

Abstract
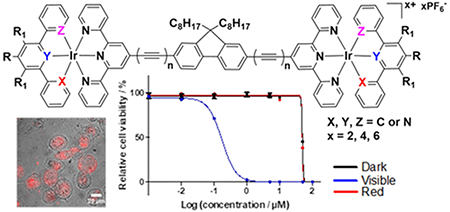
A series of cationic dinuclear iridium(III) complexes (Ir1 - Ir5) bearing terpyridine-capped fluorenyl bridging ligand and different polypyridyl or cyclometalating terminal tridentate ligands were synthesized, characterized, and evaluated for their photophysical and photobiological activities. The influence of the bridging and terminal ligands on the photophysical properties of the complexes was investigated by UV-vis absorption, emission, and transient absorption spectroscopy, and simulated by TDDFT calculations. All of the complexes displayed strong bridging-ligand localized visible 1π,π* absorption and red- or near-infrared (NIR) phosphorescence as well as broad triplet excited-state absorption across both visible and NIR wavelengths. These triplet states were assigned as predominantly 3π,π* for Ir1 (τ = 3.1 μs) and Ir4 (τ = 48 μs), and predominantly 3CT (charge transfer) for Ir2, Ir3 and Ir5 (τ = 1.7–2.7 μs). Complexes Ir1 – Ir5 acted as in vitro photodynamic therapy (PDT) agents toward human SK-MEL-28 melanoma cells when activated with visible light, with submicromolar photocytotoxicity and phototherapeutic indices (PIs) ranging from 20 to almost 300. The in vitro PDT effects with visible light did not correlate with singlet oxygen (1O2) quantum yields or DNA photocleaving capacity probed under cell-free conditions. All of the Ir(III) complexes phosphoresced brightly when associated with compromised cells (with or without a light treatment) and exhibited photoactivated cellular uptake, highlighting the theranostic potential of this new class of Ir(III) complex photosensitizers.
Graphical Abstract
Dinuclear iridium(III) complexes bearing terpyridine-capped fluorenyl bridging ligand and different polypyridyl or cyclometalating terminal tridentate ligands exhibited in vitro photodynamic therapeutic effects toward SK-MEL-28 melanoma cells upon visible light activation, with submicromolar photocytotoxicity and phototherapeutic indices ranging from 20 to almost 300. They phosphoresced brightly when associated with compromised cells and exhibited photoactivated cellular uptake, highlighting the theranostic potential of these complexes. Both the bridging and terminal ligands impacted the photophysical and photobiological activities significantly.
INTRODUCTION
In recent decades transition-metal complexes have come to the forefront in the search for new chemical entities in drug discovery and biological chemistry.1 Platinum (Pt)-based metal complexes have been investigated extensively for cancer therapy,2 with cisplatin being arguably the most successful anticancer drug to date. Nevertheless, there is continued focus on developing alternatives to cisplatin3,4 and other nonselective cytotoxic agents in an effort to reduce the systemic side effects associated with conventional chemotherapy approaches. Ruthenium (Ru)-containing compounds have been widely studied as alternatives to the Pt-derived drugs, with a few (e.g., NAMI-A, KP1019, and IT-139) entering clinical trials5–7 but none in the clinic to date. With selectivity being a key consideration for any new drug, Ru(II) coordination complexes and other transition-metal complexes are also being considered for targeted modalities such as photodynamic therapy (PDT).8–11
PDT has been known for over a hundred years12 yet remains underexploited in mainstream cancer therapy despite its precise spatiotemporal selectivity. In its narrowest definition, PDT involves activation of an otherwise nontoxic photosensitizer with photons of appropriate energy to produce a triplet excited state that can relax through singlet oxygen (1O2) sensitization or the production of other reactive oxygen species (ROS).13 Collectively, cytotoxic ROS destroy tumors and tumor vasculature, and can even invoke an antitumor immune response under the right conditions. Porphyrin-based organic compounds (and the related chlorins, bacteriochlorins, and phthalocyanines) have traditionally served as ROS-generating photosensitizers for PDT.14,15 However, transition-metal complex photosensitizers have the potential to both (i) expand the scope of PDT to include oxygen-independent mechanisms of action, and thus improve PDT efficacy in hypoxic tissue, and (ii) broaden the wavelength range that can be used to include deeper tissue-penetrating near-infrared (NIR) light. One recent example is our Ru(II) complex TLD1433,16 currently in clinical trials for treating bladder cancer with PDT (ClinicalTrials.gov Identifier: NCT03053635), and its transferrin conjugate Rutherrin™.17
TLD1433 incorporates a special π-expansive ligand derived from imidazo[4,5-f][1,10]phenanthroline appended to an α-terthienyl unit, which imparts a long-lived triplet intraligand excited state (3IL) of significant π,π* character that is lower in energy than the triplet metal-to-ligand charge transfer (3MLCT) state that typically dominates the photophysical dynamics of Ru(II) polypyridyl complexes. The reduced intersystem crossing (ISC) rates characteristic of spin-forbidden 3π,π* transitions centered on the organic moiety are responsible for the much longer intrinsic triplet lifetimes in these constructs that are known as metal-organic dyads.18,19 The implication is that slow ISC back to the ground state from 3IL states provides ample opportunity for requisite bimolecular processes that constitute the phototoxic effects of PDT. We have demonstrated very potent in vitro PDT effects from a variety of Ru(II) dyads with low-lying, long-lived 3IL states, including both contiguously-fused π-extended azaaromatic ligands20 as well as ligands tethered to π-extended aromatic chromophores.16,21–23 More recently, we have incorporated π-expansive ligands into Ir(III) metal complexes to extend intrinsic excited state lifetimes for both reverse saturable absorption (RSA) and PDT applications.24,25
Compared to the large number of Ru(II) systems that have been explored, investigations on Ir(III) complexes for in vitro PDT have been emerged in recent years.24–44 While Ir(III) complexes derived from diimine ligands (N∧N) may fall short of the optimal absorption window for PDT, we have demonstrated that incorporation of two cyclometalating ligands (C∧N) to form biscyclometalated Ir(III) complexes extends the ground-state absorption spectrum significantly, and that installation of a π-extended N∧N ligand alongside the two C∧N ligands extends the lifetime through population of 3IL states. The result is nanomolar PDT potency with phototherapeutic indices (PIs) greater than 400.25 We also showed that it was possible to use π-extended C∧N ligands without compromising the potent in vitro PDT effects in Ir(III)-based systems that act as near-infrared-emitting theranostic agents.24
The large luminescence quantum yields for certain Ir(III) complexes combined with their high yields for triplet state formation and good photostability make these metal complexes very attractive for photobiological applications such as PDT. In addition, their excitation and emission energies, as well as other photophysical and biological/chemical properties, can be systematically tuned via minor structural changes to a highly versatile and modular architecture. Despite these attributes, Ir(III) complexes studied to date30 still fall short of some of the best Ru(II)-based in vitro PDT agents. The purpose of the present study is to use rational design principles to improve the water-solubility and in vitro PDT effects within the Ir(III) class of photosensitizers. Specifically, installing two Ir(III) centers in a single complex might simultaneously amplify photocytotoxicity and increase water solubility.
The solubility of organometallic complexes in aqueous solution can be improved by increasing the number of charges in the complex, which should be applicable to multinuclear iridium(III) complexes with C∧N and/or N∧N ligands.45 Since the photophysical and biological properties of metal complexes bearing tridentate ligand(s) can be readily tuned by modification of the 4′-position of the tridentate ligand(s), and because the bis-tridentate ligand coordination prevents the formation of stereoisomers upon complexation with transition metals, tridentate ligands are chosen for this Ir(III) study.46,47 Our previous work with bis-terpyridyl dinuclear Pt(II) complexes showed that the fluorenyl bridging group imparted these systems with intense absorption in the visible region (400-500 nm) and reasonably long-lived triplet excited states.48 These desirable properties led us to investigate fluorenyl-linked Ir(III) systems as in vitro PDT agents.
Herein, we report the synthesis, characterization, and photophysical/photobiological properties of stable dinuclear Ir(III) complexes (Chart 1) of +2, +4, or +6 charges, with the charge determined by the identities of the metal coordinating atoms of different terminal tridentate ligands. Fluorene was chosen as the central bridging group for the two Ir-tpy components because it is a rigid π-conjugated linker expected to enhance molar extinction coefficients in the visible spectral region. Complexes Ir1 and Ir3 – Ir5 incorporate 9,9-dioctyl-2,7-di(terpyridyl)-9H-fluorene (L1) as the bridging ligand, with variation at the terminal tridentate ligands: 4′-phenyl-2,2′:6′,2″-terpyridine (N∧N∧N), 1,3-dipyridyl-4,6-dimethylbenzene (N∧C∧N), 4,6-diphenyl-2,2′-bipyridine (C∧N∧N), or 2,4,6-triphenyl-pyridine (C∧N∧C). Complex Ir2 uses 9,9-dioctyl-2,7-bis(2-phenylethynyl)-9H-fluorene (L2) as the bridging ligand to extend the π-conjugation length, which is anticipated to facilitate intraligand charge transfer (ILCT) transitions that fall in the PDT window and to also increase visible wavelength absorption.
Chart 1.
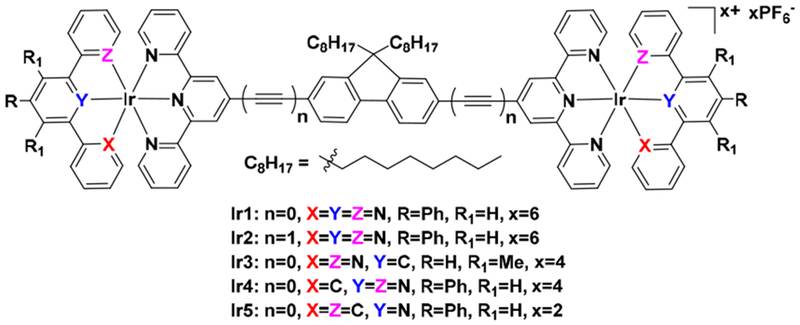
The molecule structure of target dinuclear Ir(III) complexes
EXPERIMENTAL SECTION
Synthesis and Characterizations.
Chemicals and solvents were purchased from Sigma-Aldrich or Alfa Aesar and used as received unless otherwise noted. The bridging ligands (L1 and L2) were synthesized following our previously reported procedures.48 4′-Phenyl-2,2′:6′,2″-terpyridine (N∧N∧N),49 1,3-dipyridyl-4,6-dimethylbenzene (N∧C∧N),50 4,6-diphenyl-2,2′-bipyridine (C∧N∧N),51 2,4,6-triphenylpyridine (C∧N∧C),52 4′-phenyl-2,2′:6′,2″-terpyridine-IrCl3 (N∧N∧N-IrCl3),53 and {[1,3-dipyridyl-4,6-dimethylbenzene]IrCl2}2 (N∧C∧N Ir-dimer)54 were synthesized according to established methods. Silica gel (60 Å, 230–400 mesh) and Al2O3 (activated, neutral) were purchased from Sorbent Technology. Complexes Ir1 – Ir5 were characterized by 1H NMR, high resolution electrospray ionization mass spectrometry (ESI–MS), and elemental analysis. 1H NMR spectra were recorded on Bruker-400 or Varian Oxford–500 spectrometers. ESI–MS analyses were collected using a Waters Synapt G2-Si Mass Spectrometer. Elemental analyses were performed by NuMega Resonance Laboratories, Inc. (San Diego, California). The synthetic schemes for complexes Ir1 – Ir5 are outlined in Scheme 1.
Scheme 1.
