

Summary
CD4+ T helper (Th) differentiation is regulated by diverse inputs including the Vitamin A metabolite retinoic acid (RA). RA acts through its receptor RARα to repress transcription of inflammatory cytokines but is also essential for Th-mediated immunity, indicating complex effects of RA in Th specification and the outcome of the immune response. We examined the impact of RA on the genomewide transcriptional response during Th differentiation to multiple subsets. RA effects were subset-selective, and were most significant in Th9 cells. RA globally antagonized Th9-promoting transcription factors and inhibited Th9 differentiation. RA directly targeted the extended Il9 locus and broadly modified the Th9 epigenome through RARα. RARARα activity limited murine Th9-associated pulmonary inflammation, and human allergic inflammation was associated with reduced expression of RA target genes. Thus, repression of the Th9 program is a major function of RA-RARα signaling in Th differentiation, arguing for a role for RA in IL-9 related diseases.
Keywords: ATAC-seq, ChIP-seq, Enhancers, Interleukin 9, Interleukins, Vitamin A, Retinoic acid, RNAseq, T helper cells, Th9 cells
Graphical Abstract
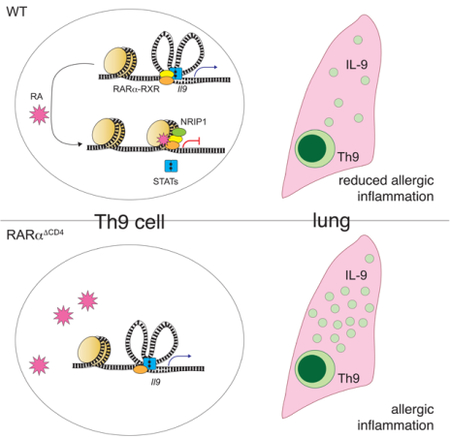
Schwartz et al. examine the impact of retinoic acid (RA) on the genome-wide transcriptional response during differentiation of CD4+ T cells to different Th subsets, revealing a role for RA in repressing the Th9 differentiation program. Their findings argue for the importance of this dietary metabolite in immune homeostasis and in Th9-associated inflammatory disease.
Introduction
CD4+ T cells are critical orchestrators of immune responses, with different T helper (Th) subsets directing responses to specific pathogens (Abbas et al., 1996; Dong and Flavell, 2000; O’Shea and Paul, 2010). CD4+ fate decisions are influenced by environmental immunomodulators – including cytokines, microbiota, and metabolites – that signal through diverse receptors (Josefowicz and Rudensky, 2009; Miossec et al., 2009). Retinoic acid (RA), a metabolite synthesized from dietary Vitamin A by cell-specific dehydrogenases, has essential roles in human health that include broad effects on hematopoietic lineage decisions and immune cell function (Hall et al., 2011b; Phan et al., 2017). In mucosal and lymphoid tissues, RA synthesized by dendritic cells acts on CD4+ T cells, signaling through nuclear receptors (RARs) to modulate Th differentiation and function (Benson et al., 2007; Mucida et al., 2007; Raverdeau and Mills, 2014). Inflammatory stimuli promote RA synthesis and increase RAR activity in Th cells (Iwata et al., 2004; Manicassamy et al., 2009; Pino-Lagos et al., 2011).
RA’s role in Th-mediated immunity is complex, as illustrated by its numerous, seemingly contradictory effects (Larange and Cheroutre, 2016). RA is important for CD4+ effector (Teff) cell responses in some contexts, including defense from infection and response to immunization; but RA ameliorates CD4+ dependent autoimmune disease and suppresses T effector (Teff) cytokines in other situations (Elias et al., 2008; Hall et al., 2011a; Kwok et al., 2012; Xiao et al., 2008). Several potential explanations are proposed for these inconsistencies, including concentration-dependent RA effects; context-dependent RA effects that vary in different Th subsets in different tissues; and indirect RA-mediated suppression of Teff differentiation via induction of the transcriptional regulator Foxp3, a key mediator of peripheral tolerance (Hall et al., 2011b; Maynard et al., 2009; Mucida et al., 2007; Takahashi et al., 2012; Xiao et al., 2008). In recent years, various models of RAR deficiency have been used to investigate the molecular mechanisms underlying RA’s effects on Th cells (Brown et al., 2015; Hall et al., 2011a; Reis et al., 2013), leading to important insights regarding the role of RA-RAR signaling in Th differentiation and function. Yet RA-RAR signaling has primarily been studied in the context of Th1 and Th17 responses, and its role in other Teff lineages is less characterized (Brown et al., 2015; Elias et al., 2008; Hall et al., 2011a; Kwok et al., 2012). This is particularly true for Th9 cells, which have recently emerged as important modulators of atopy, autoimmunity, and cancer (Kaplan et al., 2015). The factors governing Th9 specification are not as well understood as those influencing the differentiation of other Th subsets (Kaplan, 2017). Nonetheless, Th9 cells promote allergic inflammation and reduce barrier immunity at mucosal surfaces, which are RArich microenvironments (Larange and Cheroutre, 2016), suggesting that RA-RAR signaling might have a prominent role in the differentiation and function of this subset.
