

Abstract
Aims:
Peroxisome proliferator-activated receptor γ (PPARγ) agonists reduce blood pressure and vascular injury in hypertensive rodents. Pparγ inactivation in vascular smooth muscle cells (VSMC) enhances vascular injury. Transgenic mice overexpressing endothelin (ET)-1 selectively in the endothelium (eET-1) exhibit endothelial dysfunction, increased oxidative stress and inflammation. We hypothesized that inactivation of the Pparγ gene in VSMC (smPparγ−/−) would exaggerate ET-1-induced vascular injury.
Methods and results:
eET-1, smPparγ−/− and eET-1/ smPparγ−/− mice were treated with tamoxifen for 5 days and studied 4 weeks later. SBP was higher in eET-1 and unaffected by smPparγ inactivation. Mesenteric artery vasodilatory responses to acetylcholine were impaired only in smPparγ−/−. Nω-Nitro-L-arginine methyl ester abrogated relaxation responses, and the Ednra/Ednrb mRNA ratio was decreased in eET-1/smPparγ−/−, which could indicate that nitric oxide production was enhanced by ET-1 stimulation of endothelin type B receptors. Mesenteric artery media/ lumen was greater only in eET-1/smPparγ−/−. Mesenteric artery reactive oxygen species increased in smPparγ−/− and were further enhanced in eET-1/smPparγ−/−. Perivascular fat monocyte/macrophage infiltration was higher in eET-1 and smPparγ−/− and increased further in eET-1/smPparγ−/−. Spleen CD11b+ cells were increased in smPparγ−/− and further enhanced in eET-1/smPparγ−/−, whereas Ly-6Chi monocytes increased in eET-1 and smPparγ−/− but not in eET-1/smPparγ−/−. Spleen T regulatory lymphocytes increased in smPparγ−/− and decreased in eET-1, and decreased further in eET-1/smPparγ−/−.
Conclusion:
VSMC Pparγ inactivation exaggerates ET-1-induced vascular injury, supporting a protective role for PPARγ in hypertension through modulation of pro-oxidant and proinflammatory pathways. Paradoxically, ET-1 overexpression preserved endothelial function in smPparγ−/− mice, presumably by enhancing nitric oxide through stimulation of endothelin type B receptors.
Keywords: hypertension, inflammation, oxidative stress, small arteries, vascular injury
INTRODUCTION
Endothelin (ET)-1 is a powerful vasoconstrictor peptide produced by endothelial cells that acts on underlying vascular smooth muscle cells (VSMC) to cause contraction and proliferation in VSMC through activation of endothelin type A (ETA) and endothelin type B (ETb) receptors. ET-1 also binds on endothelial ETB receptors and promotes vasorelaxation by release of nitric oxide (NO), and prostacyclin [1]. ET-1 contributes to cardio-vascular disease by causing endothelial dysfunction and vascular remodeling. Transgenic mice constitutively over-expressing human ET-1 selectively in the endothelium (eET-1) exhibit increased vascular remodeling, oxidative stress and inflammation in the absence of significant blood pressure (BP) changes [2], whereas mice with inducible endothelial overexpression of human ET-1 present persistent elevation of BP [3].
Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated transcription factor that regulates adipogenesis and insulin sensitivity. PPARγ ligands include free fatty acids (FA) and thiazolidinedione antidiabetic drugs such as pioglitazone or rosiglitazone. Although PPARγ is largely expressed in adipocytes, it is also present in endothelial cells and VSMC. In humans, Pparγ-dominant negative mutations are associated with diabetes mellitus, severe insulin resistance and hypertension [4], whereas treatment with PPARγ agonists improve insulin sensitivity and may moderately reduce BP [5]. In hypertensive rodents, PPARγ agonists pioglitazone or rosiglitazone reduced BP and improved vascular remodeling and endothelial dysfunction [6-8]. Heterozygous knockin mice expressing a dominant negative Pparγ mutant exhibit cerebral vascular dysfunction and remodeling, suggesting that PPARγ is important for vascular homeostasis [9]. Mice with endothelial cell-selective deletion of Pparγ have normal BP at baseline and elevated BP under a high-fat diet [10]. Overexpression of a dominant-negative Pparγ mutant in VSMC caused hypertension, endothelial dysfunction and hypertrophic remodeling of cerebral arteries [11]. Similarly, VSMC deletion of Pparγ exacerbated angiotensin Il-induced vascular remodeling and endothelial dysfunction [12]. Interestingly, the antiatherogenic but not antihypertensive effect of pioglita-zone was lost in mice with deletion of VSMC Pparγ [13].
There is evidence from in-vitro studies that PPARγ activation may counteract ET-1 effects. First, PPARγ agonists rosiglitazone and troglitazone dose-dependently inhibit production of ET-1 in endothelial cells by interfering with AP-1-binding activity [14,15]. Rosiglitazone also inhibits ET-1-induced nuclear factor of kappa light polypeptide gene enhancer in B cells (NFĸB) activation of vascular cell adhesion molecule 1, intercellular adhesion molecule 1 and cyclooxygenase-2 expression in rat VSMC [16] and blunts ET-1-induced vasoconstriction ex vivo by upregulating endothelial ETB receptor expression [17]. In vivo, there is only indirect evidence of a protective role for PPARγ in ET-1-induced vascular damage. In DOCA-salt hypertensive rats, in which hypertension is in part ET-1-dependent, rosiglitazone prevented BP elevation, endothelial dysfunction, vascular remodeling, reactive oxygen species (ROS) production and vascular production of ET-1 [7]. Thus, it is still unclear whether PPARγ modulates ET-1-induced vascular damage in vivo. In addition, the cells responsible for the vascular protective of PPARγ activation in models of enhanced ET-1 are still unknown.
We hypothesized that inactivation of the Pparγ gene in VSMC (smPparγ−/−) would exaggerate ET-1-induced vascular injury. To test this hypothesis, we evaluated endothelial function and vascular remodeling in a model of endothelial cell-restricted ET-1 overexpression with tamoxifen-inducible VSMC-restricted Pparγ deletion. We also investigated vascular oxidative stress and inflammation, as these processes play an important role in causing vascular injury.
MATERIALS AND METHODS
Experimental design
The study was approved by the Animal Care Committee of the Lady Davis Institute for Medical Research and McGill University, and followed recommendations of the Canadian Council for Animal Care. eET-1 were previously described [2]. An inducible tissue-specific Cre/loxP approach was used to ablate Pparγ in VSMC [12]. Mice with exon 2 of Pparγ flanked by LoxP sites crossed with smCreERT2 mice expressing a tamoxifen-inducible Cre recombinase fused with a modified estrogen receptor ligand-binding domain (CreERT2), selectively in VSMC, were crossed with eET-1 mice. Ten-to-11-week-old male smCreERT2, smCreERT2/ eET-1, smCreERT2/PparγFlox/Flox and smCreERT2/eET-1/ PparγFlox/Flox mice were treated for 5 days with tamoxifen (1 mg/day, subcutaneously) suspended in miglyol 812 (generously provided by Azelis Canada Chemicals Ltd, Scarborough, Ontario, Canada) and studied 4 weeks later. The experimental groups mentioned above will be herein referred to as control (Ctrl), eET-1, smPparγ−/− and eET-1/ smPparγ−/−, respectively. In a subset of animals, BP was determined by telemetry [3]. Mice mice were anesthetized with 3% isoflurane mixed with O2 at 1 l/min (depth of anesthesia confirmed by rear foot squeezing), the nonsteroidal anti-inflammatory drug carprofen (20 mg/kg) was administered subcutaneously to minimize the postoperative pain, then surgically instrumented with PA-C10 BP telemetry transmitters (Data Sciences International; Saint Paul, Minnesota, USA). Mice were allowed to recover for 7 days, and carprofen was administered as above once a day for the first 3 days. BP was determined before (baseline) and at weekly intervals after tamoxifen treatment for 5 weeks.
Collection of tissues
At the end of the protocol, mice were weighed, anesthetized with isoflurane as above and blood collected by cardiac puncture on EDTA for ET-1 determination by ELISA (R&D Systems Inc., Minneapolis, Minnesota, USA). The mesenteric artery vascular bed was dissected, and other tissues and tibia harvested in ice-cold phosphate-buffered saline (PBS). Tissues were weighed, and tibia length was determined. Spleen was used for monocyte and T-cell profiling. Second-order mesenteric arteries were used for assessment of endothelial function and vessel mechanics by pressurized myography. Segments of mesenteric artery were embedded in Clear Frozen Section Compound (VWR International; Edmonton, Alberta, Canada) for determination of ROS generation, expression of monocyte chemotactic protein-1 (MCP-1) or evaluation of tissue infiltration and polarization of monocyte/macrophages by immunofluorescence.
