

Abstract
Eicosapentaenoic acid (EPA) in fish oil is known to improve hepatic steatosis. However, it remains unclear whether such action of EPA is actually caused by peroxisome proliferator-activated receptor α (PPARα) activation. To explore the contribution of PPARα to the effects of EPA itself, male wild-type and Ppara-null mice were fed a saturated fat diet for 16 weeks, and highly (>98%)-purified EPA was administered in the last 12 weeks. Furthermore, the changes caused by EPA treatment were compared to those elicited by fenofibrate (FF), a typical PPARα activator. A saturated fat diet caused macrovesicular steatosis in both genotypes. However, EPA ameliorated steatosis only in wild-type mice without PPARα activation, which was evidently different from numerous previous observations. Instead, EPA inhibited maturation of sterol-responsive element-binding protein (SREBP)-l in the presence of PPARα through down-regulation of SREBP cleavage-activating protein and site-1 protease. Additionally, EPA suppressed fatty acid uptake and promoted hydrolysis of intrahepatic triglycerides in a PPARα-independent manner. These effects were distinct from those offenofibrate. Although fenofibrate induced NAPDH oxidase and acyl-coenzyme A oxidase and significantly increased hepatic lipid peroxides, EPA caused PPARα-dependent induction of superoxide dismutases, probably contributing to a decrease in the lipid peroxides. These results firstly demonstrate detailed mechanisms of steatosis- ameliorating effects of EPA without PPARα activation and ensuing augmentation of hepatic oxidative stress.
Keywords: β-Oxidation, Fatty acid uptake, SCAP, S1P, Superoxide dismutase
1. Introduction
Recent lifestyle alterations, such as increased consumption of saturated fats and decreased physical activity, have raised the prevalence of obesity, metabolic syndrome, and nonalcoholic fatty liver disease(NAFLD) [1,2]. Nonalcoholic steatohepatitis (NASH) is the progressive type of NAFLD and may develop into cirrhosis, liver cancer, and ultimately death [1–4]. Since NAFLD is also associated with a high susceptibility to atherosclerosis and ischemic heart disease [3,5], the increased prevalence of NAFLD is becoming a pressing issue worldwide. Thus, establishment of strategies to treat and prevent NAFLD and related metabolic disturbances is required.
Eicosapentaenoic acid (EPA) is one of the major components of n-3 polyunsaturated fatty acids (PUFA) preferentially contained in fish oil. From the first report of high EPA levels in the diet and blood of the Greenland Inuit [6], who rarely exhibit atherosclerotic diseases, numerous epidemiological and clinical studies have been undertaken to show the efficacy of n-3 PUFA and EPA on reducing serum triglyceride (TG) concentrations and preventing cardiovas-cular events [7–9]. Some data on the steatosis-ameliorating effect of n-3 PUFA have also been obtained [10,11], creating the intriguing possibility that EPA might be beneficial for the treatment of NAFLD.
It has been considered that n-3 PUFA exhibited TG-reducing effects through regulation of peroxisome proliferator-activated receptor α (PPARα) and sterol regulatory element-binding protein (SREBP)-1, which control hepatic fatty acid (FA) catabolism and synthesis, respectively [12]. PPARα is a nuclear receptor expressed primarily in the liver and is involved in not only FA/TG metabolism, but also cell proliferation and inflammatory response [13]. SREBP is a transcription factor belonging to the basic helix-loop-helix leucine zipper family. Depending on various intracellular signals, SREBP is cleaved by several proteases and the N-terminus region is transferred into the nucleus as a mature protein where it regulates transcription of several target genes. SREBP-1 plays an important role in the development of NAFLD, and insulin and synthetic agonists of liver X receptor α (LXRα) enhance its transcription [14].
Several studies have demonstrated an activating effect of EPA on PPARα [15–20]. However, because most of these studies were in vitro experiments [15–17] and used crude fish oil [18–20], it has not been determined whether EPA alone can activate PPARα in vivo as well. Additionally, since fish oil is reported to lower serum TG levels even in PPARα-null (Ppara−/−) mice [21], it remains unclear whether such action occurs via PPARα activation. In order to clarify the contribution of PPARα to the effects of EPA in vivo, highly (>98%)- purified EPA was administered to wild-type and Ppara−/− mice fed a saturated fat diet, and the expression of genes and proteins involved in hepatic FA/TG metabolism was investigated. Furthermore, the changes caused by EPA treatment were compared to those elicited by fenofibrate (FF), a typical PPARα activator.
2. Materials and methods
2.1. Mice and treatment
Ppara−/− mice on a 129/Sv genetic background (129S4/SvJae) were generated as described previously [22]. Male 8-week-old wildtype and Ppara−/− mice weighing 24 ± 5 g (n = 30 in each genotype) were maintained in pathogen-free conditions at constant humidity and temperature with a light/dark cycle of 12 h. At the beginning of this study, mice in each genotype were randomly divided into 5 groups (n = 6/group) and pair-fed a diet. As a control, one group was treated with a standard diet for 16 weeks composed of 20.0% (per weight basis) casein, 53% corn starch, 10% sucrose, 7% olive oil, 5% cellulose, 3.5% mineral mix, 1% vitamin mix, and 0.25% choline. The other4groups were fed a saturated fat diet (Oriental Yeast Co. Ltd.,Tokyo, Japan) in which all fat contained in the standard diet was completely hydrogenated to eliminate the effects of naturally contained PUFA. We chose 16 weeks as a saturated fat diet feeding period, since our preliminary experiments demonstrated that this protocol could induce obvious hepatic steatosis not only in Ppara−/− mice but also in wild-type mice. Body weight and food intake were recorded every day. After 4 weeks on the saturated fat diet, administration of the test agents was initiated in the 4 groups. One group was given highly (>98%)-purified EPA ethyl ester (ethyl all-cis-5, 8, 11, 14, 17-icosapentaenoate) (Mochida Pharmaceutical Co., Ltd, Tokyo, Japan) (1000 mg/kg of body weight/day) for 1 week, one group was given highly (>98%)-purified EPA ethyl ester at the same dose for 12 weeks, and another group was administered FF (Wako Pure Chemicals Industries, Osaka, Japan) (25 mg/kg of body weight/day) for 12 weeks. EPA and FF were dissolved in Arabic gum, mixed, and administered once a day at 10 a.m. by gastric gavage. The last test group was given the same amount of Arabic gum as a vehicle for 12 weeks. After the administration periods, the mice were anesthesized and sacrificed in a fasting state for collection of livers and blood. All experiments were conducted in accordance with the animal study protocols outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and approved by the Animal Studies Committee at Shinshu University School of Medicine.
2.2. Immunoblot analysis
Preparation of hepatocyte nuclear fractions was carried out as described previously [23,24]. Protein concentration was measured colorimetrically with a BCA™ Protein Assay kit (Pierce, Rockford, IL, USA). Whole liver lysates (20–80 μg of protein) or nuclear fractions (80 μg of protein) were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis[23–28]. A sample from one mouse in each group was loaded into each electrophoresis assay and all samples were examined (n = 6/group). After electrophoresis, proteins were transferred to nitrocellulose membranes and incubated with primary antibody followed by alkaline phosphatase-conjugated goat anti-rabbit IgG. The antibodies against FA-metabolizing enzymes were described previously [28], and those against other proteins were purchased commercially. Suppliers and dilutions of primary antibodies are summarized in Supplementary Table 1. Actin or histone H1 was used as the loading control. Band intensities were measured densitometrically, normalized to those of actin or histone H1, and subsequently expressed as fold-changes of those of control wild-type mice fed a saturated fat diet. For confirmation of data reproducibility, immunoblot analysis using the same samples was done twice. Overall, the data on 12 band intensities were obtained from each mouse group for each target molecule and subjected to statistical analysis.
2.3. Analysis of mRNA
Total liver RNA was extracted using an RNeasy Mini Kit (Qiagen, Tokyo, Japan), and cDNA was generated by SuperScript II reverse transcriptase (Gibco BRL, Paisley, Scotland). Quantitative reverse transcription-polymerase chain reaction (RT-PCR) was performed as described elsewhere [29,30] with the primer pairs listed in Supplementary Table 2. Measured mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and subjected to statistical analysis.
