

Abstract
A pathway for cystic fibrosis transmembrane conductance regulator (CFTR) degradation is initiated by Hsp27, which cooperates with Ubc9 and binds to the common F508del mutant to modify it with SUMO-2/3. These SUMO paralogues form polychains, which are recognized by the ubiquitin ligase, RNF4, for proteosomal degradation. Here, protein array analysis identified the SUMO E3, protein inhibitor of activated STAT 4 (PIAS4), which increased wild-type (WT) and F508del CFTR biogenesis in CFBE airway cells. PIAS4 increased immature CFTR threefold and doubled expression of mature CFTR, detected by biochemical and functional assays. In cycloheximide chase assays, PIAS4 slowed immature F508del degradation threefold and stabilized mature WT CFTR at the plasma membrance. PIAS4 knockdown reduced WT and F508del CFTR expression by 40–50%, suggesting a physiological role in CFTR biogenesis. PIAS4 modified F508del CFTR with SUMO-1 in vivo and reduced its conjugation to SUMO-2/3. These SUMO paralogue-specific effects of PIAS4 were reproduced in vitro using purified F508del nucleotide-binding domain 1 and SUMOylation reaction components. PIAS4 reduced endogenous ubiquitin conjugation to F508del CFTR by ∼50% and blocked the impact of RNF4 on mutant CFTR disposal. These findings indicate that different SUMO paralogues determine the fates of WT and mutant CFTRs, and they suggest that a paralogue switch during biogenesis can direct these proteins to different outcomes: biogenesis versus degradation.
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR) is the basis of the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA)-stimulated anion conductance at the apical membranes of secretory epithelial cells in the airways, intestines, pancreas and other systems (Frizzell and Hanrahan, 2012). As a member of the ABC transporter family, CFTR is composed of two membrane spanning domains (MSD1 and MSD2), two nucleotide-binding domains (NBD1 and NBD2), and a unique and unstructured regulatory (R) domain. The R domain contains sites whose kinase-mediated phosphorylation enables CFTR channel gating via ATP binding and hydrolysis at the NBDs. The omission of phenylalanine at position 508 of NBD1, F508del, is found in ∼90% of cystic fibrosis (CF) patients on at least one allele, defining the most common mutation causing CF. Impaired folding of F508del CFTR elicits its near-complete disposal by endoplasmic reticulum (ER) quality control mechanisms and results in severe CF due largely to a marked reduction in apical membrane channel density. Significant amounts of wild-type (WT) CFTR are also degraded by most cells (Ward et al., 1995), highlighting the complex folding landscape that even WT CFTR must traverse.
Approximately 2000 mutations of the CFTR gene, many quite rare, have been proposed as CF disease causing, while correction of the folding defect of F508del CFTR provides the greatest potential for enhancing the quality of life and life expectancy of CF patients. To date, the discovery of small molecules, for example, VX-809 (lumakaftor), which corrects 10–15% of F508del CFTR function in vitro (Van Goor et al., 2011), has provided only limited improvement in clinical studies (Clancy et al., 2012). The combination of lumakaftor with the gating potentiator, VX-770 (ivakaftor), has shown the potential to benefit a significant number of F508del patients, and their combination (Orkambi) has been approved for F508del homozygotes (Konstan et al., 2017). Nevertheless, efforts to uncover the checkpoints in CFTR biogenesis where most F508del CFTR is lost to degradation pathways can be expected to identify targets whose modulation would further improve efficacy.
CFTR biogenesis is surveyed by molecular chaperones that monitor the protein's conformational state. The core chaperone systems, Hsp70, Hsp90, and the Hsp40 cochaperones, limit CFTR aggregation to facilitate its productive folding (Strickland et al., 1997; Meacham et al., 1999), yet these chaperones can also target CFTR mutants for degradation when the native fold is not achieved (Cyr et al., 2002; Amaral, 2004). Unstable conformations of CFTR remain bound to chaperones, for example, a prolonged association with Hsp70/Hsp90 recruits the ubiquitin ligase complex, Ubc5-CHiP, resulting in CFTR ubiquitylation and degradation by the 26S proteasome (Jensen et al., 1995; Ward et al., 1995; Meacham et al., 2001; Sun et al., 2006; Younger et al., 2006). In contrast to the ATP-dependent core chaperone systems, small heat shock proteins (sHsps) represent a class of ATP-independent protein stabilizers. Generally, sHsps interact with intermediate, foldable protein conformations, for example, during cell stress, and their clients are often refolded in association with ATP-dependent chaperones once stress conditions are relieved (Rajaraman et al., 1996; Treweek et al., 2000; Lindner et al., 2001).
We recently identified a new sHsp-mediated pathway that promotes the selective degradation of F508del CFTR, while having little effect on the WT protein (Ahner et al., 2013). This pathway is initiated by binding of the small heat shock protein, Hsp27, to the F508del mutant, which recruits the SUMO E2 enzyme, Ubc9, to bring about the posttranslational modification (PTM) of F508del CFTR by the small ubiquitin-like modifier, SUMO. SUMO modification occurs on transcription factors, membrane receptors, ion channels, and so on (Benson et al., 2017). Tandem mass spectrometry has identified SUMOylation sites overlapping with other posttranslational modifications such as ubiquitylation, acetylation, and methylation. SUMO knockout is embryonic lethal, and it has been estimated that a significant fraction of the mammalian proteome is subject to SUMO modification (Hendriks and Vertegaal, 2016).
Five SUMO paralogues are now considered to be functionally significant in mammalian systems, SUMO-1 through -5 (Owerbach et al., 2005). SUMO-4 can be difficult to conjugate to substrates due to a proline residue that restricts its activation by SUMO isopeptidase (Owerbach et al., 2005). SUMO-4 and -5 have limited tissue distributions and have been studied less extensively that the other paralogues (Liang et al., 2016). This work will focus on SUMO-1 and SUMO-2/3. The latter two paralogues differ by only four amino acids and generally behave in a functionally similar manner; SUMO-2/3 shares only 44% homology with SUMO-1. These SUMO paralogues are attached to their targets by isopeptide bonds, generally to lysine residues within the consensus motif ψKXD/E, where ψ is a hydrophobic residue, X is any amino acid, and the modified lysine is followed by an acidic residue, a phosphorylated site, or a larger electronegative region.
A strong physical interaction between Hsp27 and Ubc9, the single known E2 enzyme for SUMO conjugation, elicits primarily SUMO-2/3 modification of the CFTR mutant. The SUMO-2/3 paralogues contain a consensus sequence for SUMOylation, allowing them to form SUMO polychains. These chains are recognized by the SUMO-targeted ubiquitin ligase (STUbL), RNF4, which then ubiquitylates F508del CFTR via its RING domain, targeting it for degradation by the ubiquitin-proteasome system (UPS) (Ahner et al., 2013).
As a posttranslational modification, SUMOylation impacts proteins that participate in diverse cellular processes, including nuclear transport, the maintenance of genome integrity, transcriptional control, and the regulation of innate immune signaling (Adorisio et al., 2017). SUMO modification is present in several neurodegenerative diseases that arise from the aggregation of misfolded proteins. Importantly, this PTM can influence the stability and solubility of misfolded and/or aggregated proteins (Liebelt and Vertegaal, 2016).
The SUMOylation cascade resembles that for ubiquitin modification, consisting of a series of thiol-mediated transfer reactions, but these pathways differ in that the SUMO E2, Ubc9, can transfer SUMO directly to its client protein without intervention of an E3. The principal functions of E3 enzymes in the SUMO pathway appear to be assistance with target protein selection and catalytic transfer of the modifier (Gareau and Lima, 2010). The SUMO E3 ligases include members of the protein inhibitor of activated STAT (PIAS) proteins, an important family in signal transduction, which includes PIAS1, PIAS2 (also termed PIASx with isoforms PIASxα and PIASxβ), PIAS3, and PIAS4 (aka PIASy). Among the family members, PIAS4 has received attention due to its participation in signaling pathways that participate in oncogenesis and immune system regulation (Shuai and Liu, 2005; Rytinki et al., 2009).
In the present work, we examined the influence of PIAS4 on WT and F508del CFTR, motivated by its identification when our protein array was probed with poly-SUMO-3, which had attracted our attention also to the STUbL, RNF4 (Ahner et al., 2013). The identification of PIAS4 and the implication that it bound SUMO polychains suggested the hypothesis that it would support or enhance the Hsp27-Ubc9 mediated F508del CFTR degradation pathway described previously (Ahner et al., 2013). In contrast, PIAS4 expression in CFBE airway cell lines, transduced to stably express either F508del or WT CFTR, promoted CFTR biogenesis rather than degradation, and therefore we pursued its mechanism of action.
RESULTS
PIAS4 enhances CFTR biogenesis and F508del corrector efficacy
First, we examined the impact of overexpressing PIAS4 on CFTR protein levels. CFBE41o- airway epithelial cells stably expressing WT or F508del CFTR (hereafter, CFBE-WT or CFBE-F508del (Bebok et al., 2005)) were transfected with constructs to express PIAS4 or empty vector as control, followed by immunoblot (IB) for CFTR and PIAS4. As illustrated in Figure 1A, PIAS4 overexpression augmented the steady-state expression level of immature (band B) and mature (band C) CFTR; the increases in WT and F508del levels were approximately threefold for band B and approximately twofold for band C on average, as shown in the accompanying quantitation of the aggregate data. The relative increases in band B exceeded those in band C, that is, PIAS4 decreased the C/B band ratio. These findings suggest that the primary effect of PIAS4 is on the production and/or stability of the immature, ER-localized form of the WT and mutant proteins, a point to which we will return. Since the transfection efficiency of CFBE cells varies between 30 and 50% in our experience, the observed approximately threefold increase in band B is likely two to three times smaller than would result if the entire cell population were expressing PIAS4.