In this study, we broadly surveyed the transcriptomic effects of RA on the major Th subsets. In vitro, RA primarily had subset-selective effects and quantitatively regulated Th9 transcriptomes more than those of other effector subsets. Foxp3 was largely irrelevant for RA gene regulation in Th9 cells; instead, RA globally antagonized Th9 lineage-promoting transcription factors (TFs) consequently limiting the Th9 transcriptomic program. The RA receptor RARα directly targeted the extended Il9 locus, promoted chromatin remodeling, and impaired TF binding to regulatory regions. In vivo, treatment with RA ameliorated Th9associated lung inflammation in mice, whereas genetic deletion of RARα exacerbated disease. Accordingly, allergic inflammation in human subjects was associated with reduced expression of RA target genes. Our findings establish that RA-RARα directly represses the Th9 transcriptional program, arguing for a potential role for this essential dietary factor in controlling IL-9 related diseases such as asthma.
Results
RA has a quantitatively greater effect on Th9 and iTreg transcriptomes relative to other Th subsets
To gain insight into the RA effects on diverse Th subsets, we first assessed its effects on the transcriptomes of CD4+ T cells differentiated in vitro under neutral (Th0), effector (Th1, Th2, Th9, Th17), and regulatory (iTreg) conditions. RA regulated 1025 differentially expressed genes (DEGs) in ≥1 Th subset. Overall, the impact of RA was greater (>350 DEGs) in Th17, Th9, and iTreg cells compared to Th0, Th1, and Th2 cells (<250 DEGs) (Figure 1A). Only 326 DEGs (32%) were similarly regulated in ≥3 subsets (common): 225 induced and 101 repressed. Common RA-induced DEGs were enriched for RAR signaling (Crabp2, Stra6) and chemotaxis (Ccr9, Itga4), serving as a positive control since these genes are known to be RA-regulated (Coombes et al., 2007; Iwata et al., 2004). By contrast, common RA-repressed DEGs were highly enriched for JAK-STAT signaling. RA both induced (Nfatc1, Fos) and repressed (Cd24a, Icos) genes important for leukocyte activation, potentially offering an explanation for RA’s role as both a positive and negative regulator of Teff function (Bai et al., 2004; Dong et al., 2001) (Figure 1B, S1A, Table S1).
Figure 1: RA has a preferential effect on the transcriptomes of in vitro derived Th9- and iTreg cells.

A. Number of RA-regulated differentially expressed genes (DEGs) in different Th subsets. Isolated naïve CD4+ T cells were cultured with cytokines and antibodies to promote differentiation to the above Th subsets, in the presence of either vehicle control or 1000 nM RA. After 72h, polyadenylated mRNA was isolated and gene expression was measured by RNAsequencing. Total numbers of DEGs are shown for each Th subset (fold-change (FC) in expression with RA vs. vehicle control ≥2 or ≤−2, FDR<0.05). RA regulated 191, 184, 227, 359, 444, and 534 DEGs in Th0, Th1, Th2, Th17, Th9, and iTreg cells. B. RA-regulated genes and pathways. RA regulated a total of 1025 DEGs in ≥1 Th subset. 326 DEGs (32%) were regulated in ≥3 subsets, whereas 699 DEGs (68%) were regulated in ≤2 subsets. 428 DEGs (42%) were primarily regulated in Th9/iTreg cells. Key pathways enriched in each group of DEGs are shown, as well as representative DEGs from each pathway (http://metascape.org, (Tripathi et al., 2015)). (n=2. Heatmap displays log2FC in expression, RA vs. control, see also Figure S1)
RA regulated a larger number of DEGs (699, 68%) in a subset-selective fashion, either inducing expression in ≤2 subsets or repressing expression in ≤2 subsets. RA selectively induced several genes that promote Th2 immunity (Adam8, Sema7a), Th1 cell function (Cxcl10, Rsad2), and Th17 function (Il23a, Il22) (Mizutani et al., 2015; Naus et al., 2010). RA repressed 105 DEGs in Th1/Th17 cells: this included positive (Cysltr1, Rbpj) and negative (Ets1, Ecm1) regulators of Th17 function, consistent with RA’s variable effects on Th17 specification (Lee et al., 2015; Meyer Zu Horste et al., 2016; Moisan et al., 2007; Su et al., 2016). Subset-selective induction of genes important for host defense helps explain RA’s role in promoting CD4+ mediated immunity, despite suppression of many Teff cytokines.