2.4. Measurement of hepatic lipids and lipid peroxides
Total hepatic lipids were extracted according to the method developed by Folch et al. [31]. Lipid extracts were dissolved in distilled water, and the concentrations of TG and lipid peroxides [the sum of malondialdehyde (MDA) and 4-hydroxynonenal (4- HNE)] were colorimetrically measured using a TG C-test (Wako Pure Chemicals Industries) and LPO-586 kit (OXIS International, Portland, OR, USA), respectively [32].
2.5. Assessment of FA β-oxidation and uptake abilities
FA β-oxidation activity was measured according to a method described previously with palmitic acid as a substrate, and expressed as pmol/min/mg of liver tissue [28]. FA uptake ability was assessed as described elsewhere and expressed as a fold-change of that in control wild-type mice fed a saturated fat diet [33].
2.6. Assay of hepatic neutral lipase (NL) activity
Fresh liver samples (approximately 100 mg) were homogenized in 20 mM phosphate buffer (pH 7.5) containing 250 mM sucrose and 1 mM EDTA, sonicated, and centrifuged at 20,000 × g for 10 min at 25 °C. NL activity of the supernatant was colorimetrically measured using a MONOTEST (Boehringer Mannheim, Tokyo, Japan) after adjusting the pH of the substrate-containing buffer to 7.2 and expressed as mU/mg of supernatant protein. Triolein was used as a substrate.
2.7. Histological examination
Small blocks of liver tissue from each mouse were fixed in 10% formalin in phosphate-buffered saline and embedded in paraffin. Sections (4 mm thick) were stained with hematoxylin and eosin or Azan-Mallory method. The procedure for cytochemical staining for peroxisomal catalase was described elsewhere [24].
2.8. Other assays
Serum levels of TG, non-esterified FA (NEFA), glucose, aspartate and alanine aminotransferases (AST and ALT), and ketone bodies were determined with commercial kits purchased from Wako Pure Chemicals Industries. Serum insulin concentrations were measured using a mouse insulin ELISA KIT (AKRIN-011T, Shibayagi, Gunma, Japan).
2.9. Statistical analysis
Results are expressed as mean ± standard deviation (SD). Statistical analysis was performed using the two-tailed Student’s t- test. A probability value of less than 0.05 was considered statistically significant. All calculations were performed with SPSS version 11.0 software for Windows (SPSS Inc., Chicago, IL, USA).
3. Results
3.1. A saturated fat diet induces hepatic steatosis in mice
The phenotypic changes caused by a 16-week saturated fat diet are shown in Table 1. At the endpoint, body weight, ratios of liver-to-body weight and epididymal fat-to-body weight, serum TG and ALT concentrations, and hepatic TG contents were all significantly increased by the saturated fat diet. These changes were observed in both genotypes, but were more prominent in Ppara−/− mice. Histological analysis revealed that the saturated fat diet induced mild-to-moderate macrovesicular steatosis mainly in zone 3 in both genotypes, but ballooned hepatocytes, lobular inflammation, or Mallory hyaline were not found (Fig. 1).
Table 1.
Changes in anthropometric and biochemical parameters from a 16-week saturated fat diet.
| Genotype |
Ppara (+/+) |
Ppara (−/−) |
||
|---|---|---|---|---|
| Diet | Con (n = 6) | Sat (n = 6) | Con (n = 6) | Sat (n = 6) |
| Body weight (g) | 23.9 ± 1.9 | 28.3 ± 1.5* | 26.5 ± 2.7 | 41.0 ± 5.2**,## |
| Liver/body weight (%) | 3.8 ± 0.2 | 4.6 ± 0.4* | 4.4 ± 0.2 | 5.2 ± 0.6* |
| Epididymal fat/body weight (%) | 2.5 ± 0.3 | 3.7 ± 0.6* | 3.0 ± 1.5 | 6.1 ± 0.5**,## |
| Serum TG (mg/dL) | 61 ± 1 | 123 ± 41* | 124 ± 50 | 233 ± 49**,## |
| Serum NEFA (mEq/L) | 0.75 ± 0.30 | 1.33 ± 0.3* | 1.19 ± 0.25 | 1.54 ± 0.15# |
| Serum glucose (mg/dL) | 92 ± 23 | 89 ± 24 | 98 ± 14 | 103 ± 22 |
| Serum insulin (ng/mL) | 0.51 ± 0.09 | 1.21 ± 0.58 | 0.48 ± 0.06 | 2.24 ± 0.46** |
| Serum AST (U/L) | 129 ± 66 | 243 ± 62 | 149 ± 92 | 203 ± 46 |
| Serum ALT (U/L) | 13 ± 6 | 43 ± 16* | 18 ± 10 | 99 ± 21**,# |
| Liver TG (mg/g) | 10 ± 1 | 30 ± 3** | 17 ± 3 | 52 ± 7**,## |
Results are expressed as mean ± SD. Con, control standard diet; Sat, saturated fat diet; TG, triglyceride; NEFA, non-esterified fatty acid; AST, aspartate aminotransferase; ALT, alanine aminotransferase.
P< 0.05 compared with mice of the same genotype fed a control diet.
P< 0.01 compared with mice of the same genotype fed a control diet.
P< 0.05 compared with Ppara (+/+) mice fed the same diet.
P< 0.01 compared with Ppara (+/+) mice fed the same diet.
Fig. 1.
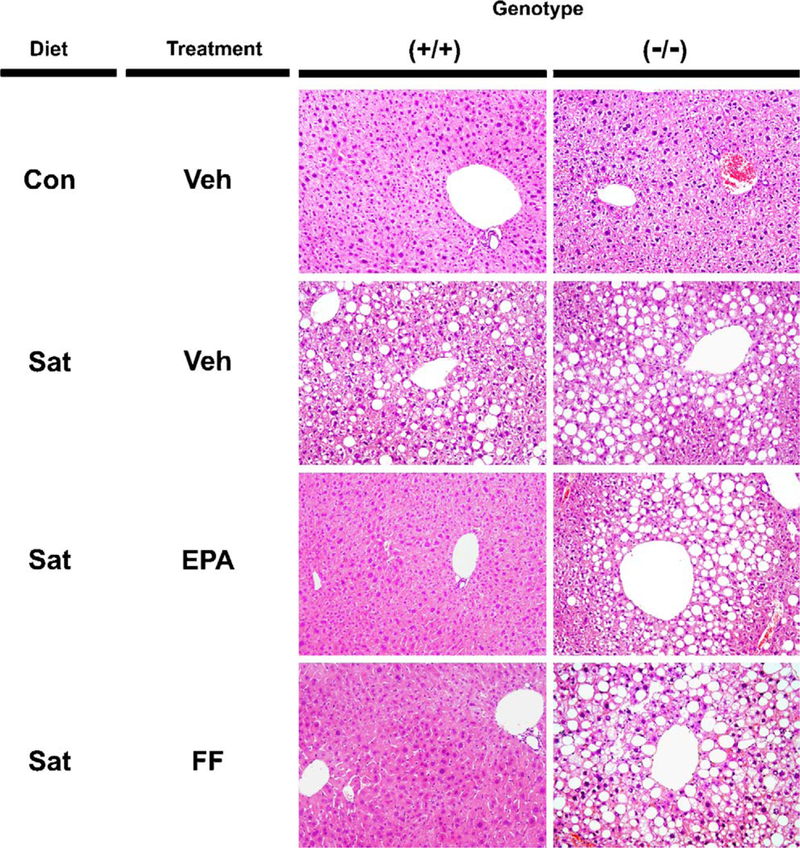
Histological findings in the livers of wild-type and Ppara−/− mice. Male 8-week-old wild-type (+/+) and Ppara (−/−) mice were fed a control standard (Con) or saturated fat diet (Sat) for 16 weeks. After 4 weeks on the saturated fat diet, treatment with highly-purified EPA or FF was initiated and continued for 12 weeks. Liver sections were stained by hematoxylin and eosin method. Original magnification, 200×. Veh, vehicle.