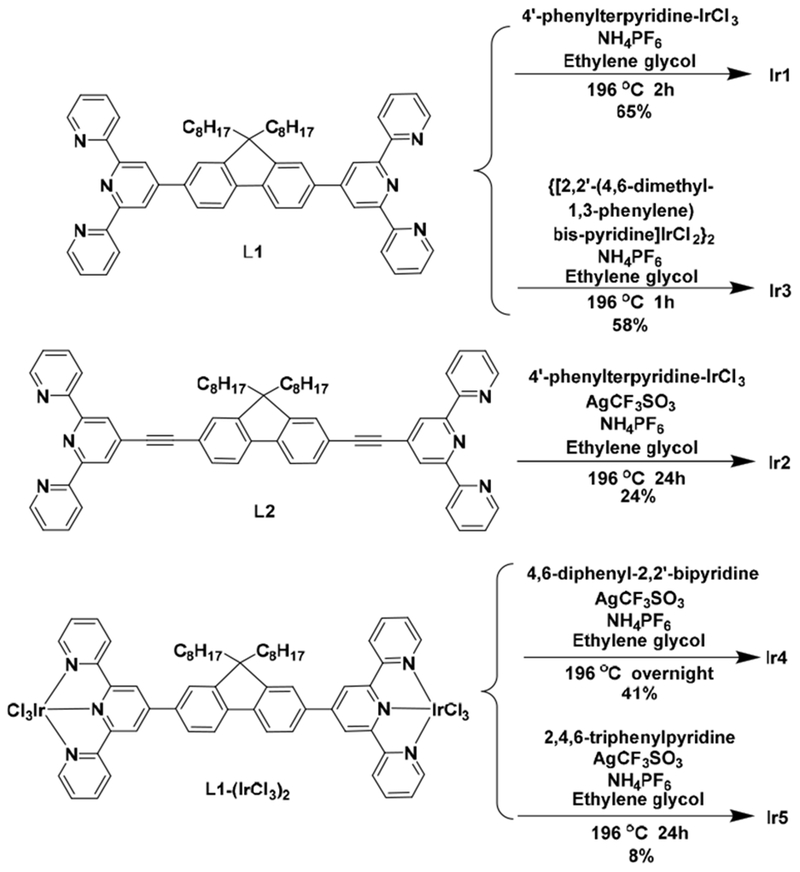
Synthetic routes for complexes Ir1 - Ir5.
L1-(IrCl3)2.
L1 (170.5 mg, 0.2 mmol) and IrCl3 3H2O (141 mg, 0.4 mmol) were combined in degassed ethylene glycol (15 mL) and heated to 160 °C with protection from light. After 30 min, the reaction mixture was cooled to room temperature and filtered. The resulting precipitate was washed with ethanol, water, and diethyl ether to give Ll-Ir(Cl3)2 as a red solid (254 mg, 88%). 1H NMR (500 MHz, d6-DMSO) δ 9.27 (d, J = 5.0 Hz, 4H), 9.18 (t, J = 8.8 Hz, 4H), 9.06 (dd, J = 11.3, 3.0 Hz, 2H), 8.98 – 8.94 (m, 2H), 8.70 (t, J = 9.8 Hz, 2H), 8.52 (dd, J = 8.2, 4.1 Hz, 2H), 8.38 (t, J = 7.8 Hz, 4H), 8.32 (d, J = 8.5 Hz, 2H), 8.06 – 8.01 (m, 4H), 2.43 (m, 4H), 0.95 – 0.69 (m, 20H), 0.48 (m, 10H).
Ir1.
4′-Phenylterpyridine-IrCl3 (60.7 mg, 0.1 mmol) and L1 (42.6 mg, 0.05 mmol) were combined in degassed ethylene glycol (10 mL) and heated to 196 °C with protection from light. After 2 h, the reaction mixture was cooled to room temperature, and saturated NH4PF6 aqueous solution (20 mL) was added to precipitate the product. The crude material was purified by column chromatography on alumina gel. Unreacted ligand was eluted first with CH2Cl2, followed by elution of the desired product with an acetone/water gradient (100:0 to 95:5 (v/v)). The product was a yellow solid (60 mg, 65%). 1H NMR (400 MHz, d6-DMSO) δ 9.76 (s, 4H), 9.67 (s, 4H), 9.50 – 9.34 (m, 4H), 9.29 (d, J = 8.2 Hz, 4H), 9.05 (s, 2H), 8.88 – 8.78 (m, 2H), 8.53-8.39 (m, 14H), 8.17 – 7.93 (m, 8H), 7.87 (t, J = 7.6 Hz, 4H), 7.78 (t, J = 7.4 Hz, 2H), 7.62 (dd, J = 12.2, 5.8 Hz, 8H), 2.61 (m, 4H), 0.89 (m, 20H), 0.62 (m, 10H). ESI-HRMS (m/z, in acetone): calcd. for [C101H90Ir2N12]6+, 309.4445; found, 309.4446. Calcd. for [C101H90Ir2N12PF6]5+, 400.3262; found, 400.3270. Calcd. for [C101H90Ir2N12P2F12]4+, 536.6489; found, 536.6503. Anal. Calcd. (%) for C101H90F36Ir2N12P6·6H2O: C, 42.80; H, 3.63; N, 5.93. Found: C, 42.61; H, 3.74; N, 5.98.
Ir2.
4′-Phenylterpyridine-IrCl3 (60.7 mg, 0.1 mmol), L2 (45 mg, 0.05 mmol), and AgOTf (77 mg, 0.3 mmol) were combined in degassed ethylene glycol (10 mL) and heated to 196 °C with protection from light. After 24 h, the mixture was cooled to room temperature, and saturated NH4PF6 aqueous solution (20 mL) was added to precipitate the product. The crude material was purified by column chromatography on alumina gel. Unreacted ligand was eluted with CH2Cl2 first, followed by elution of the desired product using an acetone/water gradient (100:0 to 95:5 (v/v)). The purified product was a dark red powder (37 mg, 24%). 1H NMR (400 MHz, d6-DMSO) δ 9.62 (t, J = 7.0 Hz, 6H), 9.22 (m, 8H), 8.48 (m, 6H), 8.32 (m, 8H), 7.92 (m, 8H), 7.77 (m, 6H), 7.71 (m, 4H), 7.51 (m, 10H), 2.51 (m, 4H), 1.02 (m, 20H), 0.67 (m, 10H). ESI-HRMS (m/z, in acetone): calcd. for [C105H90Ir2N12]6+, 317.4445; found, 317.4442. Anal. Calcd. (%) for C105H90F36Ir2N12P6·13H2O·CH2Cl2: C, 41.16; H, 3.85; N, 5.43. Found: C, 40.83; H, 3.47; N, 5.44.
Ir3.
{[2,2′-(4,6-Dimethyl-1,3-phenylene)bis-pyridine]IrCl2}2 (72 mg, 0.069 mmol) and L1 (59 mg, 0.069 mmol) were heated in degassed ethylene glycol (10 mL) at 196 °C with protection from light. After 1 h, the reaction mixture was added to water (10 mL) and filtered. Saturated NH4PF6 aqueous solution (20 mL) was added to the filtrate, and the resulting yellow precipitate was collected by centrifugation, washed with water (3×10 mL), and dried under vacuum. The crude product was purified by column chromatography on alumina gel. Unreacted ligand was eluted first with CH2Cl2, followed by elution of the desired product with an acetone/water gradient (100:0 to 95:5 (v/v)). The pure product was a yellow solid (94 mg, 58%). 1H NMR (400 MHz, d6-DMSO) δ 9.65 (td, J = 5.0, 2.5 Hz, 4H), 9.18 (t, J = 9.9 Hz, 4H), 8.90 (m, 2H), 8.71 (m, 2H), 8.47 (dd, J = 9.6, 2.6 Hz, 2H), 8.37 (d, J = 8.4 Hz, 4H), 8.30 (dd, J = 8.8, 4.6 Hz, 4H), 7.99 (ddd, J = 7.5, 5.9, 2.7 Hz, 4H), 7.77 (m, 2H), 7.68 (ddd, J = 9.7, 4.8, 2.7 Hz, 4H), 7.52 (ddd, J = 6.7, 4.1, 1.2 Hz, 4H), 7.45 (dd, J = 5.7, 1.8 Hz, 2H), 7.40 (s, 2H), 7.12 (ddd, J = 8.4, 6.5, 1.7 Hz, 4H), 2.93 (s, 12H), 2.52 (m, 4H), 0.90 (m, 20H), 0.62 (m, 10H). ESI-HRMS (m/z, in acetone): calcd. for [C95H90Ir2N10]4+, 439.1652; found, 439.1668. Calcd. for [C95H90Ir2N10PF6]3+, 633.8750; found, 633.8768. Anal. Calcd. (%) for C95H90F24Ir2N10P45H2O: C, 47.03; H, 4.15; N, 5.77. Found: C, 46.97; H, 4.32; N, 5.98.
Ir4.
L1-(IrCl3)2 (72.4 mg, 0.05 mmol), 4,6-diphenyl-2,2′-bipyridine (30.8 mg, 0.1 mmol), and AgOTf (77 mg, 0.3 mmol) were mixed in degassed ethylene glycol (10 mL) and heated to 196 °C with protection from light. After 24 h, the mixture was cooled to room temperature, and saturated NH4PF6 aqueous solution (20 mL) was added to precipitate the crude product, which was purified by column chromatography on alumina gel. Unreacted ligand was eluted first with CH2Cl2,, followed by elution of the desired product with an acetone/water gradient (100:0 to 95:5 (v/v)). The pure product was a yellow solid (38 mg, 41%). 1H NMR (400 MHz, d6-acetone) δ 10.05 (d, J = 7.2 Hz, 2H), 9.49 (d, J = 8.4 Hz, 2H), 9.30 (s, 2H), 9.00 (d, J = 15.3 Hz, 4H), 8.95 – 8.77 (m, 6H), 8.71 (s, 2H), 8.48 (dd, J = 18.4, 9.5 Hz, 8H), 8.36 (s, 2H), 8.28 (d, J = 6.3 Hz, 2H), 8.13 – 7.97 (m, 4H), 7.86 (dd, J = 10.8, 6.6 Hz, 4H), 7.75 (s, 2H), 7.61 (s, 6H), 7.48 – 7.33 (m, 2H), 7.19 (dd, J = 13.3, 4.5 Hz, 4H), 6.91 – 6.69 (m, 4H) 2.41 (m, 4H), 0.86 (m, 20H), 0.55 (m, 10H). ESI-HRMS (m/z, in acetone): calcd. for [C103H90Ir2N10PF6]3+, 655.8756; found, 655.8798. Anal. Calcd. (%) for C103H90F24Ir2N10P4·9H2O: C, 47.69; H, 4.20; N, 5.40. Found: C, 47.56; H, 4.46; N, 5.25.
Ir5.
L1-(IrCl3)2 (72.4 mg, 0.05 mmol), 2,4,6-triphenylpyridine (30.7 mg, 0.1 mmol), and AgOTf (77 mg, 0.3 mmol) were combined in degassed ethylene glycol (10 mL) and heated to 196 °C with protection from light. After 24 h, the mixture was cooled to room temperature, and saturated NH4PF6 aqueous solution (20 mL) was added to precipitate the crude product. The pure product was obtained by column chromatography on alumina gel. Unreacted ligand was eluted first with CH2Cl2,, followed by elution of the desired product with an acetone/water gradient elution (100:0 to 95:5 (v/v)). The pure product was a red solid (16 mg, 8%). 1H NMR (400 MHz, d6-DMSO) δ 9.56 (s, 4H), 9.19 – 9.09 (m, 4H), 8.95 – 8.85 (m, 4H), 8.60 (s, 4H), 8.33 (d, J = 6.6 Hz, 8H), 8.22 (s, 6H), 7.89 (s, 4H), 7.73 (s, 4H), 7.64 (s, 2H), 7.45 (s, 4H), 6.99 (s, 4H), 6.74 (s, 4H), 6.21 (s, 4H), 2.52 (m, 4H), 0.85 (m, 20H), 0.55 (m, 10H). ESI-HRMS (m/z, in acetone): calcd. for [C105H90Ir2N8]2+, 924.3283; found, 924.3291. Anal. Calcd. (%) for C105H90F12Ir2N8P2·0.6CH2Cl2: C, 57.94; H, 4.20; N, 5.12. Found: C, 57.92; H, 3.96; N, 5.27.
Photophysical Studies.
Spectrophotometric grade solvents were purchased from Alfa Aesar. The UV–vis absorption spectra of complexes Ir1 – Ir5 were collected on a Varian Cary 50 spectrophotometer, and steady-state emission measurements were carried out using a HORIBA FluoroMax 4 fluorometer/phosphorometer. The emission quantum yields of complexes Ir1 – Ir5 in argon-sparged CH3CN solution were determined by the relative actinometry method55 using [Ru(bpy)3]Cl2 (Φem = 0.097, λex = 436 nm)56 as the standard. The nanosecond transient absorption (TA) spectra and decays, triplet excited-state quantum yields, and triplet lifetimes were collected on argon-sparged (40 min) acetonitrile solutions on an Edinburgh LP920 laser flash photolysis spectrometer using the third harmonic output (355 nm) of a Nd:YAG laser (Quantel Brilliant, pulsewidth ~4.1 ns, repetition rate was set to 1 Hz). Molar extinction coefficients (εT1-Tn) for triplet excited states were determined by the singlet depletion method57 at the TA band maxima, and quantum yields for triplet excited state formation were measured according to the relative actinometry method58 using the εT1-Tn values, with SiNc in benzene as the reference (ε590 = 70,000 M−1cm−1, ΦT = 0.20).59
Singlet Oxygen Quantum Yields.
