

Abstract
Eye movements are frequently considered diagnostic markers indicating involvement of the cerebellum. Impaired amplitude of saccades (saccade dysmetria), impaired gaze holding function (horizontal or downbeat nystagmus), and interrupted (choppy) pursuit are typically considered hallmarks of cerebellar disorders. While saccade dysmetria is a frequently considered abnormality, the velocity of saccades are rarely considered part of the constellation of cerebellar involvement. Reduced saccade velocity, frequently called “slow saccades” are typically seen in a classic disorder of the midbrain called progressive supranuclear palsy. It is also traditionally diagnostic of spinocerebellar ataxia type 2. In addition to its common causes, the slowness of vertical saccades is not rare in cerebellar disorders. Frequently this phenomenology is seen in multisystem involvement that substantially involves the cerebellum. In this review we will first discuss the physiological basis and the biological need for high saccade velocities. In subsequent sections we will discuss disorders of cerebellum that are known to cause slowing of saccades. We will then discuss possible pathology and novel therapeutic strategies.
Keywords: Degenerative disorder, Dysmetria, Burst neurons, Reciprocal innervation
Background
Saccades are rapid, simultaneous movements of both eyes. Their purpose is to position the fovea at an object of interest in order to bring the object of interest more clearly into the line of sight. Saccades come in several different flavors, including visually guided (directed towards an intended target), reflexive (directed towards an unexpected target), memory guided (directed toward a remembered location) and antisaccades (equal amplitude movements in the opposite direction of a target). The complex circuitry of visually guided saccades is extensively studied, making it a useful tool for localizing and understanding neurological lesions. A variety of pathologies can alter the size, timing and speed of these saccades. In particular, characterizing changes in the horizontal and vertical velocity of visually guided saccades in various disorders holds promise as an important tool in both the diagnosis and probing treatment outcomes of these disorders. Here we review whether disorders affecting the cerebellum influence the velocity of visually guided saccades. First we will present the basic outline of anatomical and physiological principles underlying the generation of visually guided saccades. The review will then delve into discussion of various cerebellar disorders that are associated with slowing of saccades. We will then compare slow saccades in cerebellar disorders with that of brainstem and basal ganglia abnormalities. Finally we will present the physiological basis for slow saccades in cerebellar disorders.
Generation of visually guided saccades
Visually guided saccade generation begins in the cerebral hemispheres in two major pathways, both of which send input to the superior colliculus (Fig. 1) [1, 2]. In one pathway, the signal originates in the frontal eye field (FEF), supplementary eye field (SEF) and dorsolateral prefrontal cortex (dlPFC) [3–5]. In the other, input originates in the parietal eye field (posterior parietal cortex) [1, 2].
Fig. 1.
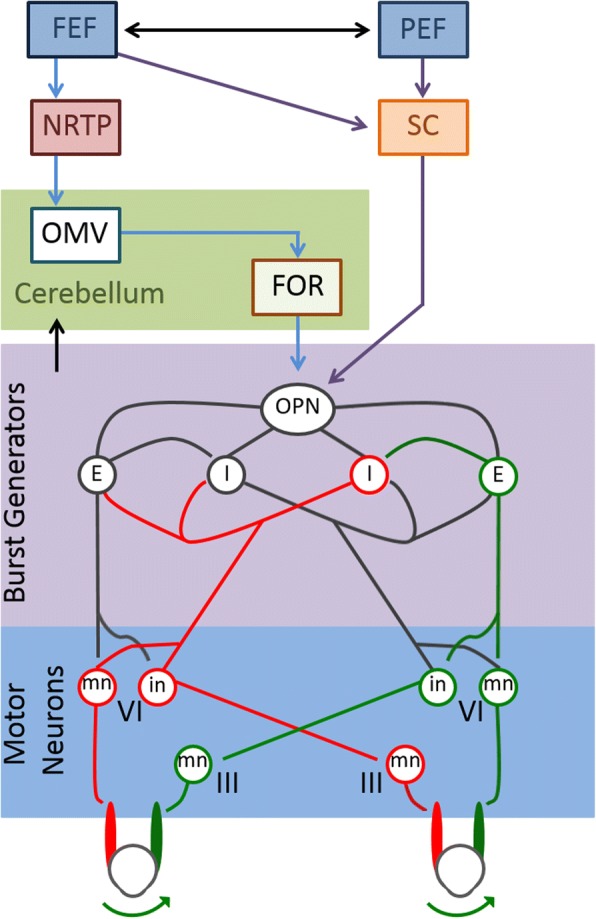
Neural circuitry involved in the generation of visually guided saccades. The frontal eye field (FEF) and parietal eye field (PEF) both send projections to the superior colliculus (SC), which in turn directly communicates with omniopause neurons (OPN) in the midline pons. The FEF also projects to the nucleus reticularis tegmenti pontis (NRTP) in the midbrain. This projects to the oculomotor vermis in the cerebellar cortex, which sends inhibitory fibers to the deep cerebellar fastigial oculomotor (FOR) nucleus. The FOR also communicates with OPN. The OPN is responsible for tonic inhibition of excitatory (E) and inhibitory (I) burst neurons, which results in steady fixation of gaze. When the OPN is inhibited during saccade generation, the excitatory and inhibitory burst neurons fire more rapidly. Excitatory burst neurons synapse on the abducens motoneurons (mn) and internuclear neurons (in) in the ipsilateral abducens nucleus. The internuclear neurons of the side receiving excitatory input project to the contralateral abducens motoneuronsmedial rectus subgroup of the oculomotor nucleus. Ipsilateral inhibitory burst neurons project to the contralateral abducens which inhibits movement of the opposing muscles. This results in the rapid, coordinated movement of gaze to a target object
The FEF, SEF and dlPFC send projections to the caudate nucleus, which in turn sends direct, inhibitory fibers to the substantia nigra pars reticulate (SNpr) [6, 7]. The SNpr maintains tonic GABAergic inhibition on the superior colliculus [8, 9]. In sending inhibitory fibers to this structure, the caudate nucleus transiently ceases the firing of inhibitory neurons, leading to timely saccade initiation [10, 11]. The caudate nucleus also sends fibers indirectly to the subthalamic nucleus via the external segment of the globus pallidus [7]. The parietal eye field (PEF) of the posterior parietal cortex projects to the FEF and the superior colliculus. In turn, PEF also receives input from the FEF [12].
The superior colliculus constitutes a shared pathway for visually and memory guided saccades [13]. It integrates excitatory inputs from the cortex and inhibitory inputs from the SNpr [14, 15]. The ventral layers of the superior colliculus consist of a motor map, containing information about eye movement parameters [16, 17]. This structure receives input from the striate, extrastriate, parietal cortex and from the frontal lobes [18, 19]. Electrophysiology studies have also confirmed the presence of neuronal populations directly involved in saccade generation, which project to the midbrain and pontine reticular formation where premotor structures involved in saccade generation are located [20, 21].
The cerebellum receives input both from the cortical eye fields (indirectly via pontine nuclei) and the superior colliculus [22–26]. The dorsal vermis is a medial cortico-nuclear zone within the cerebellum that receives fibers from the nucleus reticularis tegmenti pontis (NRTP) in the midbrain [27]. It projects to the caudal part of the fastigial nucleus, a deep cerebellar nucleus that also receives input from the super colliculus and FEF [26]. This region of the fastigial nucleus, also known as the fastigial oculomotor (FOR), sends projections to omnipause neurons (OPNs), inhibitory burst neurons (IBNs), excitatory burst neurons (EBNs), thalamus, superior colliculus and the reticular formation [28]. FOR modulates saccades by stimulating burst neurons during contralateral saccades and providing inhibition during ipsilateral saccades [29, 30].
The midline pons contains OPNs in the nucleus raphae interpositus [28, 31, 32]. Commands for saccades converge on these OPNs. The superior colliculus directly signals to the OPNs [33–35]. Frontal areas also project indirectly via the caudate nucleus and SNpr [36]. This pathway projects to the cerebellum prior to innervating the OPNs. OPNs are tonically active glycinergic neurons, which produce tonic inhibition of horizontal and vertical saccade burst generators [37, 38]. Suppression of OPNs initiates saccades, whereas electrical stimulation of these neurons stops saccades [39, 40].