FIGURE 1:

PIAS4 increases CFTR expression in CFBE airway cells. (A) Overexpression of PIAS4 increases the steady-state expression of WT and F508del CFTR. Stable CFBE-WT and -F508del cells were transiently transfected with or without Flag-PIAS4 as described under Materials and Methods. Whole cell lysates were extracted 48 h after transfection and protein expression detected by IB using the indicated antibodies. CFTR protein levels were quantified and normalized to control from three independent experiments. (*p < 0.05; **p < 0.01). (B) PIAS4 enhances the efficacy of CFTR correctors in CFBE cells stably expressing F508del CFTR. Flag-PIAS4 was expressed in CFBE-F508del cells as described in A. After 24 h, the transfected cells were treated with dimethyl sulfoxide (DMSO), 10 μM C18, or 10 μM VX-809 for 24 h, and then cells were lysed and analyzed by IB. The numbers below the CFTR blots give the band C densities relative to control (DMSO). (C) The impact of PIAS4 on CFTR expression depends on its SUMO E3 ligase activity. Flag-PIAS4 and its catalytic mutant, Flag-PIAS4-CA, were transiently transfected into CFBE-F508del cells. Whole cell lysates were examined by IB using the indicated antibodies. CFTR signals were normalized to control values in seven independent experiments (***p < 0.001).
Second, we determined whether the augmented level of immature F508del CFTR would enhance the ability of correctors to generate the mature form of the mutant protein. Experiments similar to those of Figure 1A were performed, in which CFBE-F508del cells were treated for 24 h with either vehicle, 10 µM VX-809 (Van Goor et al., 2011) or one of its developmental precursors from the same chemical series, 10 µM C18 (www.cftrfolding.org). As shown in Figure 1B, the expression of PIAS4 alone elicited a marked increase in immature F508del CFTR, together with a small increase in mature protein, noted also in Figure 1A. However, a more substantial level of F508del band C was found when PIAS4 overexpression was combined with either of the correctors. The band C densities relative to control, shown below the F508del bands, indicate that PIAS4 was as effective as the correctors at increasing the mature mutant protein level, and when they were combined, PIAS4 increased corrector efficacy approximately twofold.
Third, we performed a negative control experiment to determine whether PIAS4 was acting as a SUMO E3; that is, to assess the requirement for the catalytic cysteines of the SP-RING domain of PIAS4 in its action on F508del expression. The experiments were performed as in Figure 1A and the results shown in Figure 1C: mutation of its two catalytic cysteines (C342 and C347) to alanine eliminated the PIAS4-induced increase in F508del CFTR expression.
The hypothesis that PIAS4 normally contributes to CFTR biogenesis was further evaluated in PIAS4 knockdown experiments. Stable CFBE-WT or -F508del cell lines were transduced with short hairpin RNA (shRNA) targeting PIAS4 or with a scrambled shRNA control; these constructs were expressed using an adenovirus vector, described under Materials and Methods. After 72 h, cell lysates were prepared for CFTR IB, and the results and mean data are illustrated in Figure 2, A and B. Knockdown of the SUMO E3 produced reductions in the expression level of WT and F508del CFTR that averaged ∼40%. The shRNA of Figure 2 targeted the PIAS4 mRNA 3′-region and has been described previously (Morris et al., 2009). This finding was confirmed using a small interfering RNA targeting the 5′-region of PIAS4's mRNA (Supplemental Figure S1). These results further support the conclusion that endogenous PIAS4 plays a physiological role in promoting the biogenesis of WT and F508del CFTR.
FIGURE 2:

PIAS4 knockdown reduces CFTR expression in CFBE airway cells. (A, B) Knockdown performed in stable CFBE-WT or -F508del cells. shRNA targeting PIAS4 was transduced using a recombinant adenovirus, compared with adenotransduced scrambled shRNA. CFTR expression was evaluated by IB with the indicated antibodies after 48 h virus exposure. For all experiments here, bar graphs provide composite means ± SEM from at least four independent experiments (**p < 0.01; ***p < 0.001), and a bracket indicates the gel region used in quantitation. (C) Overexpression of PIAS4 promotes CFTR cell-surface expression. CFBE-WT cells were transduced with PIAS4 or GFP for 72 h and biotinylation assays performed; streptavidin elution was followed by IB with the indicated antibodies, as described under Materials and Methods. Surface CFTR signals were normalized to the actin loading control. (D) PIAS4 knockdown reduces CFTR surface expression. CFBE-WT cells were transduced with adenovirus vector shRNA targeting PIAS4 or scrambled shRNA for 72 h and then subjected to surface biotinylation and analysis as described in C and under Materials and Methods.
Next, we used cell-surface biotinylation to evaluate the impact of PIAS4 overexpression or knockdown on CFTR expression in the airway cell plasma membrane (PM). For these studies, CFBE cells stably expressing WT CFTR were transduced to express either PIAS4 or GFP, and then the cell surface was biotinylated as described under Materials and Methods. Cell lysates were prepared and biotinylated PM proteins pulled down using streptavidin agarose, IBs were performed for CFTR and actin, and the cell lysates were blotted for the indicated proteins. As shown in Figure 2C, the overexpression of PIAS4 increased cell-surface CFTR ∼75% on average. Actin was not detected in the pull down, a negative control indicating that labeling was restricted to proteins resident at the cell surface. Thus, the increase in WT CFTR levels in cells overexpressing PIAS4 leads also to increased CFTR density in the plasma membrane, which should lead to an increase in CFTR-mediated anion transport; those data will be presented below.
Finally, the impact of reduced PIAS4 levels on the surface expression of WT CFTR was examined by performing PIAS4 knockdown followed by cell-surface biotinylation. CFBE-WT cells were transduced to express PIAS4 or control shRNA and then subjected to surface biotinylation and streptavidin pull down after 48 h; the results are illustrated in Figure 2D. Reduced PIAS4 resulted in a ∼50% reduction in cell-surface CFTR protein, which paralleled the effects of reduced PIAS4 expression on CFTR expression levels (Figure 2, A and B). Together, the data of Figure 2 indicate that PIAS4 plays a significant role in determining the steady-state expression level of immature CFTR, as well as that of the mature WT protein that resides in the plasma membrane.
PIAS4 slows the degradation of F508del CFTR in the ER
The marked increase in immature CFTR expression, observed for both WT and F508del, suggested that PIAS4 augments the production or stability of CFTR in the endoplasmic reticulum, where the immature forms of these proteins are localized (Gregory et al., 1991; Ward and Kopito, 1994). To examine this hypothesis, we performed cycloheximide (CHX) chase experiments using CFBE-WT or CFBE-F508del stable cell lines that were transfected with constructs for PIAS4 or the vector control. Figure 3A shows the time course of immature F508del CFTR expression once protein synthesis had been interrupted in PIAS4 overexpressing or vector control cells. At the indicated times, cells were lysed and the lysates prepared for IB with the indicated antibodies. The data demonstrate that band B F508del CFTR disappeared with roughly exponential kinetics under control conditions with a t1/2 of ∼1 h, as observed in previous studies (Liang et al., 2012), whereas the t1/2 for disposal of the mutant in the PIAS4 overexpressing cells was ∼3 h. The degradation of F508del under control conditions was mediated primarily by the proteasome, as reflected by its inhibition by MG132 (unpublished data), findings that are consistent with prior studies (Ward and Kopito, 1994; Sun et al., 2008; Liang et al., 2012).
FIGURE 3:

PIAS4 stabilizes CFTR, reducing its proteasomal degradation. (A) PIAS4 slows F508del CFTR degradation. CFBE-F508del cells were transfected with or without Flag-PIAS4 as described under Materials and Methods. After 48 h, the transfected cells were treated with 100 µg/ml CHX and lysed with RIPA buffer at the indicated CHX treatment times (0, 1, 2, 4, 6 h) to follow F508del (band B) expression during inhibition of protein synthesis. Following IB, blot densities from four independent experiments were averaged to construct the relations for F508del CFTR (band B) remaining as a function of time, with values from each experiment normalized to the mean densities at the beginning of the CHX chase (t = 0). (B) PIAS4 stabilizes mature WT CFTR. Experiments performed as described in A, but with CFBE-WT cells. Time courses for expression of CFTR bands B and C relative to control are indicated. See the text for discussion. (C) The impact of PIAS4 on F508del CFTR expression is not detected at the mRNA level. Three constructs: empty vector (control), Flag-PIAS4 or Flag-PIAS2 were transfected into CFBE-F508del stable cells. After 48 h, total RNA was extracted and subjected to qPCR as described under Materials and Methods. β-Actin was used as an internal control.
