

Abstract
Building a cerebral cortex of the proper size involves balancing rates and timing of neural stem cell (NSC) proliferation, neurogenesis and cell death. The cellular mechanisms connecting genetic mutations to brain malformation phenotypes are still poorly understood. Microcephaly may result when NSC divisions are too slow, produce neurons too early or undergo apoptosis but the relative contributions of these cellular mechanisms to various types of microcephaly are not understood. We previously showed that mouse mutants in Kif20b (formerly called Mphosph1, Mpp1 or KRMP1) have small cortices that show elevated apoptosis and defects in maturation of NSC midbodies, which mediate cytokinetic abscission. Here we test the contribution of intrinsic NSC apoptosis to brain size reduction in this lethal microcephaly model. By making double mutants with the pro-apoptotic genes Bax and Trp53 (p53), we find that p53-dependent apoptosis of cortical NSCs accounts for most of the microcephaly, but that there is a significant apoptosis-independent contribution as well. Remarkably, heterozygous p53 deletion is sufficient to fully rescue survival of the Kif20b mutant into adulthood. In addition, the NSC midbody maturation defects are not rescued by p53 deletion, showing that they are either upstream of p53 activation, or in a parallel pathway. Accumulation of p53 in the nucleus of mutant NSCs at midbody stage suggests the possibility of a novel midbody-mediated pathway for p53 activation. This work elucidates both NSC apoptosis and abscission mechanisms that could underlie human microcephaly or other brain malformations.
Introduction
Human genetics is increasingly successful at linking specific gene mutations to congenital brain malformations and other neurodevelopmental disorders. However, the cellular mechanisms connecting genetic mutations to brain phenotypes are still poorly understood. There are 17 human primary microcephaly genes identified, and there are many syndromes that feature microcephaly [(1); Online Mendelian Inheritance in Man (OMIM) database]. Thus, it is a heterogeneous disorder. Known microcephaly genes encode proteins with diverse molecular functions, but many are involved in cell division.
Cell division genes may be prominent in brain malformations like microcephaly because cortical neural stem cell (NSC) divisions have several unusual features (2). NSCs are tall, thin cells that reside in the pseudostratified epithelium of the cortex, with their apical endfeet forming the ventricular surface, and their basal processes stretching to the basal lamina beneath the meninges. Their nuclei undergo interkinetic nuclear migration during the cell cycle, moving basally for S phase (DNA Synthesis phase) and to the apical membrane for Mphase (Mitosis phase) and cytokinesis. In addition, NSC divisions can produce symmetric or asymmetric daughter fates, giving rise to more NSCs, neurons, intermediate progenitors (IPs) and glia during corticogenesis. These stem cells must produce the right types and numbers of daughter cells within specific windows of time. With all these complex demands, the developing cortex is particularly vulnerable to insults to cell division.
We previously identified a mouse model of microcephaly that is recessive, perinatal lethal and relatively severe, with a brain about half as thick as normal during late gestation. It carries a recessive loss-of-function mutation in the kinesin microtubule motor gene Kif20b that causes a splicing error, frameshift and reduction of the protein to undetectable levels. These Kif20bmagoo mutant brains do not display NSC mitotic arrest or abnormal cleavage angles, which have been noted in other microcephaly mutants. Instead, Kif20b mutant brains display defects in cytokinetic abscission (3,4). Abscission is the process of severing the connection between mother and daughter cell, taking an hour or more after telophase (5). The cleavage furrow compacts the central spindle microtubules into the midbody, which mediates abscission by recruiting proteins to remodel and ‘cut’ the cytoskeleton and membrane. We showed that Kif20b protein localizes to the central spindle and midbody in human cell lines and mouse NSCs (4,6). Furthermore, Kif20b appears to facilitate changes in midbody microtubule structure as the midbody `matures’ during the abscission process, and ensures timely abscission in cell lines. Suggesting that it may accelerate cell division, Kif20b is elevated in some cancers (7–9). Interestingly, Kif20b evolved with the vertebrate lineage, so its subtle role in abscission may be important for growing bigger, more complex nervous systems.
In addition to abnormal midbodies, Kif20bmagoo mutants (called Kif20b mutant or Kif20bm/m hereafter) also display increased apoptosis in the embryonic cortex from embryonic day (E)10.5–16.5 (4). However, it was unclear whether the relatively small amount of detectable apoptosis observed could account for the severity of the microcephaly. Apoptosis appears to be relatively low in the healthy embryonic neocortex. Prevention of apoptosis by knockout of caspase-9 or caspase-3 does cause cortical overgrowth, but this appears to be because of failure of apoptosis of a small cluster of FGF8-secreting cells that is normally eliminated around E10.5, rather than failure of widespread programmed cell death (10–13). Elevated apoptosis is seen in some mouse models of brain malformations (14–20). The intrinsic or stress-induced apoptotic pathway can be triggered by environmental stresses or genetic insults, such as particular mutations (21). Bax and p53 (gene Trp53) are expressed in embryonic brain and appear to respond to damage. In response to apoptotic stimuli, Bax, a multi-BH domain-containing member of the BCL2 family, can form pores across the outer mitochondrial membrane, thereby releasing cytochrome c that can activate the caspase cascade. p53, a tumor suppressor mutated in many human cancers, is an upstream activator of Bax (22). Mouse knockouts of Bax and p53 have normal brain development with surprisingly few low penetrance defects (16,23,24).
Here, we set out to determine the relative contributions of apoptosis and abscission dysregulation to the microcephaly of the Kif20b mutant and to understand the relationship between these phenotypes. To do this, we crossed genetic mutants of the intrinsic apoptosis pathway to Kif20b mutant mice and asked whether the apoptosis, microcephaly and abscission defects in Kif20b mutants were rescued, unaffected or potentially worsened. To our surprise, we found that p53 deletion prevented apoptosis and rescued brain size and structure to a remarkable degree. A partial apoptosis inhibition by Bax deletion correlated with a lesser extent of brain size rescue. Surprisingly, deletion of even one allele of p53 is able to completely block the apoptosis and lethality in Kif20b mutants. However, Kif20b; p53 double mutant brains are still smaller than controls at birth, suggesting that Kif20b regulates cortical development through additional p53-independent mechanisms, perhaps by ensuring timely abscission. Indeed, p53 deletion does not rescue the defects in midbody structure seen in Kif20b mutant NSCs, and additional midbody defects are revealed when p53 is deleted. Our data provide the first evidence that abscission defects may cause p53 accumulation in NSCs or any cell type, through a yet-to-be identified pathway. Finally, our work informs apoptosis inhibition as a potential way to ameliorate the severity of microcephaly caused by genetic, viral or environmental insults.
Results
The intrinsic apoptotic pathway is the key driver of microcephaly in Kif20b mutant mice
To test whether the intrinsic apoptotic pathway mediates the elevated apoptosis and microcephaly in the Kif20b mutant cortex, we utilized genetic crosses to mutants in two key genes in this pathway, Bax and p53 (Trp53). Importantly, homozygous deletion mutants in Bax or p53 develop properly with normal size cortices (16,23,24). Due to the low number of true wild-type +/+ mice (1/16) produced from double heterozygote crosses, we elected to use double heterozygotes (Kif20b+/m; Bax+/− and Kif20b+/m; p53+/−) as controls in order to compare to single and double mutant littermates. First, we produced Kif20b; Bax double mutants. Bax, together with its partner Bak, increases the permeability of the mitochondrial membrane, increasing cytochrome c release (25). We examined double mutant brains at age E14.5, when apoptosis is elevated in Kif20b mutants, and the cortical plate (cp) has started to form. Interestingly, embryos carrying homozygous mutations in both Bax and Kif20b showed a partial block of the elevated apoptosis, as marked by cleaved caspase-3 (CC3), and a partial rescue of cortical thickness (Fig. 1A–J). Heterozygous deletion of Bax showed a trend for mild rescue, but this result did not reach statistical significance. Craniofacial defects observed in Kif20b mutants were not rescued by heterozygous or homozygous Bax mutation (data not shown). These data suggest that apoptosis and microcephaly are correlated, but that additional proteins are required for the full apoptotic response to Kif20b loss. For example, Bak is a partner of Bax and is partially redundant (26).
Figure 1.
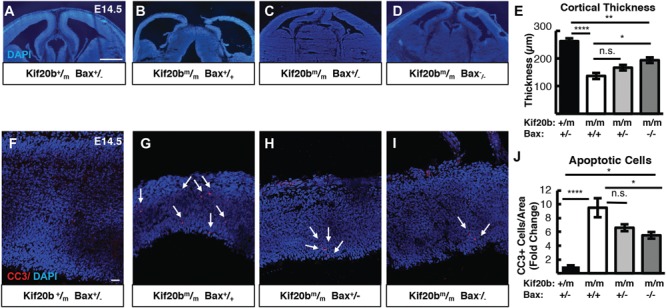
Bax deletion partially rescues microcephaly and apoptosis in Kif20b mutant mice. (A-E). Representative E14.5 cortical sections and plotted mean of cortical thicknesses show the severe reduction in Kif20b mutants is partly restored in Kif20b; Bax double mutants. (F-J) CC3 staining in E14.5 cortices show the increased apoptosis in Kif20b mutants is partially reduced in Kif20b;Bax double mutants. Arrows denote apoptotic cells. For (E, J), n = 5 Kif20b+/m; Bax+/−, 4 Kif20b ; Bax+/+, 4 Kif20b
; Bax+/+, 4 Kif20b ; Bax+/−, 5 Kif20b
; Bax+/−, 5 Kif20b ; Bax−/− mice, from a total of 8 litters. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. not significant, one-way ANOVA. Error bars are +/− s.e.m. Scale bars: A: 500 μm; E: 20 μm.
; Bax−/− mice, from a total of 8 litters. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. not significant, one-way ANOVA. Error bars are +/− s.e.m. Scale bars: A: 500 μm; E: 20 μm.