For the study of mRNA expression, the total mesenteric artery arcade was removed from the attached intestine under RNase-free conditions and stored immediately in RNAlater (Life Technologies; Burlington, Ontario, Canada). Lymph nodes, lymphatic ducts, connective tissue, nonper-ivascular fat and veins were removed under the microscope and discarded. The mesenteric artery arcade and perivascular adipose tissue (PVAT) were carefully dissected and stored in RNAlater until RNA extraction. In additional animals, VSMC were isolated from mesenteric artery and cultured for RNA extraction. Briefly, mesenteric arteries were cleaned of adipose and connective tissue, and VSMC were dissociated by the enzymatic digestion (collagenase, elastase and soybean trypsin inhibitor) of vascular arcades. The tissue was filtered and the cell suspension centrifuged and resuspended in DMEM containing heat-inactivated fetal bovine serum (FBS), HEPES, L-glutamine, penicillin and streptomycin. VSMC were cultured in DMEM containing 10% FBS and maintained at 37 °C in a humidified incubator (5% CO2/95% air). Early passage cells (passage 2) were used.
The extent of Cre activation was evaluated in reporter mice as previously described [12]. Ten-to-11-week-old male ROSA26mT-mG/mT-mG and smCreERT2/ROSA26mT-mG/mT-mG mice were treated for 5 days with tamoxifen as above and studied 4 weeks later. At the end of the protocol, mice were anesthetized with a 300-375 mg/kg intraperitoneal injection of Avertin [18]. Depth of anesthesia was confirmed by rear foot squeezing. The mice were injected intraper-itoneally with heparin (100 United States Pharmacopeia units), then perfused through the left ventricle at a constant pressure of 100mmHg for 5min with PBS to remove the blood, followed by 15-min perfusion with 4% paraformaldehyde (PFA). Tissues were collected and incubated in 4% PFA for 24 h with gentle agitation at 4 °C. Tissues were embedded in VWR Clear Frozen Section Compound (VWR International) and stored at −80 °C until used. Mesenteric arteries were stored in PBS at 4 °C until imaged.
Quantification of plasma endothelin-1
Blood samples were centrifuged at 1000 × g for 15min at 4°C to remove blood cells, followed by centrifugation at 10000 × g for 10min at 4 °C to remove platelets. Plasma samples were stored at −80 °C until tested. The concentration of ET-1 was determined in plasma on EDTA using a human ET-1 QuantiGlo ELISA Kit (R&D Systems Inc.).
Assessment of mT and mG expression
The expression of membrane-targeted tandem dimer Tomato (mT) and membrane-targeted green fluorescent protein (mG) was assessed by fluorescence microscopy imaging on intact mesenteric artery segments. Mesenteric artery segments were stained with DRAQ5 (1 μmol/l, Cell Signaling Technology, Danvers, Massachusetts, USA) for 20 min to label the nuclei and washed in PBS three times (20min each). Mesenteric arteries were mounted in Fluoromount (Sigma-Aldrich; St. Louis, Missouri, USA) and imaged using a Wave FX Spinning Disc Confocal microscope (Quorum Technologies; Guelph, Ontario, Canada).
Functional and morphological vascular studies
Second-order mesenteric artery, of average lumen size ~220 μm, were dissected, mounted on a pressurized myograph and endothelial function and vessel mechanics determined as previously described [19]. Vascular contractile properties were assessed by measuring the response to cumulative concentrations of norepinephrine (10−8 − 10−4mol/l) and ET-1 (10−13−10−6mol/l, Bachem; Torrance, California, USA). To evaluate the participation of NO to endothelium-dependent vasorelaxation and ET-1-induced contraction, the dose-response curve to each compound were determined before and after a 20-min preincubation with the NO synthase inhibitor Nω-nitro-L-arginine methyl ester (l-NAME, 10−4mol/l).
Vascular and perivascular reactive oxygen species production
Vascular and PVAT ROS production were assessed on 5-μm cryosections of mesenteric artery by measuring fluorescence after incubation with the ROS-sensitive fluorescent dye dihydroethidium (DHE, 2 μmol/l) in the dark for 1 min at 37 °C. Fluorescence was visualized and captured with a fluorescence microscope with a CY3 filter as previously described [19]. DHE fluorescence intensity per total surface area was quantified with ImageJ software (National Institute of Mental Health; Bethesda, Maryland, USA) and expressed as fold change relative to control (http://rsb.info.nih.gov/ij/).
Evaluation of monocyte chemotactic protein-1 expression and infiltration of monocyte/macrophages
Vascular inflammation was assessed by measuring the expression of MCP-1 in mesenteric artery and monocyte/ macrophage antigen-2 (MOMA-2) in mesenteric artery PVAT by immunofluorescence microscopy on 5-μm cryostat sections. Tissue cryosections were fixed in ice-cold acetone : methanol (1 : 1) mix for 10 min at room temperature (RT). Thereafter, sections were washed with PBS twice for 5 min and blocked for 1 h at RT with Tris buffered saline containing 0.1% Tween-20 (TBST), and 10% normal donkey serum (for MCP-1) or 10% normal goat serum (for MOMA-2). Sections were incubated overnight at 4 °C with goat antimouse MCP-1 antibody (1 : 50; Santa Cruz Biotechnology, Santa Cruz, California, USA) or rat antimouse MOMA-2 antibody (1 : 50; Abcam, Cambridge, Massachusetts, USA). The sections were then washed three times with TBST and blocked for 30 min at RT with TBST containing 10% normal donkey serum (for MCP-1) or 10% normal goat serum (for MOMA-2). Thereafter, sections were incubated for 1 h at RT with Alexa Fluor 568 donkey antigoat antibody (1 : 150; Invitrogen, Waltham, Massachusetts, USA) for MCP-1 or Alexa Fluor 568 goat antirat antibody (1 : 200, Invitrogen) for MOMA-2. Sections were washed afterward for three times with TBST, counterstained with 4’,6-diamidino-2-phenylindole (DAPI, 6 μmol/l, Life Technologies) and mounted with Fluoromount (Sigma-Aldrich).
Images were captured using a fluorescent microscope Leica DM2000 (Leica Microsystems, Richmond Hill, Ontario, Canada) and quantified with ImageJ software. The expression of MCP-1 was determined in mesenteric artery wall and was presented as relative fluorescence unit per μm2. Monocyte/macrophage infiltration was analyzed in mesenteric artery PVAT by determining the number of MOMA-2+ cells per mm2.
Determination of polarization of infiltrating monocyte/macrophages
Polarization of infiltrating monocyte/macrophages was determined by costaining for MOMA-2 and M1 (classically activated) macrophage marker-inducible NO synthase (iNOS) or major histocompatibility complex class II (MHC-II), or M2 (alternatively activated) macrophage markers mannose receptor (CD206) or arginase-1 (Arg-1) in 5-μm cryostat sections. Tissue cryosections were fixed in ice-cold acetone : methanol (1: 1) mix for 10 min at RT. Thereafter, sections were washed with PBS twice for 5min and blocked for 1 h at RT with TBST containing 10% normal goat serum (for iNOS and MHC-II costained with MOMA-2) or 5% bovine serum albumin, 0.4% Triton X-100 and 10% FBS (for CD206 and Arg-1 costained with MOMA-2). Sections were incubated overnight at 4 °C with rat antimouse MOMA-2 antibody (1: 50; Abcam), rabbit antimouse iNOS antibody (1 : 50; BD Biosciences; Durham, North Carolina, USA), rabbit antimouse MHC-II antibody (1: 4000; Abcam), goat antimouse CD206 antibody (1: 50; Santa Cruz Biotechnology) or goat antimouse Arg-1 antibody (1 : 50; Santa Cruz Biotechnology). The sections were then washed three times with TBST and blocked for 30 min at RT with TBST containing 10% normal goat serum (for iNOS and MHC-II costained with MOMA-2) or 5% bovine serum albumin, 0.4% Triton X-100 and 10% FBS (for CD206 and Arg-1 costained with MOMA-2). Thereafter, sections were incubated for 1 h at RT with Alexa Fluor 647 goat antirat antibody (1 : 200; Molecular Probes, Waltham, Massachusetts, USA), Alexa Fluor 594 goat antirabbit antibody (1: 200, Invitrogen) for iNOS and MHC-II or Alexa Fluor 594 donkey antigoat antibody (1: 200; Invitrogen) for CD206 and Arg-1. Sections were washed afterward for three times with TBST, counterstained with DAPI (6 μmol/l; Life Technologies) and mounted with Fluoromount (Sigma-Aldrich).
Images were captured using a Wave FX Spinning Disc Confocal Microscope (Quorum Technologies) and quantified with ImageJ software. Expression of M1 or M2 markers in infiltrating monocyte/macrophages was analyzed in mesenteric artery PVAT by determining the area of each marker within MOMA-2 stained area using color RGB thresholding, and the area of each marker expressed as percentage of MOMA-2 area.