EBNs located in several different brain structures have been described. These burst neurons are silent at all times, except during saccades [41]. In the paramedian pontine reticular formation, EBNs relay monosynaptically to oculomotor neurons for horizontal saccadic eye movements [38, 41, 42]. Vertical saccades are generated by EBNs in the rostral interstitial nucleus of the Medial Longitudinal Fasciculus, which is located in the prerubral fields of the dorsal midbrain. The firing rate of EBNs determines the saccadic eye velocity.
Dysfunction in the generation of saccades can be an important clue in determining the site of particular neurological lesions in such multisystem disorders. While pathology involving the eye itself or ocular muscles typically affects the amplitude, velocity and timing of saccades, the central pathology may selectively spare one or more parameters. Thus, understanding the kinematic properties of eye movements can localize lesions found in several neurodegenerative disorders, as well as cerebellar disorders. The readers are referred to previously published resources for the overview and practical guide for examination of the saccades [43, 44].
Disorders of cerebellum affecting saccade velocity
Spinocerebellar Ataxia type 1
Spinocerebellar ataxia type 1 is an autosomal dominant disorder that initially presents with ataxia, but subsequently affects bulbar function including speech and swallowing [45, 46]. Spasticity is also a frequently seen deficits in the spinocerebellar ataxia type 1. The saccade slowing is one of the characteristic features of spinocerebellar ataxia type 1. Although common, unlike spinocerebellar ataxia type 2 (see below), the saccade slowing is not always present in spinocerebellar ataxia type 1 [45]. In addition to slowing of saccades, dysmetria and increased laency is seen in spinocerebellar ataxia type 1 [45]. Since cerebellar degeneration along with brainstem involvement is a possible structural correlate of spinocerebellar ataxia type 1, it is believed that saccade slowing in spinocerebellar ataxia type 1 could be due to involvement of the brainstem burst generators.
Spinocerebellar Ataxia type 2
Spinocerebellar ataxia type 2 is an autosomal dominant disorder due to an unstable expansion of a polyglutamine domain within ataxin-2 [47] which is a cytoplasmic protein found in many body tissues and neurons [48]. The disorder is characterized clinically by progressive cerebellar ataxia, dysarthria, action tremor, early neuropathy, and slowing of saccades [49, 50] . Slowing of saccade velocity strongly correlates with the polyglutamine expansion, but it is inversely correlated with the severity of ataxia [51]. There is no correlation between saccade slowing and the duration of the disease, age of onset or the gender of the diseased subject [51]. Slowing of saccades can be present even before manifestation of other clinical features of the disease [52]. Therefore, saccade velocity is a sensitive and specific marker of disease activity [53] and can be used not only as a surrogate marker of disease severity [54] but also as an early marker for SCA2 [52]. Progression of slow saccades leads to complete gaze palsy in horizontal and vertical directions [55]. Quantitative brain MRI demonstrated reduction in the cerebellar, pons, midbrain, and frontal lobe volumes in SCA2 patients [56]. It is therefore possible that saccade slowing in SCA2 is not primarily a result of cerebellar dysfunction, but it is due to co-existing brainstem abnormality putatively affecting the brainstem burst generation [56, 57]. Indeed, a significant cell loss and reduced synaptic density on somata was found in the mesencephalic area that houses the EBN, which leads to adequate intensity of saccade burst [58].
Spinocerebellar Ataxia type 3
Spinocerebellar ataxia type 3, also known as Machado-Joseph disease is an autosomal dominant disorder occurring due to CAG triplet repeat expansion [48, 59–61]. It presents with progressive gait, stance, limb, and truncal ataxia, dysarthria, somatosensory deficits, dystonia and occasionally parkinsonism, dysphagia, and a varying degree of eye movement disorders [62–64]. SCA3 is clinically characterized by a higher frequency of blepharospasm, ophthalmoparesis, nystagmus, increased appendicular tone, and sensory and urinary disturbances [50, 65]. Eye movement abnormalities such as impaired smooth pursuit, optokinetic nystagmus, gaze evoked nystagmus, saccadic dysmetria and horizontal vestibulo-ocular reflex dysfunction are well known deficits of spinocerebellar ataxia type 3 [66–71]. Limitation of gaze in the vertical direction is seen which is more common in the upward direction [72]. Abduction ophthalmoplegia is one of the most common abnormalities in SCA3, however; adduction tends to be persevered [72]. The saccades in some SCA3 patients may have dynamic overshoot of the target, however, those without dynamic overshoot have saccades with low peak velocity [73]. In comparison to SCA2, the inferior olive in SCA3 is rarely affected. Diffuse atrophic changes in the nervous system are observed on MRI with marked changes in the cerebellar vermis, superior cerebellar peduncle, pontine tegmentum, and frontal lobes [74, 75]. Histopathological studies have shown a degeneration of reticulotegmental nucleus of the pons [76] and the omnipause neurons of nucleus raphe interpositus [77]. Degenerative changes in mesencephalic neurons may be responsible for the reduced saccade velocity due to deficient burst generation [77, 78].
Wernicke’s encephalopathy
Wernicke’s encephalopathy is caused by a nutritional deficiency of thiamine and is commonly seen in alcoholics. The classic triad of Wernicke’s encephalopathy is ophthalmoplegia, mental confusion, and gait ataxia [79]. Early ocular motor findings in Wernicke’s encephalopathy include gaze-evoked and upbeat nystagmus that may switch to downbeat nystagmus with convergence [80, 81]. Horizontal vestibulo-ocular response impairment occurs early, which progresses to abduction impairment, inter-nuclear ophthalmoplegia, horizontal and vertical gaze palsies, and eventually complete ophthalmoplegia [82–84]. Slowing of saccades is rarely described in Wernicke’s encephalopathy [85]. Wernicke’s disease can progress to Korsakoff’s syndrome, which manifests with a severe memory loss and psychiatric symptoms. Significant eye movement abnormalities include hypometria, slow and inaccurate saccades and impaired smooth pursuit [86, 87]. Patients with Korsakoff’s syndrome also make more directional errors on the antisaccade task [88]. Wernicke’s encephalopathy is not predominantly cerebellar, but like other neurodegenerative conditions also affects extracerebellar brainstem regions including those responsible for the saccade burst generation [81, 89]. It is therefore likely that the slow saccades that are rarely seen in Wernicke’s encephalopathy are a manifestation of impaired saccade burst generation. In atypical forms, Wernicke’s encephalogpathy may affect the substantia nigra [90]. In such instances, impaired saccades could be due to lack of tectal inhibition that is normally provided by the substantia nigra pars reticulata. In such cases, in addition to slow saccades, parkinsonism is also expected.
Syndrome of anti-GAD antibody
Glutamic acid decarboxylase (GAD) is an enzyme important in the nervous system for catalyzing the conversion of glutamic acid to γ aminobutyric acid (GABA) – an inhibitory neurotransmitter [91]. Autoantibodies directed against GAD (anti GAD-Ab) have been described in patients with insulin-dependent diabetes mellitus, stiff-person syndrome, epilepsy and in a few patients with late-onset cerebellar ataxia [92–95]. Increased muscle tone, episodic spasms, and cerebellar ataxia are known clinical manifestations [93, 96–99]. Eye movement abnormalities in patients with the syndrome of anti-GAD antibody include downbeat nystagmus, slow vertical saccades, prolonged saccade latency, loss of downward smooth pursuit, saccadic hypometria and dysmetria, impaired ocular pursuit, saccadic oscillations, and impaired cancellation of vestibulo-ocular reflex [100–103]. Case reports of periodic alternating nystagmus [104], and opsoclonus myoclonus [105] have also been described. A study in stiff-person syndrome with cerebellar degeneration described rare saccade abnormalities in the vertical direction. A downward gaze showed multiple hypometric saccades with a normal velocity profile, whereas with upward gaze there was a single saccade with abrupt slowing [102]. We also found saccade slowing in addition to opsoclonus in a patient with syndrome of anti-GAD antibody [103]. Saccade abnormalities in the patient with increased titer of anti-GAD antibody can be multifactorial. For example, frequent interruptions of ongoing saccades, but normal velocity of each “broken” saccade segment suggests impaired programming leading to frequent hypometria, a classic cerebellar phenomenology [106]. Slowing of saccades, however, could suggest involvement of burst generators. An alternate explanation for slow saccades (along with saccadic oscillations) was proposed [103] . Efficient generation of saccades requires not only robust increase in firing rate of the EBNs, latter relies on post-inhibitory rebound [107] . Excessive increase in the excitability due to glutaminergic state (due to immune mediated destruction of the gamma aminobuteric acid (GAD) and decreased degradation of glutamate to GABA) results in baseline increase in the burst neuron excitability. The latter results in an unstable reciprocally innervating circuit of burst neurons, causing opsoclonus, and results in decreased saccade velocity due to lack of the effect of post-inhibitory rebound on hyperexcitable burst neurons. Such physiology, combined, could result in slow saccades and superimposed opsoclonus [108].
