at 26 hpf, whereas shoc2 MO-injected embryos completely lacked o-dianisidine staining (Fig. 2A). At 48–72 hpf erythrocytes in control embryos were robustly stained with o-dianisidine and were prominent in the ducts of Cuvier, over the yolk sac, while in shoc2 MO-injected embryos we observed a marked decrease in the intensity of o-dianisidine staining. We then tested the specificity of MO-induced phenotypes by an mRNA rescue assay. When wild-type (WT) human shoc2 mRNA was co-injected with shoc2 MO, a rescue of shoc2 MO-induced defects in erythrocyte circulation defects was observed (Fig. 2B and C). Since the blood deficiencies were detected at the very early stages of hematopoiesis, we tested whether other blood lineages were affected in shoc2 morphants. We performed Sudan Black B staining, which detects lipids in neutrophils and monocytes (27), to examine whether shoc2 depletion affects myeloid lineage. We detected a significant reduction in the number of neutrophils in caudal hematopoietic tissue (CHT) of shoc2 morphants (93% or 105/113 injected) in comparison with control-MO-injected embryos at 72 hpf (Fig. 2D) indicating that the depletion of shoc2 causes defects in myelopoiesis. The cells positive for Sudan Black B staining were scored in five somites at the CHT region after the yolk extension (Fig. 2E). Together, these data suggest that Shoc2 is involved in general blood cell differentiation and possibly plays a role in both the primitive and definitive waves of hematopoiesis (28).
Figure 2.
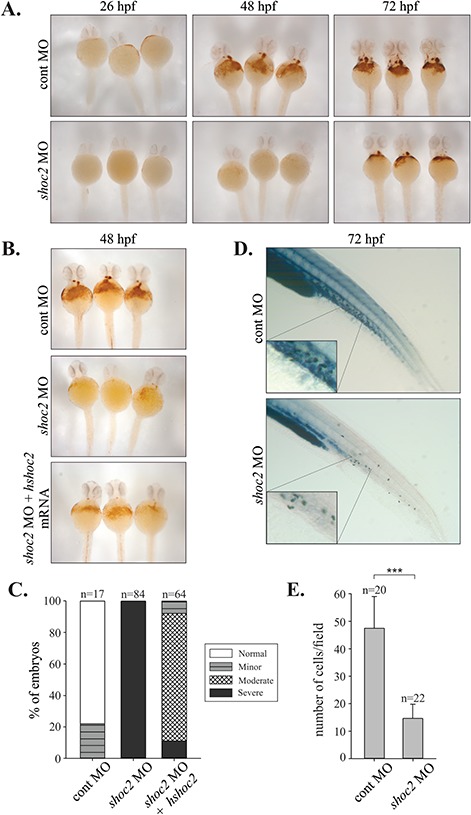
Number of erythropoietic and myelopoietic cells are affected in shoc2 MO-injected embryos. (A) o-Dianisidine staining of hemoglobin in control and shoc2 MO injected embryos at the indicated times (26, 48 and 72 hpf). In control embryos, hemoglobin-positive cells were found on the yolk sac soon after circulation starting at 26 hpf, shoc2 morphants lacked staining completely. At 48 hpf the number of o-dianisidine-positive cells was severely reduced in shoc2 morphants in comparison to control. (B) Injection of human shoc2 mRNA in shoc2 morphants. The data show rescue of hemoglobin phenotypes at 48 hpf. (C) Analysis of hemoglobin staining presented in (B). o-Dianisidine-positive area in the region of yolk and tail was assessed based on hemoglobin staining intensity. Number of embryos analyzed: 48 hpf cont-MO injected embryos, n = 17; 48 hpf shoc2 MO, 5.24 ng/embryo, n = 84; 48 hpf shoc2 MO, 5.24 ng/embryo plus 100 pg/embryo human shoc2 mRNA, n = 64. (D) Sudan Black B staining of myelopoietic cells (neutrophils) in control- and shoc2-MO-injected embryos at 72 hpf. Significant reduction in circulating neutrophils was found in shoc2 morphants (93% or 105/113 injected). Insets show neutrophils in clumps in the CHT. Three biological replicates were performed for all experiments. (E) Analysis of Sudan Black B staining presented in (D). Sudan Black B-positive cells were counted within the five somites of CHT region. Error bars represent means with SEM. ***P < 0.001 (Student’s t-test). hpf: hours post fertilization, MO: morpholino, CHT: caudal hematopoietic tissue.
Generation of zebrafish shoc2-heritable mutants
Although MO injections readily permit transient knockdown of gene expression, their efficacy is limited to the first few dpf. To gain a better understanding of the roles of Shoc2-mediated signals during development, as well as allow for a quantitative assessment of the phenotype, we employed the CRISPR/Cas9 genome-editing approach to develop stable lines of zebrafish harboring germline mutations in shoc2 (29). Using this approach, we engineered sgRNA-directing gene editing to coding exons 2 and 3 of shoc2. Neither of the sites targeted by the sgRNAs overlapped with the MO target sites (Supplementary Material, Fig. S3A). Multiple independent target-specific mutant alleles for shoc2 were identified in the F1 generation (Supplementary Material, Fig. S3B). Mutations in the target region were confirmed by both high-resolution melting analysis (HRMA) and sequencing analysis (Supplementary Material, Fig. S4A and B). Each of the introduced mutations included a deletion and/or insertion within these coding exons, introducing a frameshift and premature stop codon that would be expected to truncate the Shoc2 protein. Zebrafish mutant lines harboring null mutations in exon 2 (shoc2Δ22) and 3 (shoc2Δ14) were used in the following experiments. Both mutants carry frameshift mutations (c.357-378, del and c.671-684, del) that truncate the protein by 121 and 229 amino acids, respectively, predicting the loss of a large portion of Shoc2’s LRR region (Supplementary Material, Fig. S3B). When shoc2 mRNA was assessed in homozygous shoc2Δ22 and shoc2Δ14 mutants, a corresponding reduction in the size of the amplified fragments was detected (Supplementary Material, Fig. S3C and D). Protein expression analyses using an antibody to Shoc2 confirmed the loss of full-length Shoc2 in shoc2Δ22 and shoc2Δ14 alleles (Fig. 3C), while heterozygous Shoc2 alleles did not show decreases in Shoc2 protein (Supplementary Material, Fig. S3E). Importantly, we also observed decreased levels of pERK1/2 in shoc2Δ22 and shoc2Δ14 mutant larvae (Fig. 3C). To avoid potential off-target effects, shoc2Δ22/+ and shoc2Δ14/+ mutants were outcrossed to WT for three generations prior to the further study.
Figure 3.
