

Abstract
Children conceived using Assisted Reproductive Technologies (ART) have a higher incidence of growth and birth defects, attributable in part to epigenetic perturbations. Both ART and germline defects associated with parental infertility could interfere with epigenetic reprogramming events in germ cells or early embryos. Mouse models indicate that the placenta is more susceptible to the induction of epigenetic abnormalities than the embryo, and thus the placental methylome may provide a sensitive indicator of ‘at risk’ conceptuses. Our goal was to use genome-wide profiling to examine the extent of epigenetic abnormalities in matched placentas from an ART/infertility group and control singleton pregnancies (n = 44/group) from a human prospective longitudinal birth cohort, the Design, Develop, Discover (3D) Study. Principal component analysis revealed a group of ART outliers. The ART outlier group was enriched for females and a subset of placentas showing loss of methylation of several imprinted genes including GNAS, SGCE, KCNQT1OT1 and BLCAP/NNAT. Within the ART group, placentas from pregnancies conceived with in vitro fertilization (IVF)/intracytoplasmic sperm injection (ICSI) showed distinct epigenetic profiles as compared to those conceived with less invasive procedures (ovulation induction, intrauterine insemination). Male factor infertility and paternal age further differentiated the IVF/ICSI group, suggesting an interaction of infertility and techniques in perturbing the placental epigenome. Together, the results suggest that the human placenta is sensitive to the induction of epigenetic defects by ART and/or infertility, and we stress the importance of considering both sex and paternal factors and that some but not all ART conceptuses will be susceptible.
Introduction
Infertility and its treatment, including assisted reproductive technologies (ART) such as intracytoplasmic sperm injection (ICSI), superovulation and embryo culture, affect germ cell and embryo development at times when the epigenome undergoes dramatic remodeling (1). A number of studies in the early 2000s suggested that there was an increase in epigenetically based human imprinting disorders amongst children conceived using ART (2–6) and reviews at that time argued for biological plausibility based on emerging findings on the timing of DNA methylation events in germ cells and preimplantation embryos (7). Two main possible causes for the imprinted gene methylation defects were suggested; first, the underlying infertility of the parents, or second, the techniques used in ART. The DNA methylation abnormalities of imprinted genes were reported in the sperm of infertile men supporting the former possibility (8–12). Mouse and bovine experiments in which the use of assisted reproduction techniques can be separated from infertility supported the latter possibility with various techniques, such as superovulation, ICSI, embryo culture and embryo transfer, individually and in combination, leading to DNA methylation defects in imprinted regions in the offspring (13–19).
Since the first reports, studies of the effects of ART on imprinting in humans have reached different conclusions. A recent systematic review and meta-analysis concluded that there was an increase in imprinting disorders in children conceived through ART but, due to heterogeneity in study populations and methodologies in existing studies, there was insufficient evidence for an association between ART and the methylation of imprinted genes (20). From the published data it was suggested that ART-associated DNA methylation defects may be subtle and difficult to detect and that more controlled studies, carefully defined clinical populations and standardized methodologies would be needed to provide definitive answers.
Recent mouse and human ART studies along with advances in techniques to examine DNA methylation such as pyrosequencing, array and next generation sequencing, provide new insights and support for approaches to examine connections between human infertility, ART or their combination, in inducing epigenetic defects in ART-conceived children. Mouse and human studies using genome-wide techniques indicate that DNA methylation defects associated with ART clearly extend beyond imprinted genes to other sequences that may affect growth and development in children (21–26). The concept that only a subset of offspring, termed ‘outliers’ will be affected by ART has emerged in both mouse (19) and human (27) reports, suggesting that there may be susceptible individuals but that most conceptuses may be able to escape, compensate, eliminate or reverse early epigenetic abnormalities. The existence of epigenetic ‘outliers’ may help explain inconsistent reports of imprinted gene defects in human ART studies. In the mouse model there is evidence of cumulative effects of assisted reproduction procedures on placental development and epigenetic perturbations (14,19). In addition, our group recently showed that compromised oocyte quality could exacerbate imprinted and genome-wide DNA methylation defects associated with ART (24), providing proof of concept in a rodent model that factors influencing parental fertility and ART can interact. Finally, several mouse studies indicate that the placenta is particularly susceptible (or permissive) to the induction by ART of DNA methylation defects, suggesting that this tissue may provide a sensitive indicator of early epigenetic perturbations that may affect the embryo either directly or indirectly via alterations in placental function (13–15,19,24,28,29).
Here, we examined links between ART and epigenetic abnormalities using the Quebec-based Canadian 3D (Design, Develop, Discover) longitudinal pregnancy cohort that was specifically designed to include an ART subgroup of singleton pregnancies and from which spontaneous pregnancies could be chosen for matching in terms of factors that could affect epigenetic states such as length of gestation, birth weight, maternal and paternal age (30). A particular advantage of the Design, Develop, Discover (3D) study was that we could choose well-defined biological samples that were collected in a standardized fashion from ART and control pregnancies of individuals recruited at the same publically funded hospital over the course of 2 years. As an initial approach, we chose to examine the placenta hypothesizing that, as in mouse ART studies, this tissue may reveal evidence of epigenetic abnormalities occurring early in development associated with the use of human ART. The Illumina HumanMethylation450 (450K) BeadChip arrays were used to examine DNA methylation on a genome-wide basis, and DNA methylation alterations were validated methodologically with a second locus-specific assay, pyrosequencing and replicated in an independent sample of ART and control tissues.
Results
Demographics of the ART and control cohorts
The demographics and birth characteristics of the 44 ART pregnancies and 44 controls selected for the epigenetics study are summarized in Table 1. Overall, parameters for both groups were not statistically different including smoking status during pregnancy, baby’s sex, obstetrical complications, delivery route (vaginal or cesarean section), gestational age at delivery, paternal and maternal age and maternal body mass index (BMI). The characteristics of the ART/infertility group (referred to as ART group from here on) are shown in Table 2. In vitro fertilization (IVF) was used in 23 cases (18 by ICSI and 5 by classical IVF), 11 pregnancies took place after intrauterine insemination (IUI) and 10 pregnancies occurred spontaneously after more than 1 year of regular unprotected intercourse (clinically defined as infertility, average = 31.6 months, standard deviation = 15.4 months). For all participants in the IVF/ICSI group, embryos were transferred at the blastocyst stage, the majority during a fresh cycle (n = 20 of a total of 23 cycles).
Table 1.
Description of the Control and ART groups
| Control group n = 44 | ART group n = 44 | |
|---|---|---|
| Maternal age (years) (mean / SD) | 33/3. 8 | 34. 5/4. 3 |
| Paternal age (years) (mean / SD) | 36/5. 3 | 37. 4/7 |
| Maternal BMI (mean / SD) | 24/4. 9 | 24. 6/4. 5b |
| Smoking status during pregnancy S/SSP/NSa |
3/8/33 | 3/7/34 |
| Obstetrical issues | ||
| Normal | 33 | 32 |
| Prematurity alone | 6 | 1 |
| Gestational diabetes (GD) alone | 3 | 4 |
| Pre-eclampsia alone | 2 | 3 |
| Pre-eclampsia + prematurity | 0 | 3 |
| Pre-eclampsia + GD | 0 | 1 |
| Delivery route (Vaginal/C section) | 31/13 | 30/14 |
| Gestational age (weeks) (mean / SD) | 38. 7/1. 5 | 38. 6/1. 6 |
| Infant sex (M/F) | 23/21 | 24/20 |
aS = smoking during pregnancy, SSP = stopping smoking before pregnancy, NS = no smoking.
b3 BMI unknown.
Table 2.
Descriptive characteristics for the ART group
| IVF (overall) | 23 |
|---|---|
| IVF with ICSI | 18 |
| IVF (fresh/frozen) | 20 /3 |
| Return at blastocyst stage | 23 |
| IUI | 11 |
| Infertility with spontaneous conception | 10 |
IVF, In vitro fertilization; ICSI, intracytoplasmic sperm injection; IUI, intrauterine insemination.