The tumor suppressor p53 is upstream of Bax/Bak in the intrinsic apoptotic pathway. Therefore, we tested whether p53 deletion could fully block the apoptosis in Kif20b mutants by crossing to the Trp53 mutant (23). Strikingly, apoptosis and cortical thickness of E14.5 Kif20b m/m embryos are both fully rescued by either heterozygous or homozygous deletion of p53 (Fig. 2A–J). Thus, two functional p53 genes are required to produce the apoptosis and microcephaly triggered by Kif20b loss. Furthermore, p53 is required for the craniofacial defects of Kif20bm/m embryos: either heterozygous or homozygous p53 deletion significantly ameliorates the small eye and snout phenotypes (Supplementary Material, Fig. S1). p53 protein appears to be elevated in Kif20b mutant cortices, as immunoblots of E12.5 cortical lysates show p53 band intensity increased 50% in Kif20bm/m samples, normalized to the NSC protein beta-catenin (Supplementary Material, Fig. S2A and B). This increase was not detectable by immunohistochemistry on cortical sections (data not shown). However, immunostaining for p53 on dissociated NSC cultures shows an increase in the median nuclear-to-cytoplasmic ratio (N:C ratio) of p53 in Kif20b mutant NSCs (Nestin positive), but not in neurons or other cell types (Nestin negative) (Supplementary Material, Fig. S2C–F). Together, these data show that p53 accumulates in the nucleus of NSCs when Kif20b is lost, and p53 function is required for the excess apoptosis and microcephaly in this mutant. Moreover, the partial and full rescues of microcephaly by Bax and p53 deletion show that the amount of apoptosis and the severity of microcephaly are strongly correlated, suggesting that apoptosis is the key cellular mechanism driving microcephaly in Kif20b mutant mice.
Figure 2.
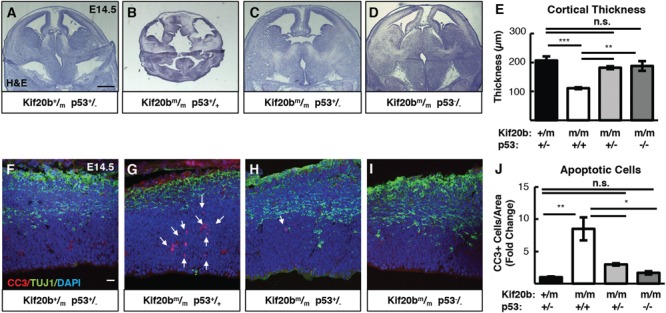
p53 deletion rescues microcephaly and apoptosis in Kif20b mutant mice. (A-E) E14.5 cortical sections and plotted mean cortical thicknesses show the severe reduction in Kif20b mutants is fully rescued by heterozygous or homozygous p53 deletion. n = 6 Kif20b+/m; p53+/−, 3 Kif20b ; p53+/+, 4 Kif20b
; p53+/+, 4 Kif20b ; p53+/−, 4 Kif20b
; p53+/−, 4 Kif20b ; p53−/− mice, from a total of 7 litters. (F-J) CC3 staining shows apoptosis in Kif20b mutants returns to control levels by heterozygous or homozygous p53 deletion. Arrows denote apoptotic cells. n = 3 Kif20b+/m; p53+/−, Kif20b
; p53−/− mice, from a total of 7 litters. (F-J) CC3 staining shows apoptosis in Kif20b mutants returns to control levels by heterozygous or homozygous p53 deletion. Arrows denote apoptotic cells. n = 3 Kif20b+/m; p53+/−, Kif20b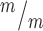 ;
p53+/+, Kif20b
;
p53+/+, Kif20b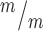 ; p53+/−, Kif20b
; p53+/−, Kif20b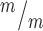 ; p53−/− mice, from a total of 6 litters. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant, one-way ANOVA. Error bars are +/− s.e.m. Scale bars: A: 500 μm; F: 20 μm.
; p53−/− mice, from a total of 6 litters. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant, one-way ANOVA. Error bars are +/− s.e.m. Scale bars: A: 500 μm; F: 20 μm.
p53 deletion restores growth of neuronal and subventricular layers in embryonic Kif20b mutant cortex
We previously showed that the reduced cortical thickness of Kif20b mutants is because of thinner neuronal layers and fewer IPs (4). To determine if p53 deletion could rescue cortical neurogenesis and structure as well as thickness, we labeled Kif20b single mutant and Kif20b; p53 double mutant E14.5 cortical sections for Pax6, Tuj1 and Tbr2, to label NSCs, neurons and IPs, respectively. In Kif20bm/m single mutant brains, as expected, the neuronal layer (nascent cp) is thin, and the axonal layer (intermediate zone, iz) is barely detected (Fig. 3A and B). As a proportion of total cortical thickness, the ventricular zone (vz) is increased, and IPs are reduced (Fig. 3D and E). Remarkably, double mutant Kif20bm/m; p53−/− embryos have cortices that appear to have normal organization, with cortical plates and intermediate zones indistinguishable from controls (Fig. 3C and G). Additionally, Kif20bm/m; p53−/− embryos display normal vz thickness and IP generation (Fig. 3F, H and I). Therefore, blocking apoptosis by p53 deletion in the cortices of Kif20bm/m brains increased cortical thickness by improving production or survival of neurons and IPs. Furthermore, the neurons and IPs in double mutant brains appear to migrate normally and create a normal-appearing structure.
Figure 3.
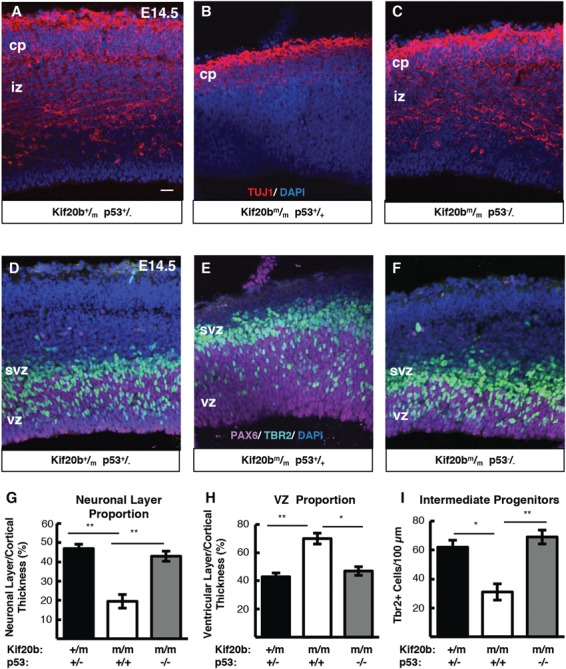
p53 deletion restores normal production of IPs and neurons in Kif20b mutant cortex. (A-C) E14.5 cortical sections labeled with neuronal-specific tubulin (Tuj1, red) show the decreased neuronal layer thickness in Kif20b mutants is rescued in Kif20b; p53 double mutants. cp, cortical plate; iz, intermediate zone. (D-F) E14.5 cortices labeled with Pax6 (NSCs, magenta) and Tbr2 (IPs, green) show the altered layer proportions in Kif20b mutant are rescued by p53 deletion. Svz, subventricular zone; vz, ventricular zone. (G) The mean neuronal layer proportional thickness is halved in Kif20b mutants but rescued by p53 co-deletion. (H) The proper NSC layer (Pax6+) proportionality is restored by p53 co-deletion. (I) The density of Tbr2+ IPs is reduced in Kif20b mutants, but rescued in Kif20b; p53 double mutants. n = 3 each Kif20b+/m; p53+/−, Kif20b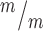 ; p53+/+, Kif20b
; p53+/+, Kif20b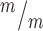 ; p53−/− mice for Tuj1 and Pax6 layer thickness. n = 6 Kif20b+/m; p53+/−, 3 Kif20b
; p53−/− mice for Tuj1 and Pax6 layer thickness. n = 6 Kif20b+/m; p53+/−, 3 Kif20b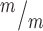 ; p53+/+, 4 Kif20b
; p53+/+, 4 Kif20b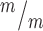 ; p53−/− mice for Tbr2+ cell counts, from a total of 6 litters. *P < 0.05, **P < 0.01, one-way ANOVA. Error bars are +/− s.e.m. Scale bars: 20 μm.
; p53−/− mice for Tbr2+ cell counts, from a total of 6 litters. *P < 0.05, **P < 0.01, one-way ANOVA. Error bars are +/− s.e.m. Scale bars: 20 μm.
p53 deletion rescues postnatal survival but not full cortical size of Kif20b mutant mice at birth
In addition to reduced brain size, Kif20b mutant mice exhibit perinatal lethality. Remarkably, while no Kif20bm/m mutant mice with wild-type p53 status survive the day of birth (postnatal day 0, P0), Kif20bm/m mice with heterozygous or homozygous p53 deletion survive postnatally at expected Mendelian ratios (Fig. 4A). Even more surprising, most Kif20bm/m; p53+/− mice and Kif20bm/m; p53−/− mice live to adulthood and are fertile. Some Kif20bm/m; p53+/− mice (∼15%) still have visible craniofacial defects including a small eye or short snout, and ∼5% have hydrocephalus, but the majority have normal facial structure. Double homozygotes (Kif20bm/m; p53−/−) have even fewer craniofacial defects, but die prematurely at ∼3–4 months of age due to spontaneous tumors, as do p53−/− single mutants (23).