The largest group of 428 DEGs (42%), was regulated primarily in Th9 and iTreg cells (Figure 1B, S1B,C). RA increased the transcription of genes related to TGF-β signaling and Th17 differentiation but repressed inflammatory genes involved in NF-κB and TNF signaling. Several genes repressed by RA in Th9 and iTreg were RA-induced in Th17 cells (Casp3, S1pr1,Cd83), whereas some RA-induced genes in Th9 and iTreg were repressed in Th17 cells (Cd101, Tigit, Ramp1); this may underlie some complexities of RA signaling. Taken together, these results indicate that RA has a major subset-specific effect on Th9 and iTreg cells.
RA represses the motif accessibility, expression, and targets of key Th9 transcription factors
Global chromatin accessibility is thought to provide a more stable view of cell state than exclusively assessing steady state mRNA expression (Shih et al., 2016). Because RA’s major effect was on Th9 cells, we hypothesized that changes in chromatin accessibility would better define the role of RA in this subset. We measured global chromatin accessibility by ATAC-seq using in vitro generated Th9 cells, including iTreg cells as a comparator because RA had similar effects in this subset. RA treatment caused the gain of 17362 ATAC peaks and loss of 6756 peaks under Th9 conditions, and the gain of 15283 peaks and loss of 6112 peaks under iTreg conditions, compared to control-treated cells (Figure 2A).
Figure 2. RA antagonizes Th9-promoting transcription factors (TFs).

A. TFs with RA-regulated motif accessibility and RA-regulated gene expression. Venn diagrams show changes in global genomic accessibility (number of ATAC peaks gained and lost) with RA treatment in Th9 and iTreg conditions. Heatmaps display RA effect on motif enrichment and gene expression for all TFs whose motif enrichment was significantly regulated by RA treatment. *FDR<0.05 B. RA effect on target genes of RA-regulated TFs. Enrichment plots display a combined Gene Set Enrichment Analysis (GSEA) enrichment score and FDR for the average, or net, effect of RA on target genesets for the following RA-regulated TFs: STAT5 (GSE77656); STAT6(GSE22801); GATA3(GSE20898); IRF4(GSE39756); NF-kB (Pahl, 1999); ATF3 (GSE61055); VDR(GSE2421); FOXP1(GSE50725); NFAT(GSE64409); JUNB (GSE98413); SMAD3(GSE19601), RUNX1(GSE6939); FOSL2, RORC, and BATF (GSE40918). A positive score (orange) indicates that RA has the same net effect as the TF on the target geneset, a negative score (blue) means that RA and the TF have the opposite net effect on the target geneset, and a neutral score indicates that RA has no consistent net effect on the geneset. The size of each data point corresponds to the size of the analyzed geneset. For full details of analysis see Supplementary Methods.
To identify transcription factors (TFs) that might mediate the actions of RA, we searched for differential motif abundance in peaks gained vs. peaks lost upon RA treatment. 52 TF binding motifs were differentially enriched in Th9-promoting conditions, and 63 motifs were differentially enriched in iTreg-promoting conditions (Figure 2A). Of these, RA increased both the expression and motif enrichment of 12 TFs in either iTreg-promoting conditions, Th9promoting conditions, or both (USF2, FLI1, FOXP1, TCF12, JUN, RORC, NFATC1, VDR, BACH2, SMAD3, CTCF, RUNX1).
To confirm the relevance of these findings we investigated RA effects on the targets of these TFs. We first identified 7 RA-induced TFs for which we could generate target gene lists using public gene expression data (TF deletion or overexpression). We then measured the average, or net, effect of RA on each target geneset, using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). RA had the same effect as RUNX1 in both Th9 and iTregpromoting conditions, inducing RUNX1-induced genes and repressing RUNX1-repressed genes. Findings were similar for SMAD3 targets, but there was no consistent RA effect on the other target genesets (Figure 2B).
