

Abstract
Chromosome translocations are catastrophic genomic events and often play key roles in tumorigenesis. Yet the biogenesis of chromosome translocations is remarkably poorly understood. Recent work has delineated several distinct mechanistic steps in the formation of translocations, and it has become apparent that non-random spatial genome organization, DNA repair pathways and chromatin features, including histone marks and the dynamic motion of broken chromatin, are critical for determining translocation frequency and partner selection.
A chromosome translocation is defined as a genome abnormality in which a chromosome breaks and either the whole or a portion of it reattaches to a different chromosome (Fig. 1a). Depending on the location of the breaks, translocations may lead to the formation of fusion genes, or may disrupt a gene or its regulatory sequences, and in this way cause gene misregulation (Fig. 1b). Complex chromosome rearrangements can also be caused by single catastrophic events, such as the recently identified phenomenon of chromothripsis1. Chromosome translocations are clinically highly relevant as they are associated with numerous human cancers as well as non-cancerous diseases such as infertility and schizophrenia2. Although chromosome translocations have long been considered mostly relevant in haematological cancers, their importance in solid tumours has recently been recognized by the identification of numerous recurrent translocations in tumours from most tissues2. Translocations play an undisputed role in the initial steps of carcinogenesis and it is estimated that they are causal in ~20% of cancers2. Beyond their role as disease agents, translocations are used as decisive diagnostic indicators, as they are easily and accurately detected using cytogenetic methods. Despite their prevalence and functional importance, our understanding of their genesis is still remarkably rudimentary.
Figure 1.
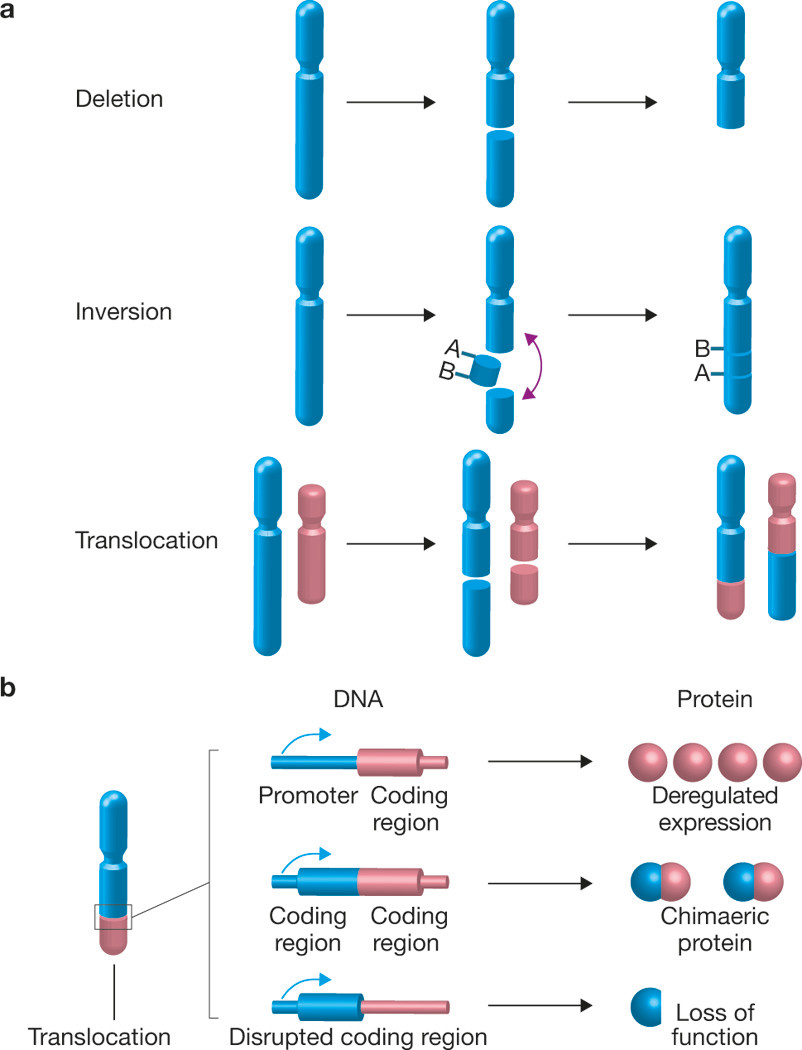
Consequences of chromosome rearrangements. (a) Chromosome breakage may lead to loss of genetic material (deletion). When two breaks occur in the same chromosome, the resulting piece of DNA can be inversed and re-inserted into the chromosome, leading to the formation of an inversion. Genomic material can also be transferred and join to a different chromosome, resulting in the formation of a chromosome translocation. (b) A translocation may provide a proliferative or survival advantage to the cell by generating a chimaeric fusion protein with oncogenic potential, through disruption of a tumour suppressor gene or by fusion of a tumour-promoting gene to a strong transcriptional promoter.
The formation of a chromosome translocation is a multistep process3. The initial event is the concomitant occurrence of double-strand breaks (DSBs) in multiple chromosomal locations (Fig. 1a). Such DSBs may arise spontaneously through replication errors, exogenous stress such as ionizing radiation and chemotherapeutic agents, or from scheduled breaks induced during development of the adaptive immune system, such as V(D)J recombination and immunoglobulin gene class-switch recombination (CSR)4. Regardless of the type of DNA damage, in response, cells will activate complex DNA repair mechanisms to restore genome integrity. If breaks occur within S phase, they may be preferentially repaired by homologous recombination using homologous sequences as templates, whereas the non-homologous end joining (NHEJ) pathway is active throughout the cell cycle and involves the simple, yet often inaccurate, ligation of the two broken chromosomes5. Although repair pathways are highly efficient, occasionally DSBs are not quickly resolved, and these are the breaks that ultimately may lead to translocations. Multiple persistent breaks may come into physical contact and illegitimate misjoining of ends located on different chromosomes may occur during the repair process, resulting in a translocation. Whereas the sequence of these basic steps is well established, it has largely been unknown how these events occur in space and time, and in the context of the three-dimensional organization of genomes.