Figure 4.
p53 deletion rescues postnatal survival but incompletely rescues cortical size at birth in Kif20b mutant mice. (A)Kif20b ; p53+/+ mice do not survive at birth at expected Mendelian ratios, but Kif20b
; p53+/+ mice do not survive at birth at expected Mendelian ratios, but Kif20b ; p53+/− and Kif20b
; p53+/− and Kif20b ; p53−/− mice survive at comparable levels to controls. (B, C) The average cortical length of Kif20b
; p53−/− mice survive at comparable levels to controls. (B, C) The average cortical length of Kif20b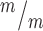 ; p53−/− mice is slightly decreased compared to controls at P0. n = 54 Kif20b+/+ or +/− p53+/+ or +/− mice, 22 Kif20b+/+ or +/− p53−/− and 5 Kif20b
; p53−/− mice is slightly decreased compared to controls at P0. n = 54 Kif20b+/+ or +/− p53+/+ or +/− mice, 22 Kif20b+/+ or +/− p53−/− and 5 Kif20b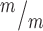 ; p53−/− mice. (D, E) Coronal sections from P0 rostral forebrains stained with hematoxylin and eosin show reduced cortical thickness and enlarged ventricles of double mutants. (F) Cortical thickness in Kif20b
; p53−/− mice. (D, E) Coronal sections from P0 rostral forebrains stained with hematoxylin and eosin show reduced cortical thickness and enlarged ventricles of double mutants. (F) Cortical thickness in Kif20b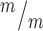 ; p53−/− mice is reduced 15–20% compared with Kif20b+/m; p53+/− controls in rostral, middle and caudal sections at P0. (G) cp and iz show reduced thickness, but not vz/svz layers. For F and G, n = 6 Kif20b+/m; p53+/−, 3 Kif20b+/m; p53−/− and 6 Kif20b
; p53−/− mice is reduced 15–20% compared with Kif20b+/m; p53+/− controls in rostral, middle and caudal sections at P0. (G) cp and iz show reduced thickness, but not vz/svz layers. For F and G, n = 6 Kif20b+/m; p53+/−, 3 Kif20b+/m; p53−/− and 6 Kif20b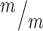 ; p53−/− brains. *P < 0.05, **P < 0.01, n.s. not significant, one-way ANOVA (C, F) and Student’s t-test (G). Error bars are +/− s.e.m. Scale bars: B: 1 mm; D: 200 μm; D’: 100 μm.
; p53−/− brains. *P < 0.05, **P < 0.01, n.s. not significant, one-way ANOVA (C, F) and Student’s t-test (G). Error bars are +/− s.e.m. Scale bars: B: 1 mm; D: 200 μm; D’: 100 μm.
The postnatal survival of Kif20b; p53 double mutant mice enabled us to investigate the requirement of Kif20b for corticogenesis without the confounding factor of excess apoptosis. Interestingly, in contrast to the full rescue of cortical thickness seen at E14.5 in Kif20bm/m; p53−/− brains, at P0 they show a 5% decrease in cortical length and a 15–20% reduction in total cortical thickness compared to heterozygous controls (Fig. 4B–F). This is not due to p53 mutation, as Kif20b+/m; p53−/− mice had normal cortical length and thickness, in agreement with previous studies [Fig. 4B, C and F; (16)]. The reduction in thickness in Kif20b; p53 double mutants is in the cortical plate and intermediate zone, but not the vz and subventricular zone (svz) (Fig. 4G). To determine whether superficial or deep layers of the cortical plate were reduced in the double mutant, we labeled sections with Cux1 for layers II–IV, Ctip2 for layers V–VI and Tbr1 for layer VI. We saw reduced thickness of all layers, with the most significant decrease in layer VI (Supplementary Material, Fig. S3A–D). Further analysis of layer VI showed that there was a loss in the absolute number of Tbr1+ cells, and that these cells were more densely packed (Supplementary Material, Fig. S3E–H). These data indicate that despite the dramatic improvements in cortical growth and postnatal survival afforded by blocking p53-dependent apoptosis, a deficit remains in neurogenesis of all cortical plate layers. Thus, while elevated apoptosis largely accounts for the microcephaly of the Kif20b mutant, proper cortical growth requires a Kif20b function that cannot be compensated by deleting p53 and preventing apoptosis.
Kif20b is required cell autonomously for midbody maturation of cortical NSCs
Previously we showed that Kif20b is expressed in germinal zones of the embryonic brain, and that Kif20b protein localizes to midbodies of dividing embryonic cortical NSCs at the ventricular surface (4). We further showed that Kif20b mutants have abnormalities in NSC midbodies; they tend to be wider and less aligned to the apical membrane. These phenotypes could be due to a cell-autonomous requirement for Kif20b during abscission or due to non-cell autonomous effects through cell–cell interactions at the apical membrane junctions, because cytokinesis in epithelia is a multicellular process (27). To further probe Kif20b’s function in embryonic NSC division, we used dissociated cortical cell cultures.
Midbodies at various stages of maturation can be detected with tubulin and Aurora Kinase B (AurKB) staining by their characteristic shapes (Fig. 5A–C). Early-stage midbodies are wide (Fig. 5A), but become thinner as the midbody matures (28). At late stages, microtubule constriction sites (abscission sites) are detectable on one or both sides of the midbody center [Fig. 5B and C, arrows; (5)]. We found in human cell lines that Kif20b is recruited to early stage midbodies, and at late stages accumulates around the constriction sites (6). Interestingly, Kif20bm/m NSC cultures have an increased frequency of wide midbodies (Fig. 5D), and fewer midbodies with at least one constriction site (Fig. 5E). These analyses show that Kif20b is required for normal NSC midbdy maturation, and that this requirement is cell autonomous.
Figure 5.
Kif20b loss cell-autonomously causes abscission defects in cortical NSCs. (A-C) Dissociated E12.5 NSC midbodies MBs at 24 h in culture labeled with alpha-tubulin (red) and AurKB (green). Early-stage MBs are wider with no constriction sites (A); late-stage MBs are thinner and have one (B) or two (C) constriction sites (arrows). (D)Kif20b MBs (white bars) have a significantly shifted width distribution, with more wide MBs compared to controls (black bars). (Medians: Control, 0.89 μm; Mutant, 0.96 μm). (E) The mean percentage of MBs with at least one constriction site (detected by tubulin) is significantly reduced in Kif20b mutant cultures. *P < 0.05, Student’s t-test; n for D, E = 141 control, 125 mutant MBs; 6 coverslips and 3 embryos each, 3 litters. (F-G) Representative images of p53 localization in control (F, F′) and Kif20b mutant (G, G’) NSCs show that p53 signal in the nucleus is increased in Kif20b
MBs (white bars) have a significantly shifted width distribution, with more wide MBs compared to controls (black bars). (Medians: Control, 0.89 μm; Mutant, 0.96 μm). (E) The mean percentage of MBs with at least one constriction site (detected by tubulin) is significantly reduced in Kif20b mutant cultures. *P < 0.05, Student’s t-test; n for D, E = 141 control, 125 mutant MBs; 6 coverslips and 3 embryos each, 3 litters. (F-G) Representative images of p53 localization in control (F, F′) and Kif20b mutant (G, G’) NSCs show that p53 signal in the nucleus is increased in Kif20b NSCs compared to control NSCs. Cells labeled with AurKB (green) to detect MBs, and 4′6-diamidino-2-phenylindole (DAPI; blue) and Nestin (white) to differentiate NSC nucleus and cytoplasm, respectively. (H)Kif20b
NSCs compared to control NSCs. Cells labeled with AurKB (green) to detect MBs, and 4′6-diamidino-2-phenylindole (DAPI; blue) and Nestin (white) to differentiate NSC nucleus and cytoplasm, respectively. (H)Kif20b NSCs at the MB stage have a significantly shifted distribution of nuclear: cytoplasmic ratios of p53, with a higher median N:C ratio. (Medians: Control, 0.83; Mutant, 0.95). K-S, Kolmogorov–Smirnov and M-W, Mann–Whitney tests. (I-J) Correlation analysis of p53 N:C ratio versus MB widths shows that MB width is significantly negatively correlated with increasing p53 N:C ratio in Kif20b
NSCs at the MB stage have a significantly shifted distribution of nuclear: cytoplasmic ratios of p53, with a higher median N:C ratio. (Medians: Control, 0.83; Mutant, 0.95). K-S, Kolmogorov–Smirnov and M-W, Mann–Whitney tests. (I-J) Correlation analysis of p53 N:C ratio versus MB widths shows that MB width is significantly negatively correlated with increasing p53 N:C ratio in Kif20b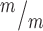 NSCs, but not controls. Mutant NSCS with thinner MBs (predicted to be late stage) are more likely to have higher p53 N:C ratios. P-values are for Pearson’s correlation test. n for F–J = 78 control and 66 Kif20b
NSCs, but not controls. Mutant NSCS with thinner MBs (predicted to be late stage) are more likely to have higher p53 N:C ratios. P-values are for Pearson’s correlation test. n for F–J = 78 control and 66 Kif20b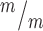 MB stage NSCS from 2 control and 2 mutant coverslips and embryos. Control denotes a combination of Kif20b+/+ and Kif20b+/m embryos. Error bars are +/− s.e.m. All cultures from E12.5 cortices and fixed after 24 h. Scale bar in A: 5 μm; F: 10 μm.
MB stage NSCS from 2 control and 2 mutant coverslips and embryos. Control denotes a combination of Kif20b+/+ and Kif20b+/m embryos. Error bars are +/− s.e.m. All cultures from E12.5 cortices and fixed after 24 h. Scale bar in A: 5 μm; F: 10 μm.
Some other mouse models of microcephaly show increased mitotic index in the cortex, because of NSC mitosis delay or arrest (14–18). We showed previously that the Kif20b mutant cortex does not have increased mitotic index (4), but here we use the dissociated cortical cell cultures to address this possibility with higher cellular resolution. We assayed whether Kif20b mutant NSCs spent more time in mitosis or abscission than control NSCs by determining the mitotic and midbdy index in cultures. Surprisingly, among cycling NSCs (Ki67+) in Kif20b mutant cultures, both the mitotic index and midbdy index were not increased but were actually slightly reduced (Supplementary Material, Fig. S4B). This could suggest that Kif20b mutant NSCs undergo mitosis and abscission more rapidly than control cells, or that more are undergoing apoptosis. To analyze relative durations of cell division phases, we categorized mitotic and midbdy stage NSCs into sub-stages by phospho-histone H3 (PH3) and AurKB appearance (Supplementary Material, Fig. S4A and C). Among mutant NSCs at some stage of cell division, the percentages in prophase or prometa/metaphase were not different, but the percentage in anaphase/early telophase was slightly increased in Kif20b mutant cultures. These data are consistent with our previous results that early steps of mitosis are not disrupted in Kif20b-depleted cells, but that cytokinesis is affected, with a small delay in furrow ingression (6).