Evaluation of gene expression
The expression of PPARγ (Pparγ), endothelial NO synthase (eNOS, Nos3), iNOS (Nos2), ETA receptor (Ednra), ETB receptor (Ednrb), superoxide dismutase 1 (Sod1), superoxide dismutase 2 (Sod2), superoxide dismutase 3 (Sod3), NADPH oxidase 1 (Nox1), NADPH oxidase 2 (Cybb), NADPH oxidase 4 (Nox4), adiponectin (Adipoq), FA-binding protein 4 (FABP4, Fabp4) and ribosomal protein S16 (Rps16) mRNA was determined in mesenteric artery, PVAT and/or cultured VSMC by reverse transcription/quantitative PCR (RT-qPCR).
As described above, mesenteric artery and PVAT were dissected and stored in RNAlater. Shortly after, fresh tissues were homogenized for 1 min with a Polytron PT 1600 E homogenizer (Brinkmann Instruments; Mississauga, Ontario, Canada) and processed for total RNA extraction using the mirVana miRNA isolation kit (Life Technologies). Cultured VSMC were lysed using Lysis/Binding Buffer and processed for total RNA extraction as above. RNA concentration was measured using a Nanodrop spectrophotometer ND-100 V3.1.2 (Thermo Fisher Scientific; Wilmington, Delaware, USA). RNA quality was assessed by determining the rRNA and mRNA profile by electrophoresis using an RNAse-free 1% agarose gel submerged in 1X TAE electrophoresis buffer (2mol/l Tris-acetate and 50 mmol/l EDTA).
After validating the RNA quality of samples, 1, 0.2 and 0.4 μg of total RNA isolated from VSMC, mesenteric artery and PVAT, respectively, were reverse-transcribed with the Quantitect RT kit (Qiagen, Foster City, California, USA). QPCR was performed using the SsoFast EvaGreen 8 Supermix (Bio-Rad Laboratories; Mississauga, Ontario, Canada) with the Mx3005P real-time PCR cycler. Oligonucleotides designed for Pparγ, Nos3, Nos2, Ednra, Ednrb, Sodl, Sod2, Sod3, Nox1, Cybb, Nox4, Adipoq, Fabp4 and Rps16 are listed in Supplemental Digital Content Table S1, http://links.lww.com/HJH/A750. The qPCR conditions were 2 min at 96 °C, followed by 40 cycles of 5 s at 96 °C and 30 s at 58 °C. Results were normalized with Rps16 and expressed as fold change over control.
Flow cytometry profiling of splenic T cells and monocytes
Profile of T cells and monocyte subtypes was determined by flow cytometry as previously [20]. Single splenocyte suspension was obtained by releasing the splenocytes by forcing pieces of spleen through a 70-μm nylon mesh cell strainer (BD Biosciences) prewet with PBS supplemented with 5% FBS (qualified, Canada origin; Life Technologies) with the back of a 3-ml syringe plunger. The cell strainer was washed with PBS/5% FBS to flush the cells through the nylon mesh. The two previous steps were repeated until only connective tissue remained in the cell strainer. Cells were centrifuged at 300 × g for 10 min at RT. Cells were resuspended in 5 ml of Red Blood Cell lysis buffer (Sigma-Aldrich Canada, Oakville, Ontario, Canada) and incubated at RT for 3 min with occasional gentle mixing to eliminate red blood cells. The mixture was diluted with 30 ml of PBS/5% FBS, filtered through a 70-μm nylon mesh cell strainer and centrifuged at 300 × g for 5 min at RT. Cells were resuspended in 2 ml of PBS/5% FBS and counted using a Z2 Coulter Counter (Beckman-Coulter; Mississauga, Ontario, Canada). Two million cells were stained with a fixable viability dye eFluor 506 (eBioscience; San Diego, California, USA) in PBS, incubated with rat antimouse CD16/CD32 Fc receptor block (clone 2.4G2, BD Biosciences), stained with specific antibodies in PBS/ 5% FBS and analyzed by flow cytometry. Antibodies are listed in Supplemental Digital Content Table S2 for profiling of T cells and Table S3 for profiling of monocytes, http://links.lww.com/HJH/A750. Flow cytometry was performed on the BD LSRFortessa cell analyzer (BD Biosciences). Fluorescence minus one controls were used to determine fluorescence background and positivity. Data analysis was performed using FlowJo software (Tree Star Inc.; Ashland, Oregon, USA).
Vascular smooth muscle cells isolation and culture
VSMC were isolated as previously described with some modification [21]. Two to three mice per VSMC isolation were used. Mice were anesthetized with isoflurane as above, and the mesenteric artery vascular beds were dissected and collected and stored in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) cell culture medium (Life Technologies) at RT until further processing for VSMC isolation. Mesenteric artery vascular beds were dissected quickly in DMEM/F12 medium to remove the fat under a dissecting microscope. Mesenteric artery vascular beds were digested in 12.5 ml of DMEM/F12 medium containing 25 mg bovine serum albumin (Probumin media grade; EMD Millipore, Billerica, Massachusetts, USA), 25 mg of collagenase type 2 (280U/mg dry weight; Worthington Biochemical Corporation; Lakewood, New Jersey, USA), 1.5 mg elastase (4.92 U/mg protein; Worthington Biochemical Corporation) and 4.5 mg soybean trypsin inhibitor (Sigma-Aldrich Canada) at 37 °C for 30 min in a 50-ml conical tube with gentle agitation in a hybridization oven (VWR International, LLC; Radnor, Pennsylvania, USA). Digestion efficiency was improved by pipetting up and down the tissue with a 1-ml Raining pipette after the first 10 min of digestion and then after every 5 min. At the end of the digestion, debris were removed by passing the digesta through a 100-μm Falcon cell strainers (VWR International) fitted on top of a 50-ml conical tube. The cell strainers were washed with 5 ml of DMEM/F12 medium. VSMC suspension was centrifuged at 300 ×g for 5 min at RT. VSMC were resuspended in 1 ml of culture medium (DMEM high glucose medium supplemented with 10% heat-inactivated FBS, 100 U/ml of penicillin - streptomycin and 2.8 mmol/l of L-glutamine (all from Life Technologies). A cell suspension aliquot was mixed 1 : 1 with a 0.4% trypan blue solution (Life Technologies), and viable cells were counted with a hemocytometer. VSMC were seeded in 25 cm2 Falcon Primaria cell culture flasks at a density of 40 000 cells/cm2 and kept in a humidified 37 °C incubator gassed with 5% CO2. Culture medium was changed every 48 h. When cells reached 70-80% confluence, they were trypsinized, washed with culture media and passaged 1 : 3 into 75 cm2 Falcon Primaria cell culture flasks (passage 1). When cells reached 70-80% confluence, they were lysed using Lysis/Binding Buffer and processed for total RNA extraction as described above.
Data analysis
Results are presented as means ± SEM. Two-way analysis of variance (ANOVA) for repeated measurements followed by a Student-Newman-Keuls post-hoc test evaluated differences among groups in concentration-response curves and BP. All other results were compared using a Student t test or two-way ANOVA followed by a Student-Newman-Keuls post-hoc test. P less than 0.05 was considered statistically significant.
RESULTS
Validation of the animal model and Pparγ deletion
We sought to determine the in-vivo efficiency of VSMC Pparγ deletion by RT-qPCR. To minimize contamination from other cell types (e.g. endothelial cells, fibroblasts and immune cells), we cultured and compared mesenteric artery VSMC from control and smPparγ−/− mice. Expression of Pparγ was ~70% lower in smPparγ−/− compared with control (Supplemental Fig. S1A, Supplemental Digital Content, http://links.lww.com/HJH/A750). The extent of Cre activation was evaluated by comparing mesenteric artery of tamoxifen-treated ROSA26mT-G/mT-mG and smCreERT2/ROSA26mT-mG/mT-mG reporter mice. Prior to CreERT2 activation, mT expression is under the transcriptional control of the CAG promoter and therefore present in all cell types [3]. mG is only expressed following CreERT2 mediated excision and is therefore a cellular marker of CreERT2 activity. Determination of mT/mG expression switch by replacement of red by green staining revealed that CreERT2 recombinase was totally activated in mesenteric artery VSMC of tamoxifen-treated smCreERT2/ ROSA26mT-mG/mT-mG mice but not ROSA26mT-mG/mT-mG mice (Supplemental Fig. S1B, Supplemental Digital Content, http://links.lww.com/HJH/A750). Using the same model, we have also previously shown that CreERT2 activation is restricted to smooth muscle cells and is not present in other tissues such as endothelial cells, renal tubules, glomeruli or cardiomyocytes [12].