The development of new experimental tools has recently enabled the cell biological delineation of the translocation process. We discuss here how broken chromosomes move, how they find their translocation partners, and how they synapse and then join to form translocations. We also discuss the role of spatial genome organization, DNA repair pathways and the epigenetic landscape in this process.
What happens when DNA breaks?
DNA lesions have the potential to be deleterious for cells. To counteract DNA damage, cells mount a rapid DNA damage response (DDR), which involves the coordinated accumulation of DNA repair factors at the site of damage, triggering cellular signalling pathways to halt the cell cycle and to initiate the repair of the lesions6. The local concentration of repair proteins around the broken chromosome ends results in the formation of cytologically detectable DNA repair foci7.
A variety of techniques have been used to probe the motion of DNA repair foci and the damaged chromatin they contain. In both the yeast Saccharomyces cerevisiae and mammalian cells, after the induction of a single DSB, the two intrachromosomal ends flanking this site remain tethered8-10. Time-lapse observations of DNA repair foci in mammalian cells indicate that their position is relatively stable over time11-12. Typically, in mammalian cells a DSB undergoes limited local motion with a mean squared displacement of ~1 μm2 h−1, comparable to that of a locus on an intact chromatin fibre11,12. Similarly, damaged chromatin is largely stationary after irradiation of human cells with ultrasoft x-rays13. However, under some circumstances, damaged DNA can undergo more extensive motion. Large-scale DNA damage induced by α-particles promotes movement over several micrometres and leads to clustering of damaged chromatin domains14. Moreover, tracking of repair foci marked by the DNA repair protein 53BP1 in human cells showed higher mobility after ionizing radiation compared to the motion of intact chromatin domains15. In the S. cerevisiae model system, breaks artificially induced by the I-Scel endonuclease exhibit increased mobility compared to intact chromosomal loci, and they explore a larger nuclear volume16,17. Intriguingly, the increased mobility of DSBs in yeast is only observed for endonuclease-induced persistent DSBs, but not for other types of damage, such as spontaneous breaks18 or breaks that arise from a protein-DNA adduct17. The ability of a DSB to explore large fractions of the volume of the yeast nucleus has been suggested to facilitate homologous pairing and repair16,17. The mobility of the unrepaired breaks in S. cerevisiae is dependent on factors that are involved in homologous recombination repair, including Rad51, Mec1 (the S. cerevisiae homologue of ATR), Rad9 and Sae2 (refs 16,17). Mec1, Rad9 and Rad53 are also involved in increased chromatin mobility of non-damaged loci after DNA damage elsewhere in the genome, suggesting a global genome-wide effect in addition to their local activity at sites of damage19. In mammalian cells, 53BP1 has been proposed to contribute to enhancing the motion and joining of deprotected telomeres in mouse cells20; however, the mobility of DSBs is not altered in human cells lacking 53BP1 (ref. 15), suggesting that distinct types of break differ in their dependence on 53BP1 for their mobility. These observations point to an involvement of major DSB repair factors in DSB motility, by mechanisms that are as yet unknown.
Spatial genome organization as a driver of translocations
The limited mobility of DSBs in mammalian cells has obvious implications for translocation formation, as physical interaction and pairing of DSBs is required for the illegitimate joining of chromosome ends. Given that genomes are non-randomly arranged in three-dimensional space21, it seems plausible that the spatial arrangement of the genome contributes to the propensity of two given chromosomes to form translocations (Fig. 2). In particular, it has been proposed that proximally positioned chromosomes undergo translocations at higher frequencies than distal chromosomes22,23. This hypothesis has been tested over the past decade using cytogenetic, genome-wide mapping and imaging approaches, and the emerging data strongly support a driving role of non-random spatial genome organization in determining the outcome of translocations.
Figure 2.
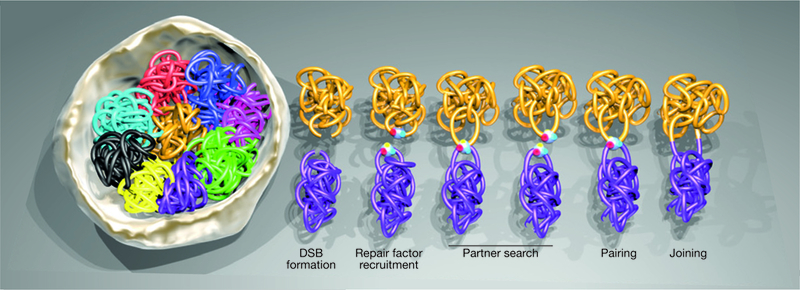
Distinct phases in the biogenesis of a translocation. The spatial arrangement of chromosomes within the 3D space of the nucleus is a key determinant of translocation frequency. In the presence of DNA damage, proximal chromosomes (magenta and gold) have a higher probability of translocating than distally located chromosomes (magenta and light blue). Following double-strand break (DSB) formation, the local concentration of repair proteins around the broken chromosome ends results in the formationof a DNA repair focus at each break site. DSBs undergo a partner search within the nuclear space driven by their locally constrained, saltatory motion. In case they encounter another DSB within their path, the two lesions can undergo cycles of transient pairing and dissociation. Although many of these synapsed breaks eventually separate, some engage in persistent pairing, making them susceptible to illegitimate joining and the formation of a translocation.