Next, we used the CHX chase protocol to evaluate the influence of PIAS4 overexpression on the kinetics of WT CFTR degradation, as shown in Figure 3B. The kinetics of immature WT CFTR during the CHX chase period is determined by two pathways: 1) the disappearance of a fraction of band B by ER associated degradation (ERAD) and 2) the conversion the remainder of band B to band C via its trafficking to the PM. The influence of PIAS4 expression on the fate of immature WT CFTR is somewhat difficult to discern since band C is more abundant than band B and it is more stable at the PM. Under control conditions, mature WT CFTR decreased to ∼50% of its initial value at 4 h, but with PIAS4 overexpression, band C was retained, decreasing by only ∼10% during the same period. Whether and how PIAS4 acts to increase CFTR stability following its exit from the ER will require additional studies. Nevertheless, increased peripheral stability is expected to contribute to the enhanced corrector action shown in Figure 1B. The PIAS4 doublet shown in Figure 3, A and B, is likely due to its posttranslational modification; for example, PKC is known to phosphorylate the RING domain of PIAS4 (Heo et al., 2016; Lear et al., 2016), and the appearance of more than one band can vary also with gel density.
In principle, PIAS4 might augment CFTR biogenesis by increasing CFTR transcription, translation or by its posttranslational modification by SUMO. The CHX chase experiments should rule out increased transcription and translation of PIAS4 as contributing to the enhanced stability of CFTR band B. To rule out a change in F508del CFTR message levels, we evaluated the influence of PIAS4 expression on CFTR mRNA levels by quantitative PCR (qPCR) the results are shown in Figure 3C. The levels of CFTR mRNA from CFBE-F508del cells did not differ between cells transfected with empty vector, PIAS4 or the PIAS2 isoform (or PIASx); the latter also had no effect on the expression level of CFTR protein (unpublished data). These findings support the action of CHX and are consistent with a posttranslational effect of PIAS4.
PIAS4 increases functional F508del CFTR at the cell surface
To determine whether the increase in immature F508del expression elicited by PIAS4 translates into increased trafficking to the plasma membrane during corrector treatment, we examined the cell-surface expression of fluorogen activating protein (FAP)-tagged F508del CFTR in CFBE cells treated with VX-809. The FAP labeling approach has been used previously to compare different correctors for their ability to mobilize F508del CFTR to the PM (Holleran et al., 2012) or to monitor its intracellular trafficking itinerary following its endocytic retrieval from the PM (Holleran et al., 2013). CFBE cells stably expressing FAP-F508del CFTR were transferred to a 96-well plate with or without recombinant adenovirus encoding PIAS4. After 24 h, the cells were incubated with 2 µM VX-809 and read for FAP activation the following day. The panels of Figure 4A show cell-surface labeling of FAP-F508del in the absence and presence of PIAS4 coexpression, respectively, on the background of the increase due to VX-809 preincubation. The accompanying bar graph, Figure 4B, provides the mean surface intensity, which was increased ∼2.3-fold by PIAS4 in four experiments, in agreement with the biochemical data that reported a 2.7-fold increase in band C of F508del (Figure 1B). Given the apparent variability in surface expression among cells, we next asked whether the increased surface intensity occurred mainly in the few very bright cells observed or was due to a general increase in the less bright cells. A plot of the relative frequency of cells quantified as a function of their fluorescence is provided in Figure 4C. The histogram shows that the fraction of cells that exhibit very low fluorescence levels (binned from 0 to 100) is decreased with PIAS4 expression (∼40% vs. ∼20%), whereas the frequency of the brighter cells was consistently higher, as anticipated from the PIAS4-induced increase in mature protein expression observed in the Western blots of Figure 1, A and B.
FIGURE 4:

PIAS4 increases functional F508del CFTR at the surface of airway cells. (A) The membrane-impermeant fluorescent dye, MG-B-Tau, was used to detect FAP-F508del CFTR in CFBE-F508del cells stably expressing the mutant CFTR and treated with 2 µM VX-809 overnight. Fluorescence imaging reveals an increase in FAP-F508del CFTR surface expression when the VX-809 treated CFBE cells were transduced with recombinant adenovirus expressing PIAS4. Representative images are shown. (B) The bar graph represents the quantified relative surface expression of FAP-F508del CFTR in CFBE cells in the presence of 2 µM VX-809 under control conditions or in CFBE cells transduced with recombinant adenovirus expressing PIAS4. The experiment was repeated four times with similar results (****p < 0.0001). (C) Histogram of cell-surface FAP-F508del CFTR expression as a function of fluorescence intensity from the data shown in B. The data were derived from four fields of view and a total of 7000–9000 individual cells analyzed for each condition. See the text for discussion. (D) Chloride currents in PIAS4 expressing CFBE41o- parental airway cells. Whole-cell patch clamp was used to monitor the small baseline Cl currents and the subsequent effects of forskolin and the inhibitor, GlyH-101. Control cells were transfected with F508del CFTR and EGFP; experimental cells transfected with F508del CFTR plus PIAS4 and EGFP. The fluorophore was used to identify expressing cells. The pipette and bath solutions contained equal concentrations of Cl, and currents were measured at a holding potential of –40 mV (**p < 0.01; ***p < 0.001; ****p < 0.0001).
We also examined the functionality of cell-surface F508del CFTR in parental CFBE41o- cells 2 d following transfection with F508del CFTR using patch clamp methods to record their whole-cell currents, as indicated under Materials and Methods and shown in Figure 4D. The pipette and bath solutions contained equal Cl concentrations and the forskolin-stimulated currents, recorded at a holding potential of –40 mV, were inhibited by GlyH-101 (Muanprasat et al., 2004). These findings indicate that mutant CFTR that traffics to the PM exhibits agonist stimulated, CFTR-mediated, Cl conductance properties.
PIAS4 drives SUMO-1 conjugation of CFTR and reduces its SUMO-2 modification
Our prior work demonstrated that the small heat shock protein, Hsp27, primarily interacted with F508del CFTR and associated with the SUMO E2 enzyme, Ubc9, to modify F508del CFTR with SUMO-2/3 (Ahner et al., 2013; Gong et al., 2016). This Hsp27/Ubc9 pathway led to the formation of SUMO polychains and their recognition by the SUMO-targeted ubiquitin ligase, RNF4, promoting mutant CFTR degradation by the proteasome (Ahner et al., 2013). However, the stabilizing effect of PIAS4 on WT and F508del CFTR suggested that an alternative pathway leads to an opposing outcome in response to intervention of the SUMO E3, which supports stabilization of immature CFTR and biogenesis of the WT and mutant proteins rather than degradation.
The hypothesis that a different SUMO paralogue modifies CFTR consequent to its interaction with PIAS4 was evaluated initially using in vitro SUMOylation experiments performed with purified SUMO pathway components and human F508del NBD1. In a previous study, we found that purified F508del NBD1 was preferentially conjugated to SUMO-2 or -3 when Hsp27 was introduced into the reaction mixture in vitro (Gong et al., 2016). This outcome was similar to the SUMO-2/3 modification of full-length F508del CFTR observed during Hsp27 overexpression in vivo (Ahner et al., 2013). As shown in Figure 5A, F508del NBD1 could be modified by SUMO-1 or -2 in vitro; its conjugation with SUMO-2 was somewhat greater than that by SUMO-1, as found previously (Gong et al., 2016). However, the addition of purified PIAS4 to the reaction mix led to increased F508del NBD1 conjugation with SUMO-1, plus a smaller but significant decrease in its modification by SUMO-2. Quantitation of the changes from all experiments showed that PIAS4 induced a ∼80% increase in SUMO-1 modification of mutant NBD1 and a ∼30% decrease in its conjugation with SUMO-2.
FIGURE 5:

PIAS4 facilitates SUMO-1 conjugation to F508del CFTR and reduces its modification by SUMO-2/3. (A) PIAS4 promotes the conjugation of NBD1-F508del to SUMO-1 in vitro and reduces that to SUMO-2/3. Purified F508del-NBD1-1S (Rabeh et al., 2012) was incubated with purified SUMOylation components for 60 min at 27°C as described under Materials and Methods and shown previously (Gong et al., 2016). The reaction mixture was resolved on 12% SDS–PAGE and NBD1 was detected by anti-CFTR (#660). Modified NBD1-F508del-1S protein signals were quantified and normalized to control values from three independent experiments. (*p < 0.05; **p < 0.001). (B) In vivo, PIAS4 induces F508del CFTR conjugation to SUMO-1, while reducing its modification by SUMO-2/3. Flag-PIAS4 was overexpressed in CFBE-F508del cells; empty vector served as control. F508del CFTR was immunoprecipitated from cell lysates and immunoblotted with antibodies against endogenous SUMO paralogues. The SUMO signals were normalized to control values in three independent experiments (*p < 0.05). (C) PIAS4 actions on F508del CFTR biogenesis or degradation were reproduced by expression of active SUMO paralogues that express the C-terminal di-glycine (G-G) motif, whose exposure by SUMO peptidase is required prior to target conjugation (Pichler et al., 2017). Parental CFBE41o- cells were transfected with F508del CFTR plus HA-SUMO-1 or -2 or myc-tagged SUMO-3, and after 48 h, the expression levels of F508del CFTR were determined by IB. The bar graph illustrates the composite data, which show that activated SUMO-1 expression reproduced the actions of PIAS4 on F508del biogenesis while activated SUMO-2/3 mimicked the action of RNF4 to promote F508del degradation; data from four experiments expressed relative to control (*p < 0.05; **p < 0.01). (D) The SUMO E3 ligase activity of PIAS4 is required for its facilitation of F508del CFTR modification by SUMO-1. CFBE-F508del cells were transiently transfected with vector, Flag-PIAS4 or the RING domain catalytic mutant, Flag-PIAS4-CA. Cell lysates were immunoprecipitated with anti-CFTR followed by IB with the indicated antibodies for SUMO-1, CFTR, or PIAS4.