Since we observed cell-autonomous defects in midbody maturation (Fig. 5A–E) as well as increased nuclear p53 in Kif20b mutant NSCs (Fig. S2E), we next tested whether these findings were correlated. Analyzing p53 immunostaining in midbody stage cells of control and Kif20b mutant NSC cultures, we noted higher N:C ratios of p53 signal in Kif20b mutant cells (Fig. 5F–H). All observed control and mutant cells in M phase (35 control and 21 mutant), as determined by chromatin appearance, did not have chromatin-associated p53. Furthermore, testing for a correlation between p53 N:C ratios and midbody width uncovered a significant negative correlation between these measurements in Kif20b mutant but not control cells. Thinner midbodies (which normally indicates a later stage of abscission) were more likely to have higher N:C p53 ratios (Fig. 5I–J). While these levels of nuclear p53 accumulation in the midbody stage cells were not as high as in some interphase Kif20b mutant NSCs (Fig. S2D and E), these data nevertheless suggest that a subset of Kif20b mutant NSCs have elevated nuclear p53 during abscission. This is notable, since total p53 is reported to normally be low during mitosis and the following G1 (Gap 1 phase), but may rise in G1 under stress conditions (29). Whether midbody maturation defects could lead to p53 accumulation in the nucleus, or conversely whether p53 accumulation can lead to midbody defects, is unknown. DNA damage is known to activate p53 through a defined pathway, but we did not detect any increase in DNA damage in Kif20b mutant NSCs over controls (Supplementary Material, Fig. S5), suggesting that a different pathway causes nuclear p53 accumulation in Kif20b mutant NSCs.
Kif20b loss activates apoptosis cell autonomously in proliferating cortical NSCs
An attractive hypothesis is that defective midbody maturation of dividing NSCs in Kif20b mutants causes p53 to accumulate in some NSCs, starting in abscission and continuing afterward, eventually activating the intrinsic pathway of apoptosis. Supporting the idea that apoptosis predominantly occurs in NSCs in Kif20b mutant brains, we previously observed that the greatest-fold increase in apoptosis over control levels was seen at E10.5, when the cortex consists only of NSCs, as IPs and neurons have not yet been produced; the relative increase in apoptosis declined through E16.5 (4). At E14.5 in Kif20bm/m; p53+/+ mutant samples, 74% of apoptotic cells are found in the proliferative zones and 67% do not express neuronal tubulin (Supplementary Material, Fig. S6). However, these in vivo analyses do not distinguish whether apoptosis is triggered cell autonomously or by cell–cell interactions, and whether it is associated with cell division. Therefore, we used dissociated NSC cultures to distinguish these possibilities. Indeed, in cultures of Kif20b mutant cells after 1 day in vitro, apoptotic cells are increased more than 2-fold over controls (Fig. 6A–C, arrows, CC3+ cells), suggesting that apoptosis is triggered cell autonomously. In cultures from Kif20b; p53 double mutant mice, the rate of apoptosis is similar to controls, showing that the elevated apoptosis in isolated Kif20bm/m NSCs is also p53-dependent (Fig. 6D). Furthermore, neurons (TuJ1+) do not display increased apoptosis, while NSCs (Nestin+) do, and cycling NSCs (Ki67+) show a more pronounced increase (Fig. 6E–F).
Figure 6.
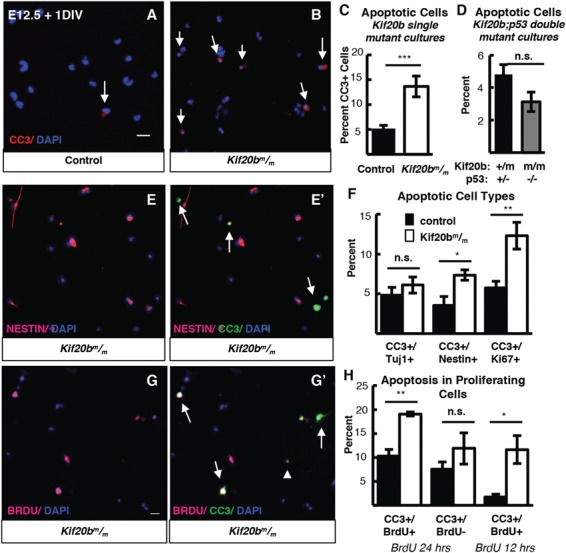
Kif20b loss causes apoptosis in proliferating cortical NSCs. (A, B) Representative fields of control (left) and Kif20b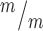 (right) dissociated cortical cultures labeled with CC3 (red, arrows) for apoptotic cells. (C) The mean percentage of apoptotic cells in Kif20b
(right) dissociated cortical cultures labeled with CC3 (red, arrows) for apoptotic cells. (C) The mean percentage of apoptotic cells in Kif20b cortical cultures is increased (>2-fold) above controls. n = 13 control, 11 mutant coverslips; 8 and 7 brains, 7 litters. (D) Apoptosis is not increased in Kif20b; p53 double mutant cultures. n = 4 Kif20b+/m; p53+/−, 3 Kif20b
cortical cultures is increased (>2-fold) above controls. n = 13 control, 11 mutant coverslips; 8 and 7 brains, 7 litters. (D) Apoptosis is not increased in Kif20b; p53 double mutant cultures. n = 4 Kif20b+/m; p53+/−, 3 Kif20b ; p53−/− coverslips; 3 and 2 embryos, 2 litters. (E) Representative fields labeled with Nestin (NSCs, pink) (E,E’) and CC3 (green) (E’). Arrows in E’ indicate cells co-positive for Nestin and CC3. (F) The percent of apoptotic neurons (CC3+/Tuj1) is not increased in Kif20b mutant cultures, but the percent of apoptotic NSCs and proliferating NSCs (CC3+/Nestin+; CC3+/Ki67+) are significantly increased. Tuj1/CC3 analysis: n = 7 control, 6 mutant coverslips; 4 and 3 brains, 4 litters. Nestin/CC3 analysis: n = 7 control, mutant coverslips; 4 brains each, 3 litters. Ki67/CC3 analysis: n = 6 control, 5 mutant coverslips; 3 brains each, 3 litters. (G) Representative fields labeled with BrdU (cells that incorporated BrdU, pink, G, G’) and CC3 (green,
G’). Arrows in G’ indicate cells co-positive for BrdU/CC3, and arrowhead indicates a cell negative for BrdU but positive for CC3. (H) Apoptosis is associated with proliferation in Kif20b mutant NSCs. Significantly more BrdU+ cells are apoptotic (CC3+) in Kif20b mutant cultures than control cultures, but BrdU− cells show no increase in apoptosis, when BrdU was in the bath for the 24 h culture period. When BrdU is given for the last 12 h before fixing at 24 h, the percent of apoptotic BrdU+ cells are also increased in Kif20b mutant cultures compared to control. For 24 h experiment, n = 3 control and mutant coverslips from 3 embryos and 2 litters. For 12 h experiment, n = 4 control and mutant coverslips from 3 embryos and 2 litters. Control denotes a combination of Kif20b+/+ and Kif20b+/m embryos. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. Error bars are +/− s.e.m. All cultures from E12.5 cortices and fixed after 24 h. Scale bars in A, E, H: 20 μm.
; p53−/− coverslips; 3 and 2 embryos, 2 litters. (E) Representative fields labeled with Nestin (NSCs, pink) (E,E’) and CC3 (green) (E’). Arrows in E’ indicate cells co-positive for Nestin and CC3. (F) The percent of apoptotic neurons (CC3+/Tuj1) is not increased in Kif20b mutant cultures, but the percent of apoptotic NSCs and proliferating NSCs (CC3+/Nestin+; CC3+/Ki67+) are significantly increased. Tuj1/CC3 analysis: n = 7 control, 6 mutant coverslips; 4 and 3 brains, 4 litters. Nestin/CC3 analysis: n = 7 control, mutant coverslips; 4 brains each, 3 litters. Ki67/CC3 analysis: n = 6 control, 5 mutant coverslips; 3 brains each, 3 litters. (G) Representative fields labeled with BrdU (cells that incorporated BrdU, pink, G, G’) and CC3 (green,
G’). Arrows in G’ indicate cells co-positive for BrdU/CC3, and arrowhead indicates a cell negative for BrdU but positive for CC3. (H) Apoptosis is associated with proliferation in Kif20b mutant NSCs. Significantly more BrdU+ cells are apoptotic (CC3+) in Kif20b mutant cultures than control cultures, but BrdU− cells show no increase in apoptosis, when BrdU was in the bath for the 24 h culture period. When BrdU is given for the last 12 h before fixing at 24 h, the percent of apoptotic BrdU+ cells are also increased in Kif20b mutant cultures compared to control. For 24 h experiment, n = 3 control and mutant coverslips from 3 embryos and 2 litters. For 12 h experiment, n = 4 control and mutant coverslips from 3 embryos and 2 litters. Control denotes a combination of Kif20b+/+ and Kif20b+/m embryos. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. Error bars are +/− s.e.m. All cultures from E12.5 cortices and fixed after 24 h. Scale bars in A, E, H: 20 μm.