Early studies using fluorescence in situ hybridization (FISH) to map the spatial position of frequent translocation partners indicated strong correlation between the spatial proximity of, chromosomes or genes and their translocation frequencies24-32. Translocating chromosomes were often found at the nuclear periphery25 and in closer spatial proximity than nontranslocating chromosomes26. Translocation frequency was also positively correlated with the degree of intermingling of adjacent chromosomes in lymphocytes27. These observations on entire chromosomes were extended to individual genes28-31,33. For example, the distance of the MYC gene to its three Burkitt’s lymphoma translocations partners IGH, IGK and IGL correlates with the observed translocation frequency in patients29. In line with the established notion of tissue- and cell-type-specific, non-random organization of genomes2,21,34, correlations between tissue-specific chromosome location and tissue-specific translocations have also been observed24,35. Similar correlations are also found in S. cerevisiae, whose genome is less stringently spatially organized, but overlapping nuclear territories were nevertheless found to recombine more efficiently than sequences located in spatially distant locations36.
These cytological studies were confirmed by genome-wide mapping approaches37,38. Sequencing of junctions of translocations after experimentally inducing DSBs at the c-myc or the Igh loci in mouse B lymphocytes demonstrated that translocations occur predominantly between the DSB and nearby regions on the same chromosome37,38. Chromosome conformation capture techniques, which allow mapping of chromatin interactions at a genome-wide scale, showed that the most frequent translocation partners of experimentally induced DSBs in transformed pro-B cells were found in cis along single chromosomes containing the induced DSBs, as well as within other chromosomes and subchromosomal domains in a manner directly related to pre-existing spatial proximity — although a low frequency of distal translocation partners were found as well39. In activated B lymphocytes, both the number and proximity of DNA breaks, induced by the activation- induced cytidine deaminase (AID), which is responsible for IgH class switch recombination and somatic hypermutation, have been implicated in influencing translocation frequency40,41
Imaging approaches have recently enabled insights beyond these correlative studies. By fluorescently tagging individual DSBs on separate chromosomes in a cell-based model system to visualize translocations42, it has become possible to directly track the fate of DSBs and to fully describe the events leading to a translocation in individual living cells42. This approach showed that more than 80% of translocating breaks originated from locations within 2.5 μm of each other42. This distance corresponds reasonably well with the observed limited motion of DSBs11-13, demonstrating that translocations are formed predominantly between proximal chromosome breaks42. Intriguingly, however, a minority of translocations do seem to form from DSBs located more than 5 μm apart, indicating that formation of translocations over long ranges does occur, and raising the possibility that not all translocations form by the same mechanism42.
Partner search in 3D space
After chromosomes have suffered breaks, how do the ends find their partners in 3D space? Time-lapse microscopy of individual DSBs in living cells shows that DSBs undergo a non-directional, locally restricted saltatory motion42 (Fig. 2). Interestingly, translocating breaks were found to undergo faster motion than non-translocating breaks42. The molecular basis for this difference is unknown.
A key question is whether the motion of chromosome loci and DSBs represents an active and directed process, or occurs by passive diffusion. Whereas the vast majority of studies on chromatin motion find that chromatin loci of various types undergo predominantly locally constrained passive diffusional motion, a few report active chromatin motion. The first evidence for active chromatin movement was the observation of linear, directed motion of a tagged chromosome site targeted with a transcriptional activator from the nuclear periphery to the interior43. This motion was sensitive to perturbation of actin and nuclear myosin I. Similarly, the long-range motion of the U2 small nuclear RNA gene locus towards Cajal bodies during transcriptional activation in human cells seemed to occur along a linear trajectory and was dependent on actin44. It remains to be seen whether the actin and myosin dependence of this motion is due to global alterations of nuclear structure or interference with chromatin remodelling activities, some of which use actin as a cofactor45. The lack of linear motion of DSBs suggests that their movements are not directed; however, energy-dependent chromatin remodelling activities may affect DSB motion46.
The mobility of chromosome breaks directly influences the probability of two breaks to synapse and thus undergo a translocation. Several factors that influence chromosome dynamics have recently been identified. In S. cerevisiae, association of telomeres with the nuclear envelope and tethering of centromeres to the membrane-associated spindle pole body constrain chromosome motion47-49. Similarly, the association of chromosome regions with the nucleolus or the nuclear periphery limits the motility of loci in mammalian cells50-51. These observations suggest that the likelihood of translocation may be affected by chromosomal location, particularly of loci in proximity to chromosomal tethering points such as the nuclear envelope or nuclear bodies.
It also seems that the mobility of both intact chromatin loci and DSBs may be cell-cycle-dependent. Although experiments using chimaeric versions of core histones fused to photoactivatable or photobleachable proteins to mark chromatin domains revealed similar mobilities of chromosomal regions from middle G1 to late G2 phase51,52, a two-fold increase in the mobility of chromosome territories was evident in early G1 phase52. Furthermore, increased chromatin mobility of a fluorescently tagged gene array during the first hours of G1 phase in human cells has been reported53 and tagged chromosomal foci in S. cerevisiae display a decrease in motion during replication54. Tracking of decondensed euchromatic chromatin domains in early S phase uncovered higher motility than condensed heterochromatic domains during mid or late S phase in human cells15,55. These observations point to a higher mobility of chromosome loci in early G1 phase, which decreases as cells progress through S phase. Similarly, studies that monitored the mobility of DSB-containing chromatin by tracking 53BP1-containing ionizing-radiation-induced foci (IRIF) in human cells demonstrated decreased motion of DSBs during S phase compared to G1 and G2 (ref. 15). Despite these cell-cycle differences in chromatin motion, the percentage of cells with synapsed DSBs in different cell cycle phases remains constant, suggesting that the variability in mobility over the cell cycle does not affect the probability of DSB pairing42.
DSB pairing
The synapsis of DSBs within the nucleus is a fundamental step in the formation of translocations. Insights into pairing of breaks comes from studies in S. cerevisiae in which a chromosome break associates with its intact homologous counterpart in order to use it as a template for homologous recombination when the sister chromatid is not available. Rad51is involved in the formation of nucleoprotein filaments on singlestranded DNA, and drives the search for homology56-58. Homologous chromosomes also pair at sites of ionizing- or endonuclease-induced DSBs in human cells59. The contact between the homologous chromosomes is centred around the location of the DSB and requires the activity of the ATM kinase and ongoing transcription59.