Physiological parameters
Four weeks after tamoxifen treatment, SBP was increased from 109 ± 2 to 123 ± 5mmHg in eET-1 compared with control (Fig. 1). VSMC Pparγ gene disruption did not significantly increase SBP in control mice nor did it exaggerate BP elevation in eET-1 mice. ET-1 overexpression decreased body, heart, liver and total kidney weight to a similar extent in control and smPparγ−/−; however, tibia length was unchanged. smPparγ−/− exhibited a decrease of 24% in spleen weight, further decreased by 16% by ET-1 overexpression (Supplemental Table S4, Supplemental Digital Content, http://links.lww.com/HJH/A750). Plasma ET-1 levels were six-fold higher in eET-1 mice compared with control (Supplemental Fig. S2, Supplemental Digital Content, http://links.lww.com/HJH/A750). VSMC Pparγ deletion did not alter plasma ET-1 levels in control or eET-1 mice.
FIGURE 1.
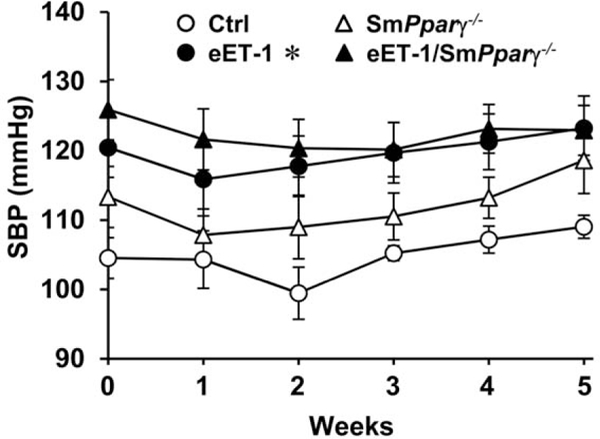
Endothelin-1 overexpression increased SBP. SBP was assessed by telemetry for 24 h a week before tamoxifen treatment and at weekly intervals thereafter for 5 weeks in control, eET-1, smPparγ−/− and eET-1/smPparγ−/− mice.Week 0 refers to blood pressure acquired the week prior to tamoxifen treatment, and week 1 refers to blood pressure acquired immediately after the last day of 5 days of tamoxifen treatment. Each point represents the average SBP over a consecutive 24-h period. Data are presented as means ± SEM, n = 4-5. *P< 0.05 vs Ctrl. Statistical differences were determined by comparing the blood pressure curves between groups.
Endothelin-1 overexpression preserved endothelium-dependent relaxation in smPparγ−/− mice
There was no difference in the vasoconstrictor responses of mesenteric artery to norepinephrine or the endothelium-independent relaxation responses to the NO donor sodium nitroprusside between the groups (Fig. 2a and d). Endothelium-dependent relaxation responses to acetylcholine were impaired by 37% in smPparγ−/− but not in eET-1 and eET-1/smPparγ−/− compared with control (Fig. 2b). The contribution of NO to endothelium-dependent relaxation determined using the NOS inhibitor L-NAME showed that vasodilatory responses to acetylcholine were decreased to a similar level in all groups (Fig. 2c). This suggested that normalization of endothelium-dependent relaxation was mediated by increased production of NO.
FIGURE 2.
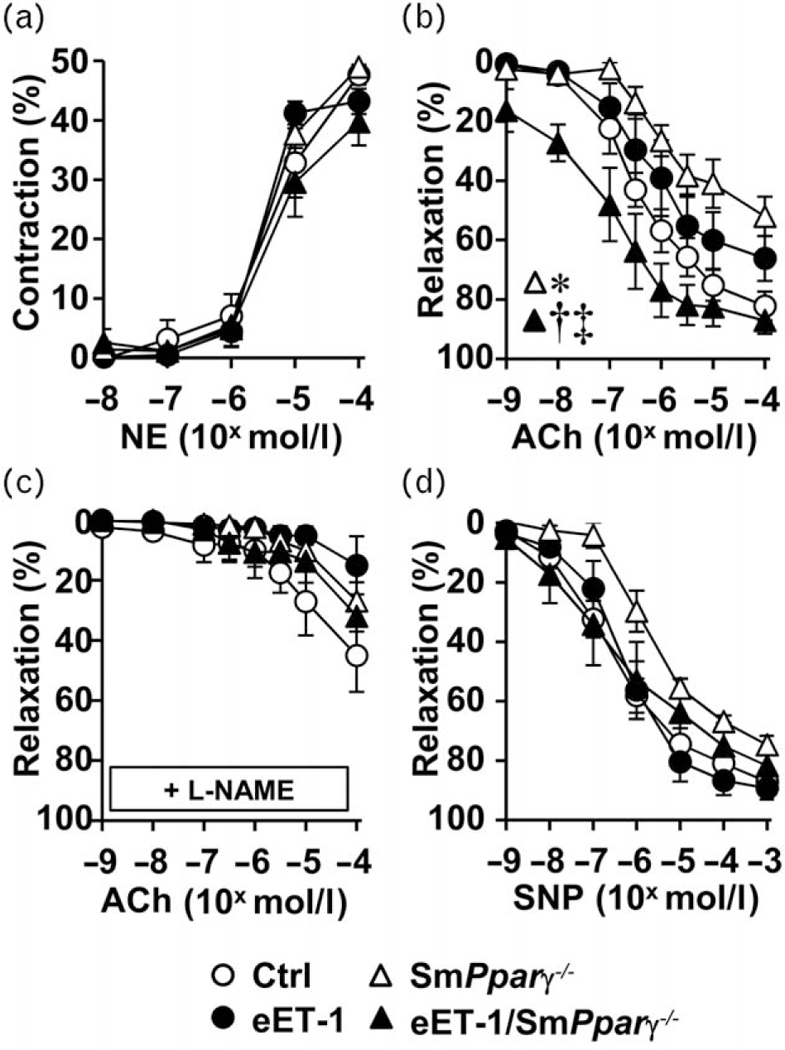
Endothelin-1 overexpression preserved endothelium-dependent relaxation in smPparγ−/− mice. Contractile responses to norepinephrine (a), endothelium-dependent relaxation responses to acetylcholine in the absence (b) and presence of Nω-nitro-L-arginine methyl ester (c) and endothelium-independent relaxation response to sodium nitroprusside (d) of small mesenteric arteries were determined in control, eET-1, smPparγ−/− and eET-1/smPparγ−/− mice. Values are means ± SEM, n = 7-10. *P< 0.05 vs Ctrl, †P< 0.001 vs smPparγ −/− and ‡P< 0.01 vs eET-1. Statistical differences were determined by comparing the whole concentration-response curves between groups.
Endothelin-1 overexpression corrected the enhanced endothelin-1 contraction induced by Pparγ deletion
Mesenteric artery from smPparγ−/− exhibited enhanced contraction to ET-1 compared with control (Fig. 3a). Although ET-1 overexpression did not alter the contractile response to ET-1 in control mice, it blunted the increased contractility to ET-1 caused by VSMC Pparγ deletion. Blockade of NO generation with L-NAME increased the contractile response to ET-1 in control, eET-1 and eET-1/ smPparγ−/−, but not in smPparγ−/− (Fig. 3b).
FIGURE 3.
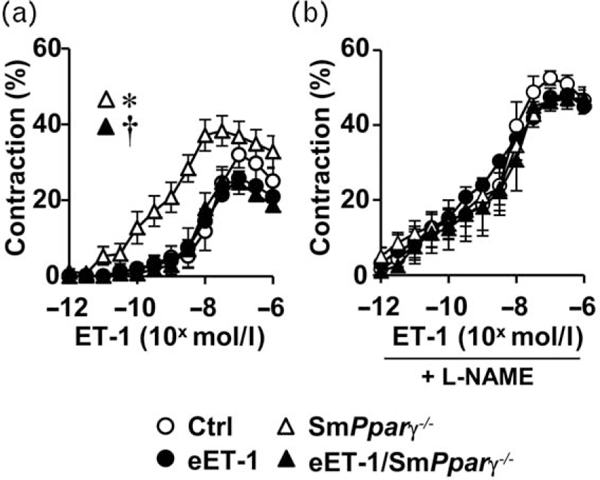
Endothelin-1 overexpression corrected vascular smooth muscle cells Pparγ gene deletion induction of enhanced endothelin-1 contraction. Contractile responses to endothelin-1 were determined in small mesenteric arteries of control, eET-1, smPparγ−/− and eET-1/smPparγ−/− mice in the absence (a) and presence of the nitric oxide synthase inhibitor N^-nitro-L-arginine methyl ester (b). Data are presented as means± SEM, n = 6-7. *P< 0.05 vs Ctrl and †P< 0.05 vs smPparγ−/−. Statistical differences were determined by comparing the whole concentration-response curves between groups.
Effect of vascular smooth muscle cell Pparγ deletion and endothelin-1 overexpression on vascular remodeling and stiffness
Media/lumen ratio at 45 mmHg was increased 20% in eET-1/smPparγ−/− compared with control (Supplemental Fig. 3A, Supplemental Digital Content, http://links.lww.com/HJH/A750). Media cross-sectional area was similar between groups (Supplemental Fig. 3B, Supplemental Digital Content, http://links.lww.com/HJH/A750). Mesenteric artery stiffness was increased to the same extent in eET-1, smPparγ−/− and eET-1/smPparγ−/− compared with control, as indicated by a similar leftward displacement of the stress-strain curves and a 16% decrease in strain at 140mmHg (Supplemental Fig. 3C, Supplemental Digital Content, http://links.lww.com/HJH/A750).