Differential DNA methylation analysis defines a subset of ART samples
The placental DNA from each participant was bisulfite modified and analyzed using the Illumina Infinium Human Methylation450 BeadChip. All 88 samples passed quality control assessment and were used for downstream analysis (see Materials and Methods). Principal component analysis (PCA) was performed on all probes for all 88 samples in the ART and control groups (Fig. 1). A subset of samples (n = 15) was determined to be outliers from the rest of the sample group as described in the methods; the clinical features of this group are shown in Supplementary Material, Table 1. In order to account for the outlier group in our differential DNA methylation analysis between ART and control, we used the limma R package to perform regression analysis of DNA methylation level as the variable dependent on a range of factors and confounders: the ART status, outlier status, sex, delivery route, maternal and paternal age, smoking status and term versus preterm delivery. We found no statistically significant DNA methylation differences in ART versus control groups, after accounting for other factors and after adjustment for multiple testing using either false discovery rate or Bonferroni correction (threshold set at P < 0.05). However, as many as 84 270 CpG sites showed significant differences in DNA methylation attributable to the outlier group status (P < 0.05 after Bonferroni correction) with DNA methylation differences as large as 30% (Supplementary Material, Fig. 1). Only 62 CpG sites showed significant DNA methylation differences due to sex (P < 0.05 after Bonferroni correction).
Figure 1.
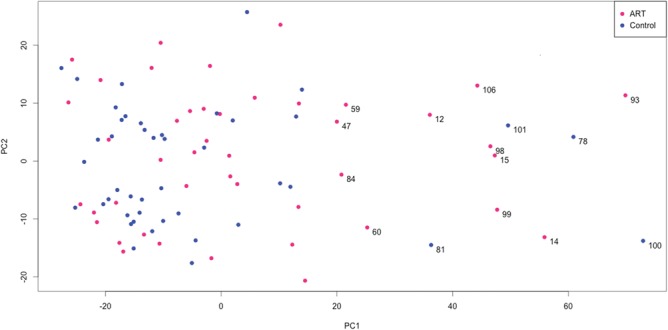
PCA of the whole placenta dataset. PCA was done on all 44 ART and 44 controls over all 414 320 CpGs. A threshold along PC1 was selected using a change-point detection algorithm to determine outliers. Outlier IDs are labeled as follows: 11 in ART, 4 in control. Red circles represent ART and blue circles represent controls.
DNA methylation outliers were enriched in placentas of female and ART pregnancies
Since we were unable to identify differentially methylated CpG sites in our ART group, due to the strong effect of the outlier group, we set out to test the outlier samples against several clinical attributes to identify associations with specific clinical criteria. We found that placenta samples from female born infants and from pregnancies conceived with ART were statistically enriched (P < 0.05) in this outlier group (Fig. 2 and Supplementary Material, Table 2).
Figure 2.
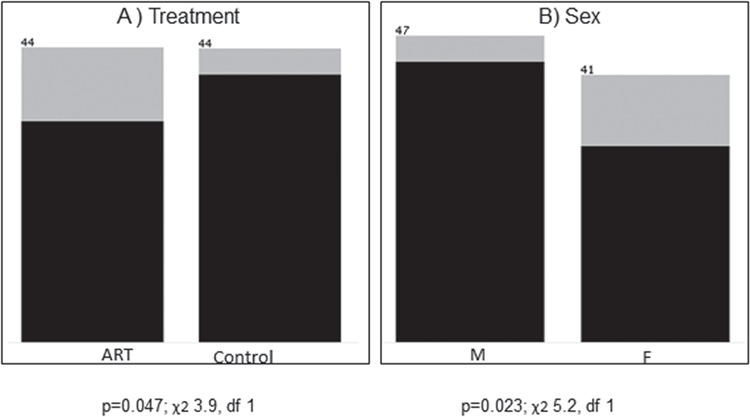
Association with outlier DNA methylation profile. Clinical attributes, ART status (A) and sex (B) are ranked by association with outlier DNA methylation. For each attribute value, number of data samples is shown above each bar. Light gray represents the portion of outliers within each clinical attribute and black represents the non-outliers. P-values of chi-square test (χ2), no correction, df = degrees of freedom, missing values excluded.
Identification of placental samples with alteration in imprinted genes
Epigenetic alterations at imprinted genes have previously been associated with the use of ART. Therefore, we extracted from the Illumina 450k array, 602 CpG sites that overlap known differentially methylated regions of imprinted loci in the genome (Supplementary Material, Table 3). Using PCA, we identified a subset of the ART and control samples that grouped together and separate from the remaining samples (Fig. 3A). This group overlaps strongly (14 out of 15 samples) with the group identified in Figure 1 indicating that the outlier group methylation profile contributes to alteration in genomic imprinting in these samples. Next we generated a heatmap on the same samples and ordered the samples based on the principal component 1 identified in Figure 3A. As displayed in Figure 3B, there was an enrichment of samples from the outlier group that were grouped together along PC1 based on their DNA methylation profiles at imprinted loci. Several imprinted regions showed loss of DNA methylation in the outlier group compared to the remaining samples and in the ART group compared to controls including for SGCE on chromosome 7, the KCNQ1OT1 differentially methylated regions (DMR) (also known as KvDMR) on chromosome 11 and the BLCAP/NNAT DMR on chromosome 20 (Supplementary Material, Fig. 2). Furthermore, our data showed that the GNAS cluster on chromosome 20 showed loss of DNA methylation across a large number of probes in the outlier group and in the ART group in general. In order to validate the importance of loss of DNA methylation at the GNAS cluster in ART, we selected an imprinted region within the GNAS cluster overlapping GNAS1A using bisulfite pyrosequencing as previously described (22). The pyrosequencing data validated the array data where, first, most of the outlier samples, both in ART and control groups, were less methylated at GNAS1A compared to the rest of the samples and second, ART samples were statistically significantly less methylated than controls (Fig. 4A).
Figure 3.
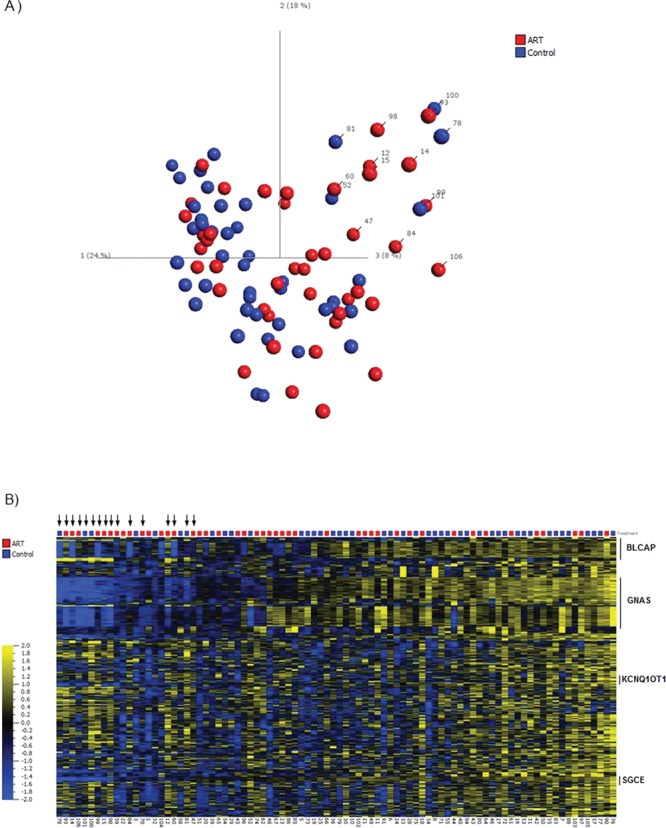
(A) PCA analysis of imprinted regions. We selected 602 CpG sites that overlap known differentially methylated regions associated with imprinted genes or domains. Red color represents ART and blue color represents controls. (B) Heatmap analysis of imprinted regions. Heatmap was generated using DNA methylation levels for 602 CpG sites overlapping known differentially methylated regions associated with imprinted genes or domains. Samples are sorted based on principal component 1 (PC1) as per Figure 3A. Variables are sorted based on gene symbols. Sample annotation is displayed at the bottom of the heatmap. Black arrows represent outlier samples identified in Figure 1. Red color represents ART and blue color represents controls. For the heatmap, data are normalized for visualization (mean = 0, variance = 1).