Although DSBs that are repaired by NHEJ do not synapse with homologous templates, some DSBs are able to persistently pair, making them susceptible for translocation. After DSBs are formed, they undergo a partner search within the nuclear space driven by their locally constrained, random motion42 (Fig. 2). When encountering another DSB within their volume of motion, the two lesions undergo cycles of transient pairing and dissociation. Although many of these pairs eventually separate, some DSBs engage in persistent pairing, making them susceptible for illegitimate joining and translocation formation42. Importantly, single-cell tracking experiments show that the two intrachromosomal ends generated by the DSB move in unison to the area of pairing with the other chromosome ends, suggesting that the two intrachromosomal breaks do not separate before translocation and the choice of the chromosomal partners involved in the joining takes place after the chromosomal partners are in close proximity42 (Fig. 2). The observed coordinated motion of the two DSBs explains the inherent reciprocity of translocation formation, as it would be highly unlikely that both broken chromosome ends find their two corresponding translocation partners if the two chromosome ends separated before congregation with other DSBs.
Are there cellular factors that promote the pairing of DSBs within the nuclear space? In S. cerevisiae, multiple DSBs coalesce into common repair centres60. These accumulations of repair proteins have been suggested to increase the efficiency of repair. On the other hand, the the association of multiple DSBs with repair centres may also aid the persistent pairing of DSBs60. Coalescence of multiple DSBs, however, is not desirable for a cell as the spatial proximity of breaks may also facilitate their synapsis and subsequent illegitimate misjoining to form a translocation. In S. cerevisiae, the appearance of a DSB within ribosomal DNA (rDNA) resulted in the transient relocalization of the lesion to an extranucleolar site to repress rDNA hyperrecombination61. Similarly, in Drosophila melanogaster, damaged heterochromatin domains are expelled from the chromosome territory, resulting in the physical separation of the repetitive heterochromatic sequences from homologous sequences on the same chromosome, and thus reducing the possibility of inaccurate recombination62. Moreover, both in yeast and in mammalian cells, the DNA damage marker phospho-histone H2AX (γ-H2AX) is found at the periphery of heterochromatic regions, indicating decondensation of heterochromatin or the movement of breaks to the periphery63,64. Congregation of individual repair foci in common repair centres is not the norm in mammalian cells, and has only been observed sporadically12. Typically, and again in contrast to yeast60, an individual repair focus forms around each DSB and, unless the DSBs come into close spatial proximity of each other, each repair focus remains separate42. During translocation formation, as individual breaks approach each other to pair, their respective repair foci coalesce into the same repair focus, which is then resolved over time — presumably as a consequence of the completion of repair towards the formation of a translocation42 (Fig. 2). Whether the observed congregation of repair foci contributes to the pairing of the DSBs and the formation of chromosome translocations by clustering DSBs and holding them in place, or whether it is merely a consequence of the pairing event, is currently unclear.
In an analogous fashion, it has been proposed that transcription factories, which contain multiple actively transcribed genes, may contribute to the formation of chromosome translocations by retaining actively transcribed genes in spatial proximity65. In support of this, the MYC gene and its frequent translocation partners IGH, IGK and IGL are thought to share a transcription factory66. Moreover, genome-wide translocation capture studies in lymphocytes revealed translocation break points to be frequently positioned near transcription start sites of active genes37,38. In addition, early replicating fragile sites67, which can be found as translocation partners in more than 50% of recurrent human diffuse large B cell lymphoma, are enriched in regions with high transcriptional activity, and their fragility positively correlates with their transcriptional activity67. Evidence supporting a potential role of transcription in the formation of translocations also comes from studies showing that transcriptional activation by androgen- receptor-dependent signalling promotes the spatial proximity of androgen-responsive genes and the formation of site-specific DSBs, and contributes to the formation of prostate-cancer-specific translocations68. Furthermore, transcriptional upregulation of translocating genes is accompanied by spatial juxtaposition in anaplastic cell lymphoma69. Taken together, these observations suggest that active transcription influences the probability of translocation formation by retaining potential translocation partners in close physical proximity within shared transcription factories (Fig. 3).
Figure 3.
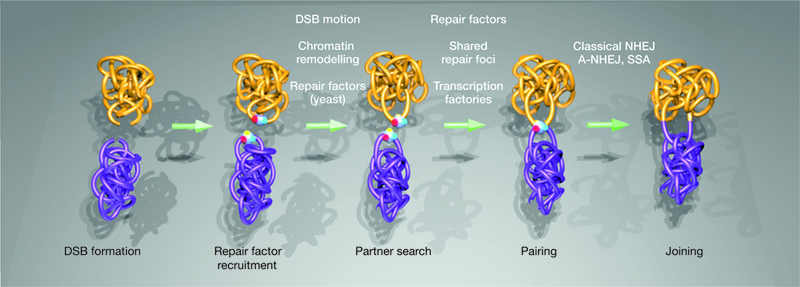
Factors influencing distinct steps in the formation of chromosome translocations. The mobility of chromosome breaks can directly influence the probability of chromosomal breaks meeting in the nuclear space. The action of major repair factors and chromatin remodellers has been shown to be an important regulator of the motion of DSBs in S. cerevisiae, and may directly contribute to the formation of translocations. The synapsis of the breaks
One protein that has been demonstrated to influence the synapsis of DSBs is the repair factor Mre11. Using single-cell analysis and high-throughput microscopy, inhibition of Mre11 by the mirin compound70 or by knockdown42, but not inhibition of the major DDR kinases ATM, ATR and DNAPK, resulted in a decrease in the percentage of cells with paired DSBs and a concomitant decrease in the fraction of cells with translocations42. These experiments support the notion that Mre11, as part of the MRN (Mre11-Rad50-Nbs1) complex71, acts as a DNA-end bridging factor, facilitating the synapsis of DSBs and translocation formation. In addition, the α-particle-induced clustering of chromosome domains marked by γ-H2AX is affected in cells from ataxia telangiectasia-like disorder (ATLD) patients14, which have reduced levels and function of the Mre11 protein72. These studies demonstrate that pairing of DSBs is an important step in the formation of translocations, and identify Mre11 as a key regulator of this event (Fig. 3). Furthermore, several studies have implicated Mre11 as the DNA-end tethering factor during classic and alternative NHEJ and in class switch and V(D)J recombination73-79.