3.2. EPA ameliorates hepatic steatosis in a PPARa-dependent manner
FF treatment for 12 weeks decreased body weight and serum/liver TG levels in wild-type mice only, but significantly increased serum ALT levels in both genotypes (Fig. 2). When EPA was administered for 1 week, there were no remarkable changes in either genotype. However, EPA treatment for 12 weeks significantly decreased liver-to-body weight ratio, serum levels of TG, NEFA, and ALT, and hepatic TG content in wild-type mice without decreases in body weight or epididymal fat weight (Fig. 2). Histologically, EPA markedly attenuated macrovesicular steatosis in wild-type mice only (Fig. 1). These results confirmed that EPA ameliorated saturated fat diet-induced steatosis and hypertriglyceridemia preferentially in wild-type mice.
Fig. 2.
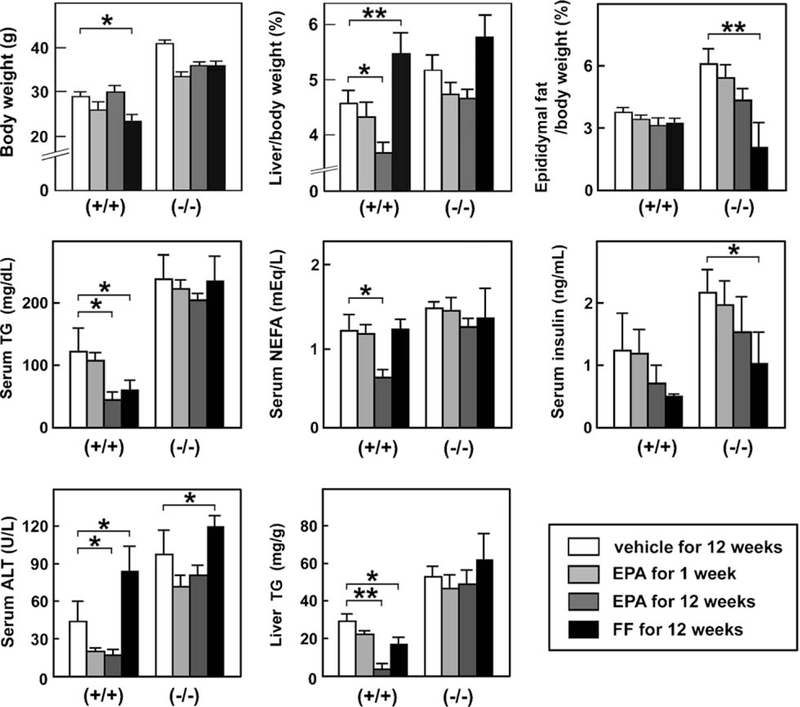
Effects of EPA and FF on anthropometric and biochemical parameters. Wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were treated with a vehicle for 12 weeks, highly-purified EPA for 1 week or 12 weeks, or FF for 12 weeks. Results are expressed as mean ± SD (n = 6/group). *P < 0.05; **P < 0.01.
3.3. EPA does not enhance mitochondrial β-oxidation
To investigate the precise mechanism of the PPARα-depen-dent steatosis-attenuating effect of EPA, we first analyzed hepatic β-oxidation pathway. FF markedly increased the expression of representative mitochondrial β-oxidation enzymes [long-chain acyl-CoA synthase (LACS), carnitine pal-mitoyl-CoA transferase-1 (CPT-I), and medium-chain acyl-CoA dehydrogenase (MCAD)] at the protein and mRNA levels (Fig. 3A and B), enhanced mitochondrial β-oxidation activity (Fig. 3C), and elevated serum ketone body concentrations (Fig. 3D) in wild-type mice only. However, these changes were not observed in the EPA-treated mice (Fig. 3), suggesting that the effect of EPA on hepatic steatosis was not derived from enhancement of mitochondrial β-oxidation.
Fig. 3.
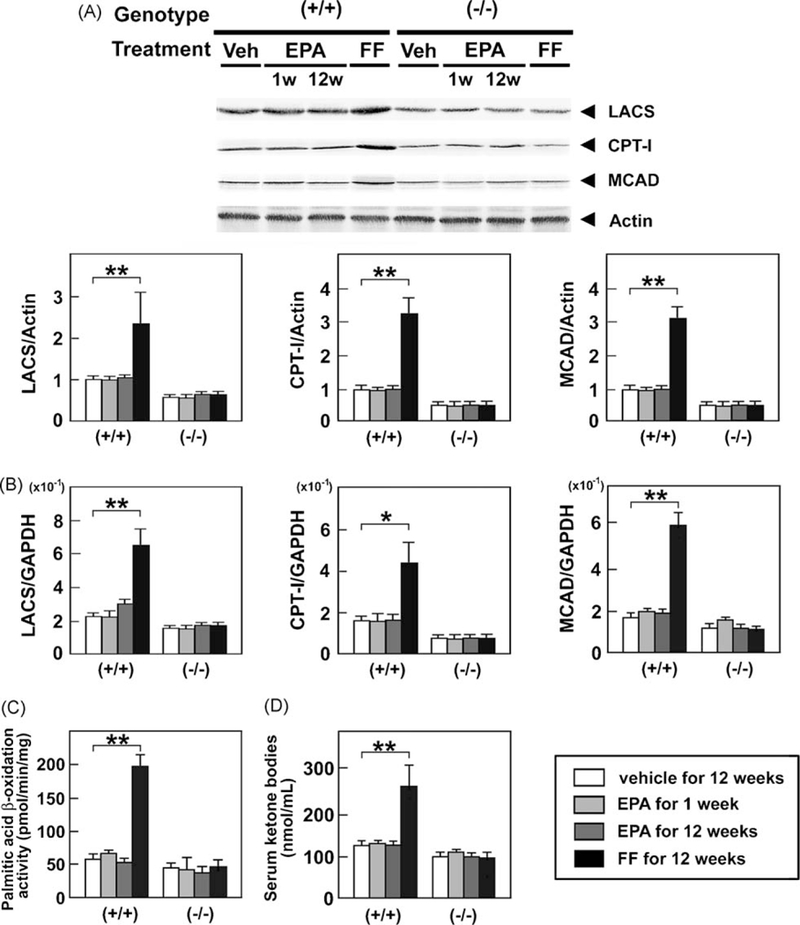
Immunoblot analysis of LACS, CPT-I, and MCAD. Wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were treated with a vehicle for 12 weeks, highly-purified EPA for 1 week or 12 weeks, or FF for 12 weeks, and whole liver lysates (20 μg of protein) obtained from mice were loaded into each well. Band intensities were measured densitometrically, normalized to those of actin, and subsequently normalized to those in control wild-type mice [(+/+)Veh]. Results are expressed as mean ± SD (n = 6/group). **P < 0.01. (B) Hepatic mRNA levels of LACS, CPT-I, and MCAD. Wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were treated with a vehicle for 12 weeks, highly-purified EPA for 1 week or 12 weeks, or FF for 12 weeks, and mRNA levels were determined by quantitative RT-PCR. Measured mRNA levels were normalized to those of GAPDH. Results are expressed as mean ± SD(n = 6/group). *P < 0.05; **P < 0.01. (C) Changes in mitochondrial β-oxidation activity in the liver. Wild-type (+/+) and Ppara (−/−)mice fed a saturatedfat diet were treated with a vehicle for 12 weeks, highly-purified EPAfor 1 week or 12 weeks, or FF for 12 weeks, and β-oxidation activity was measured usingpalmitic acid as a substrate. Results are expressed as mean ± SD (n = 6/group). **P < 0.01. (D) Serum concentrations of ketone bodies. Figure presentation is identical to that in Fig. 2.