Can we differentiate various forms of cerebellar disorders based on slow saccades?
While slow-saccades (or their absence) are instrumental, when present, to point out genetic disorders such as SCA 1,2,3, or 6; immune disorders such as syndrome of antiGAD antibody, or infectious disease such as Whipple’s disease; just by itself, the slow-saccades are not able to separate these disorders. Examination of other eye movements, and other co-existing movement disorders are critical in separating one phenomenology from the other even in their classic presentation.
Determinants of slow eye velocity in cerebellar disorders
High velocities of saccades depend on an abrupt increase in the excitability of burst neurons due to prompt cessation of the inhibition (the post inhibitory rebound [109]). The sustained inhibition of the excitatory burst neurons is achieved by sustained inhibition of the omnipause neurons, cessation of such inhibition (when saccade is desired) leads to an abrupt increase in excitatory burst neuron firing and rapid velocity of saccades [107–110]. Malfunction of omnipause neurons can cause saccade slowing, but it typically affects vertical and horizontal directions. In contrast, abnormal increases in excitatory burst neuron activity (as expected in syndrome of increased titers of anti-GAD antibody) also reduces the efficacy of omnipause inhibition – hence a slow saccade [103]. Impaired excitability of excitatory burst neurons can lead to deficient burst generation, resulting in slow saccade velocity. Such deficits can be expected in patients with SCA2, SCA3, and Wernicke’s encephalopathy. Maintanence of the saccadic efficiency requires minimizing movement accuracy and learning from the end point error; a key function of the cerebellum. Therefore, disruption of cerebellar function may affect kinematics of saccades including matrix and velocity [57, 111].
It is possible that slowing of saccades in chronic cerebellar conditions has a multifactorial etiology. The structural etiology of slowing, degenerative loss of saccade burst neurons, is one possibility for inefficient velocity command generation. The principles of neuroeconomics determine the basic underpinnings of the second explanation of slow saccade. Disorders of cerebellum, in the presence of uncertainty of destination gaze stability (increased endpoint variability), challenges the brain to optimize saccade accuracy. The optimization takes place in the form of sacrificing the rapidity of movements, hence the slow saccades. Goal directed movements, such as saccades, can occur in infinite possible trajectories onset location to the destination. However, the brain optimizes the trajectory position and speed of voluntary movements (here saccades) to minimize the time-accuracy tradeoff [112] . In other words, the dynamics of eye velocity profiles are determined to make the fastest yet most accurate eye movement and minimal transit time [113]. It was proposed that minimizing the variance of the eye position in the presence of biological and constant noise is the key determinant of trajectory planning of the movement. Minimizing the variability in the eye position in the presence of biological noise is a key determinant of saccade trajectory planning. The noise in the final common pathway that determines the activity of motor neurons leads to deviation of trajectories from the desired path. The deviations are accumulated over the duration of the movement leading to variability in desired position. The noise is independent of the control signal that originally generates the movement [114], however the accumulated error is rapidly minimized by making rapid movement. Rapid movement however requires larger control signals, hence increased variability in the final position. As a consequence the inaccurate movement leads to dysmetria requiring corrective movements [115]. One answer to assure accuracy is low control signals, which leads to slow movements. Thus signal dependent noise imposes trade-off between the movement speed, duration, and the accuracy. It was proposed that the temporal profile of neural command is designed to determine minimal variability in desired position; the velocity of saccade is therefore adjusted accordingly. The end-point variability is inherently higher in patients with cerebellar disorders due to variety of co-existing deficits such as dysmetria, nystagmus, and saccadic intrusions affecting the final gaze position. In the presence of increased endpoint variability, to achieve the best possible endpoint accuracy, the brain compromises on saccade velocity. As a result, the velocity of visually guided saccade is reduced in disorders of cerebellum that frequently causes chronic gaze holding disorders.
Multisystem neurodegenerative disorders affecting visually guided saccade velocity
Slowing of saccades can be seen in various conditions that are not prominently cerebellar. For example, visually guided saccades in Parkinson’s disease (PD) patients may have increased prevalence of hypometria compared to healthy controls [116]. Patients with asymmetric PD demonstrated asymmetric hypometria of visually guided saccades and had increased hypometria on the more symptomatic side [117]. This hypometria does not necessarily result in reduced velocity in saccades, but rather increased the time to reach the target by creating a “staircase” trajectory of the saccade in which the gaze is corrected toward the target [118, 119]. Patients with PD have higher variability of peak velocity of visually guided saccades, but saccade slowing is seen only in advanced PD [120].
In contrast, slowing of visually guided saccades in either the horizontal or vertical direction may be an important early finding in several of the atypical parkinsonian syndromes. Progressive supranuclear palsy (PSP), a tauopathy that features postural instability, supranuclear gaze and bulbar palsies and axial rigidity, causes saccadic slowing and hypometria (Fig. 2) [121–123]. This change in saccades is more prominent in the vertical axis and with disease progression may result in complete vertical gaze palsy in which vertical saccades are lost [124]. Horizontal saccades are often hypometric early in the course of the disease, and become slower as the disease progresses [125]. Saccades in PSP are not only slow but they also have irregular trajectory [121]. PSP can be distinguished from PD and atypical parkinsonism by a characteristic change in oblique saccades, which have prominent curvature and a relatively faster horizontal component [120].
Fig. 2.

The visually guided saccades from healthy subject and PSP patient are compared. Panel a illustrates normal visually guided vertical saccade from a healthy subject. Panels d,g depict two examples of visually guided vertical saccades from the same PSP subject. Panel j depicts eye positions during horizontal saccade. In panels (a,d,g,j) the eye position is plotted on the y-axis while x-axis depicts corresponding time in seconds. Black line represents the vertical eye position, while green traces depict horizontal eye position. Grey dashed line is the baseline, i.e. the straight-ahead position, while blue dashed line depicts the desired position. The arrows depict interruption in the saccades, in one type of interruption (blue arrow) the eyes continue to move at slower velocity during interruption, while in other type (green arrow) the slower eye movement in the opposite direction. The third type of interruption (red arrow) is where the eye movements completely stop during the interruption. Panels b,e, h,k depict eye velocity. Panel B depicts eye velocity of normal visually guided saccade recorded from the healthy subject, while panels (e,h) depict vertical eye velocity during vertical saccade in PSP. Green line in panel K illustrates normal horizontal eye velocity during horizontal saccade in PSP. In these subplots the eye velocity is plotted on y-axis while x-axis illustrates corresponding time. Red arrow illustrates interruption in saccade when eye velocity was zero, green arrow is when eye moved at slower velocity in the opposite direction, blue arrow is when eyes moved in the same direction at slower velocity. Panels c,f,i,l depict trajectories of horizontal and vertical saccades. Panel C shows the normal saccade from the healthy subject, while panel (f,i) are vertical saccades in PSP, and panel l is the horizontal saccade in PSP. In these plots the green dot depicts the start point, while the red dot is the stop point; grey dashed line is the desired path of the eye movement. Vertical saccades have curved and serpentine path depicting the irregularity in the trajectory. Such curvature is also present in the horizontal saccade but to much lesser extent (adapted from Shaikh et al. [121])
Neimann-Pick type C disease is another example featuring vertical supraneuclear gaze palsy. This disorder, typically caused by mutations in the NPC1 or NPC2 gene, can lead to paralysis of vertical saccades. It is believed that slowing of vertical saccades in Neimann-Pick type C disease is due to selective involvement of the burst generators at the rostral insterstitial nucleus of Cajal [126].