Singlet oxygen quantum yields (ΦΔ) were determined for the PF6− salts of the complexes directly from sensitized singlet oxygen emission centered at 1268 nm using a PTI Quantamaster equipped with a Hamamatsu R5509-42 near-infrared PMT. The metal complexes were prepared in spectroscopic-grade CH3CN at 5 μM, and the measurements were made under ambient oxygen concentration (21%). ΦΔ was calculated relative to the standard [Ru(bpy)3](PF6)2 (ΦΔ = 0.56 in aerated CH3CN)60 according to Eq 1, where I, A, and η represent integrated emission intensity, absorbance at the excitation wavelength, and refractive index of the solvent, respectively. Values calculated for ΦΔ were reproducible to within <5%.
| Eq1 |
DFT Calculations.
Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations on Ir(III) complexes were performed using the Gaussian09 quantum software package.61 The basis sets used in all calculations were LANL2DZ62–64 for Ir(III) and 6-31G*65–69 for other non-metal atoms. Unlike the previous Ir(III) complexes studied,25,70,71 complexes Ir1 - Ir5 have two metal centers connected by fluorenylbisterpyridyl ligand, which makes long-range interactions critical for the description of both geometry and excited state properties. Therefore, here we used the long-range corrected hybrid function, ωB97XD, which was designed to capture long-range atom-atom dispersion.72 Implicit solvent effects were incorporated by the conductor-like polarizable continuous model (CPCM)73,74 simulating the effects of acetonitrile. The long aliphatic side chains on fluorene were replaced with butyl groups to reduce the computational cost. This reduction does not affect the optical properties of the complexes in the visible spectral region, since octyl groups do not contribute any electronic transitions in this energy range.
The absorption spectra for the Ir(III) complexes were generated by broadening the lowest 100 singlet vertical excitations computed by TDDFT75,76 using the functional and basis sets described above. To broaden the vertical excitation to generate spectra in terms of molar absorptivity units (L·mol−1·cm−1) the method described by Bjorgaard and co-workers was followed.77 The shape of the spectra generated by broadening the vertical excitation computed by TDDFT quantitatively agreed well, except for about ~0.6 eV blue-shift, which is expected for the ωB97XD functional applied to conjugated systems78 (see Table S1 of Supporting Information for the energy differences of the experimental and theoretical transitions of the first absorption band). To align the theoretical spectra with the experimental spectra, all transitions energy were red-shifted by −0.55 eV To characterize the type of excitation for the Ir(III) complexes, natural transition orbitals (NTOs)79 were generated using Gaussian09 software. NTOs allow for representing an excitation as the electron and hole pair, while preserving the many-body nature of the excited states. Due to the high symmetry of these dinuclear complexes, multiple transition densities that only differ on which metal center electronic density is localized contribute to some excitations. Therefore, only uniquely representative NTOs are shown for those excitations and those states are indicated by “*” in Tables 2 and S2. The visualization of the NTOs were done utilizing Visual Molecular Dynamics (VMD)80 with isosurface of 0.02.
Table 2.
Natural Transition Orbitals (NTOs) for Low Energy Transitions of Ir1 - Ir5. For Transitions with Quasi-Degenerate Transition Orbitals, Only One Pair of Transition Densities Are Shown and Are Indicated By *.
| Sn | Hole | Electron | |
|---|---|---|---|
| Ir1 | S1 340 nm f = 2.637 |
||
| S5 303 nm f = 0.109 |
 |
 |
|
| Ir2 | S1 381nm f = 3.730 |
||
| S2* 336 nm f = 0.079 |
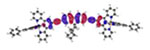 |
 |
|
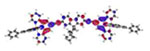
| Ir3 | S1 333 nm f = 0.010 |
 |
 |
| S3 328 nm f = 2.189 |
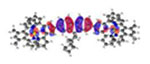 |
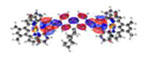 |
|
| S7 311 nm f = 0.368 |
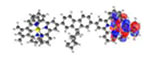 |
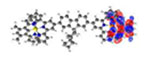 |
|
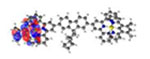
| Ir4 | S1 351 nm f = 0.003 |
||
| S2 350 nm f = 0.004 |
 |
 |
|
| S3* 340 nm f = 2.002 |
 |
||
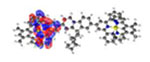
| Ir5 | S1* 395 nm f = 0.933 |
 |
|
| S6* 372 nm f = 0.050 |
 |
||
Photobiological Activity Studies.
The experimental details for cell culture, cytotoxicity and photocytotoxicity studies, confocal microscopy, and DNA mobility-shift assays are the same as those described in our previous published work24,25 and are presented in the Supporting Information.
RESULTS AND DISCUSSION
Electronic Absorption.
The experimental UV–vis absorption spectra of complexes Ir1 - Ir5 were recorded in acetonitrile (Figure 1a), and the absorption band maxima and molar extinction coefficients are listed in Table 1. The absorption follows Beer’s law in the solutions used in our study (i.e. 5×10−6 to 1×10−4 mol·L−1), indicating that no ground-state aggregation was formed in the tested concentration range. The strong absorption bands in the range of 250-350 nm and 350-500 nm are predominantly assigned to 1π,π* transitions localized on the terminal tridentate ligands, and the bridging ligand, respectively. These assignments are supported by the NTOs corresponding to the major transitions contributing to these bands (Supporting Information Table S2 and Table 2). Attribution of the absorption bands of 350-500 nm to the bridging ligand localized 1π,π* transition is in line with that revealed in the dinuclear Ir(III) complexes with trisbidentate ligands and diethynylaryl substituted diketopyrrolopyrrole bridging ligand.81,82 However, NTOs of Ir1 – Ir4 in Table 2 show that some charge transfer transitions, i.e. 1MLCT, 1LLCT (ligand-to-ligand charge transfer) or 1ILCT (intraligand charge transfer) contributed to the 350-500 nm bands as well. Contributions of the charge transfer configurations to the 350-500 nm absorption bands are partially reflected by the insignificant but noticeable negative solvatochromic effects (Figure S3 in Supporting Information), especially in Ir2 that has more 1ILCT character (see NTOs in Table 2). In contrast, the charge transfer transitions in Ir5 became more distinguishable and energetically separated from the bridging ligand localized 1π,π* transition. This is clearly evidenced by the appearance of the new absorption band at 520 nm in Ir5.
Figure 1.
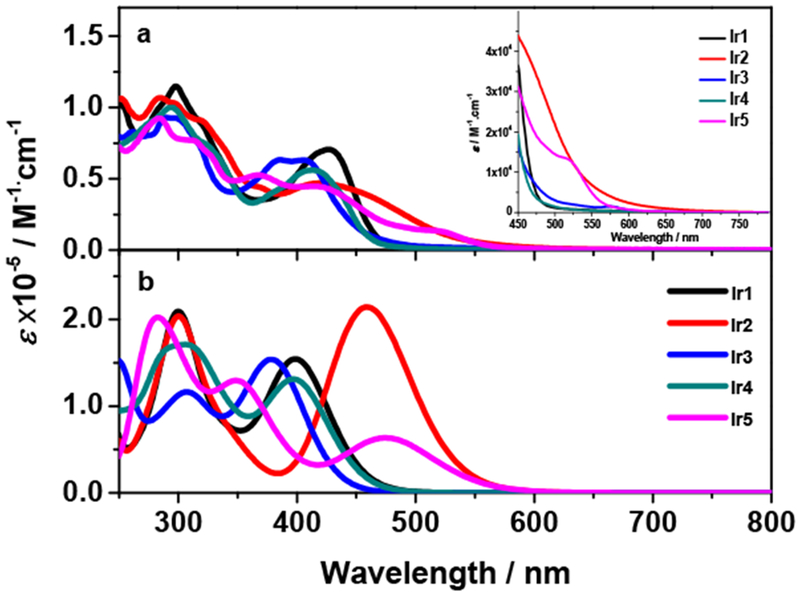
Experimental (a) and theoretical (b) UV–vis absorption spectra of Ir1 – Ir5 at room temperature in acetonitrile. The inset in panel (a) is the expansion of the spectra in the region of 450–800 nm. The theoretical spectra were computed using ωB97XD with mixed basis set. A redshift of 0.55 eV for the theoretical spectra in panel (b) was applied for better comparison with the experimental spectra.
Table 1.
Electronic Absorption, Emission, and Triplet Excited-State Absorption Parameters, as well as Singlet Oxygen Quantum Yields for Complexes Ir1 - Ir5
| λabs/nm (log ε)a | λem/nm (τem/μs); Φemb | λT1-Tn/nm (τTA/μs; log εT1-Tn); ΦTc | ΦΔd (λex/nm) | |
|---|---|---|---|---|
| Ir1 | 251 (5.01), 297 (5.06), 427 (4.85) | 583 (3.57); 0.024 | 498 (3.09; -), 765 (3.05; 4.62); 0.44 | 0.28 (430) |
| Ir2 | 251 (5.03), 285 (5.03), 364 (4.65), 420 (4.68) | 576 (1.99); 0.003 | 385 (1.72; -), 640 (1.75; 5.04); 0.03 | 0.26 (468) |
| Ir3 | 260 (4.92), 289 (4.97), 386 (4.80), 405 (4.80) | 608 (1.47); 0.025 | 513 (0.03 (17%), 1.72 (83%); -), 770 (0.03 (15%), 1.75 (85%)); 4.70); 0.14 | 0.04 (411) |
| Ir4 | 294 (5.00), 321 (4.88), 413 (4.75) | 578 (53.3); 0.22 | 498 (48.6; -), 800 (48.3; 4.64); 0.28 | 0.38 (418) |
| Ir5 | 283 (4.97), 312 (4.89), 368 (4.72), 416 (4.65), 520 (4.13) | 619 (1.92); 0.045 | 681 (2.68; 4.80), 787 (2.68; -); 0.07 | 0.22 (418) |
Absorption band maxima (λabs) and molar extinction coefficients (ε / L·mol−1·cm−1) of the UV-vis absorption in acetonitrile at room temperature.
Emission band maxima (λem), lifetimes (τem), and quantum yields (Φem) measured in acetonitrile (c = 1 × 10−5 mol·L−1) at room temperature with Ru(bpy)3Cl2 (in degassed acetonitrile; Φem = 0.097, λex = 436 nm) as the reference.
Nanosecond TA band maxima (λT1-Tn), triplet excited-state lifetimes (τTA), triplet molar extinction coefficients (εT1-Tn / L·mol−1·cm−1), and quantum yields for triplet state formation (ΦT) measured in acetonitrile at room temperature with SiNc (in degassed benzene; ε590 = 70,000 L·mol−1·cm−1, ΦT = 0.20) as the reference. λex = 355 nm.
Singlet oxygen quantum yields in acetonitrile. Values are correct to within ±5%.
Comparison of the absorption spectra of Ir1 and Ir2 revealed that incorporation of the C≡C bonds to the bridging ligand caused a broadening and a red-shift of the bridging ligand localized 1π,π* absorption band due to the extended π-conjugation. Replacing the terminal terpyridyl ligands in Ir1 by N∧C∧N (1,3-dipyridyl-4,6-dimethylbenzene) ligands in Ir3 induced a blue-shift of the 350-500 nm absorption band and incorporated more terminal ligands based 1π,π* transition and 1LLCT/1MLCT character to this band (see NTOs for Ir3 in Table 2); while changing the terminal ligands to C∧N∧N (4,6-diphenyl-2,2′-bipyridine) ligands in Ir4 only caused a slight blue-shift of this band with respect to that in Ir1. In contrast, when the terminal ligands were changed to C∧N∧C (2,4,6-triphenylpyridine) ligands in Ir5, the transition energies, intensities, and the shape of the low-energy absorption bands changed pronouncedly from those of Ir1. This can be attributed to the distinct nature of the lowest energy optical transition in these two complexes. As the NTOs in Table 2 indicated, the stronger σ-donating ability of the phenyl rings on the C∧N∧C ligand delocalized the hole of the S1 transition mainly to the 2,6-diphenyl rings and to the metal d orbitals, while the electron was predominantly on the terpyridyl ligands. Thus, the lowest-energy optical transition in Ir5 is predominantly the 1LLCT/1MLCT transition, which is in contrary to the bridging ligand localized 1π,π* transition in Ir1. The drastic change of the dominant optical transitions accounts for the different features of the low-energy absorption bands in Ir5 with respect to that in Ir1.
Photoluminescence.