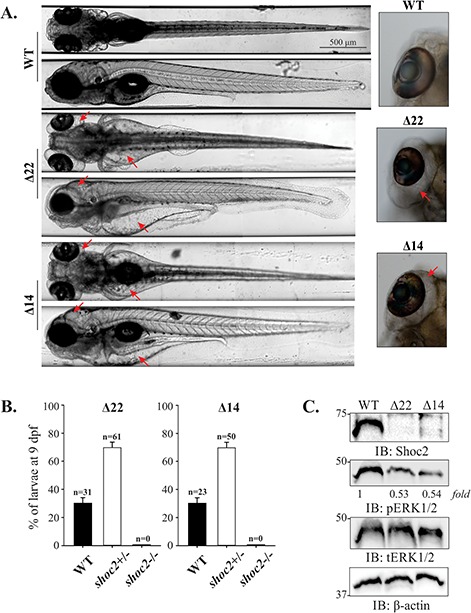
Late pleiotropic phenotype of shoc2Δ22 and shoc2Δ14 crispants. (A) Severe edema between the trunk and the yolk sac and yolk extension, heart and kidney and underinflated yolk of 6 dpf shoc2Δ22 and shoc2Δ14 crispants are indicated with red arrows. Insets show severe eye edema of shoc2Δ22 and shoc2Δ14 crispants. Larvae are shown in dorsal or lateral view. Larvae were positioned and imaged live with the vertebrate automated screening technology platform (Union Biometrica, Holliston, MA) (55). Larvae were anaesthetized with 0.2 mg/mL tricaine prior to being loaded into the sample reservoir. Dorsal and lateral images were acquired at a > 70% minimum similarity from the pattern recognition algorithm. High-resolution frontal and posterior images were stitched together using Adobe Photoshop CS6. For each larva shown, 3.5-fold magnified views of the eye region are displayed. (B) PCR analysis of genomic DNA of shoc2Δ22 and shoc2Δ14 inbred larvae at 9 dpf. Progeny of three independent inbreeding experiments using heterozygous shoc2Δ22 and shoc2Δ14 fish were genotyped. All survival larvae were genotyped as either WT or heterozygous carriers of the shoc2Δ22- and shoc2Δ14-crispant allele. Three biological replicates were performed for both alleles. (C) Immunoblot analysis of WT, shoc2Δ22 and shoc2Δ14 larvae. Larvae were harvested for immunoblotting at 6 dpf. Protein expression was analyzed using specific antibodies.β-actin was used as a loading control.
Zebrafish heterozygous for either shoc2 mutant alleles were viable, fertile and displayed no overt phenotypes. When heterozygous adults were in-crossed, no severe morphological defects were detected in the progeny prior to 5 dpf. However, at later stages of development, pleiotropic effects on larval morphology that became progressively more severe were observed in the shoc2−/− larvae. At 6 dpf, 90–100% of shoc2Δ22 and shoc2Δ14 mutant larvae developed edema of the heart cavity, and along the trunk, yolk sac and yolk extension, and around the eyes, and became lethargic (Fig. 3A). The swim bladders of shoc2Δ22 and shoc2Δ14 larvae were underinflated and we observed a small but significant increase of 1.25-fold in the heart beat of the severely edemic 6 dpf larvae when compared to WTs, the likely result of the increased edema at this stage (Supplementary Material, S1 Movie, A and B). Further quantitative survival studies indicated that at 6 dpf, shoc2Δ22 and shoc2Δ14 larvae began to die, with none surviving beyond 9 dpf. All of WT and heterozygous larvae survived to 10 dpf and displayed the expected Mendelian ratio for homozygous WT versus heterozygous genotypes (1:2) (Fig. 3B). Similar severe edema was observed for the compound mutant of shoc2Δ22 and shoc2Δ14 (Fig. S5). Because the observed defects manifested themselves at 5–6 dpf, we hypothesize that the presence of maternally inherited shoc2 allow shoc2 crispants to develop somewhat normally during the first few dpf. Taken together, these data demonstrate that loss of Shoc2 is detrimental to the embryonic development of zebrafish.
Effects of Shoc2 mutants on neural crest development
Closer examination of shoc2Δ22 and shoc2Δ14 nulls indicated possible changes in craniofacial morphology. We found that at 6 dpf, shoc2Δ22 and shoc2Δ14 mutants were shorter in length than WT larvae, displayed smaller eye surface area and a narrow spinal cord and had markedly abnormal craniofacial skeletons (Fig. 4A, B and D). Morphological analyses of Alcian blue-stained samples revealed that shoc2Δ22 mutants exhibited a prominent defect in Meckel’s cartilage, which in shoc2Δ22 mutants curved downwards and did not extend as far anteriorly as that of WT larvae. Additionally, we found that the ceratohyal cartilages of shoc2 mutants were smaller and shifted farther posteriorly or were missing entirely. To quantitatively evaluate the abnormality of the cartilage tissues, we analyzed the positional relationships between the Meckel’s cartilage and the two ceratohyal cartilages, the length of the ceratohyal arch, the angle between ceratohyal arches and the total length of craniofacial cartilage structures (Fig. 4C). Significant changes were found in all of these parameters in shoc2 mutants, indicating that Shoc2-mediated signaling is required for proper signal transmission during the development of the craniofacial structures.
Figure 4.
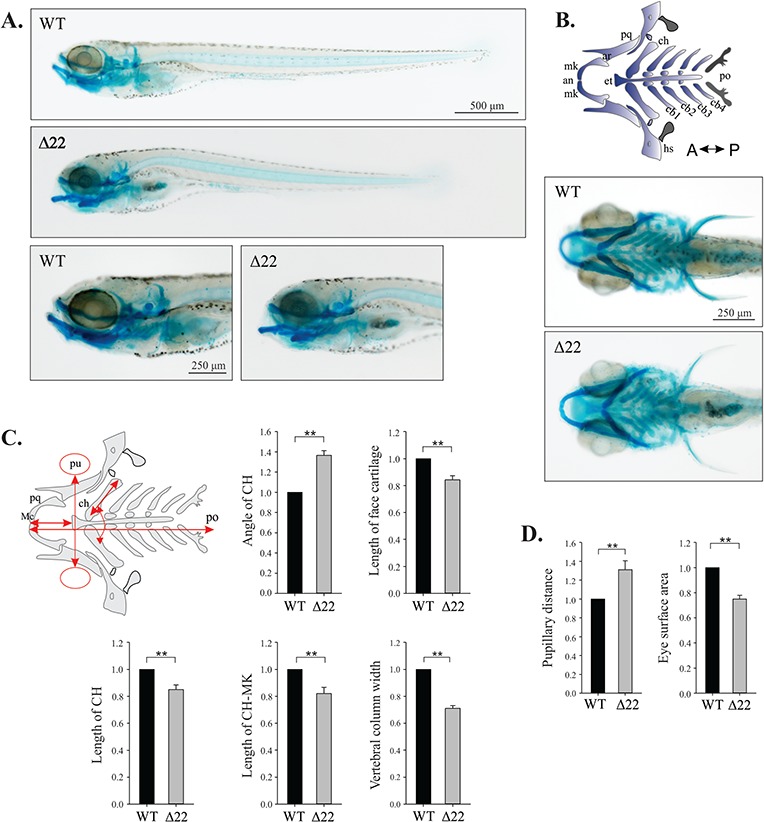
shoc2 loss leads to defects in craniofacial cartilage specification and differentiation. (A) Lateral and ventral views of a 6 dpf WT and shoc2Δ22 crispant larvae detected by Alcian blue staining. Mutant larvae show significant changes in head cartilage. (B) Schematic representation of the different head cartilage elements in ventral view of 6 dpf. Anterior limit (an), articulation (ar), ceratobranchial pairs 1 to 4 (cb1–4), ceratohyal (ch), ethmoid plate (et), hyosymplectic (hs), Meckel’s cartilage (mk), palatoquadrate (pq), posterior limit (po). (C) Schematic representation of parameters quantified; length of ceratohyal (CH) and CH to Meckel’s cartilage (mk), angle of CH, pupillary distance (pu), eye surface area, width of vertebral column and length of total face cartilage (red arrows) were measured for morphometry. Y-axes on graphs indicate the fold change of shoc2Δ22 crispants compared to WT. (D) Measured distance between the eyes and the eye surface area of 6 dpf WT and shoc2Δ22 crispant larvae. Three biological replicates were performed for all experiments (n = 20/group). (**P < 0.05, Student’s t- test). Error bars represent means with SEM. A: anterior, P: posterior.