Vascular smooth muscle cell Pparγ inactivation exaggerated endothelin-1-induced oxidative stress
VSMC Pparγ gene disruption alone increased ROS generation 1.8-fold in mesenteric artery, which was further enhanced 0.7-fold in eET-1/smPparγ−/− (Fig. 4). PVAT ROS generation tended to be higher in eET-1 and smPparγ−/−,whereas it was increased three-fold in eET-1/smPparγ−/− compared with control. Expression of Sod1 and Sod2 mRNA was similar in all groups, whereas Sod3 mRNA expression was increased similarly in eET-1, smPparγ−/− and eET-1/ smPparγ−/− compared with control (Supplemental Fig.S4A-C, Supplemental Digital Content, http://links.lww.com/HJH/A750). Nox4 mRNA expression tended to increase in eET-1/smPparγ−/−, whereas Cybb (NADPH oxidase 2) mRNA expression tended to increase in eET-1 (Supplemental Fig. S4D and E, Supplemental Digital Content, http://links.lww.com/HJH/A750).
FIGURE 4.

Vascular smooth muscle cell Pparg gene inactivation exaggerated endothelin-1-induced oxidative stress in mesenteric arteries. Reactive oxygen species generation was determined (a and b) by dihydroethidium staining in small mesenteric arteries and perivascular adipose tissue of control, eET-1, smPparγ−/− and eET-1smPparγ−/− mice. (a) Representative dihydroethidium-stained sections of mesenteric artery from four to seven mice per group are presented. Data are presented as means_SEM, n=4-7. *P<0.05 vs Ctrl, †P<0.05 vs smPparγ−/− and ‡P<0.05 vs eET-1.
Effect of vascular smooth muscle cell Pparg gene ablation and endothelin-1 overexpression on inflammation
MCP-1 expression in mesenteric artery increased similarly in smPparγ−/− and eET-1/smPparγ−/− compared with control (Fig. 5b). Monocyte/macrophage infiltration in mesenteric artery PVAT was _two-fold higher in eET-1 and smPparγ−/− compared with control and increased further two-fold in eET-1/smPparγ−/− (Fig. 5d). Polarization of infiltrating monocyte/macrophages was evaluated by the proinflammatory (M1) and anti-inflammatory (M2) markers in MOMA-2+ cells. There was no difference between groups in expression of Ml markers iNOS and MHC-II, or the M2 markers CD206 and Arg-1 (Supplemental Fig. S5, Supplemental Digital Content, http://links.lww.com/HJH/A750). The percentage of spleen CD11b+ cells (pan-macrophages) was increased 2.3-fold in smPparγ−/− and further increased 1.5-fold in eET-1/smPparγ−/− (Supplemental Fig. S6B, Supplemental Digital Content, http://links.lww.com/HJH/A750). The spleen fraction of proinflammatory Ly-6Chi monocytes was increased 1.7-fold and 1.9-fold in eET-1 and smPparγ−/−, respectively, but not in eET-1/smPparγ−/− (Supplemental Fig. S6C, Supplemental Digital Content, http://links.lww.com/HJH/A750). Spleen CD3+, CD4+, CD8+ and CD4+CD69+ T cells were similar in all groups (Supplemental Fig. S7A-C, Supplemental Digital Content, http://links.lww.com/HJH/A750). The percentage of spleen anti-inflammatory CD4+CD25+FOXP3+ T regulatory lymphocytes (Treg) was increased 1.4-fold in smPparγ−/− compared with control (Supplemental Fig. S7D, Supplemental Digital Content, http://links.lww.com/HJH/A750). ET-1 overexpression alone decreased the spleen fraction of CD4+CD25+FOXP3+ cells by 26%, with an additional 38% decrease in eET-1/smPparγ−/−.
FIGURE 5.
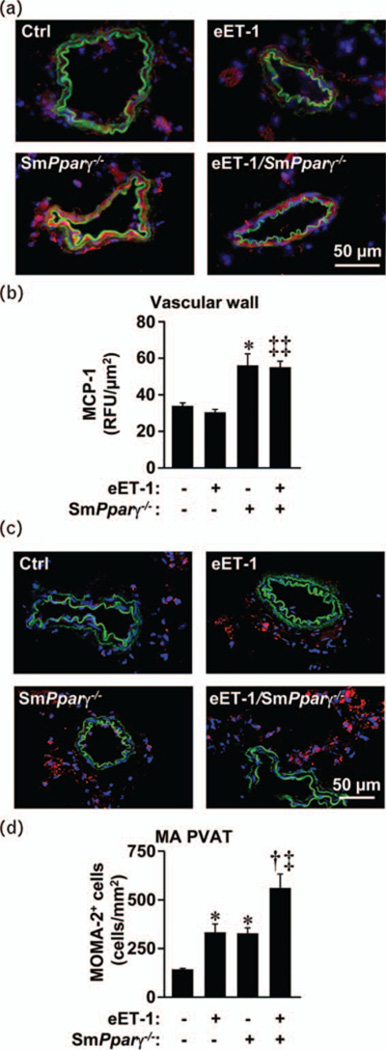
Vascular smooth muscle cell Pparg gene inactivation increased inflammation in mesenteric arteries. Monocyte chemoattractant protein-1 expression (relative fluorescence units, b) and monocyte/macrophage infiltration (monocyte/ macrophage antigen-2+cells, d) were determined by immunofluorescence in small mesenteric arteries of control, eET-1, smPparγ−/− and eET-1/smPparγ−/− mice. (a) Representative monocyte chemotactic protein-1 fluorescence images (red fluorescence) of mesenteric artery sections from five to seven mice per group are presented. (c) Representative monocyte/macrophage antigen-2 fluorescence images (red fluorescence) of mesenteric artery sections from five to seven mice per group are presented. Green and blue represent elastin autofluorescence and 4’,6-diamidino-2-phenylindole fluorescence, respectively. Data are presented as means±SEM, n=5-7. *P<0.01 vs Ctrl, †P<0.01 vs smPparγ−/− , ‡P<0.01 and ‡‡P<0.001 vs eET-1.
Effect of vascular smooth muscle cell Pparγ gene ablation and endothelin-1 overexpression on gene expression
To better understand changes to vascular reactivity, we dissected the mesenteric artery arcade and evaluated the mRNA expression level of several genes. Nos2 (iNOS) mRNA expression was increased two-fold in mesenteric artery of smPparγ−/− (Fig. 6a). ET-1 overexpression increased Nos2 mRNA expression 3.7-fold in mesenteric artery, effect blunted in eET-1/smPparγ−/− Expression of Nos3 (eNOS), the major contributor of vascular NO, was increased 1.2-fold only in eET-1 mice (Fig. 6b). ET-1 overexpression alone increased ETA and ETB receptor expression (Fig. 6c and d). VSMC Pparγ inactivation increased ETA and ETB receptor expression to a similar extent in control and eET-1 mice. The ratio of Ednra/Ednrb was decreased by 30% in eET-1/smPparγ−/− compared with smPparγ−/− (Fig. 6e). VSMC Pparγ inactivation alone increased Nox1 mRNA expression (Fig. 6f). ET-1 overexpression increased Nox1 expression in control mice, although it did not further increase expression in smPparγ−/− mice.
FIGURE 6.