DNA repair pathways in translocation formation
As the formation of a translocation requires the joining of chromosome breaks, the DNA repair machinery, which prevents the formation of translocations by rapidly repairing lesions, is, paradoxically, also a key player in the formation of any translocation (Fig. 3). It is therefore an obvious question whether the distinct pathways that mediate DSB repair, or the specific proteins of the repair machinery, influence the frequency of translocations.
In mice, dramatic genomic instability and increased frequency of chromosome translocations occur when key factors of the NHEJ pathway, such as Ku70, Ku80, DNAPKcs, XRCC4 or LIGIV, are absent80-82. This is not simply due to an increase in DNA damage because of deficiencies in repair; in the absence of the NHEJ factor Ku70, higher numbers of chromosome translocations between RAG (recombination-activating gene)-generated DSBs and an endonuclease-generated DSB on a different chromosome are observed, suggesting that NHEJ factors suppress the formation of translocations per se83. Furthermore, translocation frequencies between endonuclease-induced DSBs on different chromosomes are increased in the absence of Ku80 or DNAPKcs (ref. 42) or in the absence of XRCC4-LIGIV (ref. 84). Given that the ligation of the translocation partners is an absolute requirement for a translocation to form, these findings suggest that, in the absence of NHEJ factors, non-canonical joining pathways promote their ligation83.
Early studies in S. cerevisiae provided evidence for the presence of an alternative pathway that mediates joining of breaks in the absence of Ku70 or Ku80 (refs 85-87). This alternative mode of repair, termed alternative NHEJ (A-NHEJ), relies on microhomologies between the partners, and is believed to be active when classical NHEJ is not functional. Importantly, this pathway may account for the observed increased formation of chromosome translocations in mice lacking the core NHEJ proteins80. In support of this notion, mice lacking Ku70, Ku80, XRCC4 or LIG4 in a p53-null background develop tumours with translocations featuring junctions with microhomology88,89, and translocations and microhomology usage between IgH and Myc are increased in mouse B lymphocytes after deletion of Ku70 and LIG4 (ref. 90). As translocations can form in cells lacking LIG4, which is the major ligase in NHEJ, other ligases must be present to mediate the joining of the breaks. Depletion of either LIG1 or LIG3 in mammalian cells reduces the use of microhomology-mediated end-joining in cell-free extracts91, and cells deficient in LIG3 show decreased translocation frequency and decreased usage of microhomology92, suggesting that the ligation step in the A-NHEJ pathway is mediated by LIG3. In addition, whereas LIG1 loss has no effect on translocation frequency, co-depletion of LIG3 and LIG1 reduces translocations to a higher extent than loss of LIG3 alone, suggesting that LIG1 can act as a backup ligase for LIG3 (ref. 92). Similarly, cells deficient for the CtBP-interacting protein (CtIP) show decreased frequency of translocations and reduced microhomology usage, indicating that CtIP is also involved in the formation of translocations93. These findings suggest that the A-NHEJ pathway is a critical contributor to translocation frequency, but it is still not clear whether A-NHEJ is a translocation-prone pathway that is active in the presence of classical NHEJ or whether its dominance in the absence of classical NHEJ is merely a reflection of the behaviour of persistent unrepaired DSBs, which have a longer time window to pair with other DSBs and to translocate. In support of the latter, inhibition of DNAPKcs kinase activity substantially increases the frequency of translocations42. DNAPKcs is autophosphorylated at numerous sites94 and regulates, among other functions, its own dissociation from broken chromosome ends95. As impairment of DNAPKcs kinase activity and its autophoshorylation decreases its exchange rate and retains it at DSBs96, it is likely that the reduced turnover of DNAPKcs at broken chromosome ends decreases the propensity of intrachromosomal joining by extending the time frame in which DSBs can meet and translocate with breaks on different chromosomes. Further insight into how the repair machinery may deal with chromosome lesions comes from the study of telomeres, which are naturally occurring chromosome ends. When telomeres are uncapped by loss of the shelterin component TRF2, chromosome ends are recognized by the DNA repair machinery as DSBs and are processed by an NHEJ reaction to promote end-to-end chromosome fusions97. Importantly, these structures can cause translocations and new breaks in subsequent divisions, and in the absence of p53, their presence positively correlates with a high frequency of epithelial cancer98.
In addition to NHEJ pathways, homologous-recombination-based pathways have been implicated in the numerical variation of copies of segments in the genome, known as copy number variation, and the formation of nonreciprocal translocations99. A replication-based mechanism termed microhomology-mediated break-induced replication (MMBIR) uses a different site with partial homology to the original template, resulting in a daughter strand containing sequences from two chromosomes and, thus, a translocation99,100. Moreover, an alternative homologous repair pathway, termed single-strand annealing (SSA)101, can repair DSBs between two repeated sequences in a process that can also generate translocations102-104. However, when identical Alu repeats were introduced adjacent to inducible DSB sites in a controlled engineered system, the frequency of translocations was not altered102, suggesting that the presence of homology per se is not a driver of translocations between breaks in heterologous loci102,105. Similarly, classical NHEJ and alternative end-joining pathways1,106,107 predominate in forming complex translocations during chromothripsis, but replication-dependent processes108, such as replication fork stalling and template switching109 or MMBIR (refs 100,110), may also be involved in their aetiology. Collectively, these findings suggest that NHEJ pathways and SSA are dominant in the formation of chromosome translocations between breaks from within heterologous loci. In agreement with this notion, chromosome translocations between endonuclease-induced DSBs on different chromosomes form with the same probability in different phases of the cell cycle42, in line with NHEJ being the major repair pathway in the formation of translocations throughout the cell cycle111-114.