Intramembranous (dermal) and cartilage bones appear during early larval development. By 6 dpf, three ossified teeth and a number of pharyngeal arches and other ossified bone structures can be visualized by Alizarin Red S staining, which detects calcific deposition by cells of an osteogenic lineage (Fig. 5A) (30). Thus, we next tested whether bones form normally in homozygous shoc2Δ22 and shoc2Δ14 mutant larvae. Alizarin Red S staining demonstrated a strong reduction in calcification in craniofacial bones. We found that parasphenoid, notochord and branchiostegal rays 1 and 2 were completely missing and opercle, ceratobranchial and cleithrum bones were significantly deformed/delayed in shoc2Δ22 and shoc2Δ14 null mutants (Fig. 5A and B; Supplementary Material, Fig. S6A). Together, these data suggest that Shoc2 also contributes to the formation or remodeling of bone.
Figure 5.
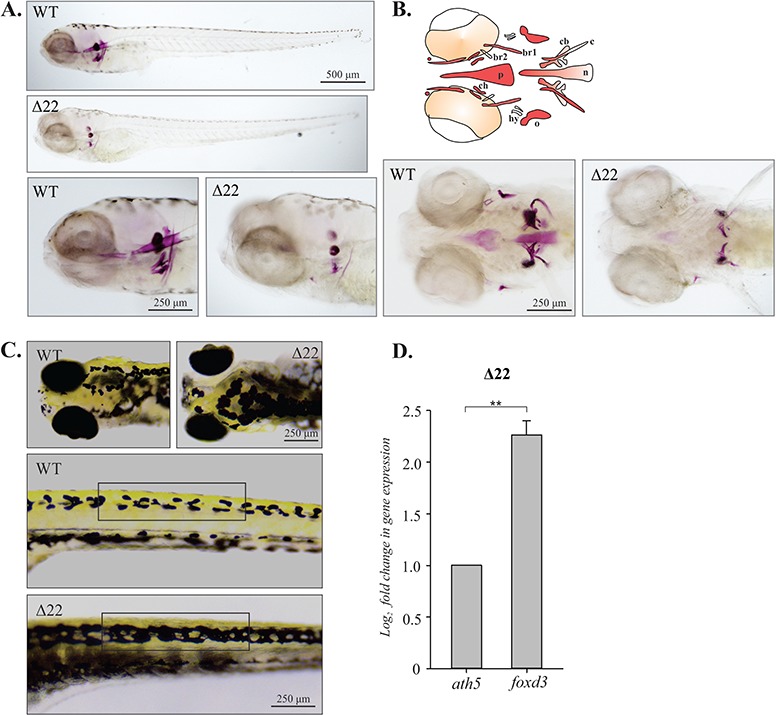
Bone development is impaired in shoc2Δ22-crispant larvae. (A) Lateral view of a 6 dpf WT and shoc2Δ22-crispant larvae detected by Alizarin Red S staining. Mutant larvae show significant differences in cranial bone formation. (B) Schematic representation of the different cranial bone elements detectable at 6 dpf: branchiostegal ray 1 and ray 2 (br1 and br2), notochord (n), opercle (o), parasphenoid (p) and ceratobranchial 5 (cb). Ventral view of 6 dpf WT and shoc2Δ22 larvae stained with Alizarin Red S. Detected changes include defects in ossification of parasphenoid, ceratohyal, branchiostegal rays, opercle, ceratohyal and notochord. Three biological replicates were performed for all experiments (n = 30/group). (C) Dorsal (head) and lateral (trunk) views of 6 dpf larvae showing melanophores in WT and shoc2Δ22 crispants. Unlike WT larvae, shoc2Δ22 crispants presented with closed gaps in pigmentation pattern of head and lateral stipe (see brackets) melanophores. (D) Total RNA from 6 dpf WT and mutant larvae and levels of foxd3 mRNA expression quantified by qPCR. The data are presented as the fold change of the mRNA levels in WT larvae versus the mRNA levels in mutant larvae. ath5 is a control mRNA. The results represent an average of three biological replicas. Error bars indicate means with SEM. **P < 0.05 (Student’s t-test).
Interestingly, mutations in shoc2 also affected the pigmentation pattern of shoc2 null larvae. At 6 dpf, WT larvae displayed a distinct uniform pattern of head and the lateral stripe melanophores (Fig. 5C, brackets). In contrast, shoc2Δ22 and shoc2Δ14 null larvae lost the regularity of melanophore patterning and presented with overlapping lentigines. Pigment deficiencies of shoc2Δ14 mutants were comparable to shoc2Δ22 allele mutants (Supplementary Material, Fig. S6B). The abnormal appearance of the melanophores in mutant larvae can be attributed to defects in patterning of regeneration and metamorphic melanophores, suggesting abnormal migration or differentiation of this lineage. Alternatively, it may be a result of a defective visual background adaption response due to the impairments in eye development. Collectively, these data suggest a defect in the development of the neural crest, which was corroborated by the findings that expression of the neural crest pluripotency marker foxd3 was increased in shoc2 null larvae (Fig. 5D). Additionally, we detected high shoc2 expression in the sox10:RFP positive cells isolated from the Tg(sox10:RFP) transgenic fish (Supplementary Material, Fig. S9A), indicating that Shoc2 may regulate neural crest specification in a cell-autonomous manner. Given the only known function of Shoc2 is to accelerate ERK1/2 signals, we conclude that altered expression of foxd3 and neural crest cell development are the consequence of impaired Shoc2 function and as a consequence-decreased ERK1/2 activity.
Effects of Shoc2 mutants on hematopoiesis
Considering the blood phenotypes induced by the Shoc2 MO injections, we set to examine circulating blood in shoc2Δ22 and shoc2Δ14 null larvae. Consistent with our observations in the shoc2 morphants, o-dianisidine staining revealed a significant decrease in erythropoietic cells in shoc2Δ22 compared to WT larvae (Fig. 6A). Relative concentrations of hemoglobin were evaluated by the intensity of red color and scored in a.u.i. from 0 to 5, 0 being the weakest and 5 the strongest. The erythrocytes were scored in the ventral side of the head of the WT and shoc2Δ22 null larvae (Fig. 6B). To determine whether numbers of macrophages and neutrophils were affected, histochemical staining for myeloperoxidase (mpx, an enzyme characteristic of neutrophil primary granules) was performed (Fig. 6C). The mpx-positive cells located in five somites after the yolk extension were scored. Mpx-enzymatic activity assays detected a significant reduction in numbers of neutrophils in shoc2Δ22 and shoc2Δ14 null larvae when compared to the WT larvae (Fig. 6D; Supplementary Material, Fig. S6C). The results of staining for mpx and hemoglobin indicate that loss of Shoc2 abrogates differentiation of cells of different blood lineages. The observed changes were not likely due to the excessive apoptosis, since acridine orange staining (which visualizes apoptotic cells) showed no significant differences in shoc2Δ22 null larvae when compared to WT (data not shown).
Figure 6.