3.4. EPA suppresses FA uptake in a PPARα-independent manner
FF markedly increased the levels of liver fatty acid-binding protein (L-FABP), fatty acid translocase (FAT), and fatty acid transport protein (FATP) and lowered apolipoprotein CIII (apoClll) in wild-type mice only (Fig. 4A and B). In contrast, EPA significantly increased the levels of mRNA encoding apoClll (Fig. 4B), and strongly decreased the expression of L-FABP, FAT, and FATP in a PPARα-independent manner (Fig. 4A). A reduction in the latter two proteins was also confirmed by quantitative RT-PCR analysis (Fig. 4B). Furthermore, EPA significantly suppressed palmitic acid uptake into hepatocytes (Fig. 4C). Both agents did not affect the mRNA levels of hepatic TG lipase (HTGL) (Fig. 4B).
Fig. 4.
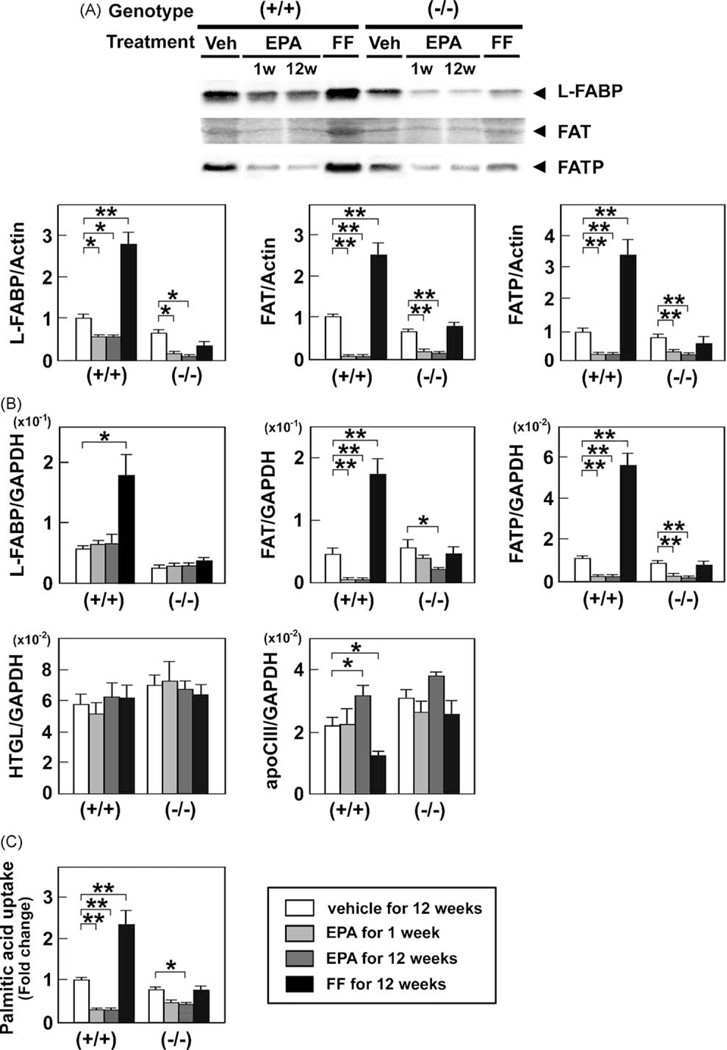
Effects of EPA and FF on the hepatic FA uptake system. (A) Immunoblot analysis of L-FABP, FAT, and FATP. The same samples used in Fig. 3A were loaded. P < 0.05; **P < 0.01. (B) Hepatic mRNA levels of molecules associated with FA uptake. The same samples in Fig. 3B were adopted. *P < 0.05; **P < 0.01. (C) Changes in palmitic acid uptake ability into the liver. Wild-type (+/+)and Ppara (−/−)mice fed a saturated fat diet were treated with a vehicle for 12 weeks, highly-purified EPA for 1 week or 12 weeks, or FF for 12 weeks, and FA uptake activity was assessed as described in Methods. Results are expressed as mean ± SD (n = 6/group). *P < 0.05; **P < 0.01.
3.5. EPA suppresses de novo lipogenesis in a PPARα-dependent manner
FF increased the expression of acetyl-CoA carboxylase (ACC) in wild-type mice, but had no impact on the expression of fatty acid synthase (FAS) or glycerol-3-phosphate acyltransferase (GPAT) (Fig. 5A and B). On the other hand, EPA markedly decreased the expression of these three enzymes in wild-type mice (Fig. 5A and B). These results showed that EPA inhibited de novo lipogenesis in a PPARα-dependent fashion.
Fig. 5.
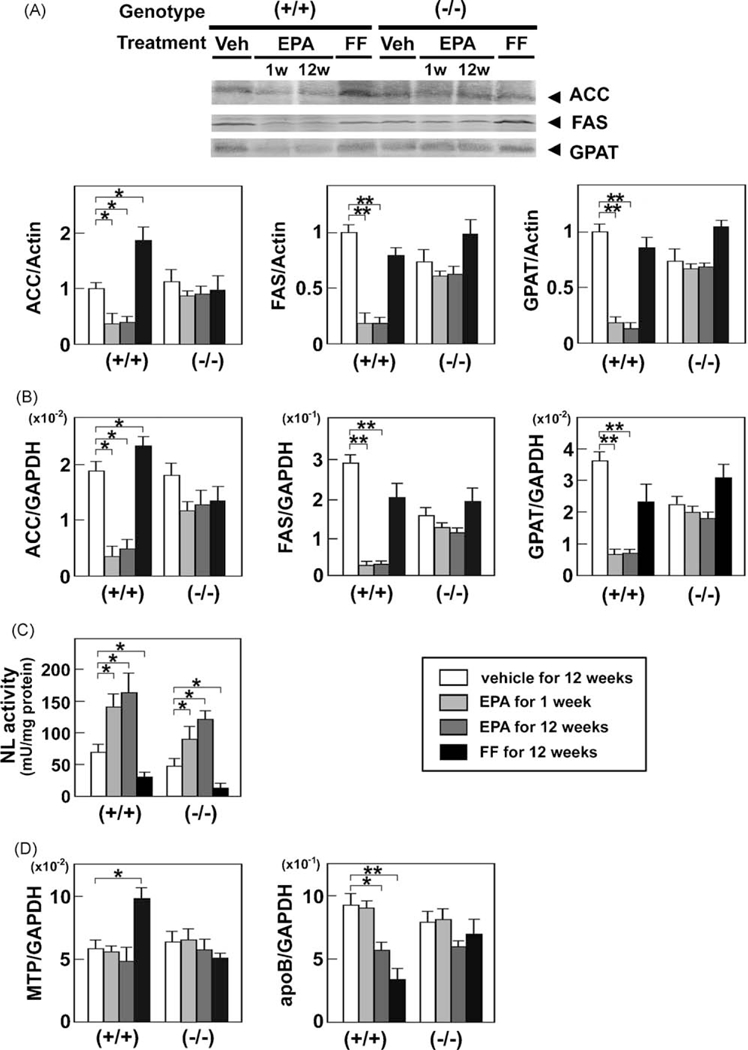
Effects of EPA and FF on hepatic de novo lipogenesis and the TG hydrolysis and secretion pathway. (A) Immunoblot analysis of ACC, FAS, and GPAT. Whole liver lysates (40–80 μg of protein) obtained from wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were loaded into each well. Band intensities were measured densitometrically, normalized to those of actin, and subsequently normalized to those in control wild-type mice [(+/+) Veh]. Results are expressed as mean ± SD(n = 6/group). *P < 0.05; **P < 0.01. (B) Hepatic mRNAlevels oflipogenic enzymes. The same samples in Fig. 3B were used. *P < 0.05; **P < 0.01. (C) Changes in hepatic NL activity. Wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were treated with a vehicle for 12 weeks, highly-purified EPA for 1 week or 12 weeks, or FF for 12 weeks, and hepatic NL activity was determined as described in Methods. Results are expressed as mean ± SD (n = 6/group). *P < 0.05. (D) Hepatic mRNA levels of molecules associated with TG secretion. The same samples in Fig. 3B were used. *P < 0.05; **P < 0.01.
3.6. Effects of EPA on the hepatic TG degradation and secretion pathway
In contrast to FF, EPA significantly enhanced NL activity in both genotypes (Fig. 5C), indicating that it strongly facilitated hydrolysis of intrahepatic TG. EPA did not affect the expression of microsomal TG transfer protein (MTP) or apoB (Fig. 5D).