Chromatin structure and histone modifications in translocation formation
DSB repair occurs in the context of higher-order chromatin structure115-117 and various chromatin components, including chromatin modifiers, remodellers, histone chaperones and histone variants, play an active role in the DDR, fine-tuning damage signalling and modulating the outcome of repair118. It is therefore likely that chromatin components influence the formation of translocations at multiple steps.
Circumstantial evidence suggests that chromatin properties may affect the formation of translocations by enhancing the susceptibility of genome regions to breakage. Several translocation breakpoints have been mapped to within, or close to, transcriptionally active genome regions37,38,68,69,119, and breaks generated in the presence or absence of AID are enriched for H3K4me3, H3 acetylation, and H3K36me3 associated with active transcription38. In addition, specific histone modifications have been implicated in the mechanisms that promote the formation of DSBs during V(D)J and CSR recombination in lymphocytes. The interaction of RAG-2 with H3K4me2 or H3K4me3, through its plant-homeodomain (PHD) domain, is required for recombination of extrachromosomal and endogenous immunoglobulin gene segments120,121. Moreover, a set of ectopic AID-targeted genes is enriched for H3K4me3 in the vicinity of their break sites, and translocations between these genes are found in tumours122. In prostate cancer cell lines, the frequently translocated TMPRSS2-ERG region is enriched in active marks such as H3K4me3, H3K36me3 and H3 acetylation, which are depleted in rearrangements of ETS-negative prostate tumors123. Moreover, in the presence of androgens and ionizing radiation in prostate cancer cells, H3K79me2 and H4K16 acetylation marks are enriched close to the TMPRSS2-ERG breakpoint regions, and overexpression of the H3K79 methyltransferase DOT1L substantially increases the frequency of translocations68. These data may point to an enrichment of open chromatin marks within translocation breakpoints.
Chromatin properties may also affect the motion of chromatin and DSBs and thus their propensity to translocate46. Tethering the viral transactivator VP16 to a heterochromatic transgene array in mammalian cells promotes chromatin decompaction and induces long-range motion43. Similar experiments in S. cerevisiae show increased motion of non-telomeric124 or silent telomeric loci47. Although these studies cannot distinguish the effect of chromatin relaxation from that of transcriptional upregulation on chromatin motion, other studies have shown that an increase in transcription alone does not necessarily lead to a higher mobility. As an example, tethering of the Gal4 activation domain to a promoter results in an increase in transcription without influencing the mobility of the locus, suggesting that changes in transcription are not sufficient to drive increased locus mobility124. These results indicate that it is the changes in chromatin structure that determine mobility. In support of this, targeting of the chromatin remodeller IN080 promotes mobility in the absence of transcriptional changes124. Importantly, the increased mobility is dependent on the catalytic subunit of IN080 (ref. 124). Moreover, targeting the actin-related protein 8 (ARP8), which is required for the remodelling activity of the IN080 complex, also leads to increased mobility of chromatin, and its deletion partially decreases the motion of an endonuclease-induced DSB124. Reduced motion of 53BP1 foci was observed in human cells treated with histone deacetylase or histone acetyltrasferase inhibitors, tentatively pointing to a role of chromatin properties in DSB motion15. These findings support the idea that chromatin remodelling regulates the motion of chromosomes and DSBs, and in this way contributes to the translocation process.
Future perspectives
Chromosome translocation is a prominent phenomenon that we have come to take for granted because of its ubiquitous prevalence and clinical importance. Due to the experimental difficulties in probing the biogenesis of translocations, the elucidation of the mechanisms leading to their formation has lagged behind their cytological characterization and application in diagnostic applications. Recent work has delineated distinct steps in the formation of translocations, and several powerful experimental systems have been developed to probe them. These studies create a conceptual and experimental framework to now dissect the molecular mechanisms involved in the formation of chromosome translocations. A caveat with most current studies in any organism is the reliance on artificial means to induce breaks experimentally. The development of systems that allow tracking of endogenous breaks at defined regions, as are now being pursued by use of recombineering technology, will be an important step forward. It will be of prime importance and interest to uncover the effect of histone modifiers, chromatin remodellers and higher-order chromatin structure on the individual steps that govern the formation of translocations. Translocation-capture sequencing techniques and experimental systems to integrate key mechanistic steps of the formation of translocations will serve as valuable tools in this effort. The delineation of distinct steps in the translocation process will also provide the framework for more clinically relevant efforts. In particular, it will be important to determine how various components of the DNA damage response affect individual steps of the process and modulate translocation frequency. Such studies may help in the design of cancer therapies with improved efficacy, and may be particularly useful in minimizing the occurrence of secondary translocation-initiated tumors, a major problem in chemotherapy. Chromosome translocations are beautifully complex yet threateningly dangerous. Elucidating their biogenesis will unravel some of the beauty of the basic biology that underpins their formation, and is likely to also generate new tools to combat cancer.
ACKNOWLEDGEMENTS
The Misteli laboratory is supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, Center for Cancer Research. We dedicate this Review to the memory of Janet Rowley, who boldly founded and thoughtfully guided the field of chromosome translocation biology.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Vassilis Roukos, National Cancer Institute, Bethesda, Maryland 20892, USA..
Tom Misteli, National Cancer Institute, Bethesda, Maryland 20892, USA..