Figure 4.
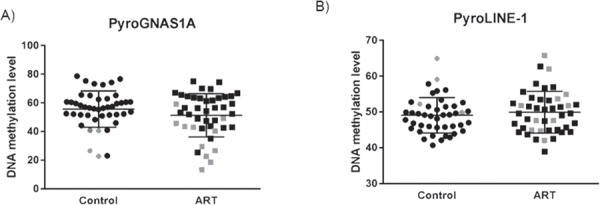
Pyrosequencing validation of targeted imprinted region and LINE-1 elements. We selected an imprinted region GNAS1A (A) for pyrosequencing validation in 44 ART and 44 controls. The scatter plot is showing the distribution of the DNA methylation levels across the different samples. Outliers represent the samples that deviate in their DNA methylation values from the overall DNA methylation levels within each group of samples. (B) Pyrosequencing analysis of the repeat elements LINE-1 in controls and ART samples. Gray color represents outliers in both ART and controls.
Global analysis of DNA methylation
In order to identify if there was any global defect in DNA methylation in our placental cohort, we performed genome-wide DNA methylation analysis of all CpG sites in both ART and controls as well as in the outlier versus non-outlier groups separately for ART and controls. As shown in Supplementary Material, Figure 3A, we did not observe any genome-wide DNA methylation differences when ART was compared to controls. Differences in DNA methylation were observed only in the outlier groups both in the ART and control cohorts (Supplementary Material, Fig 3B).
Next, we did pyrosequencing analysis of the long interspersed nuclear elements (LINE-1). Approximately, half a million LINE-1 elements exist in the genome and they are normally heavily methylated in these repetitive regions. Thus, analyzing the methylation of repetitive elements can serve as a surrogate marker for global genomic DNA methylation. Our data did not show any significant differences in the DNA methylation at these LINE-1 elements in ART versus controls (Fig. 4B) either with the outlier group (unpaired t-test, P = 0.46) or without it (unpaired t-test, P = 0.43, data not shown).
DNA methylation differences between in vitro and in vivo ART groups
Next, we examined whether the ART-associated differences in DNA methylation could be attributed to the method used prior to conception. We divided the ART group into two main sub-groups. The first sub-group was made up of samples collected from pregnancies conceived in vivo (n = 21) defined here as samples from individuals with clinically defined infertility conceiving spontaneously and pregnancies conceived with the use of intrauterine insemination. The second in vitro sub-group (n = 23) was defined here to include samples from pregnancies conceived with the use of IVF and/or ICSI. Differential DNA methylation analysis between these two groups identified a subset of 436 CpG sites that were significantly different (q < 0.05, after False Discovery Rate (FDR) correction) and mostly showing increased DNA methylation in the in vitro group compared to in vivo group (Supplementary Material, Table 4). Both unsupervised hierarchical clustering of the 436 CpG sites (Fig. 5A) as well as PCA (Fig. 5B) identified a subset of the in vitro group that clustered separately from the remaining samples (n = 14 on the right side of Fig. 5A). Further analysis of the clinical attributes of these groups identified an association/enrichment of male factor etiology for infertility and increased paternal age in the separate cluster of the in vitro group compared to the in vivo and the remaining in vitro group (Table 3).
Figure 5.
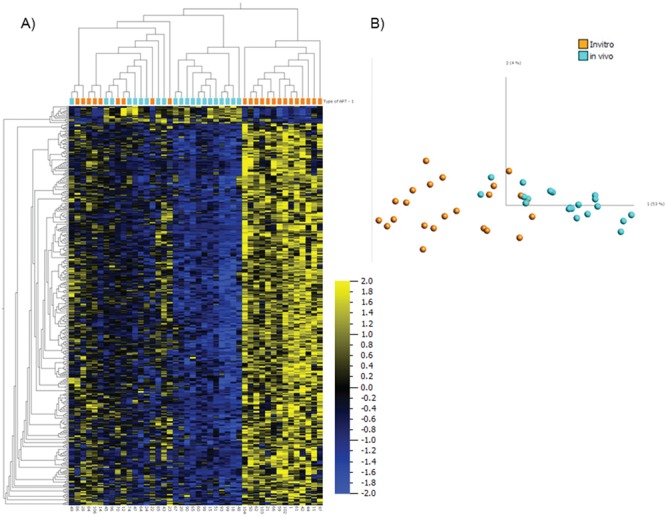
Comparison between in vivo and in vitro groups of ART. Heatmap (A) and PCA graph (B) using the discriminating 436 variables between 21 in vivo and 23 in vitro samples at q = 0.05. Orange color represents in vitro group and turquoise color represents in vivo group. For the heatmap, data are normalized for visualization (mean = 0, variance = 1).
Table 3.
Description of the ART in vivo and in vitro sub-groups
|
n = 14 (in vitro) |
n = 5 (in vitro) |
n = 21 (in vivo) |
|
|---|---|---|---|
| IVF cycles type (Fresh/Frozen) | 11/3 | 5/0 | N/A |
| ART technique (ICSI/IVF) | 11/3 | 3/2 | N/A |
| Assisted hatching | 1 | 0 | N/A |
| Number of blastocyst transfers | 14 | 5 | N/A |
| Aetiology for infertilitya | |||
| Male | 8 | 1 | 3 |
| Male + female | 1 | 0 | 2 |
| Tubal | 2 | 1 | 0 |
| PCOS | 0 | 0 | 3 |
| Unexplained | 3 | 3 | 3 |
| Infertility with spontaneous conception | N/A | N/A | 10 |
| Ejaculated sperm / TESAb | 13/1 | 5/0 | 21 |
| Smoking status during pregnancy NS/SSP/Sc |
14/0/0 | 3/1/1 | 16/5/0 |
| Baby sex (M/F) | 8/6 | 2/3 | 11/10 |
| Obstetrical issues | |||
| Normal | 9 | 4 | 18 |
| Prematurity alone | 1 | 0 | 1 |
| Preeclampsia alone | 0 | 1 | 1 |
| Gestational diabetes (GD) alone | 1 | 0 | 1 |
| Pre-eclampsia + prematurity | 2 | 0 | 0 |
| Pre-eclampsia + GD | 1 | 0 | 0 |
| Maternal fever during labor | 0 | 1 | 2 |
| Delivery route (Vaginal/C section) | 8/6 | 4/1 | 14/7 |
| Gestational age (weeks) (mean / SD) | 38/1. 7 | 38. 8/1. 3 | 38. 9/1. 5 |
| Paternal age (years) (mean / SD) | 39. 8/6. 3 | 36. 8/6 | 36. 6/7. 8 |
| Paternal age > 40 yearsd | 8 | 1 | 5 |
| Maternal age (years) (mean / SD) | 33/3. 8 | 34. 8/3. 5 | 34. 4/3. 5 |
| Maternal BMI (mean / SD) | 25. 03/4. 8 | 22/2. 96 | 25. 1/5 |
| Maternal BMI > 25 | 5/12e | 1 | 10/20f |
aAetiology for infertility: male + mixed versus no-male factor p = 0,36 (Chi square).
bTESA = Testicular sperm aspiration.
cNS = non-smoking; SSP = stopping smoking before pregnancy, S = smoking during pregnancy.
dPaternal age > 40 years: p = 0,45 (Chi square).
e2 BMI unknown.
f1 BMI unknown.
Validation of in vitro versus in vivo group DNA methylation using targeted bisulfite pyrosequencing
The top statistically significant differentially methylated CpGs included sites mapping to the promoter region of miR-663A (cg03940098, q = 0.01; cg18291941, q = 0.03), on 20p11.1, SLITRK5 (cg09083627, q = 0.01; cg15778745, q = 0.04), SLIT and NTRK-like family, member 5 on 13q31.2 and an uncharacterized protein-coding gene WBSCR17 (cg21135135, q = 0.04), Williams–Beuren syndrome chromosome region 17 on 7q11.22. These findings were further validated using bisulfite pyrosequencing on the same samples (n = 44) (Fig. 6), and the two methods demonstrated significant differences between the in vivo and in vitro groups.