Next, we asked whether differential paralogue modifications of full-length F508del CFTR in vivo are also influenced by overexpression of PIAS4; the results are shown in Figure 5B. CFBE-F508del cells were transfected with PIAS4 or the corresponding empty vector. After immunoprecipitation (IP) of CFTR, the pull downs were blotted for either SUMO-1 or SUMO-2/3. The input blots in the left panel show the increase in F508del expression observed with coexpression of PIAS4, as in prior figures. Following IP of mutant full-length CFTR, the SUMO paralogue IBs indicate that F508del CFTR was primarily modified by SUMO-1 during PIAS4 coexpression, and this was accompanied by a reduction in modification of the mutant by SUMO-2/3.
While the experiments in Figure 5B relied on endogenous SUMO paralogues to depict their differential effects on F508del CFTR fate, similar patterns could also be produced by expressing preactivated paralogues that expose the C-terminal G-G motif that is required prior to target protein conjugation. The parental CFBE41o- cell line was transfected with F508del CFTR and epitope-tagged active SUMO paralogues, and their impact on the expression levels of the mutant protein was determined one day later by IB. As illustrated in Figure 5C, the expression of active SUMO-1 elicited an ∼75% increase in F508del band B relative to control, thus mimicking the biogenic effect seen with PIAS4 overexpression. Contrasting with this result were the effects of active SUMO-2 and -3, which produced decreases of ∼30–40% in expression of immature F508del versus control, paralleling the actions of Hsp27/RNF4 in promoting mutant protein degradation.
Finally, to verify that SUMOylation of F508del CFTR could be attributed to the E3 catalytic activity of PIAS4, its ability to SUMO-1 modify F508del CFTR was assessed using mutant PIAS4 in which the catalytic cysteines of the SP-RING domain were mutated to alanine. As seen in Figure 5D, these C/A mutations suppressed the ability of F508del CFTR to co-IP the SUMO-1 modified protein, and to increase F508del expression level relative to catalytically active PIAS4, as shown previously in Figure 1C. Together, these findings indicate that PIAS4 can alter the balance between posttranslational SUMO paralogue modifications of FL F508del CFTR and NBD1.
PIAS4 reduces F508del ubiquitylation and the impact of RNF4
The above findings indicate that PIAS4 modifies FL F508del CFTR in vivo similarly to that of F508del NBD1 in vitro, increasing its conjugation with SUMO-1. The modification of FL F508del or NBD1 by SUMO-1 was associated also with a reduction in the conjugation of mutant CFTR with SUMO-2/3 (Figure 5, A and B). Since this PTM can lead to formation of SUMO polychains and interaction with the STUbL, RNF4, a reduction in ubiquitin modification of the mutant would be anticipated.
The results of this experiment are shown in Figure 6A. CFBE-F508del stable airway cells were transfected with either PIAS4 or empty vector as control. Both PIAS4 and control cells were incubated with proteasome inhibitor MG132 for 5 h to block rapid degradation of the ubiquitylated F508del CFTR, which would otherwise occur. Following F508del CFTR IP, the pull downs were blotted for CFTR and ubiquitin, revealing an increased CFTR level in the IP as well as a ∼50% reduction in the level of F508del ubiquitylation in response to PIAS4 overexpression; this effect was somewhat variable, as reflected by the SEM that characterizes the aggregate data.
FIGURE 6:

PIAS4 protects F508del CFTR from proteasomal degradation. (A) PIAS4 expression decreased the ubiquitylation of F508del CFTR. CFBE-F508del stable cells were transfected with Flag-PIAS4 or empty vector. After 48 h, cells were treated with 50 µM MG132 for 5 h. Cell lysates were immunoprecipitated with CFTR antibody and probed for ubiquitin by IB. The ubiquitin signals were normalized to control values from three independent experiments (*p < 0.05). (B) PIAS4 obviates RNF4-mediated F508del CFTR degradation. Flag-PIAS4, with or without Flag-RNF4, was overexpressed in CFBE-F508del cells. After 48 h, cells were lysed and subjected to IB with the indicated antibodies. The CFTR signals were quantified and normalized to control from five independent experiments. (*p < 0.05).
In principle, the effects of PIAS4 and RNF4 would antagonize one another since RNF4 increased F508del ubiquitylation (Ahner et al., 2013), whereas PIAS4 reduced F508del ubiquitin modification (Figure 6A). Therefore, we asked whether the decrease in F508del CFTR expression induced by RNF4 coexpression could be offset by overexpression of PIAS4. CFBE-F508del stable cells were transfected with PIAS4, RNF4, or their combination, and the steady-state levels of F508del CFTR were determined after 48 h. The composite results are provided in Figure 6B. PIAS4 overexpression increased immature F508del CFTR levels 2.1-fold, while expression of RNF4 reduced F508del band B levels ∼60%. When these posttranslational modifiers were combined, PIAS4 reversed the prodegradation impact of RNF4. This outcome likely results from competition between the two SUMO paralogues that mediate the actions of PIAS4 and RNF4, with the impact of PIAS4 on mutant CFTR fate being more significant.
Finally, we examined the influence of four predicted SUMOylation sites (K377, K447, K1199, and K1468) on F508del CFTR expression levels following their mutation to arginine. These sites were predicted by the software GPS-SUMO, found at: http://sumosp.biocuckoo.org. They lie in the MSD1-NBD1 linker, NBD1, NBD2, and the CFTR C-terminus. The 4KR mutant of F508del CFTR was expressed in CFBE41o- cells and their response to PIAS4 coexpression was examined. As shown in Figure 7, PIAS4 increased the expression of F508del by nearly fourfold in the positive control, but mutation of the four predicted SUMOylation sites virtually eliminated the ability of PIAS4 to augment CFTR biogenesis. Expression of the 4KR mutant also reduced the ability of F508del to be modified by SUMO-1 or SUMO-2/3 (Supplemental Figure S2). These data are consistent with the hypothesis that the pathways directing biogenesis versus degradation of CFTR by different SUMO paralogues are competitive and that they likely target the same sites on CFTR for SUMO modification.
FIGURE 7:
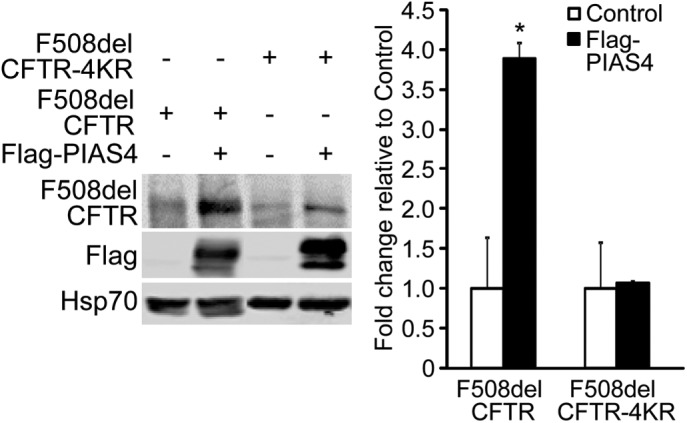
PIAS4 stabilizes F508del CFTR by modifying four consensus SUMOylation sites. F508del CFTR with K/R mutations at predicted SUMOylation sites (K377, 447, 1199, and 1468) is refractory to the impact of PIAS4 on mutant CFTR biogenesis. F508del CFTR or F508del CFTR-4KR were transfected with Flag-PIAS4 or vector control into CFBE41o- parental airway cells. After 48 h, whole cell lysates were prepared and F508del CFTR expression levels detected by IB with the indicated antibodies. Data from four independent experiments (*p < 0.05).
DISCUSSION
SUMOylation targets a variety of cellular processes and has been implicated in numerous diseases, including diseases of protein misfolding (Gareau and Lima, 2010; Srikanth and Verma, 2017). Sometimes referred to as the twins, SUMO-2 and -3 are 97% identical; and as no antibody can distinguish these paralogues, they are usually identified as SUMO-2/3. SUMO-1 has 46% identity with SUMO-2/3, and these three paralogues are widely expressed. SUMO modification is covalent and can be reversed by SUMO-specific peptidases.