Slowing of saccades in the vertical and horizontal planes may also be seen in dementia with Lewy bodies [127], cortico-basal degeneration [120, 128] and Huntington’s disease [129–131]. Other changes in saccade parameters and eye movements can also be used to distinguish between these syndromes and may be more characteristic of specific diseases.
The slow saccades are not pathognomonic of mesencephalic disorders, they can be seen in focal, degenerative, or systemic/immune deficits affecting the cerebellum. Cerebellar influence on saccade velocity could be direct, but can be due to altered cerebellar modulation of saccade burst generators or co-existing brainstem involvement in the disorders that primarily affects the cerebellum. Although slowing of saccades provides critical information about the pathophysiology of underlying disease processes, the slowing by itself is not sufficient for diagnostic classification. The latter requires collateral information such as coexisting neurological signs and deficits.
Conclusion
The classic ocular motor markers of the cerebellar disorder include impairment in amplitude (dysmetria), gaze-holding deficit (nystagmus), and deficit in ocular pursuit. Reduced saccade velocity is considered a usual manifestation of midbrain or multisystem impairment in neurodegenerative disorders such as progressive supranuclear palsy or spinocerebellar ataxia type 2. Decreased velocities of vertical saccades is not rare in cerebellar disorders, it is frequent phenomenology seen in multisystem involvement that substantially involves the cerebellum.
Acknowledgements
AS was supported by grants from Dystonia Medical Research Foundation (DMRF), Dystonia Coalition, American Academy of Neurology, and American Parkinson’s Disease Association.
Funding
AS was supported by grants from Dystonia Medical Research Foundation (DMRF), Dystonia Coalition, and The American Academy of Neurology (AAN).
Availability of data and materials
This is a review paper, hence not applicable.
Abbreviations
- Ab
Antibody
- dlPFC
Dorsolateral Prefrontal Cortex
- EBN
Excitatory Burst Neuron
- FEF
Frontal Eye Field
- FOR
Fastigial Ocular Motor Region
- GABA
Gamma Aminobuteric Acid
- GAD
Glutamic Acid Decarboxylase
- IBN
Inhibitory Burst Neuron
- NRTP
Nucleus Reticularits Tegmenti Pontis
- OPN
Omnipause Neurons
- PD
Parkinson’s Disease
- PEF
Parietal Eye Field
- PSP
Progressive Supranuclear Palsy
- SCA
Spinocerebellar Ataxia
- SEF
Supplementary Eye Field
- SNpr
Substantia Nigra Pars REticulata
Authors’ contributions
KJ Contributed to conceptualization, writing and editing of the manuscript. SB: Contributed to conceptualization, writing and editing of the manuscript. AS: Contributed to conceptualization, writing and editing of the manuscript.
Acknowledgements: Authors thank DMRF, Dystonia Coalition, and AAN for their support.
Authors’ information
Kelsey Jensen (kmj53@case.edu), Sinem Balta Beylergil (sxb818@case.edu), Aasef G. Shaikh (aasefshaikh@gmail.com). Neurological Institute, University Hospitals, Cleveland, OH. Department of Neurology, Case Western Reserve University, Cleveland, OH. Neurology Service, Louis Stokes Cleveland VA Medical Center, Cleveland, OH.
Ethics approval and consent to participate
Not applicable - this is a review paper.
Consent for publication
This is a review paper, hence not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kelsey Jensen, Email: kmj53@case.edu.
Sinem Balta Beylergil, Email: sxb818@case.edu.
Aasef G. Shaikh, Phone: 216-844-1000, Email: aasefshaikh@gmail.com
References
- 1.Munoz DP. Progress in Brain Research. 2002. Commentary: saccadic eye movements: overview of neural circuitry; pp. 89–96. [DOI] [PubMed] [Google Scholar]
- 2.Pierrot-Deseilligny C, Milea D, Müri RM. Eye movement control by the cerebral cortex. Curr Opin Neurol. 2004;17(1):17–25. doi: 10.1097/00019052-200402000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Fox PT, Fox JM, Raichle ME, Burde RM. The role of cerebral cortex in the generation of voluntary saccades: a positron emission tomographic study. J Neurophysiol. 1985;54(2):348–369. doi: 10.1152/jn.1985.54.2.348. [DOI] [PubMed] [Google Scholar]
- 4.Pierrot-deseilligny CH, Rivaud S, Gaymard B, Agid Y. Cortical control of reflexive visually-guided saccades. Brain. 1991;114(3):1473–1485. doi: 10.1093/brain/114.3.1473. [DOI] [PubMed] [Google Scholar]
- 5.Berman RA, Colby CL, Genovese CR, Voyvodic JT, Luna B, Thulborn KR, et al. Cortical networks subserving pursuit and saccadic eye movements in humans: an FMRI study. Hum Brain Mapp. 1999;8(4):209–225. doi: 10.1002/(SICI)1097-0193(1999)8:4<209::AID-HBM5>3.0.CO;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hikosaka O, Takikawa Y, Kawagoe R. Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol Rev. 2000;80:953–978. doi: 10.1152/physrev.2000.80.3.953. [DOI] [PubMed] [Google Scholar]
- 7.Nambu A, Tokuno H, Takada M. Functional significance of the cortico-subthalamo-pallidal “hyperdirect” pathway. Neurosci Res. 2002;43:111–117. doi: 10.1016/S0168-0102(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 8.Fisher RS, Buchwald NA, Hull CD, Levine MS. The GABAergic striatonigral neurons of the cat: demonstration by double peroxidase labeling. Brain Res. 1986;398(1):148–156. doi: 10.1016/0006-8993(86)91260-6. [DOI] [PubMed] [Google Scholar]
- 9.Francois C, Percheron G, Yelnik J. Localization of nigrostriatal, nigrothalamic and nigrotectal neurons in ventricular coordinates in macaques. Neuroscience. 1984;13(1):61–76. doi: 10.1016/0306-4522(84)90259-8. [DOI] [PubMed] [Google Scholar]
- 10.Handel ARI, Glimcher PW. Quantitative analysis of substantia nigra pars reticulata activity during a visually guided saccade task. J Neurophysiol. 1999;82(6):3458–3475. doi: 10.1152/jn.1999.82.6.3458. [DOI] [PubMed] [Google Scholar]
- 11.Hikosaka O, Wurtz RH. Visual and oculomotor functions of monkey substantia nigra pars reticulata. IV. Relation of substantia nigra to superior colliculus. J Neurophysiol. 1983;49(5):1285–1301. doi: 10.1152/jn.1983.49.5.1285. [DOI] [PubMed] [Google Scholar]
- 12.Pouget P. The cortex is in overall control of “voluntary” eye movement. Eye (Lond) 2015;29:241–245. doi: 10.1038/eye.2014.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanes DP, Wurtz RH. Interaction of the frontal eye field and superior colliculus for saccade generation. J Neurophysiol. 2001;85(2):804–815. doi: 10.1152/jn.2001.85.2.804. [DOI] [PubMed] [Google Scholar]
- 14.Baloh RW, Sills AW, Kumley WE, Honrubia V. Quantitative measurement of saccade amplitude, duration, and velocity. Neurology. 1975;25:1065–1070. doi: 10.1212/WNL.25.11.1065. [DOI] [PubMed] [Google Scholar]