Next, we wanted to ask whether NSC apoptosis was associated with cell division. Apoptotic cells with midbodies or mitotic spindles were not observed, perhaps because by the time CC3 is high and apoptotic chromatin changes are detectable, the microtubules of spindles and midbodies were already degraded. Therefore, we added Bromodeoxyuridine (BrdU) to the NSC cultures to test whether apoptosis was associated with proliferation. Indeed, when BrdU, which incorporates into DNA during replication, was present in NSC cultures during the 24-hour culture period, we found that apoptosis was specifically increased in BrdU+ but not BrdU− cells of Kif20b mutant cultures, suggesting that apoptosis occurred in cells that went through S phase (and presumably mitosis and cytokinesis) in the dish (Fig. 6G and H). To further refine the time interval between S phase and apoptosis, we added BrdU after 12 h in vitro before fixing at 24 h. Again, we found that the percentage of BrdU+ cells that were apoptotic was significantly higher in Kif20b mutant cultures than in control cultures (Fig. 6H). By contrast, when BrdU was added only 2 h before fixation, to label cells currently in S phase, no apoptotic cells were BrdU+ (0/36 control and 0/98 mutant). Given that S phase, G2 (Gap2 phase), M phase and cytokinesis combined take about 7 h to complete in NSCs at E12 (30), these data are consistent with apoptosis occurring following cell division. Together, these data show that loss of Kif20b causes cell-intrinsic, p53-dependent apoptosis, primarily in proliferative NSCs.
p53 deletion does not rescue impaired abscission in Kif20b mutant mice
The preceding data show that in Kif20b mutant brains, the NSC apoptosis and microcephaly are downstream of p53 activation. However, another important question is whether the midbody defects seen in Kif20b mutant NSCs, indicating an abnormal abscission process, are upstream or downstream of p53 activation. The correlation of increased p53 N:C ratios with midbody width in Kif20b mutant NSCs suggest that defects or delays in midbody maturation could cause p53 accumulation. Alternatively, these midbody defects could be a consequence of p53 activation, since p53 can regulate many genes and processes. To distinguish these possibilities, we tested whether abnormal midbody phenotypes observed in Kif20b mutant NSCs are rescued by p53 deletion. First, we analyzed NSC midbody index in the dissociated cultures. As Kif20bm/m NSCs in vitro are less frequently observed at the midbody stage (Supplementary Material, Fig. S4B), and we hypothesized this is due to NSC arrest or apoptosis, we predicted the midbody index phenotype would be rescued by p53 deletion. In fact, the midbody index of Kif20bm/m; p53−/− NSCs is not decreased, but instead is significantly increased above controls (Fig. 7A and B). This is consistent with the notion that some Kif20bm/m midbody-stage NSCs die, and further suggests that if these cells are prevented from dying, they take longer to complete abscission than control cells. Next, we analyzed NSC midbody structure in dissociated cultures. Similar to Kif20bm/m midbodies, Kif20bm/m; p53−/− midbodies are significantly wider than controls in vitro (data not shown), and fewer of them have constriction sites (Fig. 7C, compare with Fig. 5E). Thus, unlike apoptosis, the midbody width and constriction site phenotypes are not p53-dependent.
Figure 7.
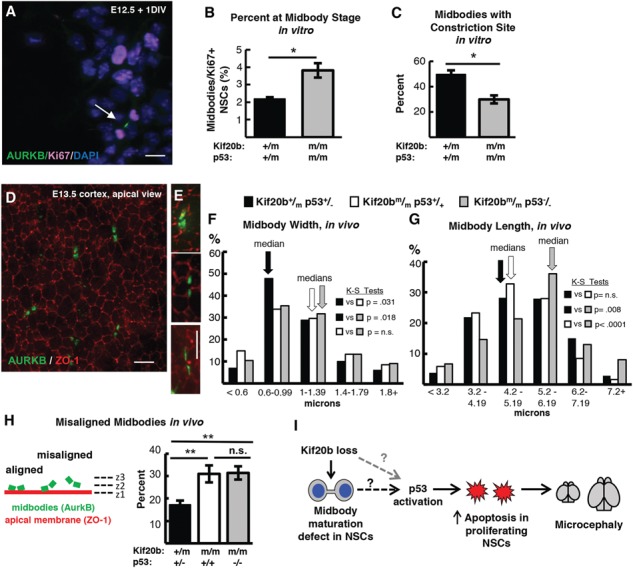
p53 deletion does not restore normal abscission of Kif20b mutant NSCs. (A) Representative image of E12.5 dissociated cortical cells, labeled with AurKB for MBs (green, arrow) and Ki67 for proliferating NSCs (pink). (B) The mean percent MB stage proliferating NSCs is significantly increased in Kif20b ; p53−/− cultures. n = 4 Kif20b+/m; p53+/−, 3 Kif20b
; p53−/− cultures. n = 4 Kif20b+/m; p53+/−, 3 Kif20b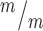 ; p53−/− coverslips; 3 embryos each, 2 litters. For comparison to Kif20b mutant cultures with wild-type p53, see Fig. S4B. (C) Of the NSCs at MB stage, the percent with a constriction site is significantly reduced in Kif20b; p53 double mutant cultures. n = 111 Kif20b+/m; p53+/−, 101 Kif20b
; p53−/− coverslips; 3 embryos each, 2 litters. For comparison to Kif20b mutant cultures with wild-type p53, see Fig. S4B. (C) Of the NSCs at MB stage, the percent with a constriction site is significantly reduced in Kif20b; p53 double mutant cultures. n = 111 Kif20b+/m; p53+/−, 101 Kif20b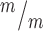 ; p53−/− MBs; 3 embryos each, 2 litters. For comparison to Kif20b
; p53−/− MBs; 3 embryos each, 2 litters. For comparison to Kif20b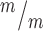 ; p53+/+ single mutant cultures, see Fig. 5E. (D) Representative image of E13.5 cortical slab, apical surface, labeled with ZO-1 (red, apical junctions) and AurKB (green, MBs). (E) NSC MBs range from short and wide (top) to long and thin (bottom). (F) The median MB width is similarly increased in Kif20b single and Kif20b;p53 double mutant cortices compared to controls. Median widths (μm) = 0.92 in Kif20b+/m; p53+/−, 1.0 in Kif20b
; p53+/+ single mutant cultures, see Fig. 5E. (D) Representative image of E13.5 cortical slab, apical surface, labeled with ZO-1 (red, apical junctions) and AurKB (green, MBs). (E) NSC MBs range from short and wide (top) to long and thin (bottom). (F) The median MB width is similarly increased in Kif20b single and Kif20b;p53 double mutant cortices compared to controls. Median widths (μm) = 0.92 in Kif20b+/m; p53+/−, 1.0 in Kif20b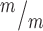 ;
p53+/+, 1.02 in Kif20b
;
p53+/+, 1.02 in Kif20b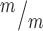 ; p53−/− cortices. (G) The median MB length is similar in Kif20b mutant and control cortices, but is significantly increased in Kif20b
; p53−/− cortices. (G) The median MB length is similar in Kif20b mutant and control cortices, but is significantly increased in Kif20b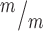 ; p53−/− cortices. Median lengths (μm) = 4.82 in Kif20b
; p53−/− cortices. Median lengths (μm) = 4.82 in Kif20b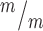 ; p53+/+, 5.11 in Kif20b+/m; p53+/−, 5.43 in Kif20b
; p53+/+, 5.11 in Kif20b+/m; p53+/−, 5.43 in Kif20b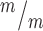 ; p53−/− cortices. n for (F, G) = 346 Kif20b+/m; p53+/−, 189 Kif20b
; p53−/− cortices. n for (F, G) = 346 Kif20b+/m; p53+/−, 189 Kif20b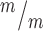 ; p53+/+ and 299 Kif20b
; p53+/+ and 299 Kif20b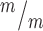 ; p53−/− MBs; 8, 5 and 7 cortical hemisphere slabs; 6, 4 and 5 embryos, 8 litters. (H) Left, Schematic of aligned and misaligned MBs of NSCS in vivo relative to the apical membrane. Right, the percent misaligned MBs is doubled in both Kif20b
; p53−/− MBs; 8, 5 and 7 cortical hemisphere slabs; 6, 4 and 5 embryos, 8 litters. (H) Left, Schematic of aligned and misaligned MBs of NSCS in vivo relative to the apical membrane. Right, the percent misaligned MBs is doubled in both Kif20b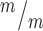 ; p53+/+ and Kif20b
; p53+/+ and Kif20b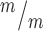 ; p53−/− cortices compared to controls. n = 8 Kif20b+/m; p53+/−, 7 Kif20b
; p53−/− cortices compared to controls. n = 8 Kif20b+/m; p53+/−, 7 Kif20b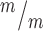 ; p53+/+, 7 Kif20b
; p53+/+, 7 Kif20b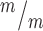 ; p53−/− cortical slabs from 6, 4, and 6 embryos, respectively, 8 litters. (I) Working model for the etiology of Kif20b microcephaly (see text also). p53 is required for NSC apoptosis and microcephaly caused by Kif20b loss but is not required for the MB defects. Thus, the MB maturation defect in the Kif20b mutant cells could signal to induce p53 accumulation through an unknown pathway (dashed black arrow with question mark). Alternatively, another cellular defect caused by Kif20b loss could induce p53 accumulation through a non-MB mediated pathway (dashed gray arrow with question mark). A–C are E12.5 cortical cultures fixed at 24 h. *P < 0.05, **P < 0.01, Student’s t-test for (B, C); K.S. test for (F, G); one-way ANOVA for (H). Error bars are +/− s.e.m. Scale bars, 10 μm.
; p53−/− cortical slabs from 6, 4, and 6 embryos, respectively, 8 litters. (I) Working model for the etiology of Kif20b microcephaly (see text also). p53 is required for NSC apoptosis and microcephaly caused by Kif20b loss but is not required for the MB defects. Thus, the MB maturation defect in the Kif20b mutant cells could signal to induce p53 accumulation through an unknown pathway (dashed black arrow with question mark). Alternatively, another cellular defect caused by Kif20b loss could induce p53 accumulation through a non-MB mediated pathway (dashed gray arrow with question mark). A–C are E12.5 cortical cultures fixed at 24 h. *P < 0.05, **P < 0.01, Student’s t-test for (B, C); K.S. test for (F, G); one-way ANOVA for (H). Error bars are +/− s.e.m. Scale bars, 10 μm.