3.7. EPA does not activate PPARα
Although EPA lowered hepatic TG levels in a PPARα-dependent manner, it did not increase the nuclear or mRNA levels of PPARα (Fig. 6A and B), induce expression of representative PPARα target genes [LACS, CPT-I, MCAD (Fig. 3), L-FABP, FAT, FATP (Fig. 4), or peroxisomal membrane protein 70 (PMP70) (Fig. 6C)], or cause hepatic peroxisome proliferation (Fig. 6D), all of which were distinct from the actions of FF. These results provide compelling evidence that the steatosis-ameliorating effect of EPA is not through PPARα activation. In addition, EPA did not influence the nuclear levels of the other PPARs: PPARδ and PPARγ (Fig. 6A).
Fig. 6.
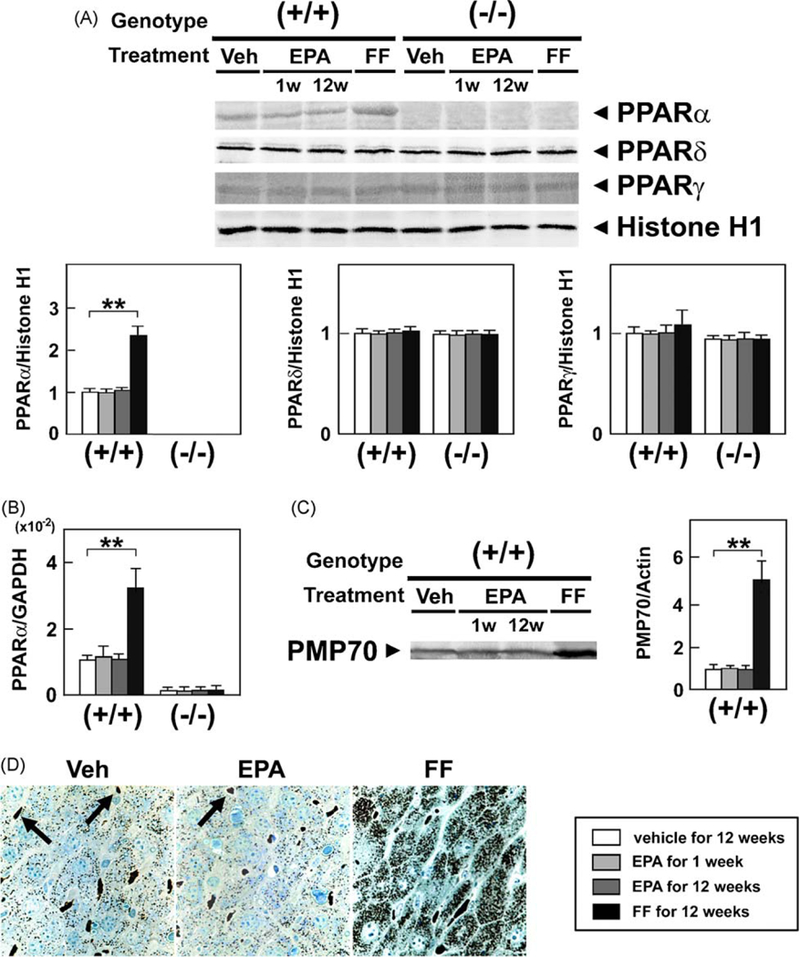
Effects of EPA and FF on hepatic PPAR. (A) Immunoblot analysis of PPAR. Hepatocyte nuclear fractions (80 μg of protein) obtained from wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were loaded into each well. Band intensities were measured densitometrically, normalized to those of histone H1, and subsequently normalized to those in control wild-type mice [(+/+) Veh]. Results are expressed as mean ± SD(n = 6/group). **P < 0.01.(B) Hepatic mRNA levels of PPARα. The same samples in Fig. 3B were used. **P < 0.01. (C) Immunoblot analysis of PMP70, a typical PPARα target. The same samples used in Fig. 3A were loaded. **P < 0.01. (D) Cytochemical staining for hepatic peroxisomes. Liver tissues were obtained from saturated fat diet-fed wild-type mice treated with a vehicle (Veh), highly-purified EPA, or FF for 12 weeks and subjected to cytochemical staining for peroxisomal catalase. Peroxisomes are seen as darkly-stained particles. Arrows indicate erythrocytes. Original magnification, 400×.
3.8. EPA suppresses SREBP-1 maturation in a PPARα-dependent manner
EPA significantly decreased the mature SREBP-1 levels only in hepatocyte nuclei of wild-type mice, but did not affect the levels of LXRα and PPARγ coactivator-1 β (PGC-1β), known to be involved in SREBP-1 regulation [34] (Fig. 7A). Decreases in mature SREBP-1 levels correlated with those in SREBP-1 target genes, including ACC, FAS, and GPAT (Fig. 5A and B). However, EPA did not lower SREBP-1 mRNA levels (Fig. 7B), suggesting that EPA modulated the expression of SREBP-1 at the post-transcriptional level. When factors associated with SREBP-1 maturation were examined, the expression of SREBP cleavage-activating protein (SCAP) and site-1 protease (S1P) were significantly decreased by EPA in a PPARα- dependent manner (Fig. 7B and C). The levels of mRNA encoding insulin-induced gene product (lnsig)-2, another SREBP-1-activat-ing molecule expressed exclusively in the liver, remained unchanged by EPA treatment (Fig. 7B). Collectively, these results demonstrated that EPA inhibited maturation of SREBP-1 in the presence of PPARα through down-regulation of SCAP and S1P.
Fig. 7.
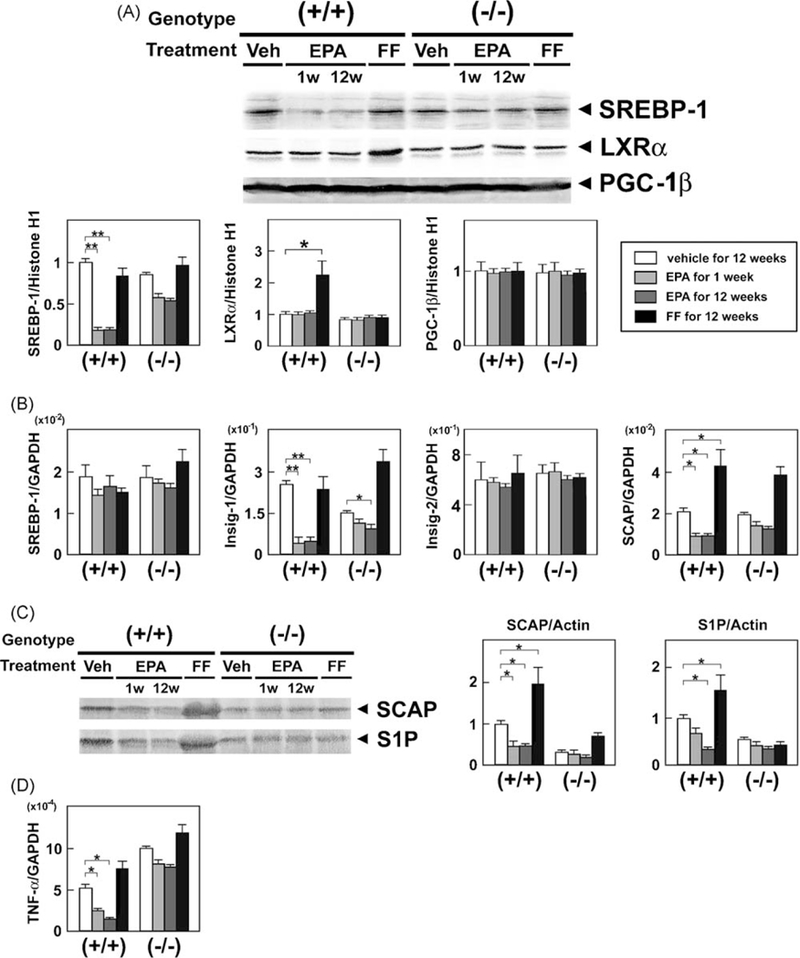
Effects of EPA and FF on hepatic SREBP-1. (A) Immunoblot analysis of SREBP-1 and its co-regulators, LXRα and PGC-1b. The same samples used in Fig. 6A were loaded. *P < 0.05; **P < 0.01. (B) Hepatic mRNA levels of SREBP-1 and its activating proteins. The same samples in Fig. 3B were used. *P < 0.05; **P < 0.01. (C) Immunoblot analysis of SCAP and S1P. The same samples in Fig. 3A were loaded. *P < 0.05. (D) Hepatic mRNA levels of TNF-α. The same samples in Fig. 3B were used. *P < 0.05.