Figure 6.
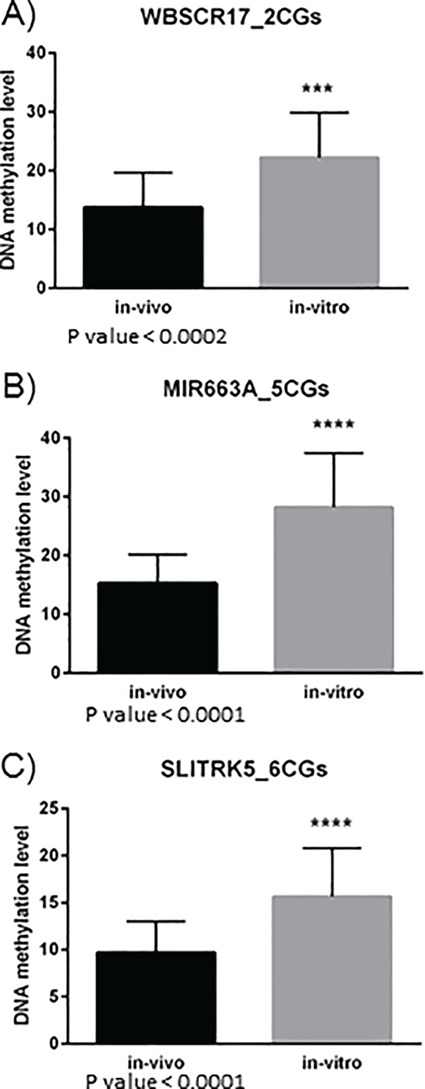
Pyrosequencing validation of three candidate regions identified between in vivo and in vitro comparison. Validation by bisulfite pyrosequencing on two CG sites overlapping WBSCR17 (A); five CG sites overlapping MIR663A including cg18291941 and cg03940098 (B) and six CG sites overlapping SLITRK5 including cg09083627 (C). Comparison was done on the same 21 in vivo and 23 in vitro samples. Statistical analysis was performed using unpaired two tailed t-test.
Testing of DNA methylation differences at candidate genes in an independent ART cohort
Next we wanted to verify that we could replicate our findings in an independent placenta cohort of samples conceived using IVF. We identified 7 in vitro cases conceived using IVF and 11 controls from placentas collected through the placental biobank at Mount Sinai Hospital in Toronto. Using available DNA methylation data from Illumina 450k array studies, we extracted the DNA methylation levels at the three candidate loci identified in our study overlapping miR-663A, SLITRK5 and WBSCR17. Since we did not have access to an in vivo group for comparison, we used the control group from the two cohorts for comparison of DNA methylation levels. As shown in Supplementary Material, Figure 4, both in the test and in the replication cohort there was an increase in DNA methylation in the in vitro group compared to the control group further validating our initial results with the ART samples from the 3D Study.
Pathway analysis of the candidate differentially methylated genes between in vivo and in vitro analysis.
We used Ingenuity Pathway Analysis to investigate which gene networks might be affected by the aberrant DNA methylation of the 200 genes overlapping the 436 CpG sites differentially methylated between in vivo and in vitro ART groups. The top three gene networks identified involved cellular assembly, function and organization; cellular and nervous system development and function; and reproductive system development and function (Supplementary Material, Figs 5–7). Prominent in the third network are genes important for oocyte function such as FOXL2 and the progesterone receptor gene (PGR) associated with establishment and maintenance of pregnancy. Other genes have important roles in cellular growth such as transforming growth factor alpha (TGFA). Also identified were several genes with neurological functions such as the glutamate ionotropic receptor AMPA type subunit 2 (GRIA2), SHANK 3; the gamma-aminobutyric acid (GABA) receptor, oxytocin (OXT) and the neuropeptide Y receptor Y5 (NPY5R). Analysis of the top functional categories of deregulated genes pointed to cellular growth and proliferation, cellular development and function and cell-to-cell signaling and connection (Table 4). The top associated diseases and disorders included developmental and neurological disorders. As for physiological system development and function, we found significant associations with embryonic and nervous system development.
Table 4.
Ingenuity pathway analysis
| Molecular and cellular functions | ||
| Name | P-value | #Molecules |
| Cellular growth and proliferation | 9.26E-03–5.55E-10 | 79 |
| Cellular development | 9.26E-03–6.93E-10 | 77 |
| Cell morphology | 9.26E-03–1.45E-05 | 35 |
| Cell-to-cell signaling and interaction | 9.26E-03–1.45E-05 | 39 |
| Cellular function and maintenance | 9.26E-03–1.45E-05 | 35 |
| Diseases and disorders | ||
| Name | P-value | #Molecules |
| Developmental disorder | 9.26E-03–5.31E-08 | 23 |
| Organismal injury and abnormalities | 9.26E-03–1.03E-07 | 98 |
| Neurological disease | 9.26E-03–3.93E-07 | 51 |
| Physiological system development and function | ||
| Name | P-value | #Molecules |
| Embryonic development | 9.26E-03–8.15E-18 | 62 |
| Organismal development | 9.26E-03–8.15E-18 | 67 |
| Nervous system development and function | 9.26E-03–6.18E-11 | 55 |
Discussion
Animal studies indicate that the placenta is susceptible to the induction of epigenetic abnormalities by ART. To date, few studies have examined the human placental DNA methylome from ART and matched control pregnancies using high density array-based approaches. Using 450K arrays we show that ART placentas are enriched for outliers with predominant DNA hypomethylation at imprinted genes, suggesting that only a subset of ART pregnancies may be susceptible to the induction of imprinting defects. Within the ART group, subgroups could be identified by DNA methylation clustering analysis, differentiated according to infertility and type of ART procedure and affecting numerous genomic regions including those involved in cellular and nervous system development and function. Together, the results indicate that the effects of ART extend beyond imprinted domains to other potentially important regions in the genome and support the use of well-characterized clinical samples and genome-wide studies to assess epigenetic abnormalities associated with infertility and ART.
For the current study ART and control pregnancies were from the same Quebec-based 3D-Study pregnancy cohort (30). In 2010 the province of Quebec implemented public funding for ART programs linked with a policy of single embryo transfer for women ≤35 years of age (31). Within 1 year of program implementation, multiple pregnancy rates, in ART pregnancies, in the province had decreased from 33.3 to 3.9% in women ≤35 years of age. Here, women were recruited between 2010–12 and thus most of the ART pregnancies in the 3D-Study cohort were singleton pregnancies. This low level of multiples amongst the ART group along with the 3D-Study design facilitated the matching of ART and control pregnancies for hospital of delivery, maternal age, gestational age at delivery, baby’s gender and maternal smoking status. As information on histories of infertility and time to pregnancy were collected, control pregnancies without a history of infertility could be selected. Other than infertility and/or ART the characteristics of the ART and control groups were very similar. By design, the ART group itself was heterogeneous in nature and divided roughly into two groups, those that were clinically infertile but subsequently conceived spontaneously with increased time to pregnancy or used IUI and those where the more invasive IVF or IVF/ICSI techniques were used. For all IVF and IVF/ICSI cases, embryos were transferred (the majority fresh) at the blastocyst stage, an important consideration since embryo culture has been associated with the induction of epigenetic abnormalities in mouse studies (13,16,29). Placental tissues were collected using standardized protocols and a specialized grid to identify sampling sites, minimizing differences between the control and ART groups that could potentially affect the DNA methylation results (32).
Our initial analysis of the 450K data did not identify differentially methylated CpGs between placentas of the ART and control groups. This is similar to the findings of a recent 450K methylation array study of air-dried blood spots from 76 children conceived by ICSI, 18 by IUI and 43 controls where general overall methylation differences between the groups were small; in this study PCA analysis did not cluster the three groups (23). Another recent 450K based study on cord blood of control and ICSI children also reported extensive overlap in methylation levels between ART and control samples and small differences in methylation between groups (26). In a genome-wide methylated DNA immunoprecipitation coupled with deep sequencing study of cord blood of twins, genome-wide significant signals in relation to method of conception (IVF versus non-IVF) were not identified; however, although only a single site differed with an FDR of 5%, 46 IVF-associated DMRs were identified at an FDR of 25% (25). Together the current study and recent equivalent genome-wide approaches suggest that heterogeneity among samples complicates the analysis of DNA methylation between control and ART pregnancies.