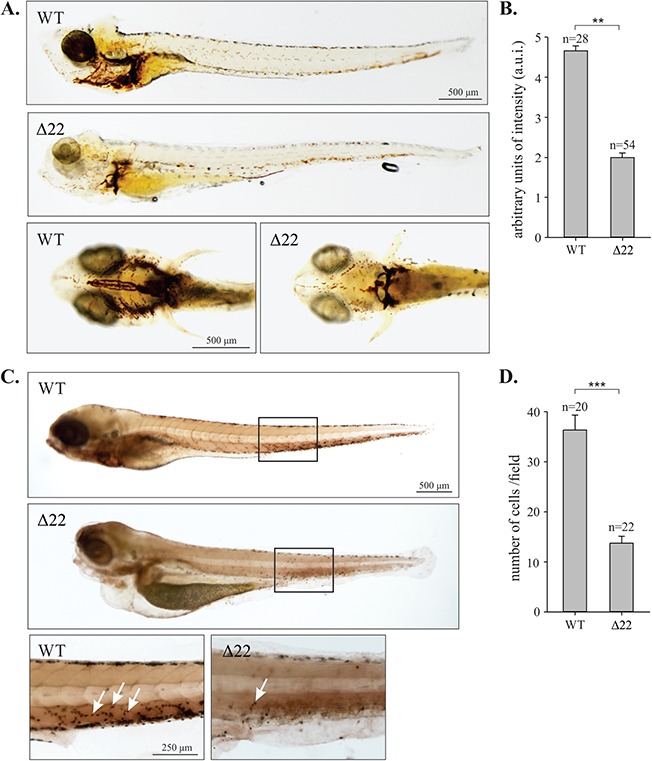
Impaired hematopoiesis in shoc2Δ22-crispant larvae. (A) o-Dianisidine staining detecting hemoglobin of erythropoietic cells in control and shoc2Δ22 larvae at 6 dpf. In control, larvae hemoglobin-positive cells were found on the yolk sac and tail while shoc2Δ22 crispants showed significantly decreased number of cells. Images are shown in lateral and ventral view. (B) Quantitative analysis of the number of erythropoietic cells in circulation. Relative intensity of hemoglobin staining was scored in arbitrary units of intensity (a.u.i.) 0 to 5, 0 being the weakest and 5 the strongest. **P < 0.05 (Student’s t-test). (C) Histochemical staining for mpx enzyme activity in WT and shoc2Δ22 at 6 dpf. Larvae are shown in lateral view with the anterior to the left. The images are representative of ≥ 20 larvae in each group. The staining was repeated with similar results for three independent experiments. Arrows point to mpx-positive cells. (D) The number of mpx-positive cells within the indicated box (five somites). Error bars represent means with SEM. ***P < 0.001 (Student’s t-test).
We next explored possible mechanisms for how Shoc2 regulates the differentiation of specific blood lineages. The transcription factors gata-1a, gata-2a and pu.1 were reported to be early key factors in the process of primitive erythropoiesis and myelopoiesis, respectively. We performed quantitative transcription analysis using RNA isolated from WT, shoc2Δ22 and shoc2Δ14 larvae. Expression levels of gata-1a, gata-2a, mpx and pu.1 were significantly lower in mutant larvae compared to WT larvae. Moreover, expression levels of other markers of blood lineages such as l-plastin (leukocytes) (31), rag-1 (B- and T-cells) (32), lck (T-cells) (33) were down-regulated approximately 2-fold in shoc2 mutants (Fig. 7A). Similar changes in expression of the relevant transcription factors were detected for shoc2Δ14 mutants (Supplementary Material, Fig. S7). The impaired blood development in shoc2-null larvae could not be due to excessive apoptosis. Therefore, our gene expression data indicate that loss of shoc2 results in dysregulation of hematopoietic gene expression programs. Moreover, we found that shoc2 is highly expressed in gata1a:dsRed cells isolated from Tg(gata1a:dsRed) transgenic embryos (Supplementary Material, Fig. S9B), showing that shoc2 is present in erythroid progenitor cells and therefore may regulate definitive hematopoiesis cell autonomously.
Figure 7.
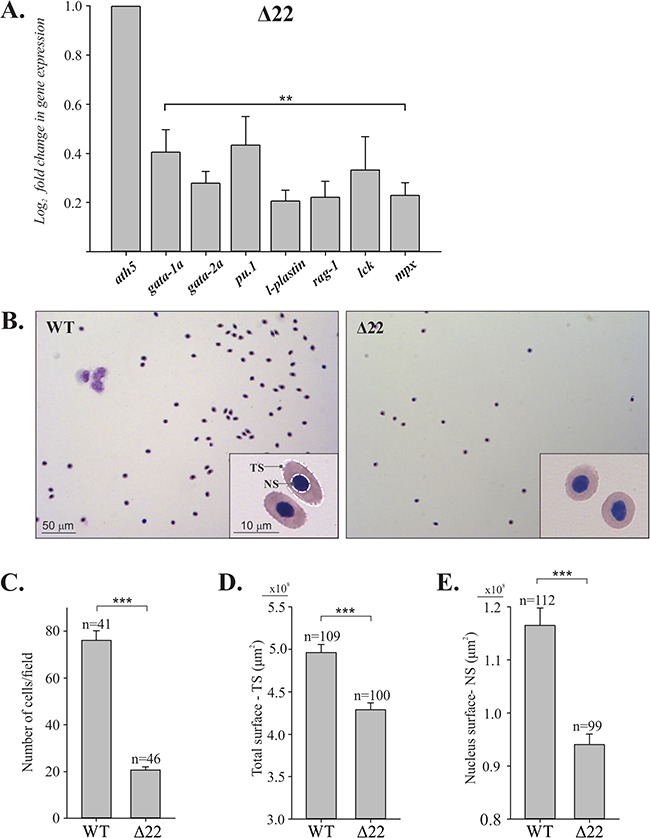
Loss of shoc2 causes a reduction in the number of circulating blood cells. (A) qPCR analysis of blood lineage markers. The total RNA was extracted from 6 dpf WT and mutant larvae and levels of mRNA expression were quantified by qPCR. The data are presented as the fold change of the mRNA levels in WT larvae versus the mRNA levels in mutant larvae. ath5 is a control mRNA. The results represent an average of three biological replicas. Error bars indicate means with SEM. **P < 0.05 (Student’s t-test). (B) Cytology of blood cells from WT and shoc2Δ22 larvae at 6 dpf. Peripheral blood cells isolated from six WT or six shoc2Δ22 larvae were cytospun onto a coated slide and stained by the May-Grünwald–Giemsa method. The shoc2Δ22 larvae had significantly lower numbers of blood cells than WT larvae. Erythrocytes from 6 dpf WT larvae show the typical elliptical morphology, while the shoc2Δ22 cells were smaller and more circular in shape. Images were taken with a Leica DFC7000 T camera with a 100× oil objective and 10× eyepiece. (C) Total number of blood cells from samples in (B). The results represent an average of three biological replicas. n represents a number of fields used for scoring. (D) Total surface area and (E) surface area of nuclei of erythrocytes from WT and shoc2 knockout cells were measured and graphed. All calculations were performed using the morphometry mask statistics modules of the Slidebook 6.0 software. The results represent an average of three biological replicas. Error bars represent means with SEM. ***P < 0.001 (Student’s t-test).
Finally, we evaluated the morphology of circulating blood cells in detail in WT and shoc2 mutants. Individual WT and mutant 6 dpf larvae were bled, and peripheral blood cells were deposited on slides by centrifugation and then stained using the May-Grünwald–Giemsa method. The classification of normal teleost blood cells in various developmental stages is well established (34). Based on their distinct cell morphologies, we detected various subtypes of blood cells in both WT and mutant blood samples (Supplementary Material, Fig. S8). As expected, the majority of circulating cells were erythroid (Fig. 7B). The erythroid cells in zebrafish, similarly to the other teleosts, are elliptical and possess compact nuclei and cytoplasm filled with hemoglobin with typical size of 7–10 μm (35). Mutations in shoc2 caused severe anemia, as significantly fewer erythrocytes were detected in the circulation (Fig. 7B and C). In addition, erythrocytes of shoc2Δ22 6 dpf larvae were spherical, had a considerably lower cell surface area as well as significantly smaller nuclei (Fig. 7D and E). We conclude that based on general changes in blood cell numbers, loss of multiple blood lineages and erythrocyte surface area, the late pleiotropic phenotype of shoc2Δ22 larvae is associated with loss of early ERK1/2 signals in hematopoiesis.