A recent study has shown that the expression of SCAP was induced by pro-inflammatory cytokines [35]. The mRNA levels of tumor necrosis factor-α (TNF-α) were decreased in EPA-treated wild-type mice only (Fig. 7D).
3.9. EPA reduces hepatic oxidative stress
Persistent PPARα activation may increase generation of reactive oxygen species (ROS) [33,36]. As expected, FF markedly increased hepatic lipid peroxide content (Fig. 8) and the expression of ROS-generating enzymes, such as NADPH oxidase (gp91phox and p47phox) and acyl-CoA oxidase (AOX), in wild-type mice only (Fig. 9). On the other hand, EPA reduced hepatic lipid peroxides in wild-type mice (Fig. 8), likely due to increased expression of manganese- and copper, zinc-superoxide dismutases (Mn- and Cu, Zn-SOD) (Fig. 9). EPA did not affect the expression of glutathione peroxidase (GPx) (Fig. 9). Thus, EPA can ameliorate fatty liver without activation of PPARα and ensuing augmentation of hepatic oxidative stress.
Fig. 8.
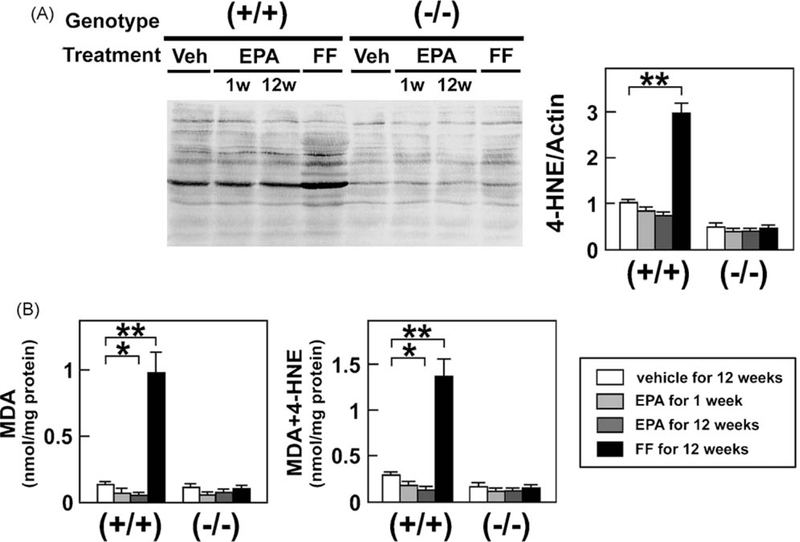
Analysis of hepatic lipid peroxide content. (A) Immunoblot analysis of 4-HNE. The same samples in Fig. 3A were loaded. **P < 0.01. (B) Hepatic levels of MDA and MDA + 4-HNE. Wild-type (+/+) and Ppara (−/−) mice fed a saturated fat diet were treated with a vehicle for 12 weeks, EPA for 1 week or 12 weeks, or FF for 12 weeks. Total lipids were extracted from mouse liver tissues and the amounts of MDA and MDA + 4-HNE were determined. Results are expressed as mean ± SD (n = 6/group). *P < 0.05; **P < 0.01.
Fig. 9.
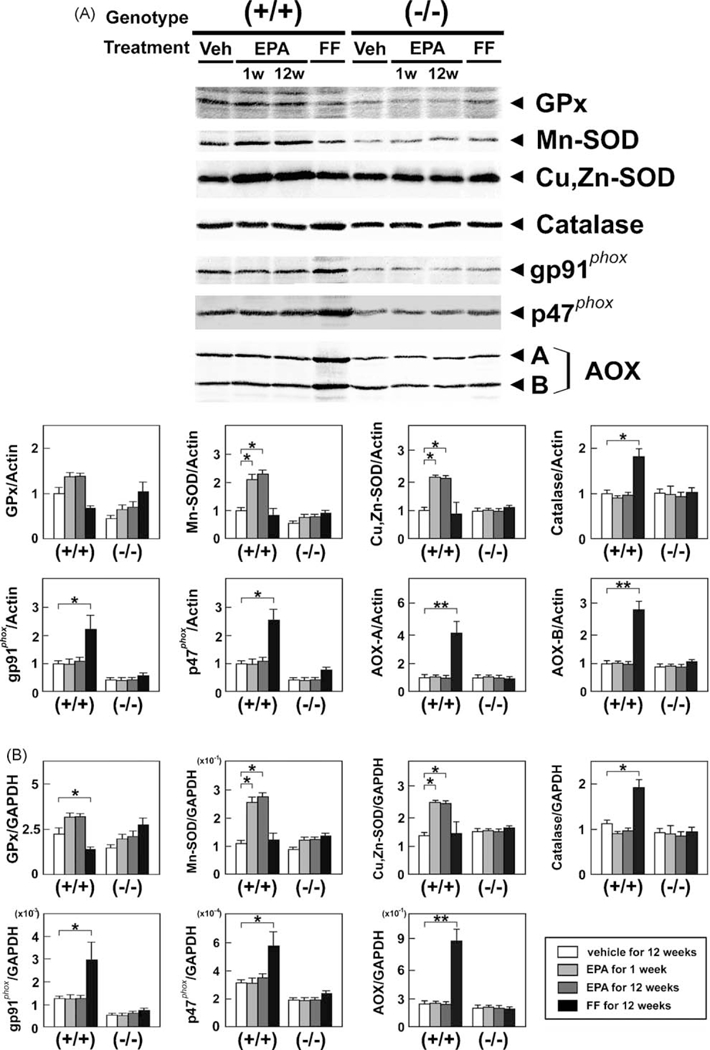
Effects of EPA and FF on hepatic ROS-scavenging and ROS-generating enzymes. (A) Immunoblot analysis of ROS-scavenging and ROS-generating enzymes. Whole liver lysates (40–80 μg of protein) were loaded into each well. The same samples used in Fig. 3A were loaded. A and B bands of AOX, full-length and truncated AOX, respectively. *P < 0.05; **P < 0.01. (B) Hepatic mRNA levels of ROS-scavenging and ROS-generating enzymes. The same samples in Fig. 3B were adopted. *P < 0.05; **P < 0.01.
4. Discussion
The present study demonstrated detailed mechanisms of steatosis-attenuating effects of highly-purified EPA in mice, which were unexpectedly unrelated to PPARα activation. They can be summarized as follows: (1) suppression of SREBP-1 processing, (2) suppression of FA uptake from the blood into hepatocytes, and (3) enhancement of intrahepatic TG hydrolysis. Interestingly, down-regulation of SCAP and S1P and the resultant suppression of SREBP- 1-mediated pathway by EPA occurred in a PPARα-dependent manner. These actions clearly differed from those of FF, a typical PPARα activator.
We uncovered that highly-purified EPA cannot singularly activate PPARa in mice. This fact is quite different from the results of the previous in vivo studies using fish oil [10,18–20]. This discrepancy probably stems from PPARα-activating properties of constituents of fish oil other than EPA. Indeed, tuna oil, in which docosahexaenoic acid is dominant, has been shown to be a much stronger PPARα activator than EPA in mice [37]. Thus, it is conceivable that pure EPA may possess pharmacological actions distinct from crude fish oil.