To explain the inconsistent results in the literature regarding differences in DNA methylation between control and ART cord blood or placentas, Ghosh and colleagues (27) proposed that individuals may differ in their susceptibility to environmental influences on epigenetic marks in early development. In support of their hypothesis they identified ‘outlier’ DNA methylation in the cord blood of a subset of children conceived by ART. ‘Outlier’ DNA hypomethylation has also been reported in a mouse ART model (19). Similarly, in the current study on placental methylation, while overall ART versus control group comparisons were not informative, PCA was able to identify an ‘outlier group’, enriched in ART pregnancies. The outlier group showed an altered DNA methylation pattern when compared to the non-outlier groups for the ART versus controls comparison. Lower DNA methylation was observed mostly for the intermediate DNA methylation ranges. Intermediate DNA methylation is characteristic of imprinted genes which are very important for placental development. Further, as altered DNA methylation at imprinted genes has been reported in several ART mouse and human studies, we identified an enrichment of the outlier samples with imprinted genes alterations.
While the magnitude of the methylation changes in imprinted genes was small, numerous CpG sites were affected in multiple imprinted genes. The major effect was imprinted gene hypomethylation, a finding also reported in placentas in human (21,33–35) and mouse ART studies (19). Our findings are biologically plausible as imprinted gene methylation must be maintained during preimplantation development, a time coinciding with the use of ARTs such as embryo culture, which in our study was predominantly through to the blastocyst stage.
Interestingly, although different analyses were used, both our study and that of Ghosh et al. (27) identified DNA methylation outliers associated with ART in similar proportions of pregnancies, 11 of 44 and 9 of 46, respectively. Despite the relatively small size of the ‘outlier’ group, it is intriguing that female sex emerged as significant; the results suggest that some female conceptuses may be more susceptible than males to the induction by ART of epigenetic abnormalities. Recent epidemiologic studies, where ART procedures would be similar to those used in our study, have reported sex skewing in humans with higher male birth rates (36,37). Both human and mouse model studies have suggested potential mechanisms underlying a skew toward males following ART. A systematic review and meta-analysis suggested that blastocyst transfer appears to be associated with a sex ratio skewed in favor of males (38). Prolonged embryo culture (i.e. the move toward transfers at 6 days rather than 3 days) is one of the factors implicated in perturbing early epigenetic patterns (1,13). The majority of the embryos in our cohort were transferred at the blastocyst stage due to Quebec’s single embryo transfer policy at the time the 3D Study was conducted. The requirement of DNA methylation for X chromosome events in female preimplantation embryos has been suggested to make them more vulnerable to conditions such as superovulation that might perturb oocyte or preimplantation embryo quality including the enzymes involved in DNA methylation (39,40). Fitting with this suggestion, in the mouse, lower levels of the DNA methylating enzyme, DNMT1o, in oocytes, more severely affected the histology and epigenetic profiles (with a preponderance of hypomethylation) of the placentas of females as compared to males (41). In another study, mouse IVF was associated with a skewed sex ratio (more males) and was attributed to impaired imprinted X chromosome inactivation (28). Together, both human and animal studies provide plausible explanations for our findings of increased placental epigenetic ‘outliers’ amongst females. As more ART clinics culture human embryos to the blastocyst stage, the potential epigenetic vulnerability of female conceptuses warrants careful attention in future epigenetic profiling studies of placentas and children.
Infertility of the parents may confound efforts to determine the contribution of techniques such as IVF and ICSI to alter epigenetic patterns in the offspring. One approach to this issue, which we took here, is to compare pregnancy outcomes between non-IVF/ICSI and IVF/ICSI-conceived conceptuses within the population seeking ART. Thus we examined placental DNA methylation in a subgroup of our samples, where parents were either clinically infertile, conceiving spontaneously with increased time to pregnancy or those using IUI, or were considered infertile and were treated with IVF/ICSI. The two types of infertile groups showed different methylation profiles genome-wide, suggesting that use of IVF/ICSI results in abnormal epigenetic outcomes. More marked, was the identification of a clear sub-group, enriched for paternal infertility and older paternal age, amongst the IVF/ICSI placentas. Both male infertility and paternal age have been linked to altered DNA methylation in sperm in 450K array based studies (42,43). The predominant type of epigenetic abnormality found in the placentas of our in vitro IVF/ICSI cohort was increased levels of DNA methylation. The results for a subset of genes were validated in the same cohort using bisulfite pyrosequencing and replicated in an independent Toronto cohort. Understanding the mechanism and basis for the observed hypermethylation and whether it is also found in the offspring or possibly identifies offspring that are more likely to have other types of epigenetic abnormalities will require further study. We suggest that epigenetic defects underlying male infertility and the techniques used in ART may interact. For instance, techniques such as superovulation or prolonged embryo culture may allow epigenetic alterations found in the sperm of infertile men to be propagated or persist and subsequently be detected in placenta. Interactions between infertility and techniques used in ART could be modeled in mouse studies, but to our knowledge, this has not been done.
Nevertheless, in comparing the in vivo versus in vitro infertile groups, there were many interesting genes and pathways identified. Many affected genes were related to cellular and embryonic growth and development including that of the nervous system. Similar pathways have been identified as enriched in placentas in a recent mouse ART study that examined DNA methylation on a genome-wide level (24). In our study, at the individual gene level, we identified SLITRK5, one of the most widely implicated genes in the pathology of obsessive compulsive disorders as well as many other neuropsychiatric disorders (44,45). MiR-663A was also identified and has been suggested to be associated with autism and different types of tumors based on multiple studies (46,47). Additionally, WBSCR17, another gene identified in our study, is a polypeptide N-acetylgalactosaminyltransferase (GalNAc-T) gene, which is designated Williams–Beuren syndrome chromosome region 17 (WBSCR17) because it is located in the chromosomal flanking region of the Williams–Beuren syndrome deletion; a range of neurodevelopmental issues including autistic traits is seen in patients with this syndrome (48,49). Further studies on genes involved in neurodevelopment and autism are warranted in the light of a recent meta-analysis suggesting that the use of ART may be associated with a higher risk of autism spectrum disorder (50). Interestingly SLITRK5, MiR-663A and WBSCR17 all have methylation values characteristic of those found in partially methylated domains (PMDs), regions of intermediate levels of DNA methylation. Genome-wide sequencing has identified large regions of PMDs in the placental methylome. Genes within PMDs are enriched in neuronal developmental genes, are repressed in placenta and have tissue-specific functions (51). Thus, alterations in DNA methylation within placental PMDs such as the increases in methylation seen between the in vitro and in vivo groups could potentially impact gene expression patterns postnatally.
While relatively small in size, our study has important strengths including the prospective nature of its design with careful matching of control and ART groups from the same study and the use of genome-wide DNA methylation analysis. We provide further support for the concept of more susceptible ART pregnancies or ‘outliers’ as well as the importance of including infertile individuals to help control for background infertility, and suggest that these factors should be considered as larger populations are studied. Our study also has limitations including those inherent in using 450K arrays (rather than a whole-genome-sequencing-based approach) that probe only a small percentage of the ∼30 million CpGs in the genome and exclude key regions such as enhancers and do not provide precise quantitative results as with sequencing, and the inability to test whether certain ART techniques are more likely than others to be associated with DNA methylation perturbations (e.g. prolonged embryo culture, frozen versus fresh embryo transfers). Important unanswered questions include whether placental epigenetic abnormalities associated with ART, such as those reported here, are tolerated by the placenta, can adversely affect normal placental function or perhaps provide an indicator of the most vulnerable conceptuses. In the context of a prospective longitudinal study such as the 3D study, epigenetic profiling of placenta at birth may provide guidance as to those children to follow more carefully later in life.