The emission of Ir1 - Ir5 was studied in different solvents at room temperature. The observed emission all exhibited large Stokes shifts with respect to the corresponding excitation wavelength, they were all long-lived (several to tens of μs), and sensitive to the presence of oxygen. Thus the emission was attributed to phosphorescence. The normalized emission spectra of Ir1 - Ir5 in acetonitrile are displayed in Figure 2, and the spectra in other solvents are given in SI Figure S4. The emission parameters are listed in Table 1 and Table S3 of SI. The emission of Ir1 and Ir4 resembled each other, both showing some vibronic structures, with much longer lifetimes and higher emission quantum yields compared to the other three complexes, and exhibiting minor solvatochromic effects. The vibronic spacing between the 580 nm and 620 nm bands is approximately 1150 cm−1 and 1090 cm−1 in Ir1 and Ir4, respectively, which is in accordance with the aromatic vibrational mode of the terpyridyl ligands. Thus, the emission of these two complexes can be assigned predominantly to the ligand localized 3π,π* state. However, the lifetime of Ir1 is one order of magnitude shorter than that of Ir4. This could be attributed to the weaker ligand field of the terpyridyl ligand with respect to that of the C∧N∧N ligand that contains the stronger σ-donating 6-phenyl ring. The nonradiative metal-centered 3d,d state is thus situated more closely to the low-lying emissive 3π,π* state and becomes thermally accessible in Ir1 compared to that in Ir4. This adds an additional decay path for the emitting 3π,π* state in Ir1 and consequently reduces its lifetime. For Ir2, Ir3, and Ir5, the emission spectra are featureless and broader, the lifetimes are less than 2 μs and the emission quantum yields are quite low, and the solvatochromic effect is more pronounced. All these characters imply charge transfer nature of the emitting states in these three complexes. Referring to the NTOs corresponding to the low-energy singlet charge transfer transitions shown in Table 2, it is reasonable to speculate that the emitting state of Ir2 could be the 3ILCT state and they are the 3LLCT/3MLCT states in Ir3 and Ir5. It appeared that either extending the π-conjugation of the bridging ligand in Ir2, or varying the terminal tridentate ligands in Ir3 and Ir5 changed the nature of the emitting state from the 3π,π* state in Ir1 to 3CT states. In addition, variation of the terminal tridentate ligands impacted the emission energies in Ir3 – Ir5 compared to that in Ir1, with a slight blue-shift of the emission in Ir4 while a salient red-shift in Ir3 and Ir5. The red-shifted emission in Ir3 and Ir5 with respect to that in Ir1 could possibly be rationalized by the stronger σ-donating ability of the phenyl rings on the terminal tridentate N∧C∧N and C∧N∧C ligands, which raised the energies of the terminal ligand and the metal d orbital based holes and thus reduced the energy gaps between the holes and electrons (likely localized on the terpyridyl motifs). Consequently, the 3LLCT/3MLCT emission energies of Ir3 and Ir5 are reduced.
Figure 2.
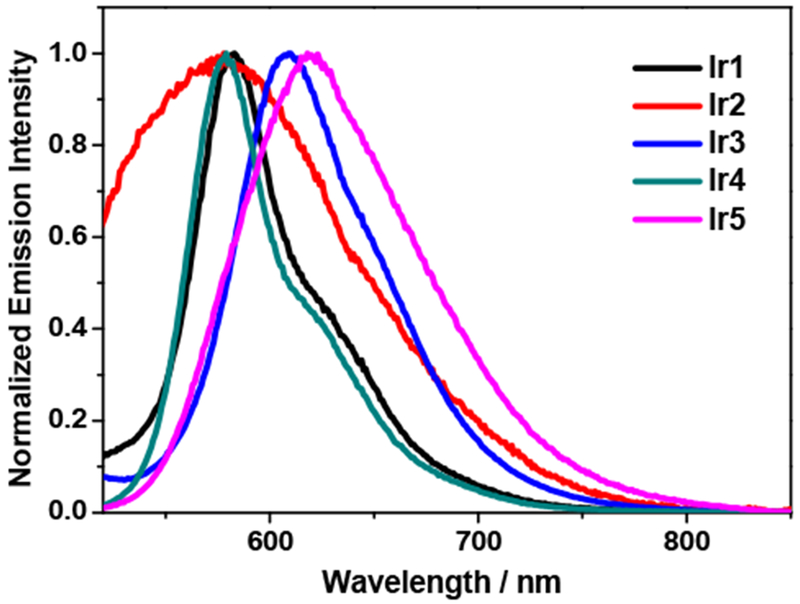
Experimental emission spectra of Ir1 (λex = 426 nm), Ir2 (λex = 420 nm), Ir3 (λex = 405 nm), Ir4 (λex = 413 nm), and Ir5 (λex = 415 nm) at room temperature in deoxygenated acetonitrile (c = 1×10−5 mol·L−1).
Transient Absorption.
To further understand the triplet excited–state characteristics, the nanosecond transient absorption (TA) studies of complexes Ir1 – Ir5 were conducted in acetonitrile solutions. The TA spectra of Ir1 – Ir5 at zero delay after excitation are presented in Figure 3 and the TA parameters are provided in Table 1. The time-resolved TA spectra of Ir1 – Ir5 are provided in Supporting Information Figure S5. The triplet lifetimes deduced from the decay of TA for Ir1 – Ir4 are similar to their emission lifetimes in acetonitrile. Therefore, we consider that the observed TA of these complexes was from the excited states that emit. In contrast, Ir3 exhibited a biexponential decay in its TA signals, with the longer lifetime being consistent with the emission lifetime. This implies that the long-lived TA signal in Ir3 could from the emitting excited state as well.
Figure 3.
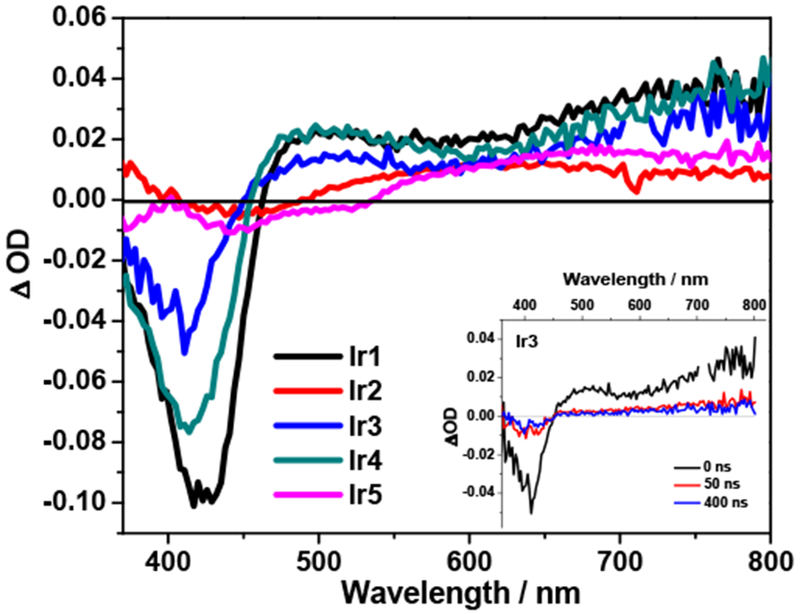
Nanosecond transient absorption (TA) spectra of complexes Ir1 – Ir5 in deoxygenated acetonitrile at zero delay after 355 nm excitation. The inset shows the TA spectra of Ir3 at different delay time after excitation. A355 nm = 0.4 in a 1-cm cuvette.
The TA spectra of Ir1 – Ir5 all possessed very broad positive absorption band(s) from the visible to the near-IR region, i.e. 463-800 nm for Ir1, 495-800 nm for Ir2, 459-800 nm for Ir3, 455-800 nm for Ir4, and 538-800 nm for Ir5. Bleaching occurred in the region corresponding to the low-energy absorption bands. Considering the similar shape of the TA spectra of Ir1 and Ir4 to that of our previously reported dinuclear platinum(II) complex with the same bridging ligand,48 and the similar lifetimes to those of emission, we tentatively attribute the contributing transient absorbing excited state predominantly to the bridging ligand localized 3π,π* state. While for Ir2 and Ir5, the transient absorbing states are likely to be the 3CT state(s), i.e. predominantly 3ILCT for Ir2 and 3LLCT/3MLCT states for Ir5. In contrast to Ir1, Ir2, Ir4 and Ir5 that exhibited monoexponential decays in their TA signals, the TA signal of Ir3 followed a biexponential decay. The short-lived transient species had a lifetime of ~30 ns and gave rise to a spectrum reminiscent to those of Ir1 and Ir4; while the long-lived species had a lifetime of ~1.7 μs, which is consistent with the lifetime obtained from the decay of emission, and the TA was much weaker and featureless. In view of the different spectral features at the shorter and longer decay time and the reminiscence of the spectra to those of Ir1/Ir4 and Ir2/Ir5, respectively, we tentatively assign the short-lived species to the high-lying bridging ligand localized 3π,π* state; while the long-lived species to the emitting 3LLCT/3MLCT state. The formation of a rapidly decaying higher excited state that subsequently leads to the lower-lying, long-lived emitting state has been reported for a mononuclear Ir(III) complex [(dpb)-Ir(tpy-ph(tBu)2]2+ that bears the same N∧C∧N ligand.83
It is noted that the measured triplet quantum yields of these complexes are not quite high, especially for Ir2, Ir3 and Ir5. This could be due to the following reasons: (i) The increased π-conjugation of the ligand would decrease the contribution of the transition metal d orbital to the frontier molecular orbitals of the complexes, which would reduce the spin-orbital coupling in the complexes and decrease the triplet quantum yield. Such a phenomenon has been reported in many Pt(II) and Ir(III) complexes.25,70,84 (ii) When a transition-metal complex is excited, especially when high-energy excitation is utilized, population of more than one triplet excited states is possible.70,85–91 However, not all of the populated triplet excited states contribute to excited-state absorption. In such a case, the calculated triplet quantum yield based on the observed TA signal could be significantly lower than the actual intersystem crossing quantum yield.
Singlet Oxygen Generation.
Production of 1O2 is known to have cytotoxic effects on cells, and thus compounds that generate 1O2 under cell-free conditions might be expected to act as in vitro PDT agents. The Ir(III) complexes Ir1 – Ir5 were assessed for singlet oxygen (1O2) sensitization in cell-free conditions through direct measurement of 1O2 emission at 1270 nm. [Ru(bpy)3](PF6)2 was used as the standard, with a reported 1O2 quantum yield (ΦΔ) of 0.56 in air-saturated CH3CN.60 The calculated ΦΔ values for all of the complexes were less than 40%. The efficiencies for 1O2 production ranged from 4% for Ir3 to 38% for Ir4, with Ir1, Ir2 and Ir5 yielding similar values (22-28%). Despite having some absorption at wavelengths longer than 500 nm, 1O2 yields were maximal with blue excitation. For N∧N∧N terminal tridentate ligands, the presence of the ethynyl groups for extending π-conjugation did not alter the singlet oxygen quantum yield as Ir1 and Ir2 gave very similar values for ΦΔ. When each terminal tridentate ligand of Ir1 had two of its nitrogens replaced with cyclometalating carbons (C∧N∧C) as in Ir5, the 1O2 yield decreased only slightly. These limited comparisons appear to indicate that substantial structural changes have little to no effect on ΦΔ. However, when only one nitrogen of each terminal tridentate ligand of Ir1 was replaced by carbon (C∧N∧N) as in Ir4, the 1O2 yield increased to almost 40%. Clearly, there are structural combinations in this family of complexes that do influence ΦΔ. The most dramatic impact on ΦΔ occurred for Ir3, where the terminal tridentate ligands were N∧C∧N with methyl substitution at R1 but no phenyl group at R. In this case, the 1O2 yield decreased by almost tenfold.
It is also worthy of noting that the ΦΔ values for Ir2, Ir4 and Ir5 are higher than the measured triplet quantum yields (ΦT, Table 1). This is not very surprising because population of excited states is wavelength dependent, which could result in different decay pathways.91 In the ΦT measurement, 355 nm excitation was used; while low-energy excitation (i.e. 411 – 468 nm) was used in the ΦΔ measurement. A 355-nm excitation in the TA measurement could populate more than one triplet excited states,70,85–91 which might not only impact the ΦT value measurement as discussed in the TA section, but could also reduce the population of the excited state that generates singlet oxygen because of the competing population of the other non-1O2-generating triplet excited states. In our previous study on the monocationic tris-bidentate Ir(N∧N)(C∧N)2 complexes, we have demonstrated that the singlet oxygen generation efficiency is wavelength dependent, with lower-energy excitation resulting in higher ΦΔ values in those Ir(III) complexes.24 We speculate the same case for the complexes studied in this work.