Hsp27/SUMO-2/3/RNF4 and F508del CFTR degradation
Numerous pathways are available for disposing of misfolded proteins. Core-glycosylated substrates that fail to fold properly such as the common CFTR mutant, F508del, undergo glucose- and mannose-trimming as an important quality control mechanism. The lectin EDEM seems to be essential for the recognition and targeting of misfolded CFTR channels to ubiquitin mediated degradation (Gnann et al., 2004). Subsequently, the ubiquitin E3, RMA1 (RNF5) monoubiquitylates F508del CFTR and gp78 mediated polyubiquitylation follows (Younger et al., 2006; Morito et al., 2008). SYVN1 (HRD1) has been shown to support RMA1/gp78 facilitated modification dependent on its catalytic activity (Ramachandran et al., 2016). While RMA1 responds to early folding defects, late in the maturation process, after translation has concluded, Csp recruits another E3 ligase, CHiP, to promote CFTR ubiquitylation (Meacham et al., 2001; Schmidt et al., 2009). As discussed above, another PTM, Hsp27, mediated SUMO-2/3 modification targets F508del CFTR to RNF4 catalyzed ubiquitylation and proteasomal degradation (Ahner et al., 2013). Whether these two pathways are completely independent and whether they target different pools of folding mutants remains to be determined. Other ubiquitin E3 ligases implicated as having the capacity to catalyze F508del degradation include SCF, NEDD4L, and RNF185 (Yoshida et al., 2002; Caohuy et al., 2009; El Khouri et al., 2013). In some cases, the evidence for the involvement of these ubiquitin E3s in misfolded CFTR disposal derives from their exogenous expression in model systems and may not reflect their physiological significance in the cell types affected by CF.
Previously, we identified an interaction between F508del CFTR and the small heat shock protein, Hsp27, which cooperated with the SUMO E2 enzyme, Ubc9, to modify the CFTR mutant with SUMO-2/3 (Ahner et al., 2013). This SUMO modification of F508del did not require the intervention of a SUMO E3 as the SUMO E2, Ubc9, can itself perform enzymatic conjugation of SUMO to target proteins. The polychains formed by SUMO-2/3 are recognized by RNF4, a SUMO-targeted ubiquitin ligase having four SUMO interacting motifs (SIMs) at its N-terminus (Tatham et al., 2008), leading to F508del ubiquitylation and degradation by the UPS. Thus, we can ask the following: why would yet another ubiquitin E3 ligase be needed to degrade F508del CFTR in addition to the eight ubiquitin E3 pathways listed above? This question prompted us to explore identification of other SUMO interacting proteins that might bind WT or F508del CFTR using a protein array probed for binding of either mono-SUMO-1 or SUMO-3 polychains; this array had previously identified RNF4 and several SUMO pathway components. The SUMO E3, PIAS4, was a prominent candidate on that list, as it is known to have two SUMOylation sites and eight SIMs.
PIAS4/SUMO-1 and CFTR biogenesis
The results of this study showed that PIAS4 stimulated the biogenesis of both WT and F508del CFTR, thus opposing the prodegradative pathway composed of Hsp27-Ubc9-SUMO-2/3-RNF4 (Ahner et al., 2013). All experiments here were performed in CFBE airway cells, either the parental line, CFBE41o- (Bruscia et al., 2002), which expresses little or no endogenous F508del CFTR, or using cells derived from this line and transduced to stably express WT or F508del CFTR (Bebok et al., 2005). The action of PIAS4 on WT CFTR was somewhat unexpected since the Hsp27/Ubc9 pathway selectively targeted F508del degradation, with no significant activity against the wild-type protein. This behavior results from detection of misfolded F508del by the sHsp, which targets a transitional (partially denatured) conformation of the mutant (Gong et al., 2016). For WT CFTR, PIAS4 increased most strongly the steady-state expression of immature CFTR (3.2-fold) while increasing the mature protein by 2.2-fold. Somewhat smaller but proportionately similar increases in F508del expression (2.8-fold for band B vs. 1.8-fold for band C; Figure 1A) were produced by PIAS4. Moreover, PIAS4 augmented the response to the class I correctors C18 and VX-809 (Okiyoneda et al., 2013) up to threefold over control (Figure 1B), and this was confirmed by cell-surface biotinylation (Figure 2, C and D) and by surface labeling of FAP-tagged F508del CFTR (Figure 4A). Currents generated by F508del CFTR that was rescued to the plasma membrane by PIAS4 were stimulated by forskolin and inhibited by GlyH-101, as detected using whole-cell patch clamp.
The physiological significance of PIAS4's impact was reflected by its knockdown, which reduced the steady-state CFTR expression levels in CFBE-WT or -F508del cells (Figure 2, A and B), as well as decreasing the cell-surface expression of WT CFTR (Figure 2D). Conversely, overexpression of PIAS4 augmented the cell-surface expression of WT CFTR nearly twofold (Figure 2C). In cycloheximide chase experiments, the increase in immature (band B) F508del was traced to a threefold slowing in the degradation rate of the mutant (Figure 3B). In contrast, the kinetics of disposal of immature WT CFTR were minimally altered by PIAS4; that is, a similar effect on ERAD did not appear to account for increased expression of WT CFTR band B. The impact of PIAS4 to stabilize WT band C could obscure an action of the SUMO E3 on WT band B, since the ER is the source of folding-competent, mature CFTR at the PM, which is more stable than its ER precursor.
Both in vitro and in vivo studies demonstrated that PIAS4 preferentially stimulated the SUMO-1 modification of F508del CFTR, while decreasing its conjugation to SUMO-2/3 (Figure 5, A and B). As SUMO-2/3 polychains are recognized by the ubiquitin E3, RNF4, we found also that PIAS4 reduced the ubiquitylation of F508del by 50% and eliminated the RNF4 induced reduction in the expression level of F508del CFTR.
While PIAS4's ability to promote CFTR biogenesis was striking, we should nevertheless ask whether PIAS4, or SUMO-1, is a physiological modulator of CFTR biogenesis. Prior studies have primarily implicated PIAS proteins in their abilities to regulate nuclear events, usually based on their modulation of gene transcription (Rytinki et al., 2009). qPCR did not detect significant PIAS4-induced changes in CFTR mRNA levels, and the actions of cycloheximide implied a posttranslational action of the E3; nevertheless, it is possible that changes in the expression of proteins other than PIAS4 contribute to the increases in immature CFTR that we observe for both WT and mutant CFTR in response to PIAS4 expression. However, other findings are consistent with a direct effect of the SUMO E3. First, when purified PIAS4 protein was added to in vitro SUMOylation reactions that included either SUMO-1 or SUMO-2, the modification of F508del NBD1 by SUMO-1 was nearly doubled, while its conjugation to SUMO-2 was significantly reduced (Figure 5A). These results were reproduced in vivo where full-length CFTR and its SUMO-1 modification were increased by PIAS4 overexpression in CFBE-F508del cells (Figure 5B). In addition, the knockdown of PIAS4 as well as expression of the F508del-4KR CFTR mutant provided evidence that this pathway, which augments ER-based CFTR biogenesis, regulates CFTR directly. The changes in CFTR expression evoked by PIAS4 were also mimicked by the expression of activated SUMO-1 versus SUMO-2 or -3, conditions under which the effects of the SUMO E3 were reproduced in the absence of exogenous PIAS4 expression. While we cannot rule out the possibility that epigenetic changes contribute to the impact of PIAS4 on CFTR biogenesis, the ability to reproduce the impact of PIAS4 in vivo, with activated SUMO paralogues and with purified proteins in vitro, make this explanation less likely.
The SUMO paralogue switch
Our findings reflect the presence of opposing pathways that determine the fate of CFTR, biogenesis versus degradation, mediated by its posttranslational, and perhaps cotranslational, conjugation with different SUMO paralogues, SUMO-1 or SUMO-2/3. A similar divergence in functional outcome based on SUMO paralogue exchange at target protein conjugation sites has been termed a “paralogue switch” (Fasci et al., 2015). This behavior has been documented for the promyelocytic leukemia protein, PML, in its chemotherapeutic response to arsenic trioxide treatment. Arsenic reorganizes PML into nuclear bodies, from which it is targeted for degradation by the STUbL, RNF4. Arsenic trioxide induces the exchange of SUMO-2, conjugated with Lys65 of PML, for SUMO-1, which also drives SUMO-2 conjugation and SUMO chain extension at the downstream site, Lys160. The resulting SUMO-2 polychains are accessed by RNF4, leading to PML ubiquitylation and its proteasomal degradation. Arsenic treatment is thought to lead to PML oxidation, altering its conformation, which is proposed to determine the timing of the signal associated with these site-specific paralogue modifications.
Although not involving a functional paralogue switch, a protease-controlled paralogue specific modification has been observed for the GTPase activating protein for Ran, RanGAP1, the first recognized SUMO substrate in mammalian cells. This GAP protein is conjugated to SUMO-1 and SUMO-2 with identical efficiency in vitro. Yet in vivo, RanGAP1 modified with SUMO-1 exhibits a higher affinity for its SUMO E3 ligase, RanBP2/Nup358, and interaction of the SUMO E2, Ubc9, with this complex provides protection from SUMO‑specific protease (SENP) activity that could otherwise remove SUMO-1. Thus, RanGAP1-SUMO-2 remains exposed and is efficiently cleaved, yielding RanGAP1-SUMO-1, the predominant form maintained in vivo (Zhu et al., 2009; Gareau et al., 2012).
In a similar protease-controlled mechanism, paralogue selectivity might be generated for WT and mutant CFTR. Cells express an excess of SUMO-2/3, which is maintained in a free pool; in contrast, the majority of cellular SUMO-1 is conjugated to substrates (Flotho and Melchior, 2013). If this is true also for epithelial cells expressing CFTR, then these findings would suggest that both WT and F508del CFTR might be conjugated to SUMO-2/3, a default process that would promote degradation unless a stable CFTR conformation is achieved. Proteases cleaving off SUMO-2/3 might effectively reverse this SUMO modification of WT CFTR, permitting a shift toward SUMO-1 and promoting WT CFTR maturation. Folding deficiencies associated with mutant CFTR might obviate its access to SUMO protease and prevent SUMO-2/3 cleavage, directing F508del CFTR towards degradation.