We next analyzed Kif20bm/m; p53−/− double mutant midbodies in vivo in E13.5 cortices. Cytokinesis is more complex in the cortical neuroepithelium than in vitro. NSC nuclei must migrate to the apical membrane to undergo mitosis and cytokinesis. During cytokinesis, the cleavage furrow ingresses asymmetrically from the basal side of the cell, forming the midbody at the apical membrane (31). To visualize NSC midbodies en face, we immunostained cortical slab whole-mounts for AurKB and the apical junction marker zona occludens-1 (ZO-1), and imaged the apical/ventricular surfaces (Fig. 7D). Similar to in vitro NSCs, some midbodies are short and wide, and others are long and thin, since midbodies narrow as they mature (see examples at Fig. 7E). In Kif20bm/m cortices, midbodies have a shifted width distribution with an increased median width compared to controls [Fig. 7F, white bars; (32)]. Interestingly, we find that the Kif20bm/m; p53−/− double mutant cortices have a strikingly similar midbody width distribution as Kif20b single mutants, with the same median (Fig. 7F, gray bars). Thus, this midbody maturation phenotype (width) in vivo is not rescued by p53 deletion. But surprisingly, we find that midbody lengths, which are similar in controls and Kif20bm/m single mutants, are significantly longer in Kif20bm/m; p53−/− double mutants than in either Kif20bm/m single mutants or controls (Fig. 7G). Exceptionally long midbodies were observed after delayed abscission in cell lines that are resistant to apoptosis (33,34). Thus, Kif20b mutant NSCs may have delayed abscission, which can manifest as longer midbodies only if apoptosis is prevented. An additional midbody phenotype we observe in vivo in Kif20b mutant cortices is that ∼30% of NSC midbodies are not aligned parallel to the apical membrane, compared to ∼15% in control brains [(4); Fig. 7H]. The cause of this phenotype is unclear, but we used the Kif20b; p53 double mutants to test whether it is p53-dependent, perhaps because of the apoptotic process in dividing or neighboring cells. Interestingly, p53 deletion did not prevent the misalignment phenotype, showing that it is not because of p53 activation or apoptosis (Fig. 7H). Taken together, these midbody analyses support the hypothesis that defective midbody maturation and alignment are primary consequences of Kif20b loss rather than secondary consequences of p53 activation. Further, the additional midbody phenotypes detected in Kif20b; p53 double mutant NSCs, namely increased midbody index and midbody length, suggest that some symptoms of midbody maturation defects from Kif20b loss are normally precluded in single mutants by p53 activation and apoptosis.
Discussion
Here we have tested the contribution of the intrinsic apoptosis pathway to the reduced cortex size in a lethal microcephaly model, the Kif20b mouse mutant. We have shown that apoptosis of cortical NSCs accounts for most of the microcephaly, but that there is a significant apoptosis-independent contribution as well. Furthermore, we showed that the excess apoptosis is cell autonomous, partially dependent on Bax and fully dependent on p53. Remarkably, heterozygous p53 deletion is sufficient to fully suppress the lethality of the Kif20b mutant, and rescues the brain size equally as well as homozygous p53 deletion. Importantly, we demonstrated that the NSC midbody maturation defects are not rescued by p53 deletion, which indicates that they are not caused by p53 activation, but may be upstream of p53. Thus, this work potentially identifies a novel midbody-initiated pathway for p53 activation, and suggests that at least some types of microcephaly, although severe, could be greatly ameliorated by inhibiting apoptosis.
The genetic and cellular experiments herein support the following working model for the etiology of microcephaly in the Kif20b mutant (Fig. 7I). Loss of Kif20b causes midbody maturation defects in some Kif20b mutant NSCs, leading to nuclear p53 accumulation by an unknown molecular pathway that could be distinct from the DNA damage pathway (black dashed line). Elevated levels of p53 then trigger the apoptotic cascade including Bax and other effectors. Apoptosis in proliferating NSCs depletes the NSC pool, thereby reducing the number of neuron and IP daughters produced, resulting in a small brain. It also remains plausible that Kif20b loss causes p53 activation and midbody defects through two separate pathways, and that p53 could be activated due to a different cellular defect (gray dashed line). We favor the former model for three reasons. First, we found the apoptosis is associated with markers of proliferation (Nestin, Ki67 and BrdU incorporation). Second, the increased nuclear accumulation of p53 in Kif20b mutant NSCs at midbody but not mitotic stage and in thinner (late stage) midbodies indicates an acute change in p53 levels or localization at this stage of division. Third, the new midbody defects that appear in Kif20b; p53 double mutants, namely increased midbod index and longer midbodies, suggest that Kif20b loss would cause abscission delay if it were not precluded by p53-dependent apoptosis. In either case, the Kif20b mutant is a tool to elucidate a potentially novel pathway to p53 activation, one that appears very sensitive in cortical NCSs.
There is likely some stochasticity to the apoptotic process, as not all Kif20bm/m NSCs have midbody defects, and only a small percentage undergo apoptosis (4); this work). It may be that apoptosis is only triggered if midbody maturation (and hence abscission) is delayed beyond a certain threshold. This would be analogous to a previous study in developing cortex showing that apoptosis likelihood increases if prophase is delayed past a threshold (35). Though both of these types of cell division delay trigger apoptosis through p53, the molecular `error sensor’ upstream of p53 is likely distinct, since midbody maturation takes place well after telophase, in the next G1 phase (36). In fact, how defects in early steps of mitosis signal to trigger p53 activation is only beginning to be elucidated, primarily in immortalized cell lines that are apoptosis-resistant (37–39). The idea that abscission delay or `stress’ could activate p53 has not been previously addressed. More work is needed to determine whether and how a midbody error sensor could directly or indirectly cause p53 accumulation in NSCs or other cell types.
p53 activates apoptosis in NSCs following diverse cellular defects
Our work furthers the evidence that p53 responds to multiple intrinsic cellular defects to acutely regulate NSC survival. For eventual treatment of human microcephalies caused by genetic mutations or viruses, it is important to determine which types of microcephaly or which phenotypes might be treatable by inhibiting apoptosis. Zika virus infection activates p53-dependent apoptosis in human NSCs (40). Some other microcephalic mouse mutants, with impaired DNA replication, mitosis or cleavage furrowing have implicated p53 in apoptosis and reduced brain size (16–18,41–43). Interestingly, inhibition of p53 in these mutants was sometimes able to increase cortical thickness, but in other cases worsened brain phenotypes (18,43,44). By contrast, Kif20bm/m embryonic brain structure and postnatal survival were well-rescued by even heterozygous deletion of p53. Heterozygous p53 deletion has not been reported to rescue other microcephaly mouse mutants. In the Brca1 mutant, dwarfism but not microcephaly was rescued by heterozygous p53 deletion (45). Thus, the Kif20b microcephaly model is more severe in reduction of brain size compared to some other models, but it is also more easily rescuable by p53 inhibition. As a key cell cycle regulator, it is also possible that p53 activation in the Kif20b mutant reduces brain size by other mechanisms in addition to apoptosis, such as by causing NSC cell cycle exit. Future work will continue to investigate such possibilities.
A p53-independent consequence of Kif20b loss accounts for the remaining cortical size deficit at birth
The deficit in cortical thickness in Kif20b; p53 double mutant mice at birth indicates that even without the excess apoptosis, Kif20b mutant NSCs and neurons cannot create a brain of normal size and structure. This could be due to abnormal abscission causing some other problem in Kif20b mutant NSCs, or due to postmitotic roles of Kif20b, or both. The midbody maturation defects that are more frequent in Kif20b mutant brains could affect whether abscission is completed on both sides of the midbody or only on one side, resulting in either midbody release into the ventricle, or midbody inheritance by one daughter cell. They could also impair daughter cell severing from the mother cell and delamination from the apical membrane in the case of neuronal daughters. The significance of the midbody misalignment increase is not clear, but here we showed that it is not caused by p53 activation or apoptosis. It is possible that Kif20b may help anchor or stabilize midbodies at the apical membrane via cell adhesion junctions until abscission is complete. Much more work is needed to understand how cytokinetic abscission mechanisms are coordinated with cell fate decisions and maintenance of neuroepithelial structure.
The reduced thickness of neuronal layers at P0 indicates a role for Kif20b in the production of neurons at all developmental timepoints. However, the most dramatic reduction was seen in layer VI neurons, which were fewer in number but also more densely packed. This may indicate that Kif20b plays a more important role in earlier stages of neurogenesis, and/or in postmitotic neurons for neurite outgrowth. We previously showed that Kif20b mutant embryonic cortical neurons in culture display a polarization defect and abnormalities in neurite growth and branching. Thus, the increased density of layer VI cells at birth may be due to reduced neuropil. A key function of Kif20b is to enable tight microtubule packing: Kif20b has microtubule crosslinking activity in vitro, and in Kif20b mutant neurons, wider neurites and gaps in microtubule bundles were noted (46,47). Therefore, Kif20b may help to organize microtubules in both NSC midbodies and neuronal axons.
Elucidating the heterogeneous mechanisms of brain malformations requires many genetic models and culture systems
Defects in cytokinetic abscission mechanisms could underlie a range of poorly understood microcephalies and related brain malformations. For example, human genetic studies in two different families showed that a very severe prenatal lethal microcephaly/anencephaly is caused by mutations in the gene encoding the key abscission protein Cep55 [(48,49); OMIM Ref #236500]. Cep55 depletion in cell lines causes more severe midbody structural defects and abscission delay than Kif20b depletion does (50). Patients with Kif20b loss-of-function mutations have not yet been identified, so it remains to be seen whether the severity of single-cell abscission phenotypes correlates with the severity of brain malformation. Cell lines such as HeLa cells can model some abscission defects, for example midbody maturation, but not others, such as midbody positioning at the apical membrane, p53 activation or apoptosis. Our studies of the Kif20b mouse model provide human geneticists with a candidate gene and cellular markers for syndromes involving peri- or prenatal lethality, microcephaly, craniofacial defects or microphthalmia. Comparing mouse models for abscission genes with other microcephalic mice as well as human phenotypes will help us understand the heterogeneous etiologies of and potential diagnosis and treatments for these devastating conditions.
Materials and Methods
Mice
Mouse colonies were maintained in accordance with NIH guidelines and policies approved by the IACUC. Embryos were harvested by cesarean section, and the morning of the vaginal plug was considered E0.5. Littermate embryos served as controls for all experiments. The Kif20bmagoo allele, as previously described (4) is maintained on both C57BL/6 and FVB/N backgrounds, and 50/50% mixed background embryos are used for experiments. Trp53tm1Tyj mice on C57BL/6 background were obtained from The Jackson Laboratory [(23); JAX stock #002101]. Mixed BL6/FVB/N background Baxtm1Sjk mice were a gift from Christopher Deppmann [(24); JAX stock #002994]. Sex of embryonic mice were not noted as sex was not a relevant biological variable for these experiments. The specific ages of embryonic mice used is noted in figure legends for each experiment.
Cortical cell cultures
Cells were dissociated from E12.5 cortices following a protocol adapted from Sally Temple’s laboratory (51). The Worthington Papain Dissociation Kit was used to dissociate cells (Worthington Biochemical Corporation, Lakewood, NJ, Cat # LK003150). Cells were cultured in DMEM with Na-Pyruvate, L-Glutamine, B-27, N2, N-acetyl-cysteine and basic fibroblast growth factor. After 24 h, cells were fixed by adding an equal volume of room temperature 8% PFA (paraformaldehyde) for 5 min to cell media, followed by removal of media and addition of −20° cold methanol for 5 min. For BrdU experiments, BrdU was added to culture media at a final concentration of 10 μm.