Discussion
In this work, we investigated the role the signaling scaffold protein Shoc2 plays in transmitting ERK1/2 signals in development. Shoc2 accelerates the activity of the Ras/ERK1/2 cascade in response to multiple stimuli (7). However, the contributions of the Shoc2 scaffolding module to biological activities controlled via the ERK1/2 kinases are not understood. We found that the highly conserved Shoc2 orthologue is expressed ubiquitously and retains its function in supporting the ERK1/2 activity in zebrafish (Fig. 1). The Shoc2-dependent function is essential for the morphogenesis of several zebrafish tissues and the absence of Shoc2 results in a number of developmental abnormalities (Figs 2 and 3). We found that shoc2 morphants and shoc2 crispants have marked defects in several neural crest lineages, most notably facial cartilage, bone and pigment cells (Figs 4 and 5). The loss of Shoc2 also leads to a profound loss of circulating blood cells, possibly due to defects in both primitive and definitive waves of hematopoiesis, as well as underdeveloped vascularization (Figs 6 and 7). Comparable to mice knockouts, our findings demonstrate that a systemic deficit of Shoc2 in zebrafish critically impairs a number of developmental functions and is lethal (36). Interestingly, unlike shoc2 knockout mice, our shoc2 CRISPR/Cas9 zebrafish knockout did not present with apparent changes in cardiac function or pericardial effusions, although further studies are needed to analyze possible subtle heart malformations. Yi and colleagues observed that conventional shoc2−/− (sur-8 in mice) knockout mice die due to an early stage embryonic lethality and partial absorption of mutant embryos at E8.5. The conditional disruption of the shoc2 gene in endothelial cells displayed multiple cardiac defects, smaller body size, subcutaneous edema in their dorsal body and fetal lung congestion and did not survive past E14.5. These shoc2-deficient mice had abnormalities in transposition of the great arteries, a ventricular septal defect and double outlets of the right ventricle and right aortic arch and defects of heart valve morphogenesis. Surprisingly, no obvious change in the level of activated ERK1/2 or two other MAP kinases, JNK and p38, were detected in shoc2 null endothelial cells, possibly indicating the non-autonomous role for Shoc2 in the endothelial cells.
A key finding of our study is that the abrogated expression of Shoc2 in zebrafish embryos results in a rapid manifestation of a robust phenotype that exhibits hallmarks of human RASopathies, further supporting the likelihood of conservation in Shoc2 signaling targets between human and zebrafish. Remarkably, severe edema, anemia and arrhythmia observed in shoc2 null larvae were reminiscent of fetal hydrops—a serious fetal condition previously reported in several cases of newborns heterozygous for the S2G missense mutation in Shoc2. In patients molecularly diagnosed with Shoc2’s NSLH mutation, fetal hydrops presented as fluid accumulation in fetal compartments, pleural and pericardial effusion, skin edema, leukocytosis and thrombocytopenia (15,18,37).
Craniofacial defects detected in Shoc2 null larvae are another well-recognized component of the pleiotropic spectrum of RASopathy phenotypes (38). The striking abnormalities in the development of cartilage, bone ossification and the patterning of pigment cells in shoc2 null zebrafish larvae indicate an essential role for Shoc2 in the specification of neural crest-derived tissues. Augmented expression of foxd3 found in shoc2 crispants provides insights into the underlying mechanisms downstream of the Shoc2-ERK1/2 module and is in-line with the canonical function of the Ras/ERK1/2 pathway in neural crest specification and migration (39). High levels of sox10 or foxD3 in embryonic stem cells and neural crest cells are shown to promote multipotentiality and the levels of these transcription factors change dramatically with cell specification (40–42). Hence, it is possible that suppression of Shoc2-mediated signals promotes the maintenance of the pluripotent progenitor cells due to increased levels of foxD3, or other related transcription factors, consequentially delaying neural crest differentiation and migration. Interestingly, lentiviral depletion of Shoc2 in neuronal progenitor cells (NPCs) resulted in misbalanced differentiation and proliferation of NPCs (43). Although these studies did not formally demonstrate a requirement for Shoc2-ERK1/2 activity in stem cell specification, they show that Shoc2 may act as an anti-differentiation factor that may regulate ERK1/2 signals to NPCs proliferation and self-renewal. However, other Shoc2-ERK1/2-dependent mechanisms, such as selective integration of signals mediated by bone morphogenetic protein, transforming growth factor-β, fibroblast growth factor or EGF cannot be excluded (44). Even though several signaling pathways are involved in the development of neural crest-derived tissues (44), our data indicate that the changes observed in shoc2 null animals attest to a lack of functional redundancy with other members of the ERK1/2 pathway and/or contribute to the expression of different sets of transcriptional downstream targets. Our data in Supplementary Figure S9A show that shoc2 is highly expressed in sox10-positive cells, indicating its cell autonomous functions in neural crest cells. Therefore, future studies in cells isolated from neural crest lineages will facilitate the identification of molecules upstream and downstream of the Shoc2-ERK1/2 module and are likely to reveal novel functions of Shoc2 in neural crest development. The exact details of how Shoc2-transmitted signals affect neural crest specification remain to be elucidated.
RASopathy patients occasionally are diagnosed with an aggressive myeloproliferative disorder resembling a rare hematologic malignancy in children—juvenile myelomonocytic leukemia (JMML) (45). Some JMML symptoms such as anemia and thrombocytopenia are reminiscent of the robust loss of circulating blood cells we observed in the Shoc2 crispants and, based on our data in Supplementary Figure S9B, is likely due to the autonomous function of Shoc2 in hematopoietic cells. Our findings that shoc2 crispants did not have a preferential loss of a particular blood lineage support our hypothesis that Shoc2-ERK1/2 signals are universally critical for the production or expansion of the progenitors at the very early stages of hematopoiesis. Based on our earlier studies demonstrating that Shoc2 transduces ERK1/2 cell motility signals (46), it is tempting to speculate that Shoc2-deficient hematopoietic progenitors are impaired in their ability to migrate to the environment that supports their maturation. Alternatively, the loss of Shoc2 may abrogate the signals potentiating maturation of these cells.
Alterations in the expression of the Shoc2 binding partners, Ras and Raf-1, or other components of the ERK1/2 pathway, such as MEK1/2, in zebrafish are found to cause phenotypes partially overlapping with those observed in shoc2 mutants (47–49). Although we did not observe the severe phenotypes in our shoc2 mutants in very early developmental stages, subtle phenotypes could have been missed, as they were not the focus of this study. Alternatively, the lack of early developmental phenotypes in our mutant lines could be attributed to the maternal expression of shoc2 from the heterozygous mother, whereas the MEK1/2 studies were carried out by overexpression of mutant mRNA (49). Yet, the robust phenotype observed for shoc2 nulls in which expression of Ras and Raf signaling proteins is unaltered suggests that the Shoc2 scaffold is critical for Ras–Raf signal transmission underscoring its role as a `master organizer’.
In summary, a novel zebrafish model was developed to identify a previously unrecognized role of the Shoc2 scaffold in vasculogenesis, in the specification of neural crest cells and in hematopoiesis. This study not only emphasizes a critical role for the Shoc2 scaffold in the morphogenesis of embryonic tissues, but also underscores the advantages of our zebrafish model for dissecting the contribution of the individual regulatory components of the ERK1/2 pathway. The zebrafish shoc2−/− model will be very instrumental in identifying the implications of the multiple Shoc2 protein interactions through in vivo structure–function analysis. Our model will further help elucidate molecular mechanisms underlying Shoc2-related pathologies, to help track the molecular changes that take place when Shoc2 is not transmitting ERK1/2 signals, as well as to determine the roles of target genes downstream of the Shoc2 scaffolding module in a number of developmental processes.
Materials and Methods
Zebrafish strains and maintenance
All zebrafish strains were bred, raised and maintained in accordance with established animal care protocols for zebrafish husbandry. Embryos were staged as previously described (22). All animal procedures were carried out in accordance with guidelines established by the University of Kentucky Institutional Animal Care and Use Committee.