The order of paralogue addition to CFTR could also be opposite to that suggested above. The modification of CFTR by SUMO-1 may occur early as a means of stabilizing nascent CFTR during its protracted domain folding/assembly process, which may take 30 min or more (Du et al., 2005). Several lines of evidence favor the concept that failure of the F508del mutant to achieve appropriate domain–domain interfaces is the limiting factor in mutant protein progression (Sharma et al., 2004; Du et al., 2005; Lewis et al., 2005; Thibodeau et al., 2005; Cui et al., 2007; Rabeh et al., 2012). SUMO modification is recognized to increase protein solubility and reduce aggregation (Butt et al., 2005; Esposito and Chatterjee, 2006). Indeed, commercial kits (e.g., www.lifesensors.com) feature SUMO fusions to enhance the production of difficult-to-express proteins (Lee et al., 2008), and a SUMO fusion construct has been used routinely to optimize the production of CFTR NBD1 and NBD2 for in vitro studies (Qu et al., 1997; Vernon et al., 2017). Likewise, our findings suggest that nature might utilize this strategy for stabilizing difficult-to-fold proteins. F508del is an intractable protein that stalls at one or more conformational transitions that must be overcome energetically and kinetically to achieve the native fold (Du and Lukacs, 2009). Thus, SUMO-1 could assist in stabilizing immature CFTR, but if this process ultimately fails, then a switch of SUMO-1 to SUMO-2/3, perhaps protease mediated, would expose the mutant to recognition by pathways that culminate in its disposal. The delineation of mechanism(s) that mediate this paralogue switch should clarify this decision point for manipulations that could therapeutically modulate CFTR fate.
MATERIALS AND METHODS
Antibodies and reagents
Monoclonal antibodies targeted CFTR NBD1, NBD2, and R-domain (#660, #596, and #217, respectively; Cystic Fibrosis Foundation Therapeutics, Bethesda, MD); other monoclonal antibodies were to the HA- or Flag-tag (Sigma-Aldrich, St. Louis, MO). Polyclonal antibodies to ubiquitin, Hsp70, SUMO-1, and SUMO-2/3 were from Enzo life Sciences (Ann Arbor, MI), antibodies to actin, PIAS4, and the myc-tag were obtained from Sigma-Aldrich (St. Louis, MO). Horseradish peroxidase–conjugated secondary antibodies, anti-mouse, and anti-rabbit were obtained from Amersham-Pharmacia Biotech (GE Healthcare Bio-Sciences, Piscataway, NJ), N-ethylmaleimide (NEM) was purchased from Thermo Scientific (Rockford, IL), complete protease inhibitor cocktail tablets were obtained from Roche Diagnostics Corporation (Indianapolis, IN), and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO) at highest grade available.
Plasmid constructs, adenoviral vector, and site-directed mutagenesis
The pCMV-Flag-hPIAS4 and pCMV-Flag-hPIASx alpha plasmids were obtained from Addgene (Cambridge, MA). The Flag-RNF4 plasmid was a generous gift of Ronald T. Hay (University of Dundee, Dundee, UK). The pCMV-Flag-hPIAS4-CA (C342/347A) and pCMV-Flag-hPIAS4-2KR (K35/128R) mutants were generated using the following primers:
5′-CCGGGCAGAGACCGCCGCCCACCTGCAG-3′ (C342A, forward),
5′-CTGCAGGTGGGCGGCGGTCTCTGCCCGG-3′ (C342A, reverse),
5′-CGCCCACCTGCAGGCCTTCGACGCCGTC-3′ (C347A, forward),
5′-GACGGCGTCGAAGGCCTGCAGGTGGGCG-3′ (C347A, reverse).
The pcDNA3.1-F508del CFTR-4KR (K377/447/1199/1468R) mutant was generated using the primers: 5′-TGGAGCAATAAACAAAATACAGGATTTCTTACAAAGGCAAGAATATAAGACATT-3′ (K377R, forward),
5′-AATGTCTTATATTCTTGCCTTTGTAAGAAATCCTGTATTTTGTTTATTGCTCCA-3′ (K377R, reverse),
5′-CCTGTCCTGAAAGATATTAATTTCAGGATAGAAAGAGGACAGTTG-3′ (K447R, forward), 5′-CAACTGTCCTCTTTCTATCCTGAAATTAATATCTTTCAGGACAGG-3′ (K447R, reverse),
5′-TTGAGAATTCACACGTGAGGAAAGATGACATCTGGCC-3′ (K1199R, forward),
5′-GGCCAGATGTCATCTTTCCTCACGTGTGAATTCTCAA-3′ (K1199R, reverse),
5′-AGCCCCAGATTGCTGCTCTGAGAGAGGAGACAG-3′ (K1468R, forward),
5′-CTGTCTCCTCTCTCAGAGCAGCAATCTGGGGCT-3′ (K1468R, reverse).
Mutants were obtained using the QuickChange lightning Site-Directed Mutagensis Kit (Agilent Technologies, Santa Clara, CA). All plasmid constructs were sequence verified.
For adenovirus production, PIAS4 was amplified by reverse transcription PCR (RT-PCR) from a CFBE cDNA library. The amplification fragment was cloned into the pAD/DEST vector (Invitrogen, Carlsbad, CA), the clones were confirmed by sequencing, and dicer-substrate interfering RNA (DsiRNA) targeting PIAS4 were synthesized from literature experience (Morris et al., 2009) and obtained from Integrated DNA Technologies (Coralville, IA). The DNA cloned into the pAD/CMV/DEST vector was then confirmed by sequencing (Genewiz, South Plainfield, NJ), and 293A cells were transfected with pAd/CMV/V5-DEST-GFP-PIAS4 or its -DsiRNA using Lipofectamine 2000 (Invitrogen). High-titer adenovirus stocks were generated from 293A cell crude homogenates using the Vivapure AdenoPACK 100 kit following the manufacturer's instructions.
Cell culture and protein expression
CFBE41o- airway cells were cultured in MEM (Invitrogen, Carlsbad, CA) and supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), and 2 mM l-glutamine, 50 U/ml penicillin, and 50 µg/ml streptomycin. For CFBE WT and F508del stable cell lines, 0.5 and 2 µg/ml Hygromycin B (InvivoGen, San Diego, CA) were added, respectively, as selection agents. All cells were maintained in a humidified chamber with 5% CO2 at 37°C.
For protein overexpression, CFBE-WT, -F508del stable cell lines (Bebok et al., 2005) or parental CFBE41o- cells grown in six-well plates were transiently transfected with the indicated expression plasmids using Lipofectamine 2000 (Invitrogen) as indicated. After 48 h, cells were rinsed with phosphate-buffered saline (PBS) and lysed in RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) with protease inhibitors. Samples were incubated for 15 min in RIPA buffer followed by several short bursts of sonication with a tip sonicator and then centrifuged at 16,000 × g for 10 min at 4°C. Cell lysates were used for immunoblot analyses.
Immunoblotting and coimmunoprecipitation assays
Equal amounts of proteins from CFBE-WT, -F508del, or parental cell lysates were resolved by SDS–PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (PerkinElmer, Boston, MA). Unbound sites were blocked for 1 h at room temperature with 5% non-fat milk powder in TBST (20 mM Tris, 150 mM NaCl, 0.01% Tween 20, pH 8.0). The blots were incubated with primary antibodies (anti-CFTR #217, 1:5000; anti-actin, 1:4000; anti-Hsp70, 1:2000; anti-Flag, 1:10,000; anti-PIAS4, 1:1000) at room temperature for 1 h. The blots were then washed three times for 10 min each with TBST and incubated for 1 h with horseradish peroxidase–conjugated secondary antibodies in TBST containing 5% non-fat milk, followed by three TBST washes. The reactive bands were visualized with enhanced chemiluminescence (PerkinElmer Life Sciences, Wellesley, MA) and exposed to x-ray film or, for some experiments, imaged and quantified on a Li-Cor Odyssey.
For coimmunoprecipitation assays, precleared cell lysates (∼1 mg of protein) were mixed with respective primary antibodies for 2 h at 4°C in lysis buffer (20 mM Tris, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, pH 8.0, containing protease inhibitors). For the detection of in vivo SUMOylation, 20 mM NEM was added to the lysis buffer. Fifty microliters of washed protein A- or G-agarose beads was added to each sample and incubated 4 h at 4°C with gentle rotation. Immunocomplexes were washed three times with lysis buffer and precipitated by centrifugation at 12,000 × g for 10 s. Then the immunocomplexes were resuspended in SDS sample buffer and subjected to immunoblotting.