- 15.Leigh RJ, Kennard C. Using saccades as a research tool in the clinical neurosciences. Brain. 2004;127:460–477. doi: 10.1093/brain/awh035. [DOI] [PubMed] [Google Scholar]
- 16.Moschovakis AK, Scudder CA, Highstein SM. The microscopic anatomy and physiology of the mammalian saccadic system. Prog Neurobiol. 1996;50:133–254. doi: 10.1016/S0301-0082(96)00034-2. [DOI] [PubMed] [Google Scholar]
- 17.May PJ. The mammalian superior colliculus: Laminar structure and connections. Prog Brain Res. 2005;151:321–378. doi: 10.1016/S0079-6123(05)51011-2. [DOI] [PubMed] [Google Scholar]
- 18.Illing RB, Graybiel AM. Convergence of afferents from frontal cortex and substantia nigra onto acetylcholinesterase-rich patches of the cat’s superior colliculus. Neuroscience. 1985;14(2):455–482. doi: 10.1016/0306-4522(85)90303-3. [DOI] [PubMed] [Google Scholar]
- 19.Sparks DL, Hartwich-Young R. The deep layers of the superior colliculus. Rev Oculomotor Res. 1989;3:213–255. [PubMed] [Google Scholar]
- 20.Schiller PH, Stryker M. Single-unit recording and stimulation in superior colliculus of the alert rhesus monkey. J Neurophysiol. 1972;35(6):915–924. doi: 10.1152/jn.1972.35.6.915. [DOI] [PubMed] [Google Scholar]
- 21.Sparks DL. Functional properties of neurons in the monkey superior colliculus: coupling of neuronal activity and saccade onset. Brain Res. 1978;156(1):1–16. doi: 10.1016/0006-8993(78)90075-6. [DOI] [PubMed] [Google Scholar]
- 22.Büttner U, Büttner-Ennever JA. Present concepts of oculomotor organization. Prog Brain Res. 2005;151:1–42. doi: 10.1016/S0079-6123(05)51001-X. [DOI] [PubMed] [Google Scholar]
- 23.Yamada J, Noda H. Afferent and efferent connections of the oculomotor cerebellar vermis in the macaque monkey. J Comp Neurol. 1987;265(2):224–241. doi: 10.1002/cne.902650207. [DOI] [PubMed] [Google Scholar]
- 24.Thielert CD, Thier P. Patterns of projections from the pontine nuclei and the nucleus reticularis tegmenti pontis to the posterior vermis in the rhesus monkey: a study using retrograde tracers. J Comp Neurol. 1993;337(1):113–126. doi: 10.1002/cne.903370108. [DOI] [PubMed] [Google Scholar]
- 25.Dicke PW, Barash S, Ilg UJ, Thier P. Single-neuron evidence for a contribution of the dorsal pontine nuclei to both types of target-directed eye movements, saccades and smooth-pursuit. Eur J Neurosci. 2004;19(3):609–624. doi: 10.1111/j.0953-816X.2003.03137.x. [DOI] [PubMed] [Google Scholar]
- 26.Noda H, Sugita S, Ikeda Y. Afferent and efferent connections of the oculomotor region of the fastigial nucleus in the macaque monkey. J Comp Neurol. 1990;302(2):330–348. doi: 10.1002/cne.903020211. [DOI] [PubMed] [Google Scholar]
- 27.Ohtsuka K, Noda H. Discharge properties of Purkinje cells in the oculomotor vermis during visually guided saccades in the macaque monkey. J Neurophysiol. 1995;74(5):1828–1840. doi: 10.1152/jn.1995.74.5.1828. [DOI] [PubMed] [Google Scholar]
- 28.May PJ, Hartwich-Young R, Nelson J, Sparks DL, Porter JD. Cerebellotectal pathways in the macaque: implications for collicular generation of saccades. Neuroscience. 1990;36(2):305–324. doi: 10.1016/0306-4522(90)90428-7. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs AF, Robinson FR, Straube A. Role of the caudal fastigial nucleus in saccade generation. I. Neuronal discharge pattern. J Neurophysiol. 1993;70(5):1723–1740. doi: 10.1152/jn.1993.70.5.1723. [DOI] [PubMed] [Google Scholar]
- 30.Kleine JF, Guan Y, Buttner U. Saccade-related neurons in the primate fastigial nucleus: what do they encode? J Neurophysiol. 2003;90(5):3137–3154. doi: 10.1152/jn.00021.2003. [DOI] [PubMed] [Google Scholar]
- 31.Büttner-Ennever JA, Cohen B, Pause M, Fries W. Raphe nucleus of the pons containing omnipause neurons of the oculomotor system in the monkey, and its homologue in man. J Comp Neurol. 1988;267(3):307–321. doi: 10.1002/cne.902670302. [DOI] [PubMed] [Google Scholar]
- 32.Büttner-Ennever JA. Mapping the oculomotor system. Prog Brain Res. 2008;171:3–11. doi: 10.1016/S0079-6123(08)00601-8. [DOI] [PubMed] [Google Scholar]
- 33.Stanton GB, Deng SY, Goldberg EM, McMullen NT. Cytoarchitectural characteristic of the frontal eye fields in macaque monkeys. J Comp Neurol. 1989;282(3):415–427. doi: 10.1002/cne.902820308. [DOI] [PubMed] [Google Scholar]
- 34.Shook BL, Schlag-Rey M, Schlag J. Primate supplementary eye field: I. comparative aspects of mesencephalic and pontine connections. J Comp Neurol. 1990;301(4):618–642. doi: 10.1002/cne.903010410. [DOI] [PubMed] [Google Scholar]
- 35.Shook BL, Schlag-Rey M, Schlag J. Primate supplementary eye field. II. Comparative aspects of connections with the thalamus, corpus striatum, and related forebrain nuclei. J Comp Neurol. 1991;307(4):562–583. doi: 10.1002/cne.903070405. [DOI] [PubMed] [Google Scholar]
- 36.Hikosaka O. GABAergic output of the basal ganglia. Prog Brain Res. 2007;160:209–226. doi: 10.1016/S0079-6123(06)60012-5. [DOI] [PubMed] [Google Scholar]
- 37.Kaneko CRS, Fuchs AF. Connections of cat omnipause neurons. Brain Res. 1982;241(1):166–170. doi: 10.1016/0006-8993(82)91240-9. [DOI] [PubMed] [Google Scholar]
- 38.Horn AK, Büttner-Ennever JA, Wahle P, Reichenberger I. Neurotransmitter profile of saccadic omnipause neurons in nucleus raphe interpositus. J Neurosci. 1994;14(4):2032–2046. doi: 10.1523/JNEUROSCI.14-04-02032.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evinger C, Kaneko CR, Fuchs AF. Activity of omnipause neurons in alert cats during saccadic eye movements and visual stimuli. J Neurophysiol. 1982;47(5):827–844. doi: 10.1152/jn.1982.47.5.827. [DOI] [PubMed] [Google Scholar]
- 40.Keller EL, Gandhi NJ, Vijay SS. Activity in deep intermediate layer collicular neurons during interrupted saccades. Exp Brain Res. 2000;130(2):227–237. doi: 10.1007/s002219900239. [DOI] [PubMed] [Google Scholar]
- 41.AKE H, Büttner-Ennever JA, Büttner U. Saccadic premotor neurons in the brainstem: Functional neuroanatomy and clinical implications. Neuro Ophthalmol. 1996;16:229–240. [Google Scholar]
- 42.Van Gisbergen JAM, Robinson DA, Gielen S. A quantitative analysis of generation of saccadic eye movements by burst neurons. J Neurophysiol. 1981;45(3):417–442. doi: 10.1152/jn.1981.45.3.417. [DOI] [PubMed] [Google Scholar]
- 43.Leigh RJ, Zee DS. The neurology of eye movements. New York: Oxford; 2015. [Google Scholar]
- 44.Termsarasab P, Thammongkolchai T, Rucker J, Frucht S. The diagnostic value of saccades in movement disorder patients: a practical guide and review. J Clin Mov Disord. 2015;2:14. doi: 10.1186/s40734-015-0025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buttner N, Geschwind D, Jen JC, Perlman S, Pulst SM, Baloh RW. Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol. 1998;55(10):1353–1357. doi: 10.1001/archneur.55.10.1353. [DOI] [PubMed] [Google Scholar]