Cytotoxicity and Photocytotoxicity.
To understand whether the photophysical properties of these Ir(III) complexes could lead to photobiological effects, the cytotoxicity profiles of Ir1 – Ir5 were assessed in SK-MEL-28 malignant melanoma cells under three conditions: (i) dark, (ii) illumination with broadband visible light, and (iii) illumination with red LEDs emitting at 625 nm. Cytotoxic and photocytotoxic activities were quantified as the effective concentration required to reduce the cell viability to 50% (EC50) under a given condition. Briefly, cells growing in log phase were dosed with nine concentrations of the complex between 1 nM and 300 μM, incubated for 16 h, and were then subjected to a sham (dark) or light treatment. The light treatments were delivered at a fluence of 100 J·cm−2 with an irradiance of 35.7 mW·cm−2 or 32.3 mW·cm−2 for visible and red light, respectively. After a 48 h incubation period, cell viability was quantified based on the ability of viable cells to reduce resazurin to resorufin. EC50 values were determined from sigmoidal fits of the dose-response curves (Figure 4, Table 3). The phototherapeutic index (PI), a measure of the therapeutic margin for in vitro PDT, was calculated as the ratio of dark to light EC50 values and determined for each complex and irradiation condition. The dark toxicities of Ir1 – Ir5 toward normal human skin fibroblasts (CCD-1064Sk) were also measured to determine any selectivity for cancer cells over normal cells. The ratio of the dark CCD-1064Sk EC50 value for a given complex and its dark SK-MEL-28 EC50 value yielded the selectivity factor (SF), where SF > 1 indicates selectivity toward the cancerous cell line. Selective activity toward the cancer cell line is not a requirement for the PDT agent as long as the dark toxicity of the photosensitizer is low and the PI is relatively large. Rather, the spatiotemporal control of the light treatment provides the selectivity known for PDT. Nevertheless, for in vivo applications, selective activity toward cancer cells over normal, healthy cells is an added benefit.
Figure 4.
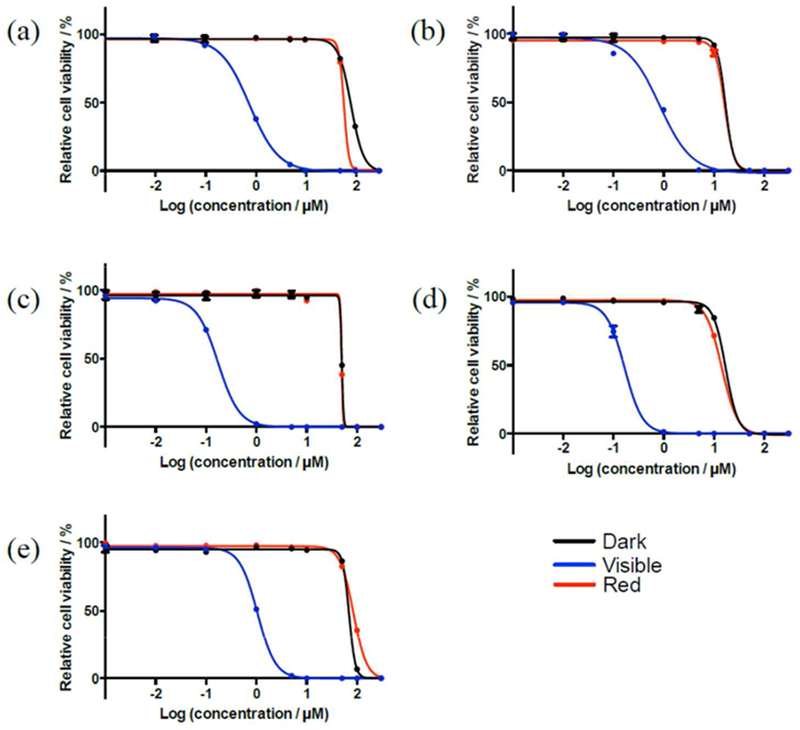
In vitro dose-response curves for complexes Ir1 (a), Ir2 (b), Ir3 (c), Ir4 (d), and Ir5 (e) in SK-MEL-28 cells treated in the dark (black) and with visible (blue) or red (red) light activation.
Table 3.
Comparison of EC50 values (μM) for SK-MEL-28 cancer cells and CCD-1064Sk normal skin fibroblasts dosed with complexes Ir1 – Ir5.
| SK-MEL-28 cells | CCD-1064Sk cells | ||||||
|---|---|---|---|---|---|---|---|
| Dark | Visa | PIb | Redc | PId | Dark | SFe | |
| Ir1 | 82.6 ± 1.5 | 0.75 ± 0.01 | 111 | 59.7 ± 0.4 | 1.4 | 102 ± 2 | 1.2 |
| Ir2 | 16.9 ± 0.8 | 0.83 ± 0.05 | 20 | 16.2 ± 0.5 | 1.0 | 32.0 ± 2.0 | 1.9 |
| Ir3 | 49.9 ± 0.1 | 0.17 ± 0.01 | 288 | 49.5 ± 0.1 | 1.0 | 49.1 ± 0.1 | 1.0 |
| Ir4 | 17.0 ± 0.7 | 0.17 ± 0.01 | 102 | 14.3 ± 0.3 | 1.2 | 31.8 ± 1.8 | 1.9 |
| Ir5 | 69.5 ± 1.0 | 1.05 ± 0.01 | 66 | 85.1 ± 1.0 | 0.82 | 142 ± 3 | 2.0 |
Vis-PDT: 16 hours drug-to-light interval followed by 100 J·cm−2 broadband visible light irradiation
PI = phototherapeutic index (ratio of dark EC50 to visible-light EC50)
Red-PDT: 16 hours drug-to-light interval followed by 100 J·cm−2 light irradiation with 625-nm LEDs
PI = phototherapeutic index (ratio of dark EC50 to red-light EC50)
SF SK-MEL-28: selectivity factor (ratio of dark CCD-1064Sk EC50 to dark SK-MEL-28 EC50).
The dark cytotoxicities of complexes Ir1 – Ir5 toward SK-MEL-28 melanoma cells ranged from 16.9 to 82.6 μM, with Ir1 being the least cytotoxic in the absence of a light trigger and Ir2 and Ir4 being the most cytotoxic (Table 3 and Figures 4–5). With the exception of Ir3 (SF = 1), the other dinuclear Ir(III) complexes exhibited some selective cytotoxicity toward the melanoma cancer cells relative to the normal human skin fibroblast cells. SF values followed the order Ir5 > Ir2 ≈ Ir4 > Ir1 > Ir3, with Ir5 exhibiting two-fold greater dark toxicity toward SK-MEL-28 cells and Ir3 showing no selectivity. The selective cytotoxicity observed for Ir2 and Ir4 was almost as great as that for Ir5 (SF = 1.9 versus SF = 2.0). Ir1 and Ir5 had dark EC50 values greater than 100 μM in the CCD-1064Sk cell line, and were thus considered to be completely nontoxic to the normal skin fibroblasts. In both cell lines, the dark toxicity was greatest for Ir2 and Ir4 and least for Ir1 and Ir5.
Figure 5.
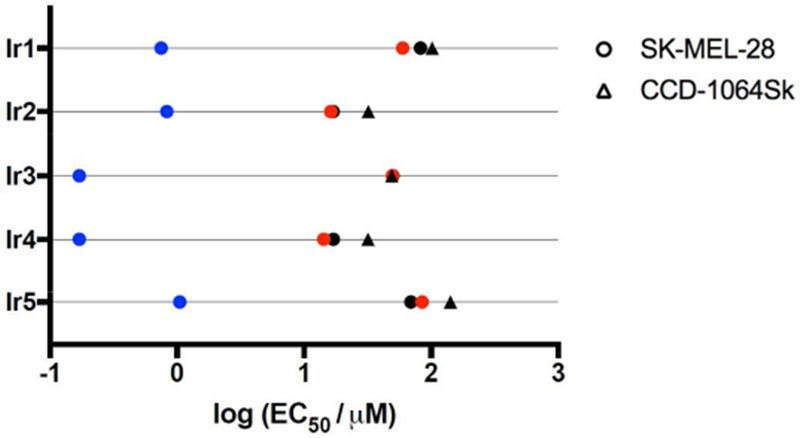
Activity plot for complexes Ir1 - Ir5 in SK-MEL-28 and CCD-1064Sk cells treated in the dark (black) and with visible (blue) or red (red) light activation.
All of the complexes in the series could be activated with visible light to become powerful phototoxins, with EC50 values ranging from 170 nM to 1 μM and PIs ranging from 20 to 288. Ir3 and Ir4 were the most phototoxic at 170 nM, while Ir1, Ir2, and Ir5 were similar (visible EC50 = 0.75–1.0 μM). Photoactivation of the complexes with red light (625 nm) did not enhance the cytotoxicity over what was observed in the dark treatment, yielding PIs close to 1.0 in all cases. The photobiological activities of Ir1 - Ir5 were tested with red light, despite their very low molar extinction coefficients in this region, because other metal complex systems with π-expansive ligands have been shown to yield potent in vitro red PDT effects even with molar extinction coefficients less than 100 M−1·cm−1.20
The presence of visible PDT effects (presumably due to the shorter wavelengths) but lack of red PDT effects suggests that direct population of the highly photosensitizing triplet states is not efficient in this class of complexes and that access to these states must be gained through 1MLCT states. The photocytotoxicity profiles in SK-MEL-28 under two irradiation conditions along with the dark cytotoxicity profiles in two cell lines are summarized in the activity plot in Figure 5.
The visible PDT effect followed the order Ir3 > Ir1 > Ir4 > Ir5 > Ir2, with Ir3 being the most promising photosensitizer based on its PI of 288 and nanomolar photocytotoxicity. Ir1 and Ir4 both had PIs greater than 100, but the dark toxicity associated with Ir4 in both cell lines limits its potential for in vivo applications. The source of the PDT effect for this series has not been established. The 1O2 quantum yields measured under cell-free conditions followed the order Ir4 > Ir1 > Ir2 > Ir5 > Ir3, with Ir1 and Ir2 being very similar. Ir3 was the poorest 1O2 generator, yet it was one of the most phototoxic complexes of the series. On the other hand, Ir4 was the best sensitizer of 1O2 and was as phototoxic as Ir3. Therefore, 1O2 may play a role in the PDT mechanism for some complexes but not others in this series, or the intracellular 1O2 quantum yields may differ from those measured under cell-free conditions. Regardless, certain members of this new series of dinuclear Ir(III) complexes have been identified as promising PDT agents for further investigation.
While the structural diversity in such a small library is somewhat limited, it was possible to identify some trends regarding structural features that affect cytotoxicity. For example, incorporation of ethynyl linkers as in Ir2 turned the relatively nontoxic complex Ir1 into one of the most potent dark cytotoxic complexes of the series (Table 3). Likewise, replacement of the terminal tridentate N∧N∧N ligands of Ir1 with C∧N∧N as in Ir4 increased the dark cytotoxicity substantially, while replacement with C∧N∧C as in Ir5 had only a very minor effect that differed between the two cell lines. In SK-MEL-28, the dark toxicity increased slightly, and in CCD-106Sk, the dark toxicity decreased slightly. The complex that departed the most structurally from the other complexes in the series and was identified as being the most promising PDT lead, Ir3, was intermediate in terms of dark cytotoxicity (EC50 ≈ 50 μM) with almost no difference between the two cell lines.
In terms of structural features affecting photocytotoxicity, the nature of the terminal tridentate ligand played some role as Ir4 (C∧N∧N) was more than six fold more phototoxic than Ir5 (C∧N∧C). While the presence of an ethynyl linker increased the dark cytotoxicity substantially in both cell lines, its presence did not impact the photocytotoxicity toward SK-MEL-28 in any significant way. The differences in dark and light-triggered cytotoxicity toward SK-MEL-28 cells alongside differences in dark cytotoxicity between normal and cancerous cells for certain members of this series indicate that even minor structural modifications can have a major impact on biological activity.
Cellular Imaging.
The phosphorescence from complexes Ir1 – Ir5 was used to probe cellular uptake by SK-MEL-28 melanoma cells with or without a light treatment (Figure 6). The excitation from a 458/488 nm argon-krypton laser matched the excitation maxima of the complexes and was used in conjunction with a 475-nm long pass filter to collect the emission from the complexes. The images were collected after a brief 1-h incubation period to ensure that some viable cells remained. Light-treated cells were illuminated with a broadband visible light (50 J·cm−2) that was 50% of the fluence used in the cellular assays in order to capture a fraction of viable cells.
Figure 6.