Apical slab preparation
Apical slabs were prepared as previously described (32). The meninges and skull were removed to expose the brain in E13.5 embryos, followed by fixation with 2% PFA for 20 min. Next, apical slabs were made by pinching off cortices, flipping so that the apical surface was upright, and trimming to flatten the slab. Slabs were fixed for another 2 min with 2% PFA followed by blocking with 5% normal goat serum (NGS) for 1 h. Primary antibodies were applied for 1 h at room temperature and then moved to 4° overnight. The next day, after 3 times, 10-minute PBS (phosphate-buffered saline) washes, secondary antibodies and DAPI were applied at a concentration of 1:200 for 30 minutes. After two more 10–minute PBS washes, slabs were coverslipped with VectaShield fluorescent mounting medium (Vector Laboratories Inc., H-1000) and imaged. z-stack depth was 8–20 μm and step size was 0.5 μm. Midbodies were considered misaligned if the two halves of the midbody were not in the same z-plane or within two adjacent z-planes.
Immunoblotting
Brain lysates were prepared with RIPA lysis buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris–HCl pH 8) with protease and phosphatase inhibitors. Protein concentration in lysates was determined by bicinchoninic acid (BCA) assay, and 60 μg total protein was loaded per lane on 4–20 gradient% polyacrylamide gels. Proteins were transferred by electroblotting onto a 0.2 μm PVDF membrane overnight at 30 mA. Membranes were blocked in 150 mM NaCl, 100 mM Tris–HCl pH 7.5 and 0.5% Tween 20 (TBST) with 5% dried milk (blocking buffer) for 1 hour. Primary antibodies were incubated with the membrane overnight at 4 °C. After three washes, LI-COR IRDye 800 CW goat anti-rabbit IgG and 680 RD goat anti-mouse secondary antibodies were applied (1:10000) in blocking buffer for 1 hour at room temperature. After washing with TBST for 5 minutes, 3 times, immune complexes were visualized using a LI-COR western blot imager.
Immunostaining
To collect cryosections for IHC, age E14.5 and P0 brains were removed from heads and fixed for 6 and 24 hours, respectively, in 4% PFA, followed by immersion in 30% sucrose in PBS overnight. Next, whole brains were embedded in OTC (Tissue-Tek, 4583) and cryosections were cut at 20 μm thickness and collected on Superfrost Plus slides (Fisher Scientific, 12–550-15). Frozen sections were stored at −80 degrees. Prior to immunostaining, cryosections were warmed to room temperature, then if antigen retrieval was needed, immersed in 10 mM citrate buffer at 95 degrees for 20 minutes. After cooling, sections were blocked in 2% NGS for 1 hour, followed by incubation with primary antibodies overnight at 4 ° C. The next day, after PBS washes sections were incubated with AlexaFluor secondary antibodies at 1:200 and DAPI at 1:100 for 30 min followed by PBS washes and coverslipping with VectaShield fluorescent mounting medium. For IF on coverslips of dissociated cortical progenitors, a similar protocol was used but with primary antibodies applied for 3 h at room temperature. Antigen retrieval was not used in dissociated progenitors except in the case of BrdU; coverslips were immersed in 0.07m NaOH pH 13 for 2 min before permeabilization. Coverslips were mounted on Superfrost Plus slides with Fluoro-Gel (Electron Microscopy Sciences, Hatfield, PA, 17985–10).
Antibodies
Antibodies used in this analysis were rabbit polyclonal anti-human CC3 (Cell-Signaling 9661S, 1:250), mouse monoclonal anti-rat beta-III-tubulin (Tuj1) (BioLegend, San Diego, CA, 801201, 1:500), rat monoclonal anti-mouse Tbr2 (eBioscience (Thermo Fisher Scientific), Waltham, MA 14–4875, 1:200), rabbit polyclonal anti-mouse Pax6 (BioLegend PRB-278P, 1:200), mouse monoclonal anti-rat Aurora B kinase (BD Biosciences 611082, 1:300), rabbit polyclonal anti-human alpha-tubulin (Thermo Scientific RB-9281-P0, 1:300), rat monoclonal alpha-tubulin (Novus Biologicals NB600–506, 1:300), rabbit monoclonal anti-human PH3 (Cell Signaling 3458, 1:200), chicken polyclonal anti-mouse Nestin (Aves Labs NES, 1:600), rat monoclonal anti-human Ki67 (eBioscience 14–5698, 1:100), mouse monoclonal anti-human p53 (MilliporeSigma, Burlington, MA, 05–224, 1:250), rabbit polyclonal anti-mouse p53 (Leica Biosystems, Wetzlar, Germany, NCL-L-p53-CM5p, 1:500), mouse monoclonal anti-human phospho-Histone H2A.X (Ser139) (Millipore 05–636 1:500), rabbit polyclonal anti-mouse Cux1 (Santa Cruz Biotechnology, Inc., Dallas, TX, (M-122) sc-13024, 1:100), rat monoclonal anti-human Ctip2 (Abcam 18465, 1:400), rabbit polyclonal anti-mouse Tbr1 (Abcam, Cambridge, MA 31940, 1:200), mouse monoclonal anti-BrdU (BD Biosciences, San Jose, CA Clone B44, 20 μl/50 μl block), rabbit polyclonal anti-human Beta-Catenin (MilliporeSigma, Burlington, MA, SAB4500545 1:1000), and polyclonal rabbit anti Zo-1 (rabbit, Invitrogen (Thermo Fisher Scientific) 61–7300,1:50). All antibodies were validated for the application used in multiple previous publications.
Imaging and statistical analysis
Images in Figures 1F–I, 2F–I and 3A–F were taken on an Olympus Confocal Fluoview FV1000. Images in Figures 5A–E, 6, 7A and S2–6 were collected on either a Zeiss Axio ImagerZ1 microscope with AxioCam MRm or a Zeiss AxioObserver fluorescent widefield inverted scope microscope. Images in Figures 5F–J and 7D–J were taken on an inverted DeltaVision with TrueLight deconvolution microscope with softWoRx Suite 5.5 image acquisition software (Applied Precision (GE Healthcare), Issaquah, WA). A Leica MZ16F microscope with DFC300FX camera was used for images in Figures 1A–E, 2A–E and 4. Control and mutant fluorescence images were collected with the same exposure times and on the same day. All image analysis was performed in ImageJ/Fuji and any changes to brightness and contrast were applied uniformly across images. Statistical analyses were performed using Excel (Microsoft) or GraphPad PRISM software. The sample sizes were pre-determined based on our lab’s previous experience with cortical development analyses and others’ published results. After obtaining pilot data, power analyses were performed if necessary to determine if the number of samples obtained was high enough for the effect size seen. NSC cultures that were unhealthy were not imaged and analyzed, but no other data was excluded from the analysis. No randomization or blinding was used as no experimental manipulation was applied other than genetic knockouts. Genotyping was performed after collection of embryos to determine genetic status. Statistical tests used are specified in each figure legend. For each sample set a statistical test of normality was performed using GraphPad PRISM software. Parametric tests were used when sample sets had a normal distribution and non-parametric tests were used when sample sets did not have a normal distribution. Variance was calculated and was typically similar between groups. N’s listed in figure legends indicate the number of coverslips, brains and litters collected for each experiment. For each brain, at least three sections were imaged, and for each coverslip, at least 5, 20× pictures or 10, 40× pictures were analyzed.
Supplementary Material
Acknowledgements
We thank Chris Deppmann for use of the Bax mutant mouse line. We are grateful to Bettina Winckler, Jing Yu, Xiaowei Lu, Ann Sutherland, Todd Stukenberg, Kodi Ravichandran, Hui Zong, and members of their laboratories, as well as Katrina McNeely for advice and discussion. We are grateful to Madison Hecht, Gabrielle Wolfe, Mackenzie Shannon, Haley Hopkinson and Adriana Ehlers for the help with cryosectioning and data analysis. Special thanks to Debra Silver and Yuanyi Feng for antibody samples and positive control sections.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (R01 NS076640 and R21 NS106162 to N.D.D.); the UVA Medical Scientist Training Program (MSTP T32 GM 7267-37); the UVA Cell and Molecular Biology Training Grant (2T32GM008136-31A1).