Furthermore, Ishii et al. [38] have reported that EPA-supple- mented diet increased the expression of PPARα in Pten-deficient mice, which is inconsistent with the present results. EPA contained in the diet is easily oxidized at room temperature and several types of oxidized EPA derivatives are generated. Sethi et al. [39] have demonstrated that oxidized EPA markedly activated PPARα and its PPARα-activating properties were approximately 2.5-fold stronger than native non-oxidized EPA and as potent as fenofibric acid in bovine aortic endothelial cells. Based on these observations, we suppose that such a discrepancy in PPARα expression may result from the difference in the amount of oxidized EPA.
In the preliminary experiments, we administered highly-purified EPA at a daily dose of 200, 400, 600, and 1000 mg/kg of body weight to wild-type mice fed a saturated fat diet for 12 weeks, and found that EPA treatment at the latter three doses was safe and effective to reduce hepatic TG contents. Down-regulation of SREBP-1 and the lack of PPARα activation were also detected by EPA administration at each dose. To confirm the absence of PPARα activation by EPA, we adopted the highest dose (1000 mg/kg of body weight/day) in this study.
A novel and unexpected finding in the present study was that EPA-induced decreases in the mature SREBP-1 protein in hepatocyte nuclei occurred by down-regulating the expression of SCAP and S1P, but not by lowering the SREBP-1 mRNA levels. This phenomenon was not observed in Ppara−/− mice, suggesting that down-regulation of SCAP and S1P by EPA are related to the presence of PPARα. The detailed molecular mechanism regarding the contribution of PPARα to SREBP-1 processing/activation system still remains unclear. However, it has been reported that TNF-α increased the expression of SCAP in the livers of casein-injected apoE knockout mice [35]. Furthermore, binding motifs for Sp1 have been detected in the promoter region of S1P gene [40]. lndeed,TNF-α-and lipid peroxide- reducing effects were observed only in EPA-treated wild-type mice (Figs. 7D and8). Therefore, we can speculate that PPARα-dependent alleviation of inflammatory stress by EPA is associated with decreases in the expression of SCAP and S1P.
EPA markedly suppressed the expression of L-FABP, FAT, and FATP and inhibited FA uptake into hepatocytes by a PPARα- independent mechanism. EPA-induced suppression of L-FABP appeared at the post-transcriptional level. L-FABP is prone to S- thiolation, N-acetylation, phosphorylation, and conformational changes, all of which are related to protein stability [41]; EPA might influence such modifications. Since the enhancement of FA uptake has also been reported in the livers of patients with NAFLD [42], its correction caused by EPA may also lead to the improvement of hepatic steatosis.
It is noteworthy that EPA significantly increased the expression of Mn- and Cu, Zn-SODs in a PPARα-dependent fashion. These increases occur via nuclear stabilization of nuclear factor-E2- related factor 2 (Nrf2) [43]. It has been documented that EPA stabilized Nrf2 by reacting directly with Keap1, a negative regulator of Nrf2, and disrupting its function [44]. Thus, EPA might affect the stabilization process of Nrf2 via PPARα. Further studies are needed to address its precise mechanism.
It is well recognized that a saturated fat-rich diet is associated with the development of NAFLD in humans, and that lower levels of hepatic n-3 PUFA predispose livers to steatosis by favoring de novo lipogenesis [12,45]. In this study, mice fed a saturated fat diet exhibited elevation of serum ALT levels, hypertriglyceridemia, and macrovesicular steatosis, which resembled the clinical features of patients with NAFLD. Furthermore, EPA treatment in these mice decreased hepatic TG levels without affecting body weight or serum insulin concentrations; these findings were also observed in NASH patients treated with highly-purified EPA [46]. Thus, the molecular mechanism of EPA action found in this study may at least in part be translated to humans.
It is known that lipotoxicity, oxidative stress, and mitochondrial dysfunction play important roles in the pathogenesis of NAFLD/ NASH [2,36]. Although FF decreased hepatic TG/FA contents, it enhanced mitochondrial β-oxidation activity and induced the expression of NADPH oxidase and AOX, which may result in production of ROS inside and outside the mitochondria. On the other hand, EPA not only down-regulated two major pathways to increase intrahepatic FA contents, but also elevated the levels of mitochondrial Mn-SOD and extra-mitochondrial Cu, Zn-SOD, which may lead to alleviation of lipotoxicity and oxidative stress. Therefore, we believe that highly-purified EPA may be useful for the treatment of NAFLD/NASH [46].
In conclusion, this study clarified that the main action of EPA in hepatic steatosis improvement was not based on the activation of PPARα. In contrast to FF, EPA significantly inhibited de novo lipogenesis and hepatic FA uptake with a reduction in hepatic oxidative stress. These data raise the possibility that EPA may be a promising candidate for various types of liver diseases associated with hepatic fat accumulation and oxidative stress, including NASH, alcoholic liver disease [32,47], and chronic hepatitis C [33]. Further studies are needed to confirm the efficacy of highly- purified EPA against these diseases.
Supplementary Material
Acknowledgements
We thank Trevor Ralph for his editorial assistance.
Financial support: The authors have declared that no financial support exists.
Abbreviations:
- ACC
acetyl-CoA carboxylase
- ALT
alanine aminotransferase
- apo
apolipoprotein
- AOX
acyl-CoA oxidase
- AST
aspartate aminotransferase
- CoA
coenzyme A
- CPT-I
carnitine palmitoyl-CoA transferase-I
- EPA
eicosapentaenoic acid
- FA
fatty acid
- FAS
fatty acid synthase
- FAT
fatty acid translocase
- FATP
fatty acid transport protein
- FF
fenofibrate
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GPAT
glycerol-3-phosphate acyltransferase
- GPx
glutathione peroxidase
- 4-HNE
4-hydroxynonenal
- HTGL
hepatic triglyceride lipase
- Insig
insulin-induced gene product
- LACS
long-chain acyl-CoA synthase
- L-FABP
liver fatty acid-binding protein
- LXR
liver X receptor
- MCAD
medium-chain acyl-CoA dehydrogenase
- MDA
malondialdehyde
- mRNA
messenger RNA
- MTP
microsomal triglyceride transfer protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NEFA
non-esterified fatty acid
- NL
neutral lipase
- Nrf2
nuclear factor-E2-related factor 2
- PGC
PPARγ coactivator
- PMP
peroxisomal membrane protein
- PPAR
peroxisome proliferator-activated receptor
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- RT-PCR
reverse transcription-polymerase chain reaction
- SCAP
SREBP cleavage-activating protein
- S1P
site-1 protease
- SD
standard deviation
- SOD
superoxide dismutase
- SREBP
sterol regulatory element-binding protein
- TG
triglyceride
- TNF
tumor necrosis factor
Footnotes
Conflicts of interest
The authors have declared that no conflict of interest exists.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bcp.2010.07.031.