- 46.Burk K, Fetter M, Abele M, Laccone F, Brice A, Dichgans J, et al. Autosomal dominant cerebellar ataxia type I: oculomotor abnormalities in families with SCA1, SCA2, and SCA3. J Neurol. 1999;246:789–797. doi: 10.1007/s004150050456. [DOI] [PubMed] [Google Scholar]
- 47.Pulst S-M, Nechiporuk A, Nechiporuk T, Gispert S, Chen X-N, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14(3):269–276. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 48.Diallo A, Jacobi H, Cook A, Labrum R, Tezenas du Montcel S. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol. 2018;17(4):327–334. doi: 10.1016/S1474-4422(18)30042-5. [DOI] [PubMed] [Google Scholar]
- 49.Wadia N. A clinicogenetic analysis of six Indian spinocerebellar ataxia (SCA2) pedigrees. The significance of slow saccades in diagnosis. Brain. 1998;121(12):2341–2355. doi: 10.1093/brain/121.12.2341. [DOI] [PubMed] [Google Scholar]
- 50.Wadia NH, Swami RK. A new form of heredo-familial spinocerebellar degeneration with slow eye movements (nine families) Brain. 1971;94(2):359–374. doi: 10.1093/brain/94.2.359. [DOI] [PubMed] [Google Scholar]
- 51.Velázquez-Pérez L, Seifried C, Santos-Falcón N, Abele M, Ziemann U, Almaguer LE, et al. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol. 2004;56(3):444–447. doi: 10.1002/ana.20220. [DOI] [PubMed] [Google Scholar]
- 52.Velázquez-Pérez L, Seifried C, Abele M, Wirjatijasa F, Rodríguez-Labrada R, Santos-Falcón N, et al. Saccade velocity is reduced in presymptomatic spinocerebellar ataxia type 2. Clin Neurophysiol. 2009;120(3):632–635. doi: 10.1016/j.clinph.2008.12.040. [DOI] [PubMed] [Google Scholar]
- 53.Rodríguez-Labrada R, Velázquez-Pérez L, Auburger G, Ziemann U, Canales-Ochoa N, Medrano-Montero J, et al. Spinocerebellar ataxia type 2: measures of saccade changes improve power for clinical trials. Mov Disord. 2016;31(4):570–578. doi: 10.1002/mds.26532. [DOI] [PubMed] [Google Scholar]
- 54.Seifried C, Velázquez-Pérez L, Santos-Falcón N, Abele M, Ziemann U, Almaguer LE, et al. Saccade velocity as a surrogate disease marker in spinocerebellar ataxia type 2. In: Annals of the New York Academy of Sciences. Hoboken: Wiley-Blackwell; 2005. p. 524–527. [DOI] [PubMed]
- 55.Klostermann W, Zühlke C, Heide W, Kömpf D, Wessel K. Slow saccades and other eye movement disorders in spinocerebellar atrophy type 1. J Neurol. 1997;244(2):105–111. doi: 10.1007/s004150050058. [DOI] [PubMed] [Google Scholar]
- 56.Politi LS, Bianchi Marzoli S, Godi C, Panzeri M, Ciasca P, Brugnara G, et al. MRI evidence of cerebellar and extraocular muscle atrophy differently contributing to eye movement abnormalities in SCA2 and SCA28 diseases. Investig Ophthalmol Vis Sci. 2016;57(6):2714–2720. doi: 10.1167/iovs.15-18732. [DOI] [PubMed] [Google Scholar]
- 57.Rufa A, Federighi P. Fast versus slow: different saccadic behavior in cerebellar ataxias. Ann N Y Acad Sci. 2011;1233(1):148–154. doi: 10.1111/j.1749-6632.2011.06126.x. [DOI] [PubMed] [Google Scholar]
- 58.Geiner S, AKE H, Wadia NH, Sakai H, Büttner-Ennever JA. The neuroanatomical basis of slow saccades in spinocerebellar ataxia type 2 (Wadia-subtype) Prog Brain Res. 2008;171:575–581. doi: 10.1016/S0079-6123(08)00683-3. [DOI] [PubMed] [Google Scholar]
- 59.Haberhausen G, Damian MS, Leweke F, Müller U. Spinocerebellar ataxia, type 3 (SCA3) is genetically identical to Machado-Joseph disease (MJD) J Neurol Sci. 1995;132(1):71–75. doi: 10.1016/0022-510X(95)90927-I. [DOI] [PubMed] [Google Scholar]
- 60.Matilla T, McCall A, Subramony SH, Zoghbi HY. Molecular and clinical correlations in spinocerebellar ataxia type 3 and Machado-Joseph disease. Ann Neurol. 1995;38:68–72. doi: 10.1002/ana.410380113. [DOI] [PubMed] [Google Scholar]
- 61.Ranum LP, Lundgren JK, Schut LJ, Ahrens MJ, Perlman S, Aita J, et al. Spinocerebellar ataxia type 1 and Machado-Joseph disease: incidence of CAG expansions among adult-onset ataxia patients from 311 families with dominant, recessive, or sporadic ataxia. Am J Hum Genet. 1995;57(3):603–608. [PMC free article] [PubMed] [Google Scholar]
- 62.Twist EC, Casaubon LK, Ruttledge MH, Rao VS, Macleod PM, Radvany J, et al. Machado Joseph disease maps to the same region of chromosome 14 as the spinocerebellar ataxia type 3 locus. J Med Genet. 1995;32(1):25–31. doi: 10.1136/jmg.32.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maruyama H, Kawakami H, Kohriyama T, Sakai T, Doyu M, Sobue G, et al. CAG repeat length and disease duration in Machado-Joseph disease: a new clinical classification. J Neurol Sci. 1997;152(2):166–171. doi: 10.1016/S0022-510X(97)00155-X. [DOI] [PubMed] [Google Scholar]
- 64.Riess O, Rüb U, Pastore A, Bauer P, Schöls L. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7:125–137. doi: 10.1007/s12311-008-0013-4. [DOI] [PubMed] [Google Scholar]
- 65.Watanabe M, Abe K, Aoki M, Kameya T, Kaneko J, Shoji M, et al. Analysis of CAG trinucleotide expansion associated with Machado-Joseph disease. J Neurol Sci. 1996;136(1–2):101–107. doi: 10.1016/0022-510X(95)00307-N. [DOI] [PubMed] [Google Scholar]
- 66.Dawson DM, Feudo P, Zubick HH, Rosenberg R, Fowler H. Electro-oculographic findings in Machado-Joseph disease 550. Neurology. 1982;32(0028–3878):1272–1276. doi: 10.1212/WNL.32.11.1272. [DOI] [PubMed] [Google Scholar]
- 67.Hotson JR, Langston EB, Louis AA, Rosenberg RN. The search for a physiologic marker of Machado-Joseph disease 534. Neurology. 1987;37(0028–3878):112–116. doi: 10.1212/WNL.37.1.112. [DOI] [PubMed] [Google Scholar]
- 68.Rivaud-Pechoux S, Dürr A, Gaymard B, Cancel G, Ploner CJ, Agid Y, et al. Eye movement abnormalities correlate with genotype in autosomal dominant cerebellar ataxia type I. Ann Neurol. 1998;43(3):297–302. doi: 10.1002/ana.410430306. [DOI] [PubMed] [Google Scholar]
- 69.Gordon CR, Joffe V, Vainstein G, Gadoth N. Vestibulo-ocular arreflexia in families with spinocerebellar ataxia type 3 (Machado-Joseph disease) J Neurol Neurosurg Psychiatry. 2003;74(10):1403–1406. doi: 10.1136/jnnp.74.10.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gordon CR, Zivotofsky AZ, Caspi A. Impaired vestibulo-ocular reflex (VOR) in spinocerebellar ataxia type 3 (SCA3): Bedside and search coil evaluation. In: Journal of Vestibular Research: Equilibrium and Orientation. 2014. p. 351–5. [DOI] [PubMed]
- 71.Ghasia FF, Wilmot G, Ahmed A, Shaikh AG. Strabismus and micro-Opsoclonus in Machado-Joseph disease. Cerebellum. 2016;15(4):491–497. doi: 10.1007/s12311-015-0718-0. [DOI] [PubMed] [Google Scholar]
- 72.Murofushi T, Mizuno M, Hayashida T, Yamane M, Osanai R, Ito K, et al. Neuro-otological and neuropathological findings in two cases with Machado-joseph disease. Acta Otolaryngol. 1995;115(S520):136–139. doi: 10.3109/00016489509125211. [DOI] [PubMed] [Google Scholar]