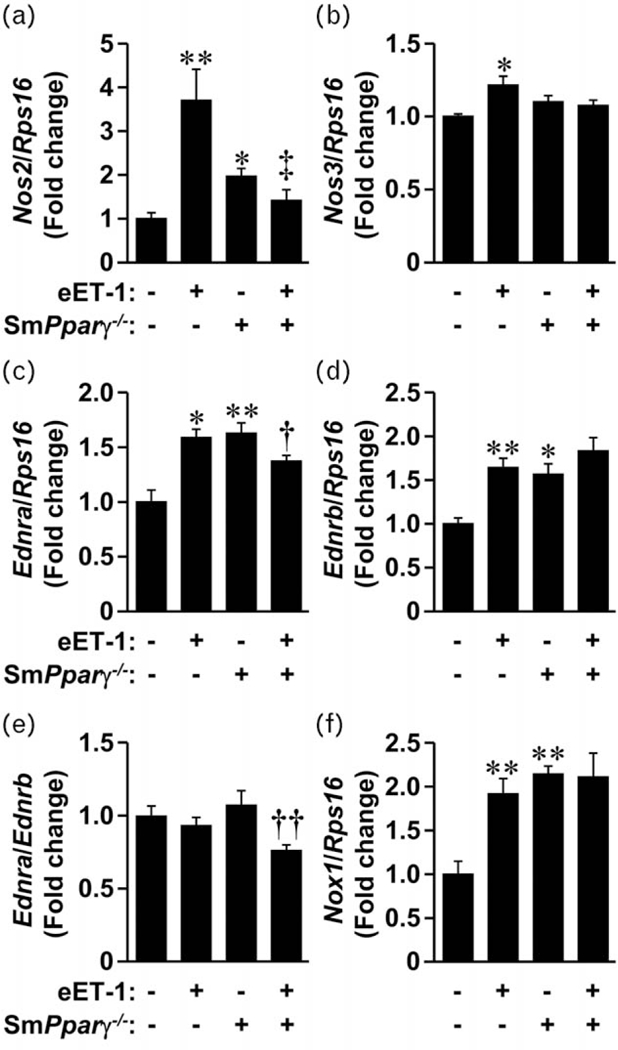
Endothelin-1 overexpression decreased the ratio of endothelin type A to endothelin type B receptors in mesenteric arteries of smPparγ−/− mice. The mRNA expression levels of inducible nitric oxide synthase (Nos2), endothelial nitric oxide synthase (Nos3), endothelin type A (Ednra) and endothelin type B (Ednrb) receptors, NADPH oxidase 1 (Nox1) and ribosomal protein S16 (Rps16) were determined by reverse transcription/quantitative PCR in small mesenteric arteries of control, eET-1, smPparγ−/− and eET-1/smPparγ−/− mice. Data are presented as mean±SEM, n=4-6. *P<0.01 vs Ctrl, **P<0.001 vs Ctrl, †P<0.05 vs smPparγ−/− , ‡‡P<0.01 vs smPparγ−/− and ‡P<0.01 vs eET-1.
DISCUSSION
These results demonstrate for the first time that Pparγ gene disruption in VSMC enhances the contractile response of vessels to ET-1 and exaggerates ET-1-induced oxidative stress, inflammation and vascular remodeling, suggesting that VSMC PPARγ counteracts ET-1-induced vascular damage. ET-1 overexpression paradoxically restored endothelium-dependent relaxation and corrected the enhanced contractile response to ET-1 in smPparγ−/− mice, despite greater vascular oxidative stress and inflammation.
Validation of smPparγ knockout model
PPARγ was first described as a nuclear receptor induced during adipocyte differentiation [22], but gained prominence when identified as the molecular target of the antidiabetic thiazolidinediones [23]. Thiazolidinediones act as high-affinity synthetic agonists of PPARγ that improve insulin sensitivity and modestly lower BP in type 2 diabetes mellitus. PPARγ has vascular protective effects that extend beyond its role in glucose and lipid metabolism. To investigate the role of VSMC PPARγ in ET-1-induced vascular injury, we used the same tamoxifen-inducible tissue-specific Cre/loxP approach that we used previously to study the role of VSMC PPARγ in angiotensin II-induced vascular disease [12]. In that study, we showed that smCreERT2 and smCreERT2/PparγFlox/Flox mice treated with vehicle, which were used as control, and smCreERT2 mice treated with tamoxifen do not have altered endothelium-dependent relaxation to acetylcholine, indicating that neither tamoxifen nor CreERT2 activation causes impairment in endothelial function. This also demonstrates that effects seen in tamoxifen-treated smCreERT2/PparγFlox/Flox mice are caused by Pparγ ablation and not by unexpected effects due to genetic differences. Given the previous results, we have not studied vehicle-treated eET-1/smPparγ−/− mice in the current study as it is unlikely that their vascular phenotype will be affected by genetic differences. We have opted for a robust experimental design study in which mice in the four groups expressed CreERT2 in VSMC and were treated with tamoxifen, ruling out effects of tamoxifen treatment and Cre activation. Furthermore, tamoxifen and its metabolites are completely eliminated from the circulation 4.2 days after injection [24], and tissues from the mice are collected 4 weeks after tamoxifen treatment.
CreERT2 recombinase efficiency was studied using smCreERT2/ROSA26mT-mG/mT-mG reporter mice. We found that tamoxifen treatment totally activated CreERT2 recom-binase in smooth muscle cells of reporter mice expressing smCreERT2. Although smooth muscle cells are present in nonvascular tissues such as the gastrointestinal tract, urinary bladder and lung bronchi, it is unlikely that smPparγ−/− in these tissues has any impact on vascular function. Using VSMC isolated from smPparγ−/− mice, we confirmed the knockout (~70%) of PPARγ expression in our model. The residual PPARγ expression could originate from other cells such as fibroblasts and endothelial cells that may have survived in the culture.
Peroxisome proliferator-activated receptor γ and the vascular system
The current study provides strong evidence that VSMC PPARγ activation has beneficial effects on the cardiovascular system independently of its insulin-sensitizing actions. Here, we show that VSMC Pparγ inactivation alone is sufficient to cause vascular injury. smPparγ−/− impaired endothelial function, enhanced the contractile response to ET-1 and increased stiffness in resistance arteries, associated with increased vascular oxidative stress and MCP-1 levels, as well as enhanced PVAT monocyte/ macrophage infiltration.
Consistent with our previous findings [12], deleterious effects of VSMC Pparγ inactivation were BP-independent. Nevertheless, Chang et al. [25] found that smPparγ−/− caused hypotension, whereas Wang et al. [26] reported increased BP in smPparγ−/− during the light but not dark cycle. Although each study utilized the Cre/LoxP recombination model, Cre expression was driven by the Sm22α promoter rather than the smMHC promoter in the first two studies. Moreover, both groups have induced smPparγ−/−- in utero, whereas we did so in adult mice, thereby avoiding potentially confounding developmental effects, which could also explain differences. Chang et al. [27] later reported that smPparγ−/− mice completely lack PVAT resulting from the transient activation of the SM22α promoter in PVAT during development.
Human ET-1 overexpression increased SBP significantly in this study, whereas in our previous study, BP rose without achieving statistical significance [2]. In a more recent mouse model with inducible human ET-1 overexpression, SBP rose 30mmHg [3], confirming the hypertensive effect of endothelial human ET-1 overexpression. Decreased body, heart, liver and total kidney weight in eET-1 mice were not due to impaired postnatal growth, as tibia length was similar in all groups. It is possible that increased endothelium-derived ET-1 in utero caused changes in fetal growth, as the Tie2 promoter has been shown to drive gene expression during embryonic development [28].
Endothelium-dependent relaxation in small arteries
In rodents and humans, PPARγ activation improves NO bioavailability by increasing NO release and through transrepression of pro-oxidant signaling pathways [29-31]. Here, we extend previous findings by demonstrating that inactivation of PPARγ in VSMC impairs endothelium-dependent responses to acetylcholine [12]. Surprisingly, ET-1 overexpression restored relaxation responses in smPparγ−/− mice despite enhanced oxidative stress, which reduces NO bioavailability and promotes endothelial dysfunction [32]. The vasodilatory responses to acetylcholine in eET-1/smPparγ−/− were largely diminished by the NOS inhibitor L-NAME, suggesting that restoration of relaxation is likely NO-dependent. Although vascular NO is mostly produced by eNOS, we did not find any changes in Nos3 expression in eET-1/smPparγ−/−. Increased Nos3 mRNA in eET-1 may explain why these mice did not present endothelial dysfunction. In eET-1/smPparγ−/−, a decrease in ETA/ETB receptor ratio (as shown here by the Ednra/Ednrb mRNA ratio) could contribute to enhanced NO production leading to the paradoxical finding of a preserved endothelial response to acetylcholine in presence of overexpression of ET-1, which results from the consequent shift in the balance of vasodilation and constriction favoring the former. This is interesting in view of evidence that a normal physiological action of ET-1 could indeed be vasodilation as demonstrated in mice with Edn1 gene deletion [33]. In situations of inflammation and oxidative stress, iNOS is upregulated and produces large amounts of NO that typically react with ROS to form ONOO¯, worsening the oxidative stress [34]. ET-1 overexpression greatly enhanced the mRNA expression of Nos2 (iNOS) in control mice but not in smPparγ−/−. The absence of iNOS-derived oxidative stress in eET-1/smPparγ−/− could have contributed to preserved endothelium-dependent relaxation. mRNA expression of adiponectin, an adipokine that improves endothelial function [35], and which was decreased in the PVAT of eET-1 and smPparγ−/− mice was partially corrected in eET-1/ smPparγ−/−, which may have contributed to preservation of vasorelaxation through its anticontractile effects [36,37]. Although NO seems to play a major role in mediating vasodilatation in eET-1/smPparγ−/−, other signaling pathways may participate in the vasodilatory response to acetylcholine. For instance, we previously showed that acetylcholine-induced vasodilation is restored in eET-1 mice deficient in apolipoprotein E and that this effect is mediated by NOS-independent activation of soluble guanylate cyclase and increased activation of 4-aminopyridine-sensitive voltage-dependent potassium channels [38]. Alternatively, it is possible that eET-1/smPparγ−/− mice have enhanced dismutation of superoxide into hydrogen peroxide, which can act as a mediator of endothelium-dependent vasorelaxation [39].