In vitro SUMOylation assays
The in vitro assay was performed using reagents purchased from Enzo Life Sciences (Ann Arbor, MI) together with F508del human NBD1 protein that contained a single solubilizing mutation, F494N, as previously described (Rabeh et al., 2012; Gong et al., 2016). In brief, 15 ng of purified NBD1 was incubated in SUMOylation buffer with a reaction mixture containing recombinant E1 (0.4 μM), Ubc9 (4 μM), a SUMO paralogue (3 μM), and Mg-ATP (2 mM), with or without purified recombinant human PIAS4 protein (15 ng), for 1 h at 27°C. After the reaction was terminated with SDS sample buffer containing 2-mercaptoethanol, reaction products were fractionated on 12% SDS–PAGE, and the gel shift resulting from SUMO modification was detected by immunoblotting using anti-CFTR NBD1 (#660).
Cycloheximide chase
CFBE-WT, -F508del, or parental cells were plated and transiently transfected as described above. After 48 h, cells were incubated with 100 μg/ml CHX for the indicated times (Figure 3), at which cells were harvested, lysed with RIPA buffer containing protease inhibitors, and subjected to immunoblot to determine the time course of CFTR decay following the inhibition of protein synthesis.
In vivo ubiquitylation assays
CFBE-F508del cells were plated and transiently transfected as described above. After 48 h, the cells were treated with 50 μM MG132 for 5 h and then lysed in lysis buffer containing protease inhibitors. Precleared lysates were mixed with CFTR antibodies (#217 or #596) for 2 h at 4°C in lysis buffer. Washed protein G agarose beads (50 μl) were added to each sample and incubated 4 h at 4°C with gentle rotation. Immunocomplexes were analyzed by immunoblotting with anti-ubiquitin antibody.
Total RNA extraction and real-time PCR
CFBE-F508del cells were transfected with empty vector, pCMV-Flag-hPIAS4, or pCMV-Flag-hPIASx alpha plasmids. After 48 h, total RNA from transfected cells was purified using the RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions; 1 μg of the total RNA was converted to cDNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Equal amounts of each single-chain cDNA were subjected to qPCR using Bio Rad iQ SYBR Supermix. The qPCR was completed using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) for 3 min at 95°C followed by 39 cycles of 10 s at 95°C and 30 s at 55°C. The primer sequences used were as follows:
CFTR: 5′-CACAGCAACTCAAACAACTGG-3′ (forward),
5′-TGTAACAAGATGAGTGAAAATTGGA-3′ (reverse);
actin: 5′-ATTGGCAATGAGCGGTTC-3′ (forward),
5′-CGTGGATGCCACAGGACT-3′ (reverse).
The Ct values were determined for each reaction (in triplicate) using Bio-Rad CFX Manager Software, and quantification was completed using the ΔΔCt method.
Expression of functional cell-surface protein
CFBE-WT or -F508del cells were subjected to cell-surface biotinylation as described (Silvis et al., 2009). Briefly, control cells or those transduced with AdPIAS4 were treated with EZ-Link Sulfo-NHS-LC-LC-Biotin (Pierce, Rockford, IL). The experiments were performed on ice with all solutions at 4°C. To biotinylate apical proteins, cells were rinsed once with PBS + CM (lacking CaCl2 + MgCl2) and then washed with PBS + CM containing 10% fetal bovine serum (FBS). The apical surface was biotinylated with 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin in borate buffer (85 mM NaCl, 4 mM KCl, and 15 mM Na2B4O7, pH 9) for 30 min with gentle agitation. Excess biotin was removed using two 10-min washes in PBS + CM with 10% FBS, followed by two washes in PBS + CM.
Cell surface CFTR was quantified by lysing cells in 700 μl biotinylation lysis buffer (BLB: 0.4% deoxycholate, 1% NP40, 50 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid [EGTA], 10 mM Tris–Cl, pH 7.4, and Complete EDTA-free Protease Inhibitor Cocktail [Roche Diagnostics]). After protein levels were determined, normalized samples were incubated overnight with 150 μl UltraLink Immobilized NeutrAvidin Protein Plus (Pierce). Precipitated proteins were washed three times with BLB, solubilized with Laemmli sample buffer, resolved by SDS–PAGE, and blotted for CFTR.
A clone of CFBE cells stably expressing a FAP-F508del CFTR construct was generated as previously described for a 293A stable cell line (Larsen et al., 2016). These FAP-CFBE-F508del expressing cells were plated in a 96-well plate in complete DMEM containing 8 µg/ml polybrene with or without addition of recombinant adenovirus encoding PIAS4. Two days postplating, the growth media was exchanged to complete DMEM containing 2 µM VX-809 and cells were transferred to a tissue culture incubator set at 27°C. The following day, FAP-F08del CFTR surface expression was detected quantitatively at 37°C by automated microscopy analysis as described previously (Larsen et al., 2016).
Whole-cell currents were recorded from transfected CFBE41o- cells as previously described for HEK293 cells (Bertrand et al., 2009). Briefly, the electronics consisted of a 200B Axopatch amplifier controlled by Clampex 8.1 software through a Digidata 1322A acquisition board (Axon Instruments). Solutions were maintained at 37°C with a flow rate of 2.0 ml/min. The imaging chamber was mounted on the stage of a Nikon Diaphot microscope equipped with standard illumination and a xenon lamp with GFP filter cube (Ex 485/Em 550) permitting identification of GFP-expressing cells. Seal resistances exceeded 8 GΩ and pipette capacitance was compensated; all experiments used a standard voltage-clamp protocol with a holding potential of −40 mV. NMDG-Cl was used for both the bath and pipette solutions to isolate chloride currents. The bath solution was (in mM) 140 NMDG-Cl, 10 HEPES, 1 MgCl2, 1.5 CaCl2, 5 glucose, pH 7.3, and the pipette solution was (in mM) 140 NMDG-Cl, 10 HEPES, 1 MgCl2, 5 glucose, 1 EGTA, pH 7.2, and also contained 1 mM Mg-ATP and 100 μM GTP. The osmolarity of the bath and pipette solutions were measured and NMDG-glutamate was added to the bath solution to generate a 25 mOsm gradient that was needed to obviate cell swelling (Worrell et al., 1989). Pipettes using thin wall borosilicate glass were pulled to tip diameters of 1–2 μm (access resistance <4 MΩ).
Statistical analysis
A p value of <0.05 was considered statistically significant. The normality of data distribution was validated by the Kolmogorov–Smirnov normality test and visual inspection of quantile-quantile (QQ) plots. Statistical analysis was performed using the paired t test with the means of at least three independent experiments if the data were normally distributed. If this condition could not be confirmed, the Wilcoxon signed-rank test was used. We used SAS (SAS Institute, Cary, NC) for statistical analyses.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK68196 and DK72506) and the Cystic Fibrosis Foundation (FRIZZE05XX0). Thanks to Marcel Bruchez of Carnegie Mellon University for supplying MG-B-Tau for the FAP experiments and to Gergely Lukacs, McGill University, Montreal, Canada, for supplying F508del NBD1 with a single solubilizing mutation.
Abbreviations used:
- ABC transporter
ATP-binding cassette transporter
- cAMP
cyclic adenosine monophosphate
- CFBE
human cystic fibrosis bronchial epithelial
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHip
C-terminus of Hsp70-interacting protein
- CHX
cycloheximide
- DMSO
dimethyl sulfoxide
- E1
SUMO-activating enzyme
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum–associated degradation
- FAP
fluorogen activating protein
- FL
full length
- GFP
green fluorescent protein
- Hsp
heat shock protein
- Hsp27
heat shock protein 27
- IB
immunoblot
- IP
immunoprecipitation
- MSD
membrane-spanning domain
- NBD
nucleotide-binding domain
- NEM
N-ethylmaleimide
- PIAS4
protein inhibitor of activated STAT 4
- PKA
protein kinase A
- PKC
protein kinase C
- PM
plasma membrane
- PTM
posttranslational modification
- R domain
regulatory domain
- RNF4
ring finger protein 4
- SENP
SUMO‑specific protease
- shRNA
short hairpin RNA
- SP-RING domain
Siz/PIAS RING domain
- STAT 4
signal transducer and activator of transcription 4
- STUbL
SUMO-targeted ubiquitin ligases
- SUMO
small ubiquitin-like modifier
- Ubc5
ubiquitin-conjugating enzyme E2
- Ubc9
SUMO E2-conjugating enzyme
- VX-770
Ivacaftor
- VX-809
lumacaftor
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-04-0252) on November 7, 2018.