- 73.Caspi A, Zivotofsky AZ, Gordon CR. Multiple saccadic abnormalities in spinocerebellar ataxia type 3 can be linked to a single deficiency in velocity feedback. Investig Ophthalmol Vis Sci. 2013;54(1):731–738. doi: 10.1167/iovs.12-10689. [DOI] [PubMed] [Google Scholar]
- 74.Murata Y, Yamaguchi S, Kawakami H, Imon Y, Maruyama H, Sakai T, et al. Characteristic magnetic resonance imaging findings in Machado-Joseph disease. Arch Neurol. 1998;55(1):33–37. doi: 10.1001/archneur.55.1.33. [DOI] [PubMed] [Google Scholar]
- 75.Tokumaru AM, Kamakura K, Maki T, Murayama S, Sakata I, Kaji T, et al. Magnetic resonance imaging findings of Machado-Joseph disease: histopathologic correlation. J Comput Assist Tomogr. 2003;27(2):241–248. doi: 10.1097/00004728-200303000-00023. [DOI] [PubMed] [Google Scholar]
- 76.Rüb U, Bürk K, Schöls L, Brunt ER, De Vos RAI, Orozco Diaz G, et al. Damage to the reticulotegmental nucleus of the pons in spinocerebellar ataxia type 1, 2, and 3. Neurology. 2004;63(7):1258–1263. doi: 10.1212/01.WNL.0000140498.24112.8C. [DOI] [PubMed] [Google Scholar]
- 77.Rüb U, Brunt ER, Gierga K, Schultz C, Paulson H, De Vos RAI, et al. The nucleus raphe interpositus in spinocerebellar ataxia type 3 (Machado-Joseph disease) J Chem Neuroanat. 2003;25(2):115–127. doi: 10.1016/S0891-0618(02)00099-6. [DOI] [PubMed] [Google Scholar]
- 78.Rüb U, Brunt ER, Deller T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease) Curr Opin Neurol. 2008;21:111–116. doi: 10.1097/WCO.0b013e3282f7673d. [DOI] [PubMed] [Google Scholar]
- 79.Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442–455. doi: 10.1016/S1474-4422(07)70104-7. [DOI] [PubMed] [Google Scholar]
- 80.Shin BS, Oh SY, Kim JS, Lee H, Kim EJ, Hwang SB. Upbeat nystagmus changes to downbeat nystagmus with upward gaze in a patient with Wernicke’s encephalopathy. J Neurol Sci. 2010;298(1–2):145–147. doi: 10.1016/j.jns.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 81.Kim K, Shin DH, Lee YB, Park KH, Park HM, Shin DJ, et al. Evolution of abnormal eye movements in Wernicke’s encephalopathy: correlation with serial MRI findings. J Neurol Sci. 2012;323(1–2):77–79. doi: 10.1016/j.jns.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 82.Cogan DG, Victor M. Ocular signs of wernicke’s disease. AMA Arch Ophthalmol. 1954;51(2):204–211. doi: 10.1001/archopht.1954.00920040206007. [DOI] [PubMed] [Google Scholar]
- 83.Cox TA, Corbett JJ, Thompson HS, Lennarson L. Upbeat nystagmus changing to downbeat nystagmus with convergence. Neurology. 1981;31(7):891–892. doi: 10.1212/WNL.31.7.891. [DOI] [PubMed] [Google Scholar]
- 84.DELAPAZ MA, CHUNG SM, JA MCCRARY. BILATERAL INTERNUCLEAR OPHTHALMOPLEGIA IN A PATIENT WITH WERNICKES ENCEPHALOPATHY. JOURNAL OF CLINICAL. Neuro-Ophthalmology. 1992;12(2):116–120. [PubMed] [Google Scholar]
- 85.Hamann KU. Slowed saccades in various neurological disorders. Ophthalmologica. 1979;178(6):357–364. doi: 10.1159/000308847. [DOI] [PubMed] [Google Scholar]
- 86.Kenyon RV, Becker JT, Butters N, Hermann H. Oculomotor function in wernicke-korsakoff’s syndrome: I. saccadic eye movements. Int J Neurosci. 1984;25(1–2):53–65. doi: 10.3109/00207458408985589. [DOI] [PubMed] [Google Scholar]
- 87.Kenyon RV, Becker JT, Butters N. Oculomotor function in wernicke-korsakoff’s syndrome: II. Smooth pursuit eye movements. Int J Neurosci. 1984;25(1–2):67–79. doi: 10.3109/00207458408985590. [DOI] [PubMed] [Google Scholar]
- 88.Van Der Stigchel S, Reichenbach RCL, Wester AJ, Nijboer TCW. Antisaccade performance in Korsakoff patients reveals deficits in oculomotor inhibition. J Clin Exp Neuropsychol. 2012;34(8):876–886. doi: 10.1080/13803395.2012.692771. [DOI] [PubMed] [Google Scholar]
- 89.Halliday GM, Ellis J, Heard R, Caine D, Harper C. Brainstem serotonergic neurons in chronic alcoholics with and without the memory impairment of korsakoff’s psychosis. J Neuropathol Exp Neurol. 1993;52(6):567–579. doi: 10.1097/00005072-199311000-00003. [DOI] [PubMed] [Google Scholar]
- 90.Kalidass B, Sunnathkal R, Rangashamanna V, Paraswani R. Atypical Wernicke’s encephalopathy showing involvement of substantia Nigra. J Neuroimaging. 2012;22(2):204–207. doi: 10.1111/j.1552-6569.2010.00545.x. [DOI] [PubMed] [Google Scholar]
- 91.Watanabe M, Maemura K, Kanbara K, Tamayama T, Hayasaki H. GABA and GABA receptors in the central nervous system and other organs. Int Rev Cytol. 2002;213:1–47. doi: 10.1016/S0074-7696(02)13011-7. [DOI] [PubMed] [Google Scholar]
- 92.Solimena M, Folli F, Denis-Donini S, Comi GC, Pozza G, De Camilli P, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med. 1988;318(16):1012–1020. doi: 10.1056/NEJM198804213181602. [DOI] [PubMed] [Google Scholar]
- 93.Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic Beta cells in stiff-man syndrome. N Engl J Med. 1990;322(22):1555–1560. doi: 10.1056/NEJM199005313222202. [DOI] [PubMed] [Google Scholar]
- 94.Abele M, Weller M, Mescheriakov S, Burk K, Dichgans J, Klockgether T. Cerebellar ataxia with glutamic acid decarboxylase autoantibodies. Neurology. 1999;52(4):857–859. doi: 10.1212/WNL.52.4.857. [DOI] [PubMed] [Google Scholar]
- 95.Vianello M, Tavolato B, Giometto B. Glutamic acid decarboxylase autoantibodies and neurological disorders. Neurol Sci. 2002;23(4):145–151. doi: 10.1007/s100720200055. [DOI] [PubMed] [Google Scholar]
- 96.Levy LM, Dalakas MC, Floeter MK. The stiff-person syndrome: an autoimmune disorder affecting neurotransmission of γ-aminobutyric acid. In: Annals of Internal Medicine. 1999. p. 522–30. [DOI] [PubMed]
- 97.Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology. 2000;55(10):1531–1535. doi: 10.1212/WNL.55.10.1531. [DOI] [PubMed] [Google Scholar]
- 98.Murinson BB. Stiff-person syndrome. Neurol. 2004;10:131–137. doi: 10.1097/01.nrl.0000126587.37087.1a. [DOI] [PubMed] [Google Scholar]
- 99.Oskarsson B, Pelak V, Quan D, Hall D, Foster C, Galetta S. STIFF EYES IN STIFF-PERSON SYNDROME. Neurol. 2008;71(5):378 LP–378380. doi: 10.1212/01.wnl.0000319725.22925.b4. [DOI] [PubMed] [Google Scholar]
- 100.Antonini G, Nemni R, Giubilei F, Gragnani F, Ceschin V, Morino S, et al. Autoantibodies to glutamic acid decarboxylase in downbeat nystagmus. J Neurol Neurosurg Psychiatry. 2003;74(7):998–999. doi: 10.1136/jnnp.74.7.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Economides JR, Horton JC. Eye movement abnormalities in stiff person syndrome. Neurology. 2005;65(9):1462–1464. doi: 10.1212/01.wnl.0000183068.42803.33. [DOI] [PubMed] [Google Scholar]
- 102.Zivotofsky AZ, Siman-Tov T, Gadoth N, Gordon CR. A rare saccade velocity profile in stiff-person syndrome with cerebellar degeneration. Brain Res. 2006;1093(1):135–140. doi: 10.1016/j.brainres.2006.03.064. [DOI] [PubMed] [Google Scholar]