Materials and Methods
3D cohort study
The 3D Study, is a prospective, longitudinal study, in which pregnant women and their partners were recruited (May 2010–August 2012) from nine sites in Quebec and followed with their children up until 2 years of age (30). The primary objective of the 3D Study was to prospectively collect quality data in order to address the intrauterine determinants of important adverse obstetrical outcomes such as intrauterine growth retardation and preterm birth as well as the links between exposure from conception to birth and patterns of early childhood development. The 3D Study recruited 2366 women in their first trimester of pregnancy (10–14 weeks), and gathered extensive medical, obstetrical, environmental and socio-demographic data on the mother–father–child triad from conception until 2 years post-partum, correcting for age in premature infants.
After recruitment, participants were seen during the second (20–24 weeks) and third (32–35 weeks) trimesters of pregnancy and at delivery. The 3D Cohort included a sub-group pregnant after using ART with the same pattern of follow-up but with a specific questionnaire on the details of the ART used to achieve pregnancy. Questionnaire data were collected prenatally and postnatally along with various biospecimens. At delivery, cord blood, placenta samples, meconium and maternal and infant hair samples were collected. Blood and urine samples were obtained from the spouse of participating women.
Women were between 18–47 years old at the time of recruitment, able to communicate in French or in English and planned to deliver in one of the participating study hospitals in Quebec. Exclusion criteria included current intravenous drug use, severe illnesses or life-threatening conditions and multiple gestation pregnancies.
ART study design
A cohort study design was used for the epigenetic studies. As part of the main 3D Study, cases (the ART or infertility group) were from a single large academic university-associated infertility clinic (the McGill University Health Centre Reproductive Centre at the Royal Victoria Hospital – Montreal – Quebec, Canada) and gave birth at the Royal Victoria Hospital. Control pregnancies without a parental history of sub-fertility gave birth at the same hospital as the cases. The groups included the following: (1) controls (naturally conceived in <6 months), (2) ART or infertility cohort (= cases) distributed as follows: (i) clinical infertility subsequently conceiving spontaneously (time to pregnancy >1 year) with no medical treatment, (ii) infertility with the use of a medically assisted reproductive technique (ovarian stimulation and artificial insemination (IUI); classical IVF; ICSI. Exclusion criteria for the cases included low birth weight, macrosomia, use of preimplantation genetic diagnosis and the use of donor sperm. Participants that were unexposed to ART were selected based on the following matching criteria: same delivery hospital as cases, maternal age (+/− 2 years) at the time of delivery, gestational age at delivery with a matching on prematurity (before or after 37 weeks of gestation and overall +/− 2 weeks), baby’s sex, maternal tobacco status (never smoked, stopped tobacco before pregnancy, continued to smoke during pregnancy). This study received approval from the institutional ethics review board of the CHU Sainte-Justine Hospital Centre (acting as the central ethics review board for the 3D cohort study) in Montreal, Quebec.
Placenta collection
A 3D Study research assistant was present at the time of placenta delivery. After rinsing the placenta to remove excess blood, the placenta was placed on the fetal side, with the cord facing upward. Biopsies were taken by using a transparent grid with a hole for the cord and other holes for additional biopsies. Each transplacental biopsy was separated into three sections (chorion, trophoblast, decidual tissue) for immediate freezing in a bath of ethanol and dry ice, and then stored at −80°C. Central (near cord insertion) biopsies taken 1 cm below the chorionic plate were used for the current study. DNA extractions were performed with the QIAamp DNA Mini Kit (QIAGEN) according to manufacturer’s instructions.
DNA methylation analyses
We profiled a total of 88 placental samples, 44 samples derived from pregnancies conceived using ART and 44 samples derived from spontaneously conceived pregnancies (for demographic information, see Table 1). All these placental samples were collected at the same institution and within a 2 year period. Control and ART samples were matched for infant sex, gestational age and maternal age. Infants born from these pregnancies had normal birth weight percentile. Control and ART DNA samples were sodium bisulfite converted using the Qiagen EZ DNA Methylation kit (Qiagen, Valencia, CA), according to the manufacturers protocol. Modified genomic DNA was then processed and analyzed on the Infinium HumanMethylation450 BeadChip from Illumina (Illumina 450K) according to the manufacturer’s protocol. The distribution of the samples on the arrays was randomized for both cases and controls. Chi-square and Wilcoxon rank-sum tests were used to compare gender and age distributions between ART and control samples, respectively.
Normalization and quality controls
We used the GenomeStudio software from Illumina to process the raw intensity data (IDAT files) for all 88 samples. Control normalization and background subtraction included in GenomeStudio were used to generate DNA methylation profiles, or beta values, for each sample at every CpG site from the ∼485 000 CpG sites. All 88 samples passed the quality controls measures and had over 485 000 CpG sites detected at a detection P-value < 0.01. Further quality controls were used to exclude probes overlapping chromosomes X and Y, probes containing single nucleotide polymorphisms [SNPs with minor allele frequency >1% that is within 5 bp of single base extension site as previously described (52,53)], and low signal probes (if more than a third of samples have detection p-value >0.01). In addition, we excluded CpG sites if their probe sequences aligned to multiple positions with ≥90% identity [see (53) for additional details]. After removing all the above probes, 414 320 probes remained for further analyses.
Detection of outliers
Outliers were detected by sorting all 88 data samples along the first principal component PC1 (Fig. 1) and computing the gap sizes between neighboring samples. A change point detection algorithm was then applied to the series of 87 gap sizes using the function cpt.mean of the R package changepoint (54). The last 15 gap sizes in the series were identified as being substantially different from the rest of the data, which was detected by both the ‘at most one change’ and pruned exact linear time algorithms (Supplementary Material, Fig. 8). Therefore, we considered the 15 data samples with the largest PC1 values to be outliers. The detected change-point gap specifies the threshold value (approximately at PC1 = 17, see Fig. 1) that separates outliers from the rest of the dataset.
Differential DNA methylation analysis
A non-parametric Mann–Whitney U test was used to identify CpGs that were differentially methylated between comparison groups. P-values for the differentially methylated CpG sites were corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure and a q-value of 0.05 was used as a threshold for statistical significance. Beta values were imported into the Qlucore Omics Explorer (QOE v2.2). For unsupervised clustering analyses, the top 1000 differentially methylated probes between comparison groups were used and representative PCA plots and heatmaps were generated. For PCA plots, data normalization in QOE was set to mean = 0, variance = 1. Statistical significance of target differentially methylated CpG sites between various comparison groups was set to false discovery rate q < 0.01.
We used probe-level regression analysis implemented in limma R package to assess the influence of various factors on the DNA methylation level at each of the 414 320 CpG sites. The independent variables used in the linear regression model were the ART status, sex, smoking, delivery route, maternal age at delivery, paternal age, preterm status and outlier status (as defined before). The P-values associated with each of the factor values were corrected for multiple testing using the strict Bonferroni method (55) and the more permissive Benjamini-Hochberg (56) FDR correction method.
Bisulfite pyrosequencing
Targeted DNA methylation analysis for the top differentially methylated probes on the Illumina 450K array was performed using pyrosequencing as previously described (22). Pyrosequencing assays containing two PCR primers and one sequencing primer were designed to target CpG sites of interest using PyroMark Assay Design Software (Qiagen). One of the PCR primers has a universal tag that anneals to the universal biotinylated primer. Genomic DNA was sodium bisulfite converted in the same manner as for the Illumina microarray and amplified using Hot-Start Taq polymerase (Qiagen). Amplicons were analyzed on a Q24 pyrosequencer (Qiagen) as specified by the manufacturer. Percent methylation was quantified as the ratio of C to C + T using the PyroMark Q24 software (Qiagen). Pyrosequencing primers are provided in Supplementary Material, Table 5. For quantification of methylation level of the LINE-1 retrotransposable element, we used the PyroMark Q24 CpG LINE-1 kit (Qiagen) which uses real-time, sequence-based Pyrosequencing® technology to quantify methylation level of three CpG sites in positions 331–318 of LINE-1 (GenBank accession number X58075).