REFERENCES
- Adorisio S, Fierabracci A, Muscari I, Liberati AM, Ayroldi E, Migliorati G, Thuy TT, Riccardi C, Delfino DV. (2017). SUMO proteins: guardians of immune system. J Autoimmun , 21–28. [DOI] [PubMed] [Google Scholar]
- Ahner A, Gong X, Schmidt BZ, Peters KW, Rabeh WM, Thibodeau PH, Lukacs GL, Frizzell RA. (2013). Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol Biol Cell , 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD. (2004). CFTR and chaperones: processing and degradation. J Mol Neurosci , 41–48. [DOI] [PubMed] [Google Scholar]
- Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, Sorscher EJ, Clancy JP. (2005). Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol , 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson M, Iniguez-Lluhi JA, Martens J. (2017). Sumo modification of ion channels. Adv Exp Med Biol , 127–141. [DOI] [PubMed] [Google Scholar]
- Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. (2009). SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol , 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruscia E, Sangiuolo F, Sinibaldi P, Goncz KK, Novelli G, Gruenert DC. (2002). Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther , 683–685. [DOI] [PubMed] [Google Scholar]
- Butt TR, Edavettal SC, Hall JP, Mattern MR. (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif , 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caohuy H, Jozwik C, Pollard HB. (2009). Rescue of DeltaF508-CFTR by the SGK1/Nedd4-2 signaling pathway. J Biol Chem , 25241–25253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, et al (2012). Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax , 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Aleksandrov L, Chang XB, Hou YX, He L, Hegedus T, Gentzsch M, Aleksandrov A, Balch WE, Riordan JR. (2007). Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol , 981–994. [DOI] [PubMed] [Google Scholar]
- Cyr DM, Hohfeld J, Patterson C. (2002). Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci , 368–375. [DOI] [PubMed] [Google Scholar]
- Du K, Lukacs GL. (2009). Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell , 1903–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Sharma M, Lukacs GL. (2005). The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol , 17–25. [DOI] [PubMed] [Google Scholar]
- El Khouri E, Le Pavec G, Toledano MB, Delaunay-Moisan A. (2013). RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J Biol Chem , 31177–31191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Chatterjee DK. (2006). Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol , 353–358. [DOI] [PubMed] [Google Scholar]
- Fasci D, Anania VG, Lill JR, Salvesen GS. (2015). SUMO deconjugation is required for arsenic-triggered ubiquitylation of PML. Sci Signal , ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotho A, Melchior F. (2013). Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem , 357–385. [DOI] [PubMed] [Google Scholar]
- Frizzell RA, Hanrahan JW. (2012). Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med , a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Lima CD. (2010). The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol , 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Reverter D, Lima CD. (2012). Determinants of small ubiquitin-like modifier 1 (SUMO1) protein specificity, E3 ligase, and SUMO-RanGAP1 binding activities of nucleoporin RanBP2. J Biol Chem , 4740–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnann A, Riordan JR, Wolf DH. (2004). Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell , 4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Ahner A, Roldan A, Lukacs GL, Thibodeau PH, Frizzell RA. (2016). Non-native conformers of cystic fibrosis transmembrane conductance regulator NBD1 are recognized by Hsp27 and conjugated to SUMO-2 for degradation. J Biol Chem , 2004–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RJ, Rich DP, Cheng SH, Souza DW, Paul S, Manavalan P, Anderson MP, Welsh MJ, Smith AE. (1991). Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol , 3886–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, Vertegaal AC. (2016). A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol , 581–595. [DOI] [PubMed] [Google Scholar]
- Heo KS, Berk BC, Abe J. (2016). Disturbed flow-induced endothelial proatherogenic signaling via regulating post-translational modifications and epigenetic events. Antioxid Redox Signal , 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran JP, Glover ML, Peters KW, Bertrand CA, Watkins SC, Jarvik JW, Frizzell RA. (2012). Pharmacological rescue of the mutant cystic fibrosis transmembrane conductance regulator (CFTR) detected by use of a novel fluorescence platform. Mol Med , 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran JP, Zeng J, Frizzell RA, Watkins SC. (2013). Regulated recycling of mutant CFTR partially restored by pharmacological treatment. J Cell Sci , 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. (1995). Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell , 129–135. [DOI] [PubMed] [Google Scholar]
- Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, Huang X, Lubarsky B, Rubin J, Millar SJ, et al (2017). Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med , 107–118. [DOI] [PubMed] [Google Scholar]
- Larsen MB, Hu J, Frizzell RA, Watkins SC. (2016). Simple image-based no-wash method for quantitative detection of surface expressed CFTR. Methods , 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear T, McKelvey AC, Rajbhandari S, Dunn SR, Coon TA, Connelly W, Zhao JY, Kass DJ, Zhang Y, Liu Y, Chen BB. (2016). Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4. J Exp Med , 1029–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CD, Sun HC, Hu SM, Chiu CF, Homhuan A, Liang SM, Leng CH, Wang TF. (2008). An improved SUMO fusion protein system for effective production of native proteins. Protein Sci , 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, et al (2005). Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem , 1346–1353. [DOI] [PubMed] [Google Scholar]
- Liang X, Da Paula AC, Bozoky Z, Zhang H, Bertrand CA, Peters KW, Forman-Kay JD, Frizzell RA. (2012). Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis. Mol Biol Cell , 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YC, Lee CC, Yao YL, Lai CC, Schmitz ML, Yang WM. (2016). SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci Rep , 26509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebelt F, Vertegaal AC. (2016). Ubiquitin-dependent and independent roles of SUMO in proteostasis. Am J Physiol Cell Physiol , C284–C296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner RA, Treweek TM, Carver JA. (2001). The molecular chaperone alpha-crystallin is in kinetic competition with aggregation to stabilize a monomeric molten-globule form of alpha-lactalbumin. Biochem J , 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. (1999). The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. Embo J , 1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. (2001). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol , 100–105. [DOI] [PubMed] [Google Scholar]
- Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata K. (2008). Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell , 1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, et al (2009). The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature , 886–890. [DOI] [PubMed] [Google Scholar]
- Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. (2004). Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol , 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, et al (2013). Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol , 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM. (2005). A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem Biophys Res Commun , 517–520. [DOI] [PubMed] [Google Scholar]
- Pichler A, Fatouros C, Lee H, Eisenhardt N. (2017). SUMO conjugation—a mechanistic view. Biomol Concepts , 13–36. [DOI] [PubMed] [Google Scholar]
- Qu BH, Strickland EH, Thomas PJ. (1997). Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem , 15739–15744. [DOI] [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, et al (2012). Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell , 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman K, Raman B, Rao CM. (1996). Molten-globule state of carbonic anhydrase binds to the chaperone-like alpha-crystallin. J Biol Chem , 27595–27600. [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Osterhaus SR, Parekh KR, Jacobi AM, Behlke MA, McCray PB., Jr (2016). SYVN1, NEDD8, and FBXO2 proteins regulate DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) ubiquitin-mediated proteasomal degradation. J Biol Chem , 25489–25504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytinki MM, Kaikkonen S, Pehkonen P, Jaaskelainen T, Palvimo JJ. (2009). PIAS proteins: pleiotropic interactors associated with SUMO. Cell Mol Life Sci , 3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BZ, Watts RJ, Aridor M, Frizzell RA. (2009). Cysteine string protein promotes proteasomal degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) by increasing its interaction with the C terminus of Hsp70-interacting protein and promoting CFTR ubiquitylation. J Biol Chem , 4168–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, Bache KG, Papsin B, Zerangue N, Stenmark H, Lukacs GL. (2004). Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol , 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Liu B. (2005). Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol , 593–605. [DOI] [PubMed] [Google Scholar]
- Silvis MR, Bertrand CA, Ameen N, Golin-Bisello F, Butterworth MB, Frizzell RA, Bradbury NA. (2009). Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol Biol Cell , 2337–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth CV, Verma S. (2017). Sumoylation as an integral Mmchanism in bacterial infection and disease progression. Adv Exp Med Biol , 389–408. [DOI] [PubMed] [Google Scholar]
- Strickland E, Qu BH, Millen L, Thomas PJ. (1997). The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem , 25421–25424. [DOI] [PubMed] [Google Scholar]
- Sun F, Mi Z, Condliffe SB, Bertrand CA, Gong X, Lu X, Zhang R, Latoche JD, Pilewski JM, Robbins PD, Frizzell RA. (2008). Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. Faseb J , 3255–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Zhang R, Gong X, Geng X, Drain PF, Frizzell RA. (2006). Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J Biol Chem , 36856–36863. [DOI] [PubMed] [Google Scholar]
- Tatham MH, Geoffroy MC, Shen L, Plechanovova A, Hattersley N, Jaffray EG, Palvimo JJ, Hay RT. (2008). RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol , 538–546. [DOI] [PubMed] [Google Scholar]
- Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. (2005). Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol , 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treweek TM, Lindner RA, Mariani M, Carver JA. (2000). The small heat-shock chaperone protein, alpha-crystallin, does not recognize stable molten globule states of cytosolic proteins. Biochim Biophys Acta , 175–188. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA , 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon RM, Chong PA, Lin H, Yang Z, Zhou Q, Aleksandrov AA, Dawson JE, Riordan JR, Brouillette CG, Thibodeau PH, Forman-Kay JD. (2017). Stabilization of a nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator yields insight into disease-causing mutations. J Biol Chem , 14147–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Kopito RR. (1994). Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem , 25710–25718. [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. (1995). Degradation of CFTR by the ubiquitin-proteasome pathway. Cell , 121–127. [DOI] [PubMed] [Google Scholar]
- Worrell RT, Butt AG, Cliff WH, Frizzell RA. (1989). A volume-sensitive chloride conductance in human colonic cell line T84. Am J Physiol , C1111–1119. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Chiba T, Tokunaga F, Kawasaki H, Iwai K, Suzuki T, Ito Y, Matsuoka K, Yoshida M, Tanaka K, Tai T. (2002). E3 ubiquitin ligase that recognizes sugar chains. Nature , 438–442. [DOI] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. (2006). Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell , 571–582. [DOI] [PubMed] [Google Scholar]
- Zhu S, Goeres J, Sixt KM, Bekes M, Zhang XD, Salvesen GS, Matunis MJ. (2009). Protection from isopeptidase-mediated deconjugation regulates paralog-selective sumoylation of RanGAP1. Mol Cell , 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.