References
- [1].Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006;43:S99–112. [DOI] [PubMed] [Google Scholar]
- [2].de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol 2008;48:S104–12. [DOI] [PubMed] [Google Scholar]
- [3].Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006;44:865–73. [DOI] [PubMed] [Google Scholar]
- [4].Nagaya T, Tanaka N, Komatsu M, Ichijo T, Sano K, Horiuchi A, et al. Develop¬ment from simple steatosis to liver cirrhosis and hepatocellular carcinoma: a 27-year follow-up case. Clin J Gastroenterol 2008;1:116–21. [DOI] [PubMed] [Google Scholar]
- [5].Vuppalanchi R, Chalasani N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology 2009;49:306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dyeberg J, Bang HO. Haemostatic function and platelet polyunsaturated fatty acids in Eskimos. Lancet 1979;2:433–5. [DOI] [PubMed] [Google Scholar]
- [7].Kromhout D, Bosschieter EB, de Lezenne Coulander C. The inverse relation between fish consumption and 20-year mortality from coronary heart disease. N Engl J Med 1985;312:1205–9. [DOI] [PubMed] [Google Scholar]
- [8].Lemaitre RN, King IB, Mozaffarian D, Kuller LH, Tracy RP, Siscovick DS. N-3 polyunsaturated fatty acids, fatal ischemic heart disease, and nonfatal myo¬cardial infarction in older adults: the Cardiovascular Health Study. Am J Clin Nutr 2003;77:319–25. [DOI] [PubMed] [Google Scholar]
- [9].Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholester-olaemic patients (JELIS): a randomized open-label, blinded endpoint analysis. Lancet 2007;369:1090–8. [DOI] [PubMed] [Google Scholar]
- [10].Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Mar-zioni M, et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-α and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol 2006;169:846–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Capanni M, Calella F, Biagini MR, Genise S, Raimondi L, Bedogni G, et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther 2006;23:1143–51. [DOI] [PubMed] [Google Scholar]
- [12].Clarke SD. Nonalcoholic steatosis and steatohepatitis. I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am J Physiol Gastrointest Liver Physiol 2001;281:G865–9. [DOI] [PubMed] [Google Scholar]
- [13].Gonzalez FJ, Shah YM. PPARα: mechanism of species differences and hepa- tocarcinogenesis of peroxisome proliferators. Toxicology 2008;246:2–8. [DOI] [PubMed] [Google Scholar]
- [14].Ferre P, Foufelle F. SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Horm Res 2007;68:72–82. [DOI] [PubMed] [Google Scholar]
- [15].Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc Natl Acad Sci U S A 1997;94:4312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T, et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem 2002;277:1705–11. [DOI] [PubMed] [Google Scholar]
- [17].Pawer A, Jump DB. Unsaturated fatty acid regulation of peroxisome prolif- erator-activated receptor α activity in rat primary hepatocytes. J Biol Chem 2003;278:35931–9. [DOI] [PubMed] [Google Scholar]
- [18].Ren B, Thelen AP, Peters JM, Gonzalez FJ, Jump DB. Polyunsaturated fatty acid suppression of hepatic fatty acid synthase and S14 gene expression does not require peroxisome proliferator-activated receptor α. J Biol Chem 1997;272:26827–32. [DOI] [PubMed] [Google Scholar]
- [19].Kim HJ, Takahashi M, Ezaki O. Fish oil feeding decreases mature sterol regulatory element-binding protein 1 (SREBP-1) by down-regulation of SREBP-1c mRNA in mouse liver. A possible mechanism for down-regulation of lipogenic enzyme mRNAs. J Biol Chem 1999;274:25892–8. [DOI] [PubMed] [Google Scholar]
- [20].Takahashi M, Tsuboyama-Kasaoka N, Nakatani T, Ishii M, Tsutsumi S, Abur- atani H, et al. Fish oil feeding alters liver gene expressions to defend against PPARα activation and ROS production. Am J Physiol Gastrointest Liver Physiol 2002;282:G338–48. [DOI] [PubMed] [Google Scholar]
- [21].Dallongeville J, Bauge E, Tailleux A, Peters JM, Gonzalez FJ, Fruchart JC, et al. Peroxisome proliferator-activated receptor α is not rate-limiting for the lipoprotein-lowering action of fish oil. J Biol Chem 2001;276:4634–9. [DOI] [PubMed] [Google Scholar]
- [22].Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, et al. Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome prolif-erators. Mol Cell Biol 1995;15:3012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tanaka N, Hora K, Makishima H, Kamijo Y, Kiyosawa K, Gonzalez FJ, et al. In vivo stabilization of nuclear retinoid X receptor a in the presence of peroxisome proliferator-activated receptor α. FEBS Lett 2003;543:120–4. [DOI] [PubMed] [Google Scholar]
- [24].Tanaka N, Moriya K, Kiyosawa K, Koike K, Aoyama T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome prolif-erator-activated receptor α in transgenic mice: implications for HCV-associ-ated hepatocarcinogenesis. Int J Cancer 2008;122:124–31. [DOI] [PubMed] [Google Scholar]
- [25].Aoyama T, Yamano S, Waxman DJ, Lapenson DP, Meyer UA, Fischer V, et al. Cytochrome P-450 hPCN3, a novel cytochrome P-450 IIIA gene product that is differentially expressed in adult human liver. CDNA and deduced amino acid sequence and distinct specificities of cDNA-expressed hPCN1 and hPCN3 for the metabolism of steroid hormones and cyclosporine. J Biol Chem 1989; 264:10388–95. [PubMed] [Google Scholar]
- [26].Aoyama T, Uchida Y, Kelley RI, Marble M, Hofman K, Tonsgard JH, et al. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem Biophys Res Commun 1993;191:1369–72. [DOI] [PubMed] [Google Scholar]
- [27].Aoyama T, Souri M, Ushikubo S, Kamijo T, Yamaguchi S, Kelley RI, et al. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J Clin Invest 1995;95:2465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor a (PPARα). J Biol Chem 1998;273:5678–84. [DOI] [PubMed] [Google Scholar]
- [29].Nakajima T, Tanaka N, Sugiyama E, Kamijo Y, Hara A, Hu R, et al. Cholesterollowering effect of bezafibrate is independent of peroxisome proliferator-activated receptor activation in mice. Biochem Pharmacol 2008;76:108–19. [DOI] [PubMed] [Google Scholar]
- [30].Nakajima T, Tanaka N, Kanbe H, Hara A, Kamijo Y, Zhang X, et al. Bezafibrate at clinically-relevant doses decreases serum/liver triglycerides via down-regu-lation of SREBP-1c in mice: a novel PPARα-independent mechanism. Mol Pharmacol 2009;75:782–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497–509. [PubMed] [Google Scholar]
- [32].Okiyama W, Tanaka N, Nakajima T, Tanaka E, Kiyosawa K, Gonzalez FJ, et al. Polyenephosphatidylcholine prevents alcoholic liver disease in PPARα-null mice through attenuation of increases in oxidative stress. J Hepatol 2009;50:1236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARa activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest 2008;119:683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metab¬olism in health and disease. J Clin Invest 2006;116:615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology 2008;48:770–81. [DOI] [PubMed] [Google Scholar]
- [36].Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, et al. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 2003;38:1529–39. [DOI] [PubMed] [Google Scholar]
- [38].Ishii H, Horie Y, Ohshima S, Anezaki Y, Kinoshita N, Dohmen T, et al. Eicosa- pentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific Pten-deficient mice. J Hepatol 2009;50:562–71. [DOI] [PubMed] [Google Scholar]
- [39].Sethi S, Ziouzenkova O, Ni H, Wagner DD, Plutzky J, Mayadas TN. Oxidized omega-3 fatty acids in fish oil inhibit leukocyte-endothelial interactions through activation of PPARα. Blood 2002;100:1340–6. [DOI] [PubMed] [Google Scholar]
- [40].Nakajima T, Iwaki K, Kodama T, Inazawa J, Emi M. Genomic structure and chromosomal mapping of the human site-1 protease (S1P) gene. J Hum Genet 2000;45:212–7. [DOI] [PubMed] [Google Scholar]
- [41].Schroeder F, Jolly CA, Cho TH, Frolov A. Fatty acid binding protein isoforms: structure and function. Chem Phys Lipids 1998;92:1–25. [DOI] [PubMed] [Google Scholar]
- [42].Greco D, Kotronen A, Westerbacka J, Puig O, Arkkila P, Kiviluoto T, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol 2008;294:G1281–7. [DOI] [PubMed] [Google Scholar]
- [43].Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem 2003;278:8135–45. [DOI] [PubMed] [Google Scholar]
- [44].Gao R, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem 2007;282:2529–37. [DOI] [PubMed] [Google Scholar]
- [45].Allard JP, Aghdassi E, Mohammed S, Raman M, Avand G, Arendt BM, et al. Nutritional assessment and hepatic fatty acid composition in non-alcoholic fatty liver disease (NAFLD): A cross-sectional study. J Hepatol 2008;48: 300–307. [DOI] [PubMed] [Google Scholar]
- [46].Tanaka N, Sano K, Horiuchi A, Tanaka E, Kiyosawa K, Aoyama T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J Clin Gastroenterol 2008;42:413–8. [DOI] [PubMed] [Google Scholar]
- [47].Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K, et al. Peroxisome proliferator-activated receptor a protects against alcohol-induced liver damage. Hepatology 2004;40:972–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.