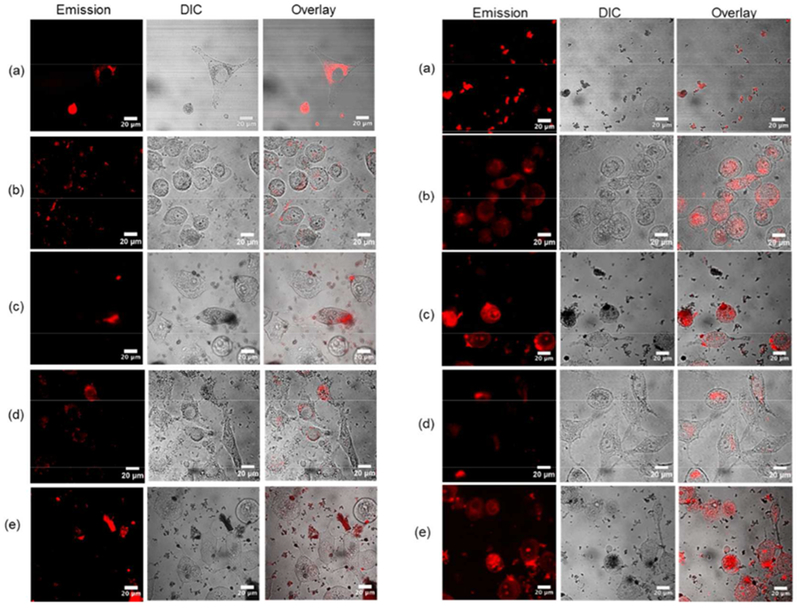
Confocal luminescence images of SK-MEL-28 cells dosed with Ir1 - Ir5 (a-e, 50 μM) in the dark (left) and with visible light (50 J·cm−2) (right).
Untreated SK-MEL-28 cells have a dendritic morphology. Treatment with the dinuclear Ir(III) complexes with or without illumination caused a conversion from dendritic to spherical morphology. The complexes showed detectable phosphorescence when associated with or in dead/dying and compromised cells with or without a light treatment. Only Ir1 appeared to be readily taken up into SK-MEL-28 cells in the dark at the observation time point. However, phosphorescence from the Ir(III) complexes in all cells was apparent after a light treatment, suggesting photoactivated uptake. For light-treated cells incubated with Ir1, it was not possible to discern subcellular localization because only cellular debris was present at the observation time point. However, Ir2 and Ir4 localized to the cytoplasm and multiple nucleoli whereas Ir3 and Ir5 were distributed throughout the cell and phosphoresced with a very intense signal by comparison.
Qualitatively, SK-MEL-28 cells treated with Ir4 with or without visible illumination appeared the most viable with healthy morphology in the imaging experiments but were the most susceptible in the cellular assays, highlighting the need to exercise caution when reconciling the cellular assay results with confocal imaging performed at different time points post-complex-delivery and post-irradiation and a different light fluence. When the conditions were similar, the imaging experiments did reflect the trends observed in the cellular assays with SK-MEL-28 but did not provide any information regarding uptake and localization since all cells were dead/dying but at slightly different stages. A quantitative comparison of the cellular uptake and induced morphological changes for the five complexes and correlations to cellular cytotoxicity or photocytotoxicity were not attempted given the need to alter incubation and illumination times to preserve some viable cells. Rather, the purpose of the imaging was to highlight the potential of these new Ir(III) complexes as theranostic agents based on their abilities to yield visible PDT effects and to be simultaneously imaged by their inherent phosphorescence.
DNA Interactions.
The ability of the Ir(III) complexes to act as DNA photocleaving agents was investigated to establish whether light-mediated DNA damage could contribute to the observed in vitro PDT effects for this class of photosensitizers. Supercoiled plasmid DNA (20 μM bases) was treated with increasing concentrations of Ir1 – Ir5 and then exposed to a visible light treatment of 14 J·cm−2 (Figure 7, lanes 3–14). The fluence is less that what was used in the cellular assays because the DNA is more susceptible to damage by the light treatment alone when not protected by the cellular environment. The photolyzed samples were then electrophoresed and compared to DNA alone with or without a light treatment (Figure 7, lanes 1 and 2) and DNA exposed to the highest concentration of the complex without a light treatment (Figure 7, lane 15). The gels were cast either with the DNA stain ethidium bromide (EB) incorporated or without EB and stained after electrophoresis (non-EB). EB gels allow detection of photocleavage not compounded by DNA unwinding; the non-EB gels allow detection of DNA unwinding in addition to photocleavage. Under the conditions employed for this gel electrophoretic mobility shift assay, undamaged supercoiled DNA (Form I) migrates the farthest in the gel, while aggregated/condensed DNA (Form IV) migrates very little from the loading well. Plasmid DNA that has undergone single-strand breaks (Form II) will relax and migrate between Forms I and IV, and plasmid DNA with frank double-strand breaks or double-strand breaks that arise from the build-up of single-strand breaks on opposing strands within about 16 base pairs (Form III) will migrate slightly faster than Form II. Forms I, II, and IV were detectable in both EB and non-EB gels. None of the complexes acted as DNA unwinders on the non-EB gel, indicating that they most likely do not act as DNA intercalators.
Figure 7.

DNA photocleavage of pUC19 DNA (20 μM) dosed with metal complex (MC) Ir1 (a), Ir2 (b), Ir3 (c), Ir4 (d), Ir5 (e) and visible light (14 J·cm−2). Gel mobility shift assays employed 1% agarose gels (0.75 μg·mL−1 ethidium bromide) electrophoresed in 1× TAE at 8 V·cm−1 for 30 min. Lane 1, DNA only (−hv); lane 2, DNA only (+hv); lane 3, 0.5 μM MC (+hv); lane 4, 1 μM MC (+hv); lane 5, 2 μM MC (+hv) lane 6, 3 μM MC (+hv) lane 7, 5 μM MC (+hv) lane 8, 8 μM MC (+hv); lane 9, 10 μM MC (+hv) lane 10, 12 μM MC (+hv) lane 11, 15 μM MC (+hv) lane 12, 20 μM MC (+hv); lane 13, 50 μM MC (+hv); lane 14, 100 μM MC (+hv); lane 15, 100 μM MC (−hv). Forms I, II and IV DNA refer to supercoiled plasmid, nicked circular plasmid, and aggregated plasmid, respectively.
All of the complexes showed some ability to photocleave DNA (Figure 7) in a cell-free environment. Qualitatively, DNA photocleaving ability appeared to increase in the order Ir5 < Ir2 ≈ Ir3 < Ir4 < Ir1. The formation of Form IV DNA and the disappearance of gel bands precluded a more quantitative comparison, but some general trends could still be discerned.
Despite its 1O2 quantum yield of 22%, Ir5 appeared to show the weakest interactions with DNA (although some strand breaks to yield detectable Form II92 were observed toward the highest concentrations). On the other hand, Ir1, with a similar ΦΔ, acted as a much more potent DNA photocleaving agent, converting a significant amount of supercoiled Form I DNA to Form II DNA at a metal complex (MC) concentration of only 1 μM and 20 μM DNA bases (Figure 7a, lane 4). At similarly low [MC]:[bases] ratios of 0.05, Ir2 – Ir5 caused no detectable strand breaks, which can be seen by comparing lane 4 for all of the complexes. Ir4, with the largest value for ΦΔ, photocleaved DNA in a concentration-dependent manner to yield Form II DNA as expected.
All of the complexes caused DNA aggregation/condensation, although Ir5 produced trace amounts of Form IV DNA only at the highest complex concentrations investigated. Interestingly, Ir5 was the only complex that did not cause the DNA gel bands to disappear. The lack of DNA staining by EB for the other complexes could be due to fluorescence quenching of the EB dye by the complex, their competition for EB intercalation sites, or their distortion of the DNA helix that prevents EB binding.
Clearly, the structural differences between the Ir(III) complexes of this small library resulted in markedly different interactions with DNA, and possibly different photophysical interactions with the EB dye. The observation that Ir5 shows marginal DNA interactions in the gel electrophoretic analysis yet acts as an in vitro PDT agent suggests that DNA may not be the intracellular target, at least for this particular complex. In fact, DNA photodamage did not correlate clearly with 1O2 quantum yields across the series, which also supports the notion that another biological target is likely involved. However, the cell-free experiment does not accurately mimic the complexity of the cellular environment and dynamic processes (e.g., uptake, efflux, metabolism, and localization), and in vitro DNA damage and 1O2 damage cannot be ruled out completely. What can be gleaned from the DNA photocleavage study is that minor structural changes in this series have profound effects on the complex interactions with biological macromolecules such as DNA, which is in agreement with their different profiles in the cellular assays and imaging studies.
CONCLUSIONS
The synthesis, photophysical and photobiological properties of a family of water-soluble cationic dinuclear iridium(III) complexes (Ir1 - Ir5) were explored. The influence of the bridging and terminal ligands on the photophysical properties of the complexes was investigated. Compared to Ir1 that had the single bond connection between the fluorenyl motif and the terpyridyl ligands on the bridging ligand, the extended π-conjugation afforded by the ethynyl connectors of the bridging ligand in Ir2 red-shifted the UV-vis absorption markedly, but the low-lying 3CT state of Ir2 accelerated nonradiative decay and resulted in weak phosphorescence. A considerable bathochromic shift also occurred in the absorption and emission of Ir5, owing to the stronger σ-donating ability of the negatively charged coordinating carbon relative to nitrogen and thus more charge transfer from the C∧N∧C ligands to the terpyridyl ligands. Complexes Ir1 – Ir5 all featured with broad positive absorption bands spanning the visible region and NIR regions in their nanosecond TA spectra. However, the triplet state TA lifetimes of Ir1 and Ir4 were much longer (3.1 μs and 48 μs, respectively) than those of Ir2, Ir3 and Ir5, implying the dominant bridging ligand-localized 3π,π* nature for the lowest triplet states in Ir1 and Ir4 rather than the 3CT states for the other three complexes. Based on photophysical properties alone, Ir4 was predicted to be the best in vitro PDT agent.
All of the Ir(III) complexes of this study exhibited photobiological effects when activated with visible light, but were inactive with single-wavelength red light (625 nm). Thus, the in vitro PDT effects with broadband visible light were attributed to the shorter wavelengths. Some of the complexes showed selective cytotoxicity toward cancerous human melanoma cells over normal human skin fibroblasts. The photobiological trends could not be readily correlated to any differences in photophysical properties despite accessible long-lived 3π,π* states often resulting in red PDT activity. But the long-lived Ir4 did not yield a red PDT effect, nor did any of the other complexes due to the lack of ground-state absorption in the red. Rather, Ir3 emerged as a promising photosensitizer for further investigation owing to its nanomolar photocytotoxicity and visible PI > 280, with Ir1 and Ir5 also having suitable profiles. This small library of just five complexes proved to be a rich source of photophysical and photobiological diversity with only minor structural modifications. They gave 1O2 quantum yields that ranged from 4 to 38%, light EC50 values from nanomolar to micromolar, dark toxicities that ranged from 32 to >140 μM, and DNA interactions that were characteristic for a particular cationic complex. For in vitro PDT applications in particular, there was a clear indication that the terminal tridentate N∧C∧N ligand performed best when combined with methyl substituents on the central cyclometalating ring and no ethynyl linkers between terminal ligands and the central fluorene unit. Thus, Ir3 will serve as the lead complex for future studies and as the parent complex of a second-generation library.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the financial support from the National Science Foundation (DMR-1411086 and CNS-1229316) for materials synthesis, characterization and computational simulation of the optical spectra. For computational resources and administrative support, authors thank the Center for Computationally Assisted Science and Technology (CCAST) at North Dakota State University. SM thanks the Natural Sciences and Engineering Council of Canada (NSERC), the Canadian Institutes of Health Research (CIHR), the Canadian Foundation for Innovation (CFI), and the Nova Scotia Research and Innovation Trust (NSRIT) as well as Acadia University and the University of North Carolina at Greensboro. SK thanks the National Energy Research Scientific Computing Center (NERSC) allocation award 86678, supported by the Office of Science of the Department of Energy under Contract No. DE-AC02-05CH11231. For partial financial support of the quantum chemistry software, SK acknowledges Sloan Research Fellowship BR2014-073.