Vascular contractile response to endothelin-1
PPARγ activation counteracts ET-1-induced vasoconstriction, as shown by Halabi et al. [11] who found that VSMC overexpression of a dominant negative Pparγ mutant caused exaggerated contraction of aorta by ET-1. Excess contractility was associated with decreased aortic ET-1 expression and no change in ETA or ETB receptor expression. More recently, Tian et al. [17] demonstrated that rosiglitazone attenuates ET-1-induced aortic contraction by upregulating endothelial ETB receptor expression. In the current study, we found that VSMC Pparγ gene inactivation enhanced contractile responses to ET-1 in mesenteric artery. The enhanced contractile responses were not due to general hypercontractility, as concentration-response curves to norepinephrine were similar between groups. ET-1 mainly causes contraction by activating ETA receptors, whereas it stimulates vasodilation largely through endothelial ETB receptor-mediated NO release. The enhanced contractile responses to ET-1 in smPparγ−/− are unlikely to be due to increased ETA mRNA expression, as eET-1 mice had similar levels with no change in contractility. Blockade of ETB-induced NO production with L-NAME increased the contractile response to ET-1 in all groups except smPparγ−/−, providing additional evidence for reduced NO bioavailability in smPparγ−/− mice. The absence of any differences between groups after L-NAME treatment also suggests that the enhanced contractile response to ET-1 was caused by reduced NO bioavailability and/or impaired ETB signaling. The enhanced responsiveness to ET-1 induced by VSMC Pparγ inactivation was lost in eET-1/smPparγ−/−, associated with a decrease in Ednra/ Ednrb, indicating a shift in the balance of vasodilation and constriction favoring the former.
Vascular smooth muscle cell Pparγ inactivation exaggerates vascular oxidative stress
The current study demonstrates for the first time that VSMC Pparγ inactivation exaggerates ET-1-induced oxidative stress, vascular remodeling and inflammation. eET-1/ smPparγ−/− mice presented increased ROS in their mesenteric artery when compared with eET-1 and smPparγ−/− mice. Mesenteric artery gene expression of the pro-oxidant enzyme Nox1 was increased to a similar extent in eET-1, smPparγ−/− and eET-1smPparγ−/− mice. It is possible that the exaggerated oxidative stress in eET-1/smPparγ−/− stems from other sources such as mitochondria. Mesenteric artery gene expression of the antioxidant enzyme Sod3 was also increased to a similar extent in eET-1, smPparγ−/− and eET-1/smPparγ−/− mice. The expression of Sod3 has been shown by many investigators to be upregulated by pro-oxidant and proinflammatory stimuli as a mechanism to counterbalance increases in oxidative stress. For example, studies have shown that IFN-γ and TNF-α upregulate Sod3 mRNA expression levels through activation of NFκB [40,41]. Likewise, increased levels of pro-oxidant peptides such as ET-1 or angiotensin II also increase Sod3 expression in VSMC [42,43].
Vascular smooth muscle cell Pparγ inactivation exaggerates vascular remodeling
Enhanced oxidative stress is a common finding in hypertension and is often associated with vascular remodeling.Eutrophic vascular remodeling was only found in eET-1/ smPparγ−/− Eutrophic remodeling may be an early manifestation of vascular disease, which can progress to more severe hypertrophic remodeling when additional factors such as increased angiotensin II, aldosterone or additional ET-1 are present. Accordingly, the milder vascular damage found in eET-1 mice and smPparγ−/− mice may not be sufficient to cause detectable remodeling of small arteries, whereas VSMC Pparγ gene inactivation in the presence of increased endothelial ET-1 expression will cause eutrophic vascular remodeling. It is unlikely that vascular remodeling in eET-1/smPparγ−/− is due to hemodynamic effects resulting from BP changes, as similar increases in BP were observed in eET-1 mice with no mesenteric artery remodeling.
Vascular inflammation
Vascular inflammation involving both innate and adaptive immune processes play important roles in the pathogenesis of hypertension [44]. Monocyte/macrophage infiltration in PVAT was increased in eET-1 and smPparγ−/− and further exaggerated in eET-1/ smPparγ−/−. MCP-1 levels in mesenteric artery were also increased in smPparγ−/−, although this was not worsened by ET-1 overexpression. Vascular inflammation caused by smPparγ−/− could increase migration of inflammatory cells from the spleen to peripheral sites, resulting in decreased spleen weight, found in mice with VSMC Pparγ gene inactivation and further enhanced by ET-1 overexpression.
To better understand cellular mediators involved in the inflammatory process, flow cytometry of splenocytes was performed. eET-1/smPparγ−/− mice exhibited reduced percentage of anti-inflammatory CD4+CD25+FOXP3+ Treg. This was not associated with changes in activated CD4+ T lymphocytes. Increases in percentages of cells expressing CD11b, marker found on many leukocytes and macrophages, in smPparγ−/− mice and enhanced in eET-1/ smPparγ−/−, may explain the infiltration of monocytes/ macrophages in the PVAT in eET-1/smPparγ−/−. As spleen proinflammatory Ly-6Chi monocytes were increased in eET-1 and smPparγ−/− but not eET-1/smPparγ−/−, monocyte/ macrophages that have infiltrated the PVAT of eET-1/ smPparγ−/− mice could derive from less-inflammatory precursors and have adopted a less-inflammatory phenotype. Accordingly, we hypothesized that there could be a greater fraction of immunomodulatory M2 macrophages than proinflammatory M1 macrophages, which may have contributed to the preserved vasodilatory response to acetylcholine seen in eET-1/smPparγ−/−. However, there were no changes in the relative expression of M1 or M2 markers.
In conclusion, these results demonstrate that inducible VSMC Pparγ inactivation exaggerates ET-1-induced vascular injury, supporting a protective role for PPARγ in hypertension through modulation of pro-oxidant and proinflammatory pathways. Paradoxically, ET-1 overexpression restored endothelium-dependent relaxation and corrected the increased contractile response to ET-1 in smPparγ−/− mice, suggesting complex interactions between ET-1 and PPARγ within the vasculature.
Supplementary Material
Reviewer’s Summary Evaluation.
Reviewer 1
This study investigated whether peroxisome proliferator-activated receptor γ (Pparγ) expressed in vascular smooth muscle (VSM) protects against vascular injury following selective endothelial cell overexpression of endothelin-1 (ET-1). Mesenteric artery oxidative stress, inflammation and remodeling in mice overexpressing endothelial ET-1 were all enhanced by inactivation of VSM PPARγ, suggesting a protective role for VSM PPARγ in these effects. Interestingly, endothelial ET-1 overexpression restored endothelium-dependent relaxation following VSM PPARγ inactivation. Novel, selective activators of VSM PPARγ may be useful therapeutic agents to protect the vasculature in conditions involving increased activity of ET-1 in the endothelium.
ACKNOWLEDGEMENTS
We are grateful to Adriana Cristina Ene and Guillem Colell Dinarès for excellent technical support and animal care.
The work was supported by Canadian Institutes of Health Research (CIHR) grants 37917 and 102606, and a CIHR First Pilot Foundation Grant 332675, a Canada Research Chair (CRC) on Hypertension and Vascular Research by the CRC Government of Canada/CIHR Program and by the Canada Fund for Innovation (CFI), all to E.L.S., and by fellowships to N.I.-K. [Société québécoise d’hypertension artérielle (SQHA) and ‘Fonds de recherche du Québec en Santé’], S.O. (CIHR Canada Graduate Scholarship-Master’s scholarship), M.T. [National Council for Research and Technology (CNPq), Brazil] and T.B. (SQHA and Richard and Edith Strauss Postdoctoral Fellowship).
The work has been presented in whole or in part at the 2015 Canadian Hypertension Congress (Toronto, Ontario, Canada), at the 14th International Conference on Endothelin (Savannah, Georgia, USA), at the 2014 High Blood Pressure Research Scientific Sessions (San Francisco, California, USA) and at the Council on Hypertension 2015 Scientific Sessions (Washington, District of Columbia, USA).