Supplementary Material
Acknowledgements
This project was conducted as part of the research program of the Integrated Network in Perinatology of Quebec and Eastern Ontario (IRNPQEO) 3D (Design, Develop, Discover) pregnancy cohort study. The authors thank all the participating families. The investigators of the 3D cohort group are William D. Fraser, Université de Sherbrooke; François Audibert, Université de Montréal; Lise Dubois, Université d’Ottawa; Pierre Julien, Université Laval; Zhong-Cheng Luo, University of Toronto; Jacques Michaud, Université de Montréal; Jean-Marie Moutquin, Université de Sherbrooke; Gina Muckle, Université Laval; Jean Séguin, Université de Montréal; Jacquetta Trasler, McGill University; Richard Ernest Tremblay, Université de Montréal; Haim Abenhaim, McGill University; Marie-Josée Bédard, Université de Montréal; Anick Bérard, Université de Montréal; Emmanuel Bujold, Université Laval; Robert Gagnon, McGill University; Isabelle Girard, McGill University; Zoha Kibar, Université de Montréal; Isabelle Marc, Université Laval; Patricia Monnier, McGill University; Marie-Noelle Simard, Université de Montréal; Jean-Charles Pasquier, Université de Sherbrooke; Michel Welt, Université de Montréal.
Conflict of Interest statement. None declared.
Contributor Information
3D cohort study group:
William D Fraser, François Audibert, Lise Dubois, Pierre Julien, Zhong-Cheng Luo, Jacques Michaud, Jean-Marie Moutquin, Gina Muckle, Jean Séguin, Jacquetta Trasler, Richard Ernest Tremblay, Haim Abenhaim, Marie-Josée Bédard, Anick Bérard, Emmanuel Bujold, Robert Gagnon, Isabelle Girard, Zoha Kibar, Isabelle Marc, Patricia Monnier, Marie-Noelle Simard, Jean-Charles Pasquier, and Michel Welt
Array DNA methylation data can be accessed from the gene expression omnibus GEO under the accession #GSE120250.
Funding
Canadian Institutes of Health Research (CIHR) (CRI 88413 to A.B., W.D.F., P.M., R.W. and J.T., FND-148425 to J.T.); The Natural Sciences and Engineering Research Council of Canada-NSERC (418734–2012 to R.W.); Mount Sinai Department of Obstetrics and Gynaecology’s Research Fund for the replication cohort (to R.W.). Bioinformatics analyses were supported in part by the Canadian Centre for Computational Genomics (C3G), part of the Genome Technology Platform (GTP), funded by Genome Canada through Genome Quebec and Ontario Genomics (to A.L.T. and M.B.); and Genome Canada through Ontario Genomics (to A.L.T., S.C., M.B., and R.W.).
References
- 1. Lucifero D., Chaillet J.R. and Trasler J.M. (2004) Potential significance of genomic imprinting defects for reproduction and assisted reproductive technology. Hum. Reprod. Update, 10, 3–18. [DOI] [PubMed] [Google Scholar]
- 2. Cox G.F., Burger J., Lip V., Mau U.A., Sperling K., Wu B.L. and Horsthemke B. (2002) Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am. J. Human Genet., 71, 162–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DeBaun M.R., Niemitz E.L. and Feinberg A.P. (2003) Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet., 72, 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gicquel C., Gaston V., Mandelbaum J., Siffroi J.P., Flahault A. and Le Bouc Y. (2003) In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am. J. Hum. Genet., 72, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maher E.R., Brueton L.A., Bowdin S.C., Luharia A., Cooper W., Cole T.R., Macdonald F., Sampson J.R., Barratt C.L., Reik W. et al. (2003) Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J. Med. Genet., 40, 62–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orstavik K.H., Eiklid K., Hagen C.B., Spetalen S., Kierulf K., Skjeldal O. and Buiting K. (2003) Another case of imprinting defect in a girl with Angelman syndrome who was conceived by intracytoplasmic semen injection. Am. J. Hum. Genet., 72, 218–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gosden R., Trasler J., Lucifero D. and Faddy M. (2003) Rare congenital disorders, imprinted genes, and assisted reproductive technology. Lancet, 361, 1975–1977. [DOI] [PubMed] [Google Scholar]
- 8. Houshdaran S., Cortessis V.K., Siegmund K., Yang A., Laird P.W. and Sokol R.Z. (2007) Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. PLoS One, 2, e1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi H., Sato A., Otsu E., Hiura H., Tomatsu C., Utsunomiya T., Sasaki H., Yaegashi N. and Arima T. (2007) Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum. Mol. Genet., 16, 2542–2551. [DOI] [PubMed] [Google Scholar]
- 10. Marques C.J., Costa P., Vaz B., Carvalho F., Fernandes S., Barros A. and Sousa M. (2008) Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol. Hum. Reprod., 14, 67–74. [DOI] [PubMed] [Google Scholar]
- 11. Hammoud S.S., Purwar J., Pflueger C., Cairns B.R. and Carrell D.T. (2010) Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertility and sterility, 94, 1728–1733. [DOI] [PubMed] [Google Scholar]
- 12. Poplinski A., Tuttelmann F., Kanber D., Horsthemke B. and Gromoll J. (2010) Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int. J. Androl., 33, 642–649. [DOI] [PubMed] [Google Scholar]
- 13. Mann M.R., Lee S.S., Doherty A.S., Verona R.I., Nolen L.D., Schultz R.M. and Bartolomei M.S. (2004) Selective loss of imprinting in the placenta following preimplantation development in culture. Development, 131, 3727–3735. [DOI] [PubMed] [Google Scholar]
- 14. Rivera R.M., Stein P., Weaver J.R., Mager J., Schultz R.M. and Bartolomei M.S. (2008) Manipulations of mouse embryos prior to implantation result in aberrant expression of imprinted genes on day 9.5 of development. Hum. Mol. Genet., 17, 1–14. [DOI] [PubMed] [Google Scholar]
- 15. Fortier A.L., Lopes F.L., Darricarrere N., Martel J. and Trasler J.M. (2008) Superovulation alters the expression of imprinted genes in the midgestation mouse placenta. Hum. Mol. Genet., 17, 1653–1665. [DOI] [PubMed] [Google Scholar]
- 16. Market-Velker B.A., Fernandes A.D. and Mann M.R. (2010) Side-by-side comparison of five commercial media systems in a mouse model: suboptimal in vitro culture interferes with imprint maintenance. Biol. Reprod., 83, 938–950. [DOI] [PubMed] [Google Scholar]
- 17. Waal E., Yamazaki Y., Ingale P., Bartolomei M.S., Yanagimachi R. and McCarrey J.R. (2012) Gonadotropin stimulation contributes to an increased incidence of epimutations in ICSI-derived mice. Hum. Mol. Genet., 21, 4460–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Z., Robbins K.M., Wells K.D. and Rivera R.M. (2013) Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics, 8, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waal E., Vrooman L.A., Fischer E., Ord T., Mainigi M.A., Coutifaris C., Schultz R.M. and Bartolomei M.S. (2015) The cumulative effect of assisted reproduction procedures on placental development and epigenetic perturbations in a mouse model. Hum. Mol. Genet., 24, 6975–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lazaraviciute G., Kauser M., Bhattacharya S., Haggarty P. and Bhattacharya S. (2014) A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum. Reprod. Update, 20, 840–852. [DOI] [PubMed] [Google Scholar]
- 21. Katari S., Turan N., Bibikova M., Erinle O., Chalian R., Foster M., Gaughan J.P., Coutifaris C. and Sapienza C. (2009) DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum. Mol. Genet., 18, 3769–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Melamed N., Choufani S., Wilkins-Haug L.E., Koren G. and Weksberg R. (2015) Comparison of genome-wide and gene-specific DNA methylation between ART and naturally conceived pregnancies. Epigenetics, 10, 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Estill M.S., Bolnick J.M., Waterland R.A., Bolnick A.D., Diamond M.P. and Krawetz S.A. (2016) Assisted reproductive technology alters deoxyribonucleic acid methylation profiles in bloodspots of newborn infants. Fertil. Steril., 106, 629–639, e610. [DOI] [PubMed] [Google Scholar]