References
- 1.Stephens PJ et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitelman F, Johansson B & Mertens F The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–45 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Meaburn KJ, Misteli T & Soutoglou E Spatial genome organization in the formation of chromosomal translocations. Semin. Cancer Biol. 17, 80–90 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudley DD, Chaudhuri J, Bassing CH & Alt FW Mechanism and control of V(D) J recombination versus class switch recombination: similarities and differences. Adv. Immunol. 86, 43–112 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Lieber MR The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukas J, Lukas C & Bartek J More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 13, 1161–1169 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Lukas C, Bartek J & Lukas J Imaging of protein movement induced by chromosomal breakage: tiny ‘local’ lesions pose great ‘global’ challenges. Chromosoma 114, 146–154 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Kaye JA et al. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr. Biol. 14, 2096–2106 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Lobachev K, Vitriol E, Stemple J, Resnick MA & Bloom K Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr. Biol. 14, 2107–2112 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Soutoglou E et al. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 9, 675–682 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruhlak MJ et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 172, 823–834 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jakob B, Splinter J, Durante M & Taucher-Scholz G Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl Acad. Sci. USA 106, 3172–3177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelms BE, Maser RS, MacKay JF, Lagally MG & Petrini JH In situ visualization of DNA double-strand break repair in human fibroblasts. Science 280, 590–592 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Aten JA et al. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 303, 92–95 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Krawczyk PM et al. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 1-25, 2127–2133 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Mine-Hattab J & Rothstein R Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 14, 510–517 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Dion V, Kalck V, Horigome C, Towbin BD & Gasser SM Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 14, 502–509 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Dion V, Kalck V, Seeber A, Schleker T & Gasser SM Cohesin and the nucleolus constrain the mobility of spontaneous repair foci. EMBO Rep. 14, 984–991 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seeber A, Dion V & Gasser SM Checkpoint kinases and the IN080 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev. 27, 1999–2008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimitrova N, Chen YC, Spector DL & de Lange T 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misteli T Beyond the sequence: cellular organization of genome function. Cell 128, 787–800 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Roukos V, Burman B & Misteli T The cellular etiology of chromosome translocations. Curr. Opin. Cell Biol. 25, 357–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misteli T Higher-order genome organization in human disease. Cold Spring Harb. Persp. Biol. 2, a000794 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikiforova MN et al. Proximity of chromosomal loci that participate in radiation- induced rearrangements in human cells. Science 290, 138–141 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Bickmore WA & Teague P Influences of chromosome size, gene density and nuclear position on the frequency of constitutional translocations in the human population. Chromosome Res. 10, 707–715 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Parada LA, McQueen PG, Munson PJ & Misteli T Conservation of relative chromosome positioning in normal and cancer cells. Curr. Biol. 12, 1692–1697 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Branco MR & Pombo A Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 4, e138 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neves H, Ramos C, da Silva MG, Parreira A & Parreira L The nuclear topography of ABL, BCR, PML, and RARa genes: evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood 93, 1197–1207 (1999). [PubMed] [Google Scholar]
- 29.Roix JJ, McQueen PG, Munson PJ, Parada LA & Misteli T Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 34, 287–291 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Lukasova E et al. Localisation and distance between ABL and BCR genes in interphase nuclei of bone marrow cells of control donors and patients with chronic myeloid leukaemia. Hum. Genet. 100, 525–535 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Roccato E et al. Proximity of TPR and NTRK1 rearranging loci in human thyrocytes. Cancer Res. 65, 2572–6576 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Stahl A et al. Structural basis for Robertsonian translocations in man: association of ribosomal genes in the nucleolar fibrillar center in meiotic spermatocytes and oocytes. Proc. Natl Acad. Sci. USA 80, 5946–5950 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikiforov YE, Koshoffer A, Nikiforova M, Stringer J & Fagin JA Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene 18, 6330–6334 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Cremer T & Cremer C Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292–301 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Parada LA, McQueen PG & Misteli T Tissue-specific spatial organization of genomes. Genome Biol. 5, R44 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agmon N, Liefshitz B, Zimmer C, Fabre E & Kupiec M Effect of nuclear architecture on the efficiency of double-strand break repair. Nat. Cell Biol. 15, 694–699 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Chiarle R et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein IA et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147, 95–106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148, 908–921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hakim 0 et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484, 69–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rocha PP et al. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol. Cell 47, 873–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roukos V et al. Spatial dynamics of chromosome translocations in living cells. Science 341, 660–664 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chuang CH et al. Long-range directional movement of an interphase chromosome site. Curr. Biol. 16, 825–831 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Dundr M et al. Actin-dependent intranuclear repositioning of an active gene locus in vivo. J. Cell Biol. 179, 1095–1103 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu JI & Crabtree GR Cell signaling. Nuclear actin as choreographer of cell morphology and transcription. Science 316, 1710–1711 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Dion V & Gasser SM Chromatin movement in the maintenance of genome stability. Cell 152, 1355–1364 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Hediger F, Neumann FR, Van Houwe G, Dubrana K & Gasser SM Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr. Biol. 12, 2076–2089 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Zimmer C & Fabre E Principles of chromosomal organization: lessons from yeast. J. Cell Biol. 192, 723–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taddei A, Schober H & Gasser SM The budding yeast nucleus. Cold Spring Harb. Perspect. Biol. 2, a000612 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chubb JR, Boyle S, Perry P & Bickmore WA Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 12, 439–445 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Wiesmeijer K, Krouwels IM, Tanke HJ & Dirks RW Chromatin movement visualized with photoactivable GFP-labeled histone H4. Differentiation 76, 83–90 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Walter J, Schermelleh L, Cremer M, Tashiro S & Cremer T Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160, 685–697 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomson I, Gilchrist S, Bickmore WA & Chubb JR The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1. Curr. Biol. 14, 166–172 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Heun P, Laroche T, Shimada K, Furrer P & Gasser SM Chromosome dynamics in the yeast interphase nucleus. Science 294, 2181–2186 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Pliss A, Malyavantham K, Bhattacharya S, Zeitz M & Berezney R Chromatin dynamics is correlated with replication timing. Chromosoma 118, 459–470 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Forget AL & Kowalczykowski SC Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ragunathan K, Liu C & Ha T RecA filament sliding on DNA facilitates homology search. eLife 1, e00067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Renkawitz J, Lademann CA, Kalocsay M & Jentsch S Monitoring homology search during DNA double-strand break repair in vivo. Mol. Cell 50, 261–272 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Gandhi M et al. Homologous chromosomes make contact at the sites of double-strand breaks in genes in somatic G0/G1-phase human cells. Proc. Natl Acad. Sci. USA 109, 9454–9459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lisby M, Mortensen UH & Rothstein R Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 5, 572–577 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Torres-Rosell J et al. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 9, 923–931 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Chiolo I et al. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jakob B et al. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 39, 6489–6499 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A & Haber JE Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J. Cell Biol. 178, 209–218 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Osborne CS Molecular pathways: transcription factories and chromosomal translocations. Clin. Cancer Res. 20, 296–300 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Osborne CS et al. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 5, e192 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barlow JH et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin C et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139, 1069–1083 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mathas S et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc. Natl Acad. Sci. USA 106, 5831–5836 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dupre A et al. A forward chemical genetic screen reveals an inhibitor of the Mre11- Rad50-Nbs1 complex. Nat. Chem. Biol. 4, 119–125 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stracker TH & Petrini JH The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stewart GS et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99, 577–587 (1999). [DOI] [PubMed] [Google Scholar]
- 73.Williams RS et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135, 97–109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dinkelmann M et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 16, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Helmink BA et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J. Exp. Med. 206, 669–679 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rass E et al. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 16, 819–824 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Xie A, Kwok A & Scully R Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 16, 814–818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Della-Maria J et al. Human Mre11/human Rad50/Nbs1 and DNA ligase Illalpha/ XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 286, 33845–33853 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moore JK & Haber JE Cell cycle and genetic requirements of two pathways of nonhomologous end joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell Biol. 16, 2164–2173 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferguson DO et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc. Natl Acad. Sci. USA 97, 6630–6633 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Difilippantonio MJ et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404, 510–514 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao Y et al. Interplp ay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404, 897–900 (2000). [DOI] [PubMed] [Google Scholar]
- 83.Weinstock DM, Brunet E & Jasin M Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat. Cell Biol. 9, 978–981 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simsek D & Jasin M Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 17, 410–416 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boulton SJ & Jackson SP Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 24, 4639–4648 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boulton SJ & Jackson SP Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 15, 5093–5103 (1996). [PMC free article] [PubMed] [Google Scholar]
- 87.Ma JL, Kim EM, Haber JE & Lee SE Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell Biol. 23, 8820–8828 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Difilippantonio MJ et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J. Exp. Med. 196, 469–480 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu C et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 109, 811–821 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Boboila C et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J. Exp. Med. 207, 417–427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liang L et al. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 36, 3297–3310 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Simsek D et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 7, e1002080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Y & Jasin M An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 18, 80–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dobbs TA, lainer JA & Lees-Miller SP A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair 9, 1307–1314 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neal JA & Meek K Choosing the right path: does DNA-PK help make the decision? Mutat. Res. 711, 73–86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Uematsu N et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 177, 219–229 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palm W & de Lange T How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Artandi SE et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406, 641–645 (2000). [DOI] [PubMed] [Google Scholar]
- 99.Hastings PJ, Lupski JR, Rosenberg SM & Ira G Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Payen C, Koszul R, Dujon B & Fischer G Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 4, e1000175 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pâques F & Haber JE Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Elliott B, Richardson C & Jasin M Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol. Cell 17, 885–894 (2005). [DOI] [PubMed] [Google Scholar]
- 103.Richardson C & Jasin M Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 405, 697–700 (2000). [DOI] [PubMed] [Google Scholar]
- 104.Haber JE & Leung WY Lack of chromosome territoriality in yeast: promiscuous rejoining of broken chromosome ends. Proc. Natl Acad. Sci. USA 93, 13949–13954 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weinstock DM, Elliott B & Jasin M A model of oncogenic rearrangements: differences between chromosomal translocation mechanisms and simple double-strand break repair. Blood 107, 777–780 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kloosterman WP et al. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep. 1, 648–655 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Chiang C et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 44, 390–397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu P et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146, 889–903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee JA, Carvalho CM & Lupski JR A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247 (2007). [DOI] [PubMed] [Google Scholar]
- 110.Hastings PJ, Ira G & Lupski JR A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5, e1000327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Takata M et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 17, 5497–5508 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weinstock DM, Richardson CA, Elliott B & Jasin M Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair 5, 1065–1074 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Mao Z, Bozzella M, Seluanov A & Gorbunova V DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7, 2902–2906 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Symington LS & Gautier J Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 (2011). [DOI] [PubMed] [Google Scholar]
- 115.Polo SE & Jackson SP Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Misteli T & Soutoglou E The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 10, 243–254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Price BD & D’Andrea AD Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Soria G, Polo SE & Almouzni G Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol. Cell 46, 722–734 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Mathas S & Misteli T The dangers of transcription. Cell 139, 1047–1049 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu Y, Subrahmanyam R, Chakraborty T, Sen R & Desiderio S A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity 27, 561–571 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Matthews AG et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450, 1106–1110 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kato L et al. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proc. Natl Acad. Sci. USA 109, 2479–2484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Berger MF et al. The genomic complexity of primary human prostate cancer. Nature 470, 214–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Neumann FR et al. Targeted IN080 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev. 26, 369–383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]