Cloning of zebrafish Shoc2 complementary DNA
Total RNA of 120 hpf zebrafish was extracted with RNAzol reagent (Bio-Rad, CA) and reverse transcribed using iScript RT Supermix according to the manufacturer’s recommendations (Bio-Rad, CA). Forward primer: 5-ATGTGCCATTGGACTGCGG-3′ and reverse primer: 5′-GTCCAAGGTAAAGGCCACCG-3′ were used to amplify Shoc2. Forward primer: 5′- ATGTCATCGACTCTGGGCAAAGATAAAGAC-3′ and reverse primer: 5′-TCAGACCATGGCGCGGTA-3′ were used to amplify Shoc2 insensitive to ATG MO. PCR products were cloned into TA vector and sequenced.
Whole-mount in situ hybridization
Embryos or larvae were treated with 0.003% 1-phenyl-2-thiourea (PTU, Sigma, St. Louis, MO) to block pigmentation. A standard protocol was used to perform whole-mount in situ hybridization (50). Briefly, probes were added to RNase-free tubes at a concentration of 2 ng/μl and hybridized overnight at 55°C. Larvae were washed and incubated with an alkaline phosphatase-conjugated antibody (Roche Diagnostics, Switzerland) at a dilution of 1:1500 on the second day. The color reaction was mediated by nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate (NBT/BCIP, Roche) on the third day. The above experiment was repeated three times.
HRMA
To isolate genomic DNA from uninjected or sgRNA/Cas9-injected individual embryos, 24 hpf de-chorionated embryos were placed into individual wells of a 96-well plate containing 20 μl of 1× ThermoPol Buffer (New England Biolabs, MA). The plate was placed in a PCR cycler at 95°C for 10 min, after which 5 μl of 10 mg/ml Proteinase K (Sigma, MO) was added to each well and the plate was incubated at 55°C for 1 h and 95°C for 10 min. HRMA was performed using a LightCycler 96 RealTime PCR System (Roche, MO) and LightCycler 480 High Resolution Melting Master (Roche, MO) according to the manufacturer’s instructions. Primers shoc2Δ22 5′-CCATCAAGGAGCTGACCCAG-3′ and 5′- TCTGACCAGCCTACCTGACT -3′, shoc2Δ14 5′-AGCGACTCTGTTGTCTTGTGTTA-3′ and 5′- AGGTTGGTGATCTGAGTGCAA -3′ were used for HRMA.
Genotyping
Genomic DNA was extracted from individual embryos or adult tail clips. Briefly, 20 μl of the ThermoPol Buffer (New England Biolabs, MA) was added to the samples and boiled for 5 min and digested with Proteinase K (10 mg/ml) for 2 h at 55°C. Proteinase K was then inactivated by boiling for 10 min after digestion. PCR was carried out in a 25 μl reaction solution containing 1 μl of 10 mm dNTP, 1 μl of 10 mm forward and reverse primer, 2.5 μl of 1× ThermoPol Buffer (New England Biolabs, MA), and 0.5 units of Taq Polymerase (New England Biolabs, MA).
Isolation of lineage-specific cells from zebrafish embryos
Homozygous gata1a:dsRed and sox10:RFP transgenic fish were crossed and 72 hpf embryos were used. Embryonic tissues were dissociated in 0.05% Trypsin–EDTA solution using a 20G syringe. Cells were then filtered through 40 μm meshes to obtain a single cell suspension and pelleted. The fluorescent positive cells were collected at the Flow Cytometry Core at the University of Kentucky.
MO and mRNA injection
All MOs were obtained from Gene Tools, LLC (Philomath, OR) and injected into one- to two-cell stage zebrafish embryos. The following MOs were used in this study: standard control MO: 5′- CCTCTTACCTCAGTTACAATTTATA-3′; shoc2 MO1: 5′- TACTGCTCATGGCGAAAGCCCCGCA-3′; shoc2 MO2: 5′- TCCAATGGCACATGGGACCCTCAGT-3′. Embryos were injected with 5.2 ng each of MOs. Both shoc2 MO1 and MO2 generated similar phenotypes. All data presented in this study were from embryos injected with shoc2 MO1. For mRNA rescue experiments, the human WT shoc2 coding sequences (with silent mutation at MO target sites) were PCR amplified from human shoc2 complementary DNA and cloned into the pGEM-T-easy vector (Promega, WI). The capped mRNAs were synthesized with the mMessage mMACHINE transcription kit (Life Technologies, CA) according to the manufacturer’s instructions. For mRNA rescue experiments, 100 pg/embryo of WT was co-injected with 5.2 ng of shoc2 MO1.
CRISPR sgRNA and Cas9 mRNA synthesis and injection
shoc2 CRISPR target sites were identified and the corresponding sgRNA oligos were designed using the ZiFiT online software package (www.zifit.partners.org/ZiFiT). Oligo pairs (100 mm) for each sgRNA were mixed with NEBuffer 4 (New England Biolabs, MA), incubated in boiling water for 5 min, followed by 2 h annealing at room temperature, and then ligated with a linearized pDR274 vector (Addgene, MA) at 16°C overnight. Recombinant plasmid was digested with DraI to remove the sgRNA template, followed by PCR amplification and purification using a QIAquick PCR Purification Kit (Qiagen, Germany). sgRNAs were generated using the MEGAscript T7 Transcription Kit (Life Technologies, CA). To generate the Cas9 mRNA, the pCS2-nCas9n plasmid (Addgene, MA) was linearized with NotI and capped Cas9 mRNA was synthesized using the mMESSAGE mMACHINE SP6 Transcription Kit (Life Technologies, CA). To confirm the quality of sgRNA and Cas9 mRNA, RNA was mixed with formamide, heated at 72°C for 5 min and run on a 1% (w/v) agarose gel. The following sgRNA and Cas9 mRNA doses were microinjected into embryos at the one-cell stage: 200 pg/embryo of shoc2 sgRNA; 300 pg/embryo of Cas9 mRNA.
Real-time quantitative polymerase chain reaction
The total RNA was isolated from the pool of five embryos at 6 dpf using PureZOL/Aurum Total RNA Isolation Kit (Bio-Rad, CA) according to the manufacturer’s instructions. Aliquots containing equal amounts of RNA were subjected to RT-PCR analysis. Quantitative PCR (qPCR) was performed using SsoAdvanced SYBR green Supermix and a Bio-Rad CFX detection system (Bio-Rad, CA). Relative amounts of RNAs were calculated using the comparative CT method (51). Sequence-specific primer sets are presented in Supplementary Material, Table S1. The values for the samples were normalized against those for the reference gene, and the results are presented as the fold change in the amount of mRNA recovered from WT and mutant embryo. The data represent the means ± SEM from three independent experiments.
Western blot analysis
Proteins were extracted from de-chorionated and de-yolked embryos/larvae and resolved by SDS-PAGE. Western blot analysis was performed as described previously (9). Quantification was performed using the densitometry analysis mode of Image Lab software (Bio-Rad, CA). Antibodies against the following proteins were used: β-actin, pERK1/2 and total ERK1/2 (tERK1/2, Santa Cruz Biotechnology, CA); Shoc2 (Genetex, CA).
Staining methods
Whole-embryo staining for erythropoietic cells was performed using o-dianisidine histochemistry according to previously described methods (25,26). PTU-treated de-chorionated embryos were stained for 30 min in the dark in o-dianisidine (0.6 mg/ml), 0.01 M sodium acetate, 0.65% H202 and 40% (vol/vol) ethanol. After staining, embryos were washed with PBS-T (Phosphate buffer saline with 0.1% Tween-20) and then postfixed in 4% paraformaldehyde (PFA) overnight in 4°C. Sudan Black B staining was performed to detect neutrophils and macrophages. Whole embryos were fixed with 4% PFA for 2 h at room temperature, rinsed in PBS, incubated with Sudan Black B solution (2 mg/ml Sudan Black B dissolved in 70% ethanol) in the dark for 30 min. Stained embryos were then washed with 70% ethanol and stored in 90% glycerol for imaging. Histochemical staining for the mpx activity of whole zebrafish embryos was performed according to the manufacturer’s instructions (Sigma-Aldrich, MO).