Footnotes
Supporting Information
The experimental details for photobiological activity studies, NTOs representing transitions contributing to the high-energy absorption bands in Ir1 – Ir5, comparison of the experimental and theoretical UV-vis absorption spectra of Ir1 – Ir5 in acetonitrile, normalized UV-vis absorption and emission spectra in different solvents, the emission parameters in different solvents, the time-resolved TA spectra of Ir1 – Ir5 in CH3CN, and the full author list for Refs. 22, 24 and 61. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Storr T; Thompson KH; Orvig C Design of Targeting Ligands in Medicinal Inorganic Chemistry. Chem. Soc. Rev 2006, 35, 534–544. [DOI] [PubMed] [Google Scholar]
- (2).Dasari S; Tchounwou PB Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol 2014, 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bergamo A; Gaiddon C; Schellens JHM; Beijnen JH; Sava G Approaching Tumour Therapy beyond Platinum Drugs: Status of the Art and Perspectives of Ruthenium Drug Candidates. J. Inorg. Biochem 2012, 106, 90–99. [DOI] [PubMed] [Google Scholar]
- (4).Dabrowiak JC Anticancer Agents Beyond Cisplatin In Metals in Medicine; John Wiley & Sons, Ltd, 2017; pp 157–216. [Google Scholar]
- (5).Trondl R; Heffeter P; Kowol CR; Jakupec MA; Berger W; Keppler BK NKP-1339, the First Ruthenium-Based Anticancer Drug on the Edge to Clinical Application. Chem. Sci 2014, 5, 2925–2932. [Google Scholar]
- (6).Schoenhacker-Alte B; Mohr T; Pirker C; Kryeziu K; Kuhn P-S; Buck A; Hofmann T; Gerner C; Hermann G; Koellensperger G; et al. Sensitivity towards the GRP78 Inhibitor KP1339/IT-139 Is Characterized by Apoptosis Induction via Caspase 8 upon Disruption of ER Homeostasis. Cancer Lett. 2017, 404, 79–88. [DOI] [PubMed] [Google Scholar]
- (7).Burris HA; Bakewell S; Bendell JC; Infante J; Jones SF; Spigel DR; Weiss GJ; Ramanathan RK; Ogden A; Von Hoff D Safety and Activity of IT-139, a Ruthenium-Based Compound, in Patients with Advanced Solid Tumours: A First-in-Human, Open-Label, Dose-Escalation Phase I Study with Expansion Cohort. ESMO Open 2017, 1, e000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mari C; Gasser G Lightening up Ruthenium Complexes to Fight Cancer? Chim. Int. J. Chem 2015, 69, 176–181. [DOI] [PubMed] [Google Scholar]
- (9).Mari C; Pierroz V; Ferrari S; Gasser G Combination of Ru(II) Complexes and Light: New Frontiers in Cancer Therapy. Chem. Sci 2015, 6, 2660–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Knoll JD; Turro C Control and Utilization of Ruthenium and Rhodium Metal Complex Excited States for Photoactivated Cancer Therapy. Coord. Chem. Rev 2015, 282–283, 110–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Poynton FE; Bright SA; Blasco S; Williams DC; Kelly JM; Gunnlaugsson T The Development of Ruthenium(II) Polypyridyl Complexes and Conjugates for in Vitro Cellular and in Vivo Applications. Chem. Soc. Rev 2017, 46, 7706–7756. [DOI] [PubMed] [Google Scholar]
- (12).Bonnett R Chemical Aspects of Photodynamic Therapy; Gordon and Breach Science Publishers, 2000. [Google Scholar]
- (13).van Straten D; Mashayekhi V; de Bruijn H; Oliveira S; Robinson D Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hamblin MR; Huang Y-Y Handbook of Photomedicine; Taylor & Francis, 2014. [Google Scholar]
- (15).Abrahamse H; Hamblin MR New Photosensitizers for Photodynamic Therapy. Biochem. J 2016, 473, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L; et al. Ru(II) Dyads Derived from Alpha-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. [Google Scholar]
- (17).Kaspler P; Lazic S; Forward S; Arenas Y; Mandel A; Lilge L A Ruthenium(II) Based Photosensitizer and Transferrin Complexes Enhance Photo-Physical Properties, Cell Uptake, and Photodynamic Therapy Safety and Efficacy. Photochem. Photobiol. Sci 2016, 15, 481–495. [DOI] [PubMed] [Google Scholar]
- (18).Ford WE; Rodgers MAJ Reversible Triplet-Triplet Energy Transfer within a Covalently Linked Bichromophoric Molecule. J. Phys. Chem 1992, 96, 2917–2920. [Google Scholar]
- (19).McClenaghan ND; Leydet Y; Maubert B; Indelli MT; Campagna S Excited-State Equilibration: A Process Leading to Long-Lived Metal-to-Ligand Charge Transfer Luminescence in Supramolecular Systems. Coord. Chem. Rev 2005, 249, 1336–1350. [Google Scholar]
- (20).Yin H; Stephenson M; Gibson J; Sampson E; Shi G; Sainuddin T; Monro S; McFarland SA In Vitro Multiwavelength PDT with 3IL States: Teaching Old Molecules New Tricks. Inorg. Chem 2014, 53, 4548–4559. [DOI] [PubMed] [Google Scholar]
- (21).Lincoln R; Kohler L; Monro S; Yin H; Stephenson M; Zong R; Chouai A; Dorsey C; Hennigar R; Thummel RP; et al. Exploitation of Long-Lived 3IL Excited States for Metal-Organic Photodynamic Therapy: Verification in a Metastatic Melanoma Model. J. Am. Chem. Soc 2013, 135, 17161–17175. [DOI] [PubMed] [Google Scholar]
- (22).Stephenson M; Reichardt C; Pinto M; Wachtler M; Sainuddin T; Shi G; Yin H; Monro S; Sampson E; Dietzek B; et al. Ru(II) Dyads Derived from 2-(1-Pyrenyl)-1H-Imidazo[4,5-f][1,10]Phenanthroline: Versatile Photosensitizers for Photodynamic Applications. J. Phys. Chem. A 2014, 118, 10507–10521. [DOI] [PubMed] [Google Scholar]
- (23).Arenas Y; Monro S; Shi G; Mandel A; McFarland S; Lilge L Photodynamic Inactivation of Staphylococcus Aureus and Methicillin-Resistant Staphylococcus Aureus with Ru(II)-Based Type I/Type II Photosensitizers. Photodiag. Photodyn. Ther 2013, 10, 615–625. [DOI] [PubMed] [Google Scholar]
- (24).Wang L; Yin H; Cui P; Hetu M; Wang C; Monro S; Schaller RD; Cameron CG; Liu B; Kilina S; et al. Near-Infrared-Emitting Heteroleptic Cationic Iridium Complexes Derived from 2,3-Diphenylbenzo[g]Quinoxaline as in Vitro Theranostic Photodynamic Therapy Agents. Dalton Trans. 2017, 46, 8091–8103. [DOI] [PubMed] [Google Scholar]
- (25).Wang C; Lystrom L; Yin H; Hetu M; Kilina S; McFarland SA; Sun W Increasing the Triplet Lifetime and Extending the Ground-State Absorption of Biscyclometalated Ir(III) Complexes for Reverse Saturable Absorption and Photodynamic Therapy Applications. Dalton Trans. 2016, 45, 16366–16378. [DOI] [PubMed] [Google Scholar]
- (26).Majumdar P; Yuan X; Li S; Guennic BL; Ma J; Zhang C; Jacquemin D; Zhao J Cyclometalated Ir(III) Complexes with Styryl-BODIPY Ligands Showing near IR Absorption/Emission: Preparation, Study of Photophysical Properties and Application as Photodynamic/Luminescence Imaging Materials. J. Mater. Chem. B 2014, 2, 2838–2854. [DOI] [PubMed] [Google Scholar]
- (27).Ye R-R; Tan C-P; He L; Chen M-H; Ji L-N; Mao Z-W Cyclometalated Ir(III) Complexes as Targeted Theranostic Anticancer Therapeutics: Combining HDAC Inhibition with Photodynamic Therapy. Chem. Commun 2014, 50, 10945–10948. [DOI] [PubMed] [Google Scholar]
- (28).He L; Li Y; Tan C-P; Ye R-R; Chen M-H; Cao J-J; Ji L-N; Mao Z-W Cyclometalated Iridium(III) Complexes as Lysosome-Targeted Photodynamic Anticancer and Real-Time Tracking Agents. Chem. Sci 2015, 6, 5409–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kando A; Hisamatsu Y; Ohwada H; Itoh T; Moromizato S; Kohno M; Aoki S Photochemical Properties of Red-Emitting Tris(Cyclometalated) Iridium(III) Complexes Having Basic and Nitro Groups and Application to pH Sensing and Photoinduced Cell Death. Inorg. Chem 2015, 54, 5342–5357. [DOI] [PubMed] [Google Scholar]
- (30).Jiang X; Zhu N; Zhao D; Ma Y New Cyclometalated Transition-Metal Based Photosensitizers for Singlet Oxygen Generation and Photodynamic Therapy. Sci. China Chem. 2016, 59, 40–52. [Google Scholar]
- (31).Zheng Y; He L; Zhang D-Y; Tan C-P; Ji L-N; Mao Z-W Mix-ligand Iridium(III) Complexes as Photodynamic Anticancer Agents. Dalton Trans. 2017, 46, 11395–11407. [DOI] [PubMed] [Google Scholar]
- (32).Liu J; Jin C; Yuan B; Chen Y; Liu X; Ji L; Chao H Enhanced Cancer Therapy by the Marriage of Metaabolic Alternation and Mitochondrial-targeted Photodynamic Therapy Using Cyclometalated Ir(III) Complexes. Chem. Commun 2017, 53, 9878–9881. [DOI] [PubMed] [Google Scholar]
- (33).Xiang H; Chen H; Tham HP; Phua SZF; Liu J-G; Zhao Y Cyclometalated Iridium(III)-Complex-Based Mocelles for Glutathione-REsponsive Targeted Chemtherapy and Photodynamic Therapy. ACS Appl. Mater. Interfaces 2017, 9, 27553–27562. [DOI] [PubMed] [Google Scholar]
- (34).Zhang P; Chiu CKC; Huang H; Lam YPY; Habtemariam A; Malcomson T; Paterson MJ; Clarkson GJ; O’Connor PB; Chao H; Sadler PJ Organoiridium Photosensitizers Induce Specific Oxidative Attack on Proteins within Cancer Cells. Angew. Chem. Int. Ed 2017, 56, 14898–14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lv W; Zhang Z; Zhang KY; Yang H; Liu S; Xu A; Guo S; Zhao Q; Huang W A Mitochondria-Targeted Photosensitizer Showing Improved Photodynamic Therapy Effects Under Hypoxia. Angew. Chem. Int. Ed 2016, 55, 9947–9951. [DOI] [PubMed] [Google Scholar]
- (36).Qiu K; Ouyang M; Liu Y; Huang H; Liu C; Chen Y; Ji L; Chao H Two-photon Photodynamic Ablation of Tumor Cells by Mitochonfria-Targeted Iridium(III) Complexes in Aggregated States. J. Mater. Chem. B 2017, 5, 5488–5498. [DOI] [PubMed] [Google Scholar]
- (37).Ouyang M; Zeng L; Qiu K; Chen Y; Li L; Chao H Cyclometalated IrIII Complexes as Mitochondria-Targeted Photodynamic Anticancer Agents. Eur. J. Inorg. Chem 2017, 1764–1771. [Google Scholar]
- (38).Xue F; Lu Y; Zhou Z; Shi M; Yan Y; Yang H; Yang S Two in One: Luminescence Imaging and 730 nm Continuous Wave Laser Driven Photodynamic Therapy of Iridium Complexes. Organometallics 2015, 34, 73–77. [Google Scholar]
- (39).Maggioni D; Galli M; D’Alfonso L; Inverso D; Dozzi MV; sironi L; Iannacone M; Collin M; Ferruti P; Ranucci E; D’Alfonso G A Luminescent Poly(amidoamine)-Iridium Complex as a New Singlet-Oxygen Sensitizer for Photodynamic Therapy. Inorg. Chem 2015, 54, 544–553. [DOI] [PubMed] [Google Scholar]
- (40).Nam JS; Kang M-G; Kang J; Park S-Y; Lee SJC; Kim H-T; Seo JK; Kwon O-H; Lim MH; Rhee H-W; Kwon T-H Endoplasmic Reticulum-Localized Iridium(III) Complexes as Efficient Photodynamic Therapy Agents via Protein Modifications. J. Am. Chem. Soc 2016, 138, 10968–10977. [DOI] [PubMed] [Google Scholar]
- (41).Li Y; Lu X-R; Li M-F; Ji L-N; Mao Z-W Cyclometalated Iridium(III) N-Heterocyclic Carbene Complexes as Potnetial Mitochondria Anticancer and Photodynamic Agents. Dalton Trans. 2017, 46, 11363–11371. [DOI] [PubMed] [Google Scholar]