References
- 1. Duerinckx S. and Abramowicz M. (2018) The genetics of congenitally small brains. Semin. Cell. Dev. Biol., 76, 76–85. [DOI] [PubMed] [Google Scholar]
- 2. Dwyer N.D., Chen B., Chou S.J., Hippenmeyer S., Nguyen L. and Ghashghaei H.T. (2016) Neural stem cells to cerebral cortex: emerging mechanisms regulating progenitor behavior and productivity. J. Neurosci., 36, 11394–11401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dwyer N.D., Manning D.K., Moran J.L., Mudbhary R., Fleming M.S., Favero C.B., Vock V.M., O'Leary D.D., Walsh C.A. and Beier D.R. (2011) A forward genetic screen with a thalamocortical axon reporter mouse yields novel neurodevelopment mutants and a distinct Emx2 mutant phenotype. Neural. Dev., 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Janisch K.M., Vock V.M., Fleming M.S., Shrestha A., Grimsley-Myers C.M., Rasoul B.A., Neale S.A., Cupp T.D., Kinchen J.M., Liem K.F., Jr., and Dwyer N.D. (2013) The vertebrate-specific Kinesin-6, Kif20b, is required for normal cytokinesis of polarized cortical stem cells and cerebral cortex size. Development, 140, 4672–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mierzwa B. and Gerlich D.W. (2014) Cytokinetic abscission: molecular mechanisms and temporal control. Dev. Cell, 31, 525–538. [DOI] [PubMed] [Google Scholar]
- 6. Janisch K.M., McNeely K.C., Dardick J.M., Lim S.H. and Dwyer N.D. (2018) Kinesin-6 KIF20B is required for efficient cytokinetic furrowing and timely abscission in human cells. Mol. Biol. Cell, 29, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanehira M., Katagiri T., Shimo A., Takata R., Shuin T., Miki T., Fujioka T. and Nakamura Y. (2007) Oncogenic role of MPHOSPH1, a cancer-testis antigen specific to human bladder cancer. Cancer Res., 67, 3276–3285. [DOI] [PubMed] [Google Scholar]
- 8. Lin W.F., Lin X.L., Fu S.W., Yang L., Tang C.T., Gao Y.J., Chen H.Y. and Ge Z.Z. (2018) Pseudopod-associated protein KIF20B promotes Gli1-induced epithelial-mesenchymal transition modulated by pseudopodial actin dynamic in human colorectal cancer. Mol. Carcinog., 57, 911–925. [DOI] [PubMed] [Google Scholar]
- 9. Liu X., Zhou Y., Liu X., Peng A., Gong H., Huang L., Ji K., Petersen R.B., Zheng L. and Huang K. (2014) MPHOSPH1: a potential therapeutic target for hepatocellular carcinoma. Cancer Res., 74, 6623–6634. [DOI] [PubMed] [Google Scholar]
- 10. Haydar T.F., Kuan C.Y., Flavell R.A. and Rakic P. (1999) The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb. Cortex, 9, 621–626. [DOI] [PubMed] [Google Scholar]
- 11. Kuida K., Haydar T.F., Kuan C.Y., Gu Y., Taya C., Karasuyama H., Su M.S., Rakic P. and Flavell R.A. (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell, 94, 325–337. [DOI] [PubMed] [Google Scholar]
- 12. Kuida K., Zheng T.S., Na S., Kuan C., Yang D., Karasuyama H., Rakic P. and Flavell R.A. (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature, 384, 368–372. [DOI] [PubMed] [Google Scholar]
- 13. Nonomura K., Yamaguchi Y., Hamachi M., Koike M., Uchiyama Y., Nakazato K., Mochizuki A., Sakaue-Sawano A., Miyawaki A., Yoshida H. et al. (2013) Local apoptosis modulates early mammalian brain development through the elimination of morphogen-producing cells. Dev. Cell, 27, 621–634. [DOI] [PubMed] [Google Scholar]
- 14. Breuss M., Fritz T., Gstrein T., Chan K., Ushakova L., Yu N., Vonberg F.W., Werner B., Elling U. and Keays D.A. (2016) Mutations in the murine homologue of TUBB5 cause microcephaly by perturbing cell cycle progression and inducing p53-associated apoptosis. Development, 143, 1126–1133. [DOI] [PubMed] [Google Scholar]
- 15. Chen J.F., Zhang Y., Wilde J., Hansen K.C., Lai F. and Niswander L. (2014) Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat. Commun., 5, 3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Insolera R., Bazzi H., Shao W., Anderson K.V. and Shi S.H. (2014) Cortical neurogenesis in the absence of centrioles. Nat. Neurosci., 17, 1528–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marjanovic M., Sanchez-Huertas C., Terre B., Gomez R., Scheel J.F., Pacheco S., Knobel P.A., Martinez-Marchal A., Aivio S., Palenzuela L. et al. (2015) CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat. Commun., 6, 7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marthiens V., Rujano M.A., Pennetier C., Tessier S., Paul-Gilloteaux P. and Basto R. (2013) Centrosome amplification causes microcephaly. Nat. Cell Biol., 15, 731–740. [DOI] [PubMed] [Google Scholar]
- 19. Stottmann R.W., Donlin M., Hafner A., Bernard A., Sinclair D.A. and Beier D.R. (2013) A mutation in Tubb2b, a human polymicrogyria gene, leads to lethality and abnormal cortical development in the mouse. Hum. Mol. Genet., 22, 4053–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yingling J., Youn Y.H., Darling D., Toyo-Oka K., Pramparo T., Hirotsune S. and Wynshaw-Boris A. (2008) Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell, 132, 474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arya R. and White K. (2015) Cell death in development: Signaling pathways and core mechanisms. Semin. Cell Dev. Biol., 39, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kastenhuber E.R. and Lowe S.W. (2017) Putting p53 in Context. Cell, 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T. and Weinberg R.A. (1994) Tumor spectrum analysis in p53-mutant mice. Curr. Biol., 4, 1–7. [DOI] [PubMed] [Google Scholar]
- 24. Knudson C.M., Tung K.S., Tourtellotte W.G., Brown G.A. and Korsmeyer S.J. (1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science, 270, 96–99. [DOI] [PubMed] [Google Scholar]
- 25. Westphal D., Dewson G., Czabotar P.E. and Kluck R.M. (2011) Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta, 1813, 521–531. [DOI] [PubMed] [Google Scholar]
- 26. Lindsten T., Ross A.J., King A., Zong W.X., Rathmell J.C., Shiels H.A., Ulrich E., Waymire K.G., Mahar P., Frauwirth K. et al. (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell, 6, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herszterg S., Pinheiro D. and Bellaiche Y. (2014) A multicellular view of cytokinesis in epithelial tissue. Trends. Cell Biol., 24, 285–293. [DOI] [PubMed] [Google Scholar]
- 28. Guizetti J., Schermelleh L., Mantler J., Maar S., Poser I., Leonhardt H., Muller-Reichert T. and Gerlich D.W. (2011) Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science, 331, 1616–1620. [DOI] [PubMed] [Google Scholar]
- 29. Yang H.W., Chung M., Kudo T. and Meyer T. (2017) Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature, 549, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takahashi T., Nowakowski R.S. and Caviness V.S.,.J. (1995) The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J. Neurosci., 15, 6046–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kosodo Y., Toida K., Dubreuil V., Alexandre P., Schenk J., Kiyokage E., Attardo A., Mora-Bermudez F., Arii T., Clarke J.D. et al. (2008) Cytokinesis of neuroepithelial cells can divide their basal process before anaphase. Embo. J., 27, 3151–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Janisch K.M. and Dwyer N.D. (2016) Imaging and quantitative analysis of cytokinesis in developing brains of Kinesin-6 mutant mice. Methods Cell Biol., 131, 233–252. [DOI] [PubMed] [Google Scholar]
- 33. Gromley A., Jurczyk A., Sillibourne J., Halilovic E., Mogensen M., Groisman I., Blomberg M. and Doxsey S. (2003) A novel human protein of the maternal centriole is required for the final stages of cytokinesis and entry into S phase. J. Cell Biol., 161, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiderhold K.N., Fadri-Moskwik M., Pan J., Nishino M., Chuang C., Deeraksa A., Lin S.H. and Yu-Lee L.Y. (2016) Dynamic phosphorylation of NudC by Aurora B in cytokinesis. PLoS One, 11, e0153455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pilaz L.J., McMahon J.J., Miller E.E., Lennox A.L., Suzuki A., Salmon E. and Silver D.L. (2016) Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron, 89, 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gershony O., Pe'er T., Noach-Hirsh M., Elia N. and Tzur A. (2014) Cytokinetic abscission is an acute G1 event. Cell Cycle, 13, 3436–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lambrus B.G., Daggubati V., Uetake Y., Scott P.M., Clutario K.M., Sluder G. and Holland A.J. (2016) A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J. Cell Biol., 214, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McKinley K.L. and Cheeseman I.M. (2017) Large-scale analysis of CRISPR/Cas9 cell-cycle knockouts reveals the diversity of p53-dependent responses to cell-cycle defects. Dev. Cell, 40, 405–420e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meitinger F., Anzola J.V., Kaulich M., Richardson A., Stender J.D., Benner C., Glass C.K., Dowdy S.F., Desai A., Shiau A.K. et al. (2016) 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J. Cell Biol., 214, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghouzzi V.E., Bianchi F.T., Molineris I., Mounce B.C., Berto G.E., Rak M., Lebon S., Aubry L., Tocco C., Gai M. et al. (2016) ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly. Cell Death Dis., 7, e2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bianchi F.T., Tocco C., Pallavicini G., Liu Y., Verni F., Merigliano C., Bonaccorsi S., El-Assawy N., Priano L., Gai M. et al. (2017) Citron kinase deficiency leads to chromosomal instability and TP53-sensitive microcephaly. Cell Rep., 18, 1674–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Houlihan S.L. and Feng Y. (2014) The scaffold protein Nde1 safeguards the brain genome during S phase of early neural progenitor differentiation. Elife, 3, e03297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murga M., Bunting S., Montana M.F., Soria R., Mulero F., Canamero M., Lee Y., McKinnon P.J., Nussenzweig A. and Fernandez-Capetillo O. (2009) A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet., 41, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mao H., McMahon J.J., Tsai Y.H., Wang Z. and Silver D.L. (2016) Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. PLoS Genet., 12, e1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu X., Qiao W., Linke S.P., Cao L., Li W.M., Furth P.A., Harris C.C. and Deng C.X. (2001) Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet., 28, 266–271. [DOI] [PubMed] [Google Scholar]
- 46. Abaza A., Soleilhac J.M., Westendorf J., Piel M., Crevel I., Roux A. and Pirollet F. (2003) M phase phosphoprotein 1 is a human plus-end-directed kinesin-related protein required for cytokinesis. J. Biol. Chem., 278, 27844–27852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McNeely K.C., Cupp T.D., Little J.N., Janisch K.M., Shrestha A. and Dwyer N.D. (2017) Mutation of Kinesin-6 Kif20b causes defects in cortical neuron polarization and morphogenesis. Neural. Dev., 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bondeson M.L., Ericson K., Gudmundsson S., Ameur A., Ponten F., Wesstrom J., Frykholm C. and Wilbe M. (2017) A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy. Clin. Genet., 92, 510–516. [DOI] [PubMed] [Google Scholar]
- 49. Frosk P., Arts H.H., Philippe J., Gunn C.S., Brown E.L., Chodirker B., Simard L., Majewski J., Fahiminiya S., Russell C. et al. (2017) A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J. Med. Genet., 54, 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao W.M., Seki A. and Fang G. (2006) Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol. Biol. Cell., 17, 3881–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Qian X., Goderie S.K., Shen Q., Stern J.H. and Temple S. (1998) Intrinsic programs of patterned cell lineages in isolated vertebrate CNS ventricular zone cells. Development, 125, 3143–3152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.