For Alcian blue staining, zebrafish larvae were fixed in 4% PFA for 2 h at room temperature and stained according to Kimmelet al. (56). Calcified structures were examined by acid-free Alizarin Red S staining. Embryos were fixed in 4% PFA for 2 h and stained in a 0.05% Alizarin Red S solution in water for 30 min in the dark on low agitation, rinsed in a 50% glycerol, 0.1% KOH solution to remove excessive staining and kept at 4°C in the same solution for imaging.
Isolation of hematopoietic cells
Anesthetized 6 dpf larvae were transferred into PBS containing 50 U/ml heparin, 1% bovine serum albumin and 0.006% tricaine (3-amino benzoic acidethylester) (Western Chemical Inc., WA). Tails were excised posterior to the yolk extension using a scalpel, and blood cells were extruded from the site of excision with the scalpel and collected using a micropipette.
Cytology of peripheral blood
For cytological analyses, blood cells collected from zebrafish larvae were transferred onto glass slides by cytospin and stained with May-Grünwald–Giemsa stain (Sigma-Aldrich, MO) following the manufacturer’s instructions.
Statistical analyses
Results are expressed as means ± SEM. The statistical significance of the differences between groups was determined using either Student’s t-test or a one-way analysis of variance (followed by the Tukey’s test). P < 0.05 was considered statistically significant. All statistical analyses were carried out using SigmaStat 13.0 (Systat Software Inc., CA).
Photography and image analysis
Images of the phenotypes of embryos or larvae and images of whole-mount in situ hybridization were captured with a Nikon DS-Ri2 digital camera mounted on a dissecting microscope (Nikon SMZ18, Japan) or an inverted microscope (Nikon Eclipse Ti-U; Nikon Instruments, NY). NIS-Elements software (Nikon, NY) was used to calculate the body lengths, eye size and areas of Alcian blue- and Alizarin Red S-positive signals. Images of mpx staining were photographed with a Leica DFC450 digital camera and May-Grünwald–Giemsa staining was imaged with Leica DFC7000 T mounted on a dissecting microscope (Leica S9D, Germany). All images were compiled in Adobe Photoshop CS6 Portable (Adobe Systems Inc., CA) and resized.
Supplementary Material
Acknowledgements
We thank Drs Jessica Blackburn, Charles Waechter and Louis Hersh for providing reagents and critical reading of the manuscript. The UK Flow Cytometry and Cell Sorting core facility is supported in part by the UK Office of the Vice President for Research, the Markey Cancer Center and a National Cancer Institute Center Core Support Grant (P30 CA177558). We are also grateful to Sara Perkins, Chris Mitchell and Lucas Vieiro Francisco for zebrafish care.
Conflict of Interest statement. None declared.
Funding
National Cancer Institute (R00CA126161 to E.G.); National Institute of General Medical Sciences (GM113087 to E.G.); National Eye Institute (EY021769 to A.C.M.): American Cancer Society (RSG-14-172-01-CSM to E.G.); American Heart Association (15PRE25090207 to H.J.).
References
- 1. Cobb M.H. (1999) MAP kinase pathways. Prog. Biophys. Mol. Biol., 71, 479–500. [DOI] [PubMed] [Google Scholar]
- 2. Corson L.B., Yamanaka Y., Lai K.M. and Rossant J. (2003) Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development, 130, 4527–4537. [DOI] [PubMed] [Google Scholar]
- 3. Nussinov R., Ma B. and Tsai C.J. (2013) A broad view of scaffolding suggests that scaffolding proteins can actively control regulation and signaling of multienzyme complexes through allostery. Biochim. Biophys. Acta, 1834, 820–829. [DOI] [PubMed] [Google Scholar]
- 4. Langeberg L.K. and Scott J.D. (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol., 16, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selfors L.M., Schutzman J.L., Borland C.Z. and Stern M.J. (1998) soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl. Acad. Sci. U.S.A., 95, 6903–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sieburth D.S., Sun Q. and Han M. (1998) SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell, 94, 119–130. [DOI] [PubMed] [Google Scholar]
- 7. Jang E.R. and Galperin E. (2016) The function of Shoc2: a scaffold and beyond. Commun. Integr. Biol., 9, e1188241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jang E.R., Jang H., Shi P., Popa G., Jeoung M. and Galperin E. (2015) Spatial control of Shoc2-scaffold-mediated ERK1/2 signaling requires remodeling activity of the ATPase PSMC5. J. Cell Sci., 128, 4428–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jang E.R., Shi P., Bryant J., Chen J., Dukhante V., Gentry M.S., Jang H., Jeoung M. and Galperin E. (2014) HUWE1 is a molecular link controlling RAF-1 activity supported by the Shoc2 scaffold. Mol. Cell. Biol., 34, 3579–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeoung M., Abdelmoti L., Jang E.R., Vander Kooi C.W. and Galperin E. (2013) Functional integration of the conserved domains of Shoc2 scaffold. PLoS One, 8, e66067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tidyman W.E. and Rauen K.A. (2016) Pathogenetics of the RASopathies. Hum. Mol. Genet., 25, R123–R132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Capalbo D., Scala M.G., Melis D., Minopoli G., Improda N., Palamaro L., Pignata C. and Salerno M. (2012) Clinical heterogeneity in two patients with Noonan-like syndrome associated with the same SHOC2 mutation. Ital. J. Pediatr., 38, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cordeddu V., Di Schiavi E., Pennacchio L.A., Ma'ayan A., Sarkozy A., Fodale V., Cecchetti S., Cardinale A., Martin J., Schackwitz W. et al. (2009). Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet., 41, 1022–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Komatsuzaki S., Aoki Y., Niihori T., Okamoto N., Hennekam R.C., Hopman S., Ohashi H., Mizuno S., Watanabe Y., Kamasaki H. et al. (2010) Mutation analysis of the SHOC2 gene in Noonan-like syndrome and in hematologic malignancies. J. Hum. Genet., 55, 801–809. [DOI] [PubMed] [Google Scholar]
- 15. Hoban R., Roberts A.E., Demmer L., Jethva R. and Shephard B. (2012) Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am. J. Med. Genet. A, 158A, 1411–1413. [DOI] [PubMed] [Google Scholar]
- 16. Hannig V., Jeoung M., Jang E.R., Phillips J.A. 3rd and Galperin E.A. (2014) A novel SHOC2 variant in Rasopathy. Hum. Mutat., 35, 1290–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gripp K.W., Zand D.J., Demmer L., Anderson C.E., Dobyns W.B., Zackai E.H., Denenberg E., Jenny K., Stabley D.L. and Sol-Church K. (2013) Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am. J. Med. Genet. A., 161A, 2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gargano G., Guidotti I., Balestri E., Vagnarelli F., Rosato S., Comitini G., Wischmeijer A., La Sala G.B., Iughetti L., Cordeddu V. et al. (2014) Hydrops fetalis in a preterm newborn heterozygous for the c.4A>G SHOC2 mutation. Am. J. Med. Genet. A., 164A, 1015–1020. [DOI] [PubMed] [Google Scholar]
- 19. Baldassarre G., Mussa A., Banaudi E., Rossi C., Tartaglia M., Silengo M. and Ferrero G.B. (2014) Phenotypic variability associated with the invariant SHOC2 c.4A>G (p.Ser2Gly) missense mutation. Am. J. Med. Genet. A, 164A, 3120–3125. [DOI] [PubMed] [Google Scholar]