- 24. Whidden L., Martel J., Rahimi S., Chaillet J.R., Chan D. and Trasler J.M. (2016) Compromised oocyte quality and assisted reproduction contribute to sex-specific effects on offspring outcomes and epigenetic patterning. Hum. Mol. Genet., 25, 4649–4660. [DOI] [PubMed] [Google Scholar]
- 25. Castillo-Fernandez J.E., Loke Y.J., Bass-Stringer S., Gao F., Xia Y., Wu H., Lu H., Liu Y., Wang J., Spector T.D. et al. (2017) DNA methylation changes at infertility genes in newborn twins conceived by in vitro fertilisation. Genome Med., 9, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El Hajj N., Haertle L., Dittrich M., Denk S., Lehnen H., Hahn T., Schorsch M. and Haaf T. (2017) DNA methylation signatures in cord blood of ICSI children. Hum. Reprod., 32, 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghosh J., Mainigi M., Coutifaris C. and Sapienza C. (2016) Outlier DNA methylation levels as an indicator of environmental exposure and risk of undesirable birth outcome. Hum. Mol. Genet., 25, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan K., An L., Miao K., Ren L., Hou Z., Tao L., Zhang Z., Wang X., Xia W., Liu J. et al. (2016) Impaired imprinted X chromosome inactivation is responsible for the skewed sex ratio following in vitro fertilization. Proc. Natl. Acad. Sci. USA, 113, 3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waal E., Mak W., Calhoun S., Stein P., Ord T., Krapp C., Coutifaris C., Schultz R.M. and Bartolomei M.S. (2014) In vitro culture increases the frequency of stochastic epigenetic errors at imprinted genes in placental tissues from mouse concepti produced through assisted reproductive technologies. Biol. Reprod., 90, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fraser W.D., Shapiro G.D., Audibert F., Dubois L., Pasquier J.C., Julien P., Berard A., Muckle G., Trasler J., Tremblay R.E. et al. (2016) 3D Cohort Study: the integrated research network in perinatology of Quebec and Eastern Ontario. Paediatr. Perinat. Epidemiol., 30, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Velez M.P., Connolly M.P., Kadoch I.J., Phillips S. and Bissonnette F. (2014) Universal coverage of IVF pays off. Hum. Reprod., 29, 1313–1319. [DOI] [PubMed] [Google Scholar]
- 32. Hogg K., Price E.M. and Robinson W.P. (2014) Improved reporting of DNA methylation data derived from studies of the human placenta. Epigenetics, 9, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rancourt R.C., Harris H.R. and Michels K.B. (2012) Methylation levels at imprinting control regions are not altered with ovulation induction or in vitro fertilization in a birth cohort. Hum. Reprod., 27, 2208–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelissen E.C., Dumoulin J.C., Daunay A., Evers J.L., Tost J. and Montfoort A.P. (2013) Placentas from pregnancies conceived by IVF/ICSI have a reduced DNA methylation level at the H19 and MEST differentially methylated regions. Hum. Reprod., 28, 1117–1126. [DOI] [PubMed] [Google Scholar]
- 35. Choux C., Binquet C., Carmignac V., Bruno C., Chapusot C., Barberet J., Lamotte M., Sagot P., Bourc'his D. and Fauque P. (2018) The epigenetic control of transposable elements and imprinted genes in newborns is affected by the mode of conception: ART versus spontaneous conception without underlying infertility. Hum. Reprod., 33, 331–340. [DOI] [PubMed] [Google Scholar]
- 36. Maalouf W.E., Mincheva M.N., Campbell B.K. and Hardy I.C. (2014) Effects of assisted reproductive technologies on human sex ratio at birth. Fertil. Steril., 101, 1321–1325. [DOI] [PubMed] [Google Scholar]
- 37. Dean J.H., Chapman M.G. and Sullivan E.A. (2010) The effect on human sex ratio at birth by assisted reproductive technology (ART) procedures—an assessment of babies born following single embryo transfers, Australia and New Zealand, 2002–2006. BJOG, 117, 1628–1634. [DOI] [PubMed] [Google Scholar]
- 38. Chang H.J., Lee J.R., Jee B.C., Suh C.S. and Kim S.H. (2009) Impact of blastocyst transfer on offspring sex ratio and the monozygotic twinning rate: a systematic review and meta-analysis. Fertil. Steril., 91, 2381–2390. [DOI] [PubMed] [Google Scholar]
- 39. Bestor T.H. (2003) Imprinting errors and developmental asymmetry. Philos. Trans. R. Soc. Lond. B Biol. Sci., 358, 1411–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weksberg R., Shuman C., Caluseriu O., Smith A.C., Fei Y.L., Nishikawa J., Stockley T.L., Best L., Chitayat D., Olney A. et al. (2002) Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum. Mol. Genet., 11, 1317–1325. [DOI] [PubMed] [Google Scholar]
- 41. McGraw S., Oakes C.C., Martel J., Cirio M.C., Zeeuw P., Mak W., Plass C., Bartolomei M.S., Chaillet J.R. and Trasler J.M. (2013) Loss of DNMT1o disrupts imprinted X chromosome inactivation and accentuates placental defects in females. PLoS Genet., 9, e1003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jenkins T.G., Aston K.I., Pflueger C., Cairns B.R. and Carrell D.T. (2014) Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet., 10, e1004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aston K.I., Uren P.J., Jenkins T.G., Horsager A., Cairns B.R., Smith A.D. and Carrell D.T. (2015) Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril., 104, 1388–1397e1381–1385. [DOI] [PubMed] [Google Scholar]
- 44. Proenca C.C., Gao K.P., Shmelkov S.V., Rafii S. and Lee F.S. (2011) Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci., 34, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song M., Mathews C.A., Stewart S.E., Shmelkov S.V., Mezey J.G., Rodriguez-Flores J.L., Rasmussen S.A., Britton J.C., Oh Y.S., Walkup J.T. et al. (2017) Rare synaptogenesis-impairing mutations in SLITRK5 are associated with obsessive compulsive disorder. PLoS One, 12, e0169994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mundalil Vasu M., Anitha A., Thanseem I., Suzuki K., Yamada K., Takahashi T., Wakuda T., Iwata K., Tsujii M., Sugiyama T. et al. (2014) Serum microRNA profiles in children with autism. Molecular autism, 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y., Xu X., Zhang M., Wang X., Bai X., Li H., Kan L., Zhou Y., Niu H. and He P. (2016) MicroRNA-663a is downregulated in non-small cell lung cancer and inhibits proliferation and invasion by targeting JunD. BMC Cancer, 16, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Francke U. (1999) Williams-Beuren syndrome: genes and mechanisms. Hum. Mol. Genet., 8, 1947–1954. [DOI] [PubMed] [Google Scholar]
- 49. Connolly S., Anney R., Gallagher L. and Heron E.A. (2017) A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur. J. Hum. Genet., 25, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu L., Gao J., He X., Cai Y., Wang L. and Fan X. (2017) Association between assisted reproductive technology and the risk of autism spectrum disorders in the offspring: a meta-analysis. Sci. Rep., 7, 46207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schroeder D.I. and LaSalle J.M. (2013) How has the study of the human placenta aided our understanding of partially methylated genes? Epigenomics, 5, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Choufani S., Shapiro J.S., Susiarjo M., Butcher D.T., Grafodatskaya D., Lou Y., Ferreira J.C., Pinto D., Scherer S.W., Shaffer L.G. et al. (2011) A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res., 21, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Y.-A., Lemire M., Choufani S., Butcher D.T., Grafodatskaya D., Zanke B.W., Gallinger S., Hudson T.J. and Weksberg R. (2013) Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics, 8, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Killick R., Eckley I.A.. (2014) changepoint: an R package for changepoint analysis. J. Stat. Softw., 58, 1–19. [Google Scholar]
- 55. Bland J.M. and Altman D.G. (1995) Multiple significance tests: the Bonferroni method. BMJ, 310, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Benjamini Y., Hochberg and Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B, 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.