- 103.Shaikh AG, Wilmot G. Opsoclonus in a patient with increased titers of anti-GAD antibody provides proof for the conductance-based model of saccadic oscillations. J Neurol Sci. 2016;362:169–173. doi: 10.1016/j.jns.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 104.Tilikete C, Veghetto A, Trouillas P, Honnorat J. Anti-GAD antibodies and periodic alternating nystagmus. Arch Neurol. 2005;62(8):1300–1303. doi: 10.1001/archneur.62.8.1300. [DOI] [PubMed] [Google Scholar]
- 105.Markakis I, Alexiou E, Xifaras M, Gekas G, Rombos A. Opsoclonus-myoclonus-ataxia syndrome with autoantibodies to glutamic acid decarboxylase. Clin Neurol Neurosurg. 2008;110(6):619–621. doi: 10.1016/j.clineuro.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 106.Goffart L, Pélisson D, Guillaume A. Orienting gaze shifts during Muscimol inactivation of caudal fastigial nucleus in the cat. II. Dynamics and eye-head coupling. J Neurophysiol. 1998;79(4):1959–1976. doi: 10.1152/jn.1998.79.4.1959. [DOI] [PubMed] [Google Scholar]
- 107.Shaikh AG, Miura K, Optican LM, Ramat S, Leigh RJ, Zee DS. A new familial disease of saccadic oscillations and limb tremor provides clues to mechanisms of common tremor disorders. Brain. 2007;130(11):3020–3031. doi: 10.1093/brain/awm240. [DOI] [PubMed] [Google Scholar]
- 108.Shaikh AG, Wong AL, Optican LM, Miura K, Solomon D, Zee DS. Sustained eye closure slows saccades. Vis Res. 2010;50(17):1665–1675. doi: 10.1016/j.visres.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Enderle JD, Engelken EJ. Simulation of oculomotor post-inhibitory rebound burst firing using a Hodgkin-Huxley model of a neuron. In: Biomedical Sciences Instrumentation. 1995. p. 53–8. [PubMed]
- 110.Miura K, Optican LM. Membrane channel properties of premotor excitatory burst neurons may underlie saccade slowing after lesions of omnipause neurons. J Comput Neurosci. 2006;20(1):25–41. doi: 10.1007/s10827-006-4258-y. [DOI] [PubMed] [Google Scholar]
- 111.Federighi P, Cevenni G, Dotti MT, Rosini F, Pretegiani E, Federico A, et al. Differences in saccade dynamics between spinocerebellar ataxia 2 and late-onset cerebellar ataxias. Brain. 2011;134(3):879–891. doi: 10.1093/brain/awr009. [DOI] [PubMed] [Google Scholar]
- 112.Fitts PM. The information capacity of the human motor system in controlling the amplitude of movement. J Exp Psychol. 1954;47(6):381–391. doi: 10.1037/h0055392. [DOI] [PubMed] [Google Scholar]
- 113.Enderle JD, Wolfe JW. Time-optimal control of saccadic eye movements. Biomed Eng IEEE Trans. 1987;BME-34(1):43–55. doi: 10.1109/TBME.1987.326014. [DOI] [PubMed] [Google Scholar]
- 114.Wolpert DM, Ghahramani Z, Jordan MI. An internal model for sensorimotor integration. Science. 1995;269(5232):1880–1882. doi: 10.1126/science.7569931. [DOI] [PubMed] [Google Scholar]
- 115.Meyer DE, Abrams RA, Kornblum S, Wright CE, Smith JEK. Optimality in human motor performance: ideal control of rapid aimed movements. Psychol Rev. 1988;95(3):340–370. doi: 10.1037/0033-295X.95.3.340. [DOI] [PubMed] [Google Scholar]
- 116.Terao Y, Fukuda H, Yugeta A, Hikosaka O, Nomura Y, Segawa M, et al. Initiation and inhibitory control of saccades with the progression of Parkinson’s disease - changes in three major drives converging on the superior colliculus. Neuropsychologia. 2011;49(7):1794–1806. doi: 10.1016/j.neuropsychologia.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Choi SM, Lee SH, Choi KH, Nam TS, Kim JT, Park MS, et al. Directional asymmetries of saccadic hypometria in patients with early Parkinson’s disease and unilateral symptoms. Eur Neurol. 2011;66(3):170–174. doi: 10.1159/000330671. [DOI] [PubMed] [Google Scholar]
- 118.Kimmig H, Haußmann K, Mergner T, Lücking CH. What is pathological with gaze shift fragmentation in Parkinson’s disease? J Neurol. 2002;249(6):683–692. doi: 10.1007/s00415-002-0691-7. [DOI] [PubMed] [Google Scholar]
- 119.Blekher T, Weaver M, Rupp J, Nichols WC, Hui SL, Gray J, et al. Multiple step pattern as a biomarker in Parkinson disease. Parkinsonism Relat Disord. 2009;15(7):506–510. doi: 10.1016/j.parkreldis.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rottach KG, Riley DE, DiScenna AO, Zivotofsky AZ, Leigh RJ. Dynamic properties of horizontal and vertical eye movements in parkinsonian syndromes. Ann Neurol. 1996;39(3):368–377. doi: 10.1002/ana.410390314. [DOI] [PubMed] [Google Scholar]
- 121.Shaikh AG, Factor SA, Juncos JL. Saccades in progressive Supranuclear palsy-maladapted, irregular, curved, and slow. Mov Disord Clin Pract. 2017;4(5):671–681. doi: 10.1002/mdc3.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Steele J, Richardson JC, Olszewski J. Progressive Supranuclear palsy. Ann Neurol. 1964;10. [DOI] [PubMed]
- 123.Williams DR, Lees AJ, Wherrett JR, Clifford SJCJ. Richardson and 50 years of progressive supranuclear palsy. Neurol. 2008;70:566–573. doi: 10.1212/01.wnl.0000286938.39473.0e. [DOI] [PubMed] [Google Scholar]
- 124.Chen AL, Riley DE, King SA, Joshi AC, Serra A, Liao K, et al. The disturbance of gaze in progressive supranuclear palsy: Implications for pathogenesis. Front Neurol. 2010;1:147. doi: 10.3389/fneur.2010.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bhidayasiri R, Riley DE, Somers JT, Lerner AJ, Büttner-Ennever JA, Leigh RJ. Pathophysiology of slow vertical saccades in progressive supranuclear palsy. Neurology. 2001;57(11):2070–2077. doi: 10.1212/WNL.57.11.2070. [DOI] [PubMed] [Google Scholar]
- 126.Salsano E, Umeh C, Rufa A, Pareyson D, Zee DS. Vertical supranuclear gaze palsy in Niemann-pick type C disease. Neurol Sci. 2012;33(6):1225–1232. doi: 10.1007/s10072-012-1155-1. [DOI] [PubMed] [Google Scholar]
- 127.Kapoula Z, Yang Q, Vernet M, Dieudonné B, Greffard S, Verny M. Spread deficits in initiation, Speed and Accuracy of Horizontal and Vertical Automatic Saccades in Dementia with Lewy Bodies. Front Neurol. 2010;1:138. doi: 10.3389/fneur.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mahapatra R, Edwards M, Schott J, Bhatia K. Review: Corticobasal degeneration. Lancet Neurol. 2004;3:736–743. doi: 10.1016/S1474-4422(04)00936-6. [DOI] [PubMed] [Google Scholar]
- 129.Leigh RJ, Newman SA, Folstein SE, Lasker AG, Jensen BA. Abnormal ocular motor control in Huntington’s disease. Neurology. 1983;33(10):1268–1275. doi: 10.1212/WNL.33.10.1268. [DOI] [PubMed] [Google Scholar]
- 130.Collewijn H, Went LN, Tamminga EP, Vegter-Van der Vlis M. Oculomotor defects in patients with Huntington’s disease and their offspring. J Neurol Sci. 1988;86(2–3):307–320. doi: 10.1016/0022-510X(88)90107-4. [DOI] [PubMed] [Google Scholar]
- 131.Lasker AG, Zee DS, Hain TC, Folstein SE, Singer HS. Saccades in Huntington’s disease: slowing and dysmetria. Neurology. 1988;38(3):427–431. doi: 10.1212/WNL.38.3.427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is a review paper, hence not applicable.