- 20. Galperin E., Abdelmoti L. and Sorkin A. (2012) Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One, 7, e36469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jindal G.A., Goyal Y., Burdine R.D., Rauen K.A. and Shvartsman S.Y. (2015) RASopathies: unraveling mechanisms with animal models. Dis. Model. Mech., 8, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B. and Schilling T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 23. Boisset J.C. and Robin C. (2012) On the origin of hematopoietic stem cells: progress and controversy. Stem Cell Res., 8, 1–13. [DOI] [PubMed] [Google Scholar]
- 24. Lawson N.D. and Weinstein B.M. (2002) In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol., 248, 307–318. [DOI] [PubMed] [Google Scholar]
- 25. Iuchi I. and Yamamoto M. (1983) Erythropoiesis in the developing rainbow trout, Salmo gairdneri irideus: histochemical and immunochemical detection of erythropoietic organs. J. Exp. Zool., 226, 409–417. [DOI] [PubMed] [Google Scholar]
- 26. Detrich H.W. 3rd, Kieran M.W., Chan F.Y., Barone L.M., Yee K., Rundstadler J.A., Pratt S., Ransom D. and Zon L.I. (1995) Intraembryonic hematopoietic cell migration during vertebrate development. Proc. Natl. Acad. Sci. U.S.A., 92, 10713–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le Guyader D., Redd M.J., Colucci-Guyon E., Murayama E., Kissa K., Briolat V., Mordelet E., Zapata A., Shinomiya H. and Herbomel P. (2008) Origins and unconventional behavior of neutrophils in developing zebrafish. Blood, 111, 132–141. [DOI] [PubMed] [Google Scholar]
- 28. Jong J.L. and Zon L.I. (2005) Use of the zebrafish system to study primitive and definitive hematopoiesis. Annu. Rev. Genet., 39, 481–501. [DOI] [PubMed] [Google Scholar]
- 29. Jao L.E., Wente S.R. and Chen W. (2013) Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. U.S.A., 110, 13904–13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walker M.B. and Kimmel C.B. (2007) A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem., 82, 23–28. [DOI] [PubMed] [Google Scholar]
- 31. Kell M.J., Riccio R.E., Baumgartner E.A., Compton Z.J., Pecorin P.J., Mitchell T.A., Topczewski J. and LeClair E.E. (2018) Targeted deletion of the zebrafish actin-bundling protein L-plastin (lcp1). PLoS One, 13, e0190353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Willett C.E., Zapata A.G., Hopkins N. and Steiner L.A. (1997) Expression of zebrafish rag genes during early development identifies the thymus. Dev. Biol., 182, 331–341. [DOI] [PubMed] [Google Scholar]
- 33. Langenau D.M., Ferrando A.A., Traver D., Kutok J.L., Hezel J.P., Kanki J.P., Zon L.I., Look A.T. and Trede N.S. (2004) In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc. Natl. Acad. Sci. U.S.A., 101, 7369–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ransom D.G., Haffter P., Odenthal J., Brownlie A., Vogelsang E., Kelsh R.N., Brand M., Eeden F.J., Furutani-Seiki M., Granato M. et al. (1996) Characterization of zebrafish mutants with defects in embryonic hematopoiesis. Development, 123, 311–319. [DOI] [PubMed] [Google Scholar]
- 35. Kulkeaw K. and Sugiyama D. (2012) Zebrafish erythropoiesis and the utility of fish as models of anemia. Stem Cell Res. Ther., 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yi J., Chen M., Wu X., Yang X., Xu T., Zhuang Y., Han M. and Xu R. (2010) Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev. Dyn., 239, 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takenouchi T., Sakamoto Y., Miwa T., Torii C., Kosaki R., Kishi K., Takahashi T. and Kosaki K. (2014) Severe craniosynostosis with Noonan syndrome phenotype associated with SHOC2 mutation: clinical evidence of crosslink between FGFR and RAS signaling pathways. Am. J. Med. Genet. A., 164A, 2869–2872. [DOI] [PubMed] [Google Scholar]
- 38. Rauen K.A. (2013) The RASopathies. Annu. Rev. Genomics Hum. Genet., 14, 355–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newbern J., Zhong J., Wickramasinghe R.S., Li X., Wu Y., Samuels I., Cherosky N., Karlo J.C., O'Loughlin B., Wikenheiser J. et al. (2008) Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc. Natl. Acad. Sci. U.S.A., 105, 17115–17120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teng L., Mundell N.A., Frist A.Y., Wang Q. and Labosky P.A. (2008) Requirement for Foxd3 in the maintenance of neural crest progenitors. Development, 135, 1615–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mundell N.A. and Labosky P.A. (2011) Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development, 138, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim J., Lo L., Dormand E. and Anderson D.J. (2003) SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron, 38, 17–31. [DOI] [PubMed] [Google Scholar]
- 43. Moon B.S., Kim H.Y., Kim M.Y., Yang D.H., Lee J.M., Cho K.W., Jung H.S. and Choi K.Y. (2011) Sur8/Shoc2 involves both inhibition of differentiation and maintenance of self-renewal of neural progenitor cells via modulation of extracellular signal-regulated kinase signaling. Stem Cells, 29, 320–331. [DOI] [PubMed] [Google Scholar]
- 44. Simões-Costa M. and Bronner M.E. (2015) Establishing neural crest identity: a gene regulatory recipe. Development, 142, 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niemeyer C.M. (2014) RAS diseases in children. Haematologica, 99, 1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeoung M., Jang E.R., Liu J., Wang C., Rouchka E.C., Li X. and Galperin E. (2016) Shoc2-tranduced ERK1/2 motility signals—novel insights from functional genomics. Cell. Signal., 28, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L., Zhu S., Gong Z. and Low B.C. (2008) K-ras/PI3K-Akt signaling is essential for zebrafish hematopoiesis and angiogenesis. PLoS One, 3, e2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Razzaque M.A., Nishizawa T., Komoike Y., Yagi H., Furutani M., Amo R., Kamisago M., Momma K., Katayama H., Nakagawa M. et al. (2007) Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet., 39, 1013–1017. [DOI] [PubMed] [Google Scholar]
- 49. Goyal Y., Jindal G.A., Pelliccia J.L., Yamaya K., Yeung E., Futran A.S., Burdine R.D., Schüpbach T. and Shvartsman S.Y. (2017) Divergent effects of intrinsically active MEK variants on developmental Ras signaling. Nat. Genet., 49, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thisse C. and Thisse B. (2008) High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc., 3, 59–69. [DOI] [PubMed] [Google Scholar]
- 51. Schmittgen T.D. and Livak K.J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc., 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- 52. Roy A., Kucukural A. and Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc., 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang J., Yan R., Roy A., Xu D., Poisson J. and Zhang Y. (2015) The I-TASSER suite: protein structure and function prediction. Nat. Methods, 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lieschke G.J., Oates A.C., Paw B.H., Thompson M.A., Hall N.E., Ward A.C., Ho R.K., Zon L.I. and Layton J.E. (2002) Zebrafish SPI-1 (PU.1) marks a site of myeloid development independent of primitive erythropoiesis: implications for axial patterning. Dev. Biol., 246, 274–295. [DOI] [PubMed] [Google Scholar]
- 55. Chang T.Y., Pardo-Martin C., Allalou A., Wahlby C. and Yanik M.F. (2012) Fully automated cellular-resolution vertebrate screening platform with parallel animal processing. Lab Chip, 12, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kimmel C.B., Miller C.T., Kruz e G., Ullmann B., BreMiller R.A., Larison K.D. and Snyder H.C. (1998) The Shaping of Pharyngeal Cartilages during Early Development of the Zebrafish. Dev Biol., 203, 245–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.