- (42).Tabrizi L; Chiniforoshan H New Cyclometalated Ir(III) Complexes with NCN Pincer and meso-Phenylcyanamide BODIPY Ligands as Efficient Photodynamic Therapy Agents. RSC Adv. 2017, 7, 34160–34169. [Google Scholar]
- (43).McKenzie LK; Sazanovich IV; Baggaley E; Bonnneau M; Guerchais V; Williams JAG; Weinstein JA; Bryant HE Metal Complexes for Two-Photon Photodynamic Therapy: A Cyclometalated Iridium Complex Induces Two-Photon Photosensitization of Cancer under Near-IR Light. Chem. Eur. J 2017, 23, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tian X; Zhu Y; Zhang M; Luo L; Wu J; Zhou H; Guan L; Battaglia G; Tian Y Localization Matters: A Nuclear Targeting Two-Photon Absorption Iridium Complex in Photodynamic Therapy. Chem. Commun 2017, 53, 3303–3306. [DOI] [PubMed] [Google Scholar]
- (45).Cui Y; Wen LL; Shan GG; Sun HZ; Mao HT; Zhang M; Su ZM Di-/Trinuclear Cationic Ir(III) Complexes: Design, Synthesis and Application for Highly Sensitive and Selective Detection of TNP in Aqueous Solution. Sens. Actuators B. 2017, 244, 314–322. [Google Scholar]
- (46).Constable EC 2,2′:6′,2″-Terpyridines: From Chemical Obscurity to Common Supramolecular Motifs. Chem. Soc. Rev 2007, 36, 246–253. [DOI] [PubMed] [Google Scholar]
- (47).Hofmeier H; Schubert US Recent Developments in the Supramolecular Chemistry of Terpyridine-Metal Complexes. Chem. Soc. Rev 2004, 33, 373–399. [DOI] [PubMed] [Google Scholar]
- (48).Ji Z; Li S; Li Y; Sun W Back-to-Back Dinuclear Platinum Terpyridyl Complexes: Synthesis and Photophysical Studies. Inorg. Chem 2010, 49, 1337–1346. [DOI] [PubMed] [Google Scholar]
- (49).Tu S; Li T; Shi F; Wang Q; Zhang J; Xu J; Zhu X; Zhang X; Zhu S; Shi D Convenient One-Pot Synthesis of 4′-Aryl-2,2′:6′,2″-terpyridines and 2,4,6-Triarylpyridines under Microwave Irradiation. Synthesis 2005, 18, 3045–3050. [Google Scholar]
- (50).Wilkinson AJ; Puschmann H; Howard JAK; Foster CE; Williams JAG Luminescent Complexes of Iridium (III) Containing N∧C∧N-coordinating Terdentate Ligands. Inorg. Chem 2006, 45, 8685–8699. [DOI] [PubMed] [Google Scholar]
- (51).Basnet A; Thapa P; Karki R; Na Y; Jahng Y; Jeong BS; Jeong TC; Lee CS; Lee ES 2,4,6-Trisubstituted Pyridines: Synthesis, Topoisomerase I and II Inhibitory Activity, Cytotoxicity, and Structure–Activity Relationship. Bioorg. Med. Chem 2007, 15, 4351–4359. [DOI] [PubMed] [Google Scholar]
- (52).Adib M; Ayashi N; Mirzaei P An Efficient Synthesis of 2,4,6-Triarylpyridines by Use of Benzyl Halides under Neat Conditions. Synlett 2016, 27, 417–421. [Google Scholar]
- (53).Chirdon DN; Transue WJ; Kagalwala HN; Kaur A; Maurer AB; Pintauer T; Bernhard S [Ir(N∧N∧N)(C∧N)L]+: A New Family of Luminophores Combining Tunability and Enhanced Photostability. Inorg. Chem 2014, 53, 1487–1499. [DOI] [PubMed] [Google Scholar]
- (54).Choi D; Kim T; Reddy SM; Kang J Synthesis and Electronic Properties of Double Pincer-type Cyclometalated Iridium Complexes. Inorg. Chem. Commun 2009, 12, 41–44. [Google Scholar]
- (55).Demas JN; Crosby GA The Measurement of Photoluminescence Quantum Yields. A Review.. J. Phys. Chem 1971, 75, 991–1024. [Google Scholar]
- (56).van Houten J; Watts RJ Temperature Dependence of the Photophysical and Photochemical Properties of the Tris(2,2′-bipyridyl) Ruthenium(II) Ion in Aqueous Solution. J. Am. Chem. Soc 1976, 98, 4853–4858. [Google Scholar]
- (57).Carmichael I; Hug GL Triplet-triplet Absorption Spectra of Organic Molecules in Condensed Phases. J. Phys. Chem. Ref. Data 1986, 15, 1–250. [Google Scholar]
- (58).Kumar CV; Qin L; Das PK Aromatic Thioketone Triplets and Their Quenching Behaviour towards Oxygen and Di-t-butylnitroxy Radical. A Laser-Flash-Photolysis Study. J. Chem. Soc., Faraday Trans. 2 1984, 80, 783–793. [Google Scholar]
- (59).Firey PA; Ford WE; Sounik JR; Kenney ME; Rodgers MAJ Silicon Naphthalocyanine Triplet State and Oxygen. A Reversible Energy-transfer Reaction. J. Am. Chem. Soc 1988, 110, 7626–7630. [Google Scholar]
- (60).DeRosa MC; Crutchley RJ Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev 2002, 233–234, 351–371. [Google Scholar]
- (61).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; et al. Gaussian 09, Revision D.01, Gaussian. Inc., Wallingford, CT, 2013. [Google Scholar]
- (62).Hay PJ; Wadt WR Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys 1985, 82, 299–310. [Google Scholar]
- (63).Hay PJ; Wadt WR Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- (64).Wadt WR; Hay PJ Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys 1985, 82, 284–298. [Google Scholar]
- (65).Clark T; Chandrasekhar J; Spitznagel GW; Schleyer PVR Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem 1983, 4, 294–301. [Google Scholar]
- (66).Francl MM; Pietro WJ; Hehre WJ; Binkley JS; Gordon MS; DeFrees DJ; Pople JA Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys 1982, 77, 3654–3665. [Google Scholar]
- (67).Gill PM; Johnson BG; Pople JA; Frisch MJ The Performance of the Becke-Lee-Yang-Parr (B-LYP) Density Functional Theory with Various Basis Sets. Chem. Phys. Lett 1992, 197, 499–505. [Google Scholar]
- (68).Hariharan PC; Pople JA The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- (69).Krishnan R; Binkley JS; Seeger R; Pople JA Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys 1980, 72, 650–654. [Google Scholar]
- (70).Liu B; Lystrom L; Kilina S; Sun W Tuning the Ground State and Excited State Properties of Monocationic Iridium (III) Complexes by Varying the Site of Benzannulation on Diimine Ligand. Inorg. Chem 2017, 56, 5361–5370. [DOI] [PubMed] [Google Scholar]
- (71).Zhu X; Lystrom L; Kilina S; Sun W Tuning the Photophysics and Reverse Saturable Absorption of Heteroleptic Cationic Iridium (III) Complexes via Substituents on the 6,6′-Bis (Fluoren-2-yl)-2,2′-biquinoline Ligand. Inorg. Chem 2016, 55, 11908–11919. [DOI] [PubMed] [Google Scholar]
- (72).Chai J-D; Head-Gordon M Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. PCCP 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (73).Barone V; Cossi M; Tomasi J Geometry Optimization of Molecular Structures in Solution by the Polarizable Continuum Model. J. Comput. Chem 1998, 19, 404–417. [Google Scholar]
- (74).Cossi M; Rega N; Scalmani G; Barone V Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- (75).Marques MA; Gross EK Time-Dependent Density Functional Theory. Annu. Rev. Phys. Chem 2004, 55, 427–455. [DOI] [PubMed] [Google Scholar]
- (76).Ullrich CA Time-Dependent Density-Functional Theory: Concepts and Applications. OUP Oxford: 2011. [Google Scholar]
- (77).Bjorgaard JA; Sifain AE; Nelson T; Myers TW; Veauthier JM; Chavez DE; Scharff RJ; Tretiak S Two-Photon Absorption in Conjugated Energetic Molecules. J. Phys. Chem. A 2016, 120, 4455–4464. [DOI] [PubMed] [Google Scholar]
- (78).Kilina S; Kilin D; Tretiak S Light-Driven and Phonon-Assisted Dynamics in Organic and Semiconductor Nanostructures. Chem. Rev 2015, 15, 5929–5978. [DOI] [PubMed] [Google Scholar]
- (79).Martin RL Natural Transition Orbitals. J. Chem. Phys 2003, 118, 4775–4777. [Google Scholar]
- (80).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (81).McCusker CE; Hablot D; Ziessel R; Castellano FN Metal Coordination Induced π-Extension and Triplet State Production in Diketopyrrolopyrrole Chromophores. Inorg. Chem 2012, 51, 7957–7959. [DOI] [PubMed] [Google Scholar]
- (82).McCusker CE; Hablot D; Ziessel R; Castellano FN Triplet State Formation in Homo- and Heterometallic Diketopyrrolopyrrole Chromophores. Inorg. Chem 2014, 53, 12564–12571. [DOI] [PubMed] [Google Scholar]
- (83).Auffrant A; Barbieri A; Barigelletti F; Collin J-P; Flamigni L; Sabatini C; Sauvage J-P Dinuclear Iridium(III) Complexes Consisting of Back-to-Back tpy-(ph)n-tpy Bridging Ligands (n = 0, 1, or 2) and Terminal Cyclometallating Tridentate N-C-N Ligands. Inorg. Chem 2006, 45, 10990–10997. [DOI] [PubMed] [Google Scholar]
- (84).Dubinina GG; Price RS; Abboud KA; Wicks G; Wnuk P; Stepanenko Y; Drobizhev M; Rebane A; Schanze KS Phenylene Vinylene Platinum(II) Acetylides with Prodigious Two-Photon Absorption. J. Am. Chem. Soc 2012, 134, 19346–19349. [DOI] [PubMed] [Google Scholar]
- (85).Liu R; Li Y; Li Y; Zhu H; Sun W Photophysics and Nonlinear Absorption of Cyclometalated 4,6-Diphenyl-2,2′-bipyridyl Platinum(II) Complexes with Different Acetylide Ligands. J. Phys. Chem. A 2010, 114, 12639–12645. [DOI] [PubMed] [Google Scholar]
- (86).Liu R; Dandu N; Li Y; Kilina S; Sun W Synthesis, Photophysics and Reverse Saturable Absorption of Bipyridyl Platinum(II) Bis(arylfluorenylacetylide) Complexes. Dalton Trans. 2013, 42, 4398–4409. [DOI] [PubMed] [Google Scholar]
- (87).Sun Y; Joyce LE; Dickson NM; Turro C Efficient DNA Photocleavage by [Ru(bpy)2(dppn)]2+ with Visible Light. Chem. Commun 2010, 46, 2426–2428. [DOI] [PubMed] [Google Scholar]
- (88).Liu Y; Hammitt R; Lutterman DA; Joyce LE; Thummel RP; Turro C Ru(II) Complexes of New Tridentate Ligands: Unexpected High Yield of Sensitized 1O2. Inorg. Chem 2009, 48, 375–385. [DOI] [PubMed] [Google Scholar]
- (89).Foxon SP; Metcalfe C; Adams H; Webb M; Thomas JA Electrochemical and Photophysical Properties of DNA Metallo-intercalators Containing the Ruthenium(II) Tris(1-pyrazolyl)methane Unit. Inorg. Chem 2007, 46, 409–416. [DOI] [PubMed] [Google Scholar]
- (90).Foxon SP; Alamiry MAH; Walker MG; Meijer AJHM; Sazanovich IV; Weinstein JA; Thomas JA Photophysical Properties and Singlet Oxygen Production by Ruthenium(II) Complexes of Benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine: Spectroscopic and TD-DFT Study. J. Phys. Chem. A 2009,113, 12754–12762. [DOI] [PubMed] [Google Scholar]
- (91).Papanikolaou PA; Tkachenko NV Probing the Excited State Dynamics of a New Family of Cu(I)-Complexes with an Enhanced Light Absorption Capacity: Excitation-Wavelength Dependent Population of States Through Branching. Phys. Chem. Chem. Phys 2013, 15, 13128–13136. [DOI] [PubMed] [Google Scholar]
- (92).Blazek ER; Peak JG; Peak MJ Singlet Oxygen Induces Frank Strand Breaks as Well as Alkali- and Piperidine-Labile Sites in Supercoiled Plasmid DNA. Photochem. Photobiol 1989, 49, 607–613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.