Abbreviations:
- Arg-1
arginase-1
- ANOVA
analysis of variance
- BP
blood pressure
- CD206
mannose receptor
- CreERT2
tamoxifen-inducible Cre recombinase fused with a modified estrogen receptor ligand-binding domain
- DHE
dihydroethidium
- eET-1
transgenic mice constitutively overexpressing human ET-1 selectively in the endothelium
- eNOS
endothelial nitric oxide synthase
- ET
endothelin
- ETA
endothelin type A
- ETB
endothelin type B
- FABP4
fatty acid binding protein 4
- FOXP3
forkhead box P3
- iNOS
inducible nitric oxide synthase
- L-NAME
Nω-nitro-L-arginine methyl ester
- MA
mesenteric artery
- MCP-1
monocyte chemotactic protein-1
- MHC-II
major histocompatibility complex class II
- MOMA-2
monocyte/ macrophage antigen-2
- NE
norepinephrine
- NO
nitric oxide
- PPARγ
peroxisome proliferator-activated receptor gamma
- PVAT
perivascular adipose tissue
- ROS
reactive oxygen species
- Rps16
ribosomal protein S16
- smPparγ−/−
inactivation of the Pparγ gene in VSMC
- SNP
sodium nitroprusside
- SOD
superoxide dismutase
- TL
tibia length
- Treg
T-regulatory lymphocytes
- VSMC
vascular smooth muscle cells
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol 2005; 43:19–29. [DOI] [PubMed] [Google Scholar]
- 2.Amiri F, Virdis A, Neves MF, Iglarz M, Seidah NG, Touyz RM, et al. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation 2004; 110:2233–2240. [DOI] [PubMed] [Google Scholar]
- 3.Rautureau Y, Coelho SC, Fraulob-Aquino JC, Huo KG, Rehman A, Offermanns S, et al. Inducible human endothelin-1 overexpression in endothelium raises blood pressure via endothelin type A receptors. Hypertension 2015; 66:347–355. [DOI] [PubMed] [Google Scholar]
- 4.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999; 402:880–883. [DOI] [PubMed] [Google Scholar]
- 5.Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366:1279–1289. [DOI] [PubMed] [Google Scholar]
- 6.Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation 2002; 105:2296–2302. [DOI] [PubMed] [Google Scholar]
- 7.Iglarz M, Touyz RM, Amiri F, Lavoie MF, Diep QN, Schiffrin EL. Effect of peroxisome proliferator-activated receptor-alpha and -gamma activators on vascular remodeling in endothelin-dependent hypertension. Arterioscler Thromb Vasc Biol 2003; 23:45–51. [DOI] [PubMed] [Google Scholar]
- 8.Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. PPAR(gamma) agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension 2004; 43:661–666. [DOI] [PubMed] [Google Scholar]
- 9.Beyer AM, Baumbach GL, Halabi CM, Modrick ML, Lynch CM, Gerhold TD, et al. Interference with PPARgamma signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension 2008; 51:867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicol CJ, Adachi M, Akiyama TE, Gonzalez FJ. PPARgamma in endothelial cells influences high fat diet-induced hypertension. Am JHypertens 2005; 18 (4 Pt 1):549–556. [DOI] [PubMed] [Google Scholar]
- 11.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, et al. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. CellMetab 2008; 7:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchesi C, Rehman A, Rautureau Y, Kasal DA, Briet M, Leibowitz A, et al. Protective role of vascular smooth muscle cell PPARgamma in angiotensin Il-induced vascular disease. Cardiovasc Res 2013; 97:562–570. [DOI] [PubMed] [Google Scholar]
- 13.Subramanian V, Golledge J, Ijaz T, Bruemmer D, Daugherty A. Pioglitazone-induced reductions in atherosclerosis occur via smooth muscle cell-specific interaction with PPAR{gamma}. Circ Res 2010; 107:953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, et al. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res 1999; 85:394–402. [DOI] [PubMed] [Google Scholar]
- 15.Satoh H, Tsukamoto K, Hashimoto Y, Hashimoto N, Togo M, Hara M, et al. Thiazolidinediones suppress endothelin-1 secretion from bovine vascular endothelial cells: a new possible role of PPARgamma on vascular endothelial function. Biochem Biophys Res Commun 1999; 254:757–763. [DOI] [PubMed] [Google Scholar]
- 16.Montezano AC, Amiri F, Tostes RC, Touyz RM, Schiffrin EL. Inhibitory effects of PPAR-gamma on endothelin-1-induced inflammatory pathways in vascular smooth muscle cells from normotensive and hypertensive rats. J Am Soc Hypertens 2007; 1:150–160. [DOI] [PubMed] [Google Scholar]
- 17.Tian J, Wong WT, Tian XY, Zhang P, Huang Y, Wang N Rosiglitazone attenuates endothelin-1-induced vasoconstriction by upregulating endothelial expression of endothelin B receptor. Hypertension 2010; 56:129–135. [DOI] [PubMed] [Google Scholar]
- 18.Aries A, Paradis P, Lefebvre C, Schwartz RJ, Nemer M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc Natl Acad Sci U S A 2004; 101:6975–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leibovitz E, Ebrahimian T, Paradis P, Schiffrin EL. Aldosterone induces arterial stiffness in absence of oxidative stress and endothelial dysfunction. J Hypertens 2009; 27:2192–2200. [DOI] [PubMed] [Google Scholar]
- 20.Li MW, Mian MO, Barhoumi T, Rehman A, Mann K, Paradis P, et al. Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 2013; 33:2306–2315. [DOI] [PubMed] [Google Scholar]
- 21.Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension 2005; 45:773–779. [DOI] [PubMed] [Google Scholar]
- 22.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med 2013; 19:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 1995; 270:12953–12956. [DOI] [PubMed] [Google Scholar]
- 24.Robinson SP, Langan-Fahey SM, Johnson DA, Jordan VC. Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab Dispos 1991; 19:36–43. [PubMed] [Google Scholar]
- 25.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, et al. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-gamma deletion leads to hypotension. Circulation 2009; 119:2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang N, Yang G, Jia Z, Zhang H, Aoyagi T, Soodvilai S, et al. Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab 2008; 8:482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C, et al. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 2012; 126:1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, et al. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A 1997; 94:3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campia U, Matuskey LA, Panza JA. Peroxisome proliferator-activated receptor-gamma activation with pioglitazone improves endothelium-dependent dilation in nondiabetic patients with major cardiovascular risk factors. Circulation 2006; 113:867–875. [DOI] [PubMed] [Google Scholar]
- 30.Calnek DS, Mazzella L, Roser S, Roman J, Hart CM. Peroxisome proliferator-activated receptor gamma ligands increase release of nitric oxide from endothelial cells. Arterioscler Thromb Vasc Biol 2003; 23:52–57. [DOI] [PubMed] [Google Scholar]
- 31.Hwang J, Kleinhenz DJ, Lassegue B, Griendling KK, Dikalov S, Hart CM. Peroxisome proliferator-activated receptor-gamma ligands regulate endothelial membrane superoxide production. Am J Physiol Cell Physiol 2005; 288:C899–C905. [DOI] [PubMed] [Google Scholar]
- 32.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am SocNephrol 2004; 15:1983–1992. [DOI] [PubMed] [Google Scholar]
- 33.Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature 1994; 368:703–710. [DOI] [PubMed] [Google Scholar]
- 34.Aktan F iNOS-mediated nitric oxide production and its regulation. Life Sci 2004; 75:639–653. [DOI] [PubMed] [Google Scholar]
- 35.Li R, Wang WQ, Zhang H, Yang X, Fan Q, Christopher TA, et al. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/ iNOS activity. Am J Physiol Endocrinol Metab 2007; 293:E1703–E1708. [DOI] [PubMed] [Google Scholar]
- 36.Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH, et al. Perivascular adipose tissue-derived adiponectin activates BK(Ca) channels to induce anticontractile responses. Am J Physiol Heart Circ Physiol 2013; 304:H786–H795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension 2009; 54:1384–1392. [DOI] [PubMed] [Google Scholar]
- 38.Mian MO, Idris-Khodja N, Li MW, Leibowitz A, Paradis P, Rautureau Y, et al. Preservation of endothelium-dependent relaxation in atherosclerotic mice with endothelium-restricted endothelin-1 overexpression. J Pharmacol Exp Ther 2013; 347:30–37. [DOI] [PubMed] [Google Scholar]
- 39.Cai H Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res 2005; 68:26–36. [DOI] [PubMed] [Google Scholar]
- 40.Brady TC, Chang LY, Day BJ, Crapo JD. Extracellular superoxide dismutase is upregulated with inducible nitric oxide synthase after NF-kappa B activation. Am J Physiol 1997; 273 (5 Pt 1):L1002–L1006. [DOI] [PubMed] [Google Scholar]
- 41.Marklund SL. Regulation by cytokines of extracellular superoxide dismutase and other superoxide dismutase isoenzymes in fibroblasts. J Biol Chem 1992; 267:6696–6701. [PubMed] [Google Scholar]
- 42.Stralin P, Marklund SL. Vasoactive factors and growth factors alter vascular smooth muscle cell EC-SOD expression. Am J Physiol Heart Circ Physiol 2001; 281:H1621–H1629. [DOI] [PubMed] [Google Scholar]
- 43.Fukai T, Siegfried MR, Ushio-Fukai M, Griendling KK, Harrison DG. Modulation of extracellular superoxide dismutase expression by angiotensin II and hypertension. Circ Res 1999; 85:23–28. [DOI] [PubMed] [Google Scholar]
- 44.Idris-Khodja N, Mian MO, Paradis P, Schiffrin EL. Dual opposing roles of adaptive immunity in hypertension. EurHeartJ 2014; 35:1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.