Conversely, RA reduced the expression and motif enrichment of 10 TFs in either iTregpromoting conditions, Th9-promoting conditions, or both (RXRα, FOSL2, IRF4, BATF, ATF3, NFκB-p52, NFkB-p65, STAT5a, STAT6, GATA3). Investigating the net effect of RA on target genes of these TFs revealed no consistent effect on BATF, FOSL2, or ATF3 targets (Figure 2B). However, RA had a net effect of repressing STAT5-induced genes and of inducing STAT5repressed genes, both in Th9 and iTreg conditions. Results were similar for STAT6, GATA3, and NF-κB targets in Th9 and iTreg conditions; and for IRF4 target genes in iTreg conditions. These TFs are major positive regulators of Th9 differentiation, implying that repression of Th9 specification might be a major function of RA (Kaplan et al., 2015; Wei et al., 2011). Consistent with this, TF accessibility was reduced for PU.1; while this may be another relevant action of RA, PU.1 was expressed at very low levels (rpkm<1).
RA suppresses a Th9 transcriptional program and limits Th9 differentiation
Our observation that RA opposed TFs important for Th9 differentiation led us to ask whether RA might impact genes differentially expressed in Th9 cells. We identified a cassette of genes differentially expressed (FC≥1.5, FDR<0.05, ANOVA) in in vitro differentiated Th9 cells relative to other subsets (Th9-high genes, n=30). Treatment with RA reduced expression of Il9 and other Th9-high genes (Figure 3A,B). Testing the overall, or net, effect of RA on this entire geneset revealed significant downregulation (Figure S1D). RA also significantly repressed expression of genes previously identified as enriched in Th9 cells (Figure S1E) (Jabeen et al., 2013). Together, these results suggest that RA represses not only Il9, but also other genes associated with a Th9 program.
Figure 3. RA represses a Th9 transcriptional program.

A. Volcano plot of gene expression in Th9 cells. Fold change in gene expression (RA vs. vehicle control) vs. FDR is shown for the transcriptomes of Th9 cells (n=2). Selected RAregulated genes (FC>2 or <−2, FDR<0.05) are highlighted: canonical RA-regulated genes (orange); and Th9-high genes (blue). B. Th9-high genes and RA effect on these genes. Heatmap depicts log2 normalized average fold change in gene expression (RA vs. vehicle control, n=2) for Th9-high genes, which were selectively expressed in Th9 cells more than in Th1, Th2, Th17, and iTreg cells (n=30) C-F. Flow cytometric analysis of Th9 cells cultured in the presence of RA vs. vehicle control. C. Representative plots of IL-9 and Foxp3 expression in Th9 cells cultured with vehicle control or 1000 nM RA. D. Bar graph summarizing IL-9 and Foxp3 expression in Th9 cells cultured with vehicle control or escalating doses of RA (n=6). E. Representative plots of IL-9 expression in Th9 cells exposed to 1000nM RA at different time points. F. Bar graph summarizing IL-9 expression (n=3) (data shown as mean ± SEM; *p<0.05,**p<0.01,***p<0.005, ****p<0.001, paired t-test). See also Figure S1.
To confirm the effect of RA on Th9 cells, we measured IL-9 protein under conditions used for Th9 differentiation, using Foxp3 as a control. Without RA, 30–50% of the cultured cells produced IL-9, and 10–20% expressed Foxp3 (Figure 3C,D). RA significantly reduced IL-9 production in a dose-dependent manner, while increasing Foxp3 expression (Figure 3C,D). Under the conditions used, few Th9 cells expressed IL-10, which was repressed by RA, indicating that this was unlikely to be a mechanism by which RA repressed IL-9 (Figure S1F,G). Exposure to RA at later time points also inhibited IL-9 production, indicating that RA also repressed IL-9 in established Th9 cells (Figure 3E,F). In summary, these results indicate that RA represses Il9 and other aspects of a Th9 transcriptional program while inducing Foxp3.
Foxp3 is dispensable for RA-mediated transcriptional regulation of Th9 cells
Because RA repressed Th9 differentiation while upregulating Foxp3, we expected that RA’s suppressive effects would be mediated by Foxp3, which globally limits Teff function (Fontenot et al., 2003). Our ability to study the interactions between RA and Foxp3, however, was constrained by major alterations in T cell homeostasis seen in Foxp3 deficiency (Clark et al., 1999). We therefore crossed Foxp3Sf mice that express a mutant (truncated) Foxp3 mRNA to Rag2-deficient, OT-II T-cell receptor transgenic mice (Rag2−/− OT-II mice, Figure S2A). The Rag2−/− OT-II Foxp3Sf progeny failed to produce functional Foxp3 protein, but preserved a population of naïve CD4+ T cells and remained healthy (Figure S2B,C).
We next differentiated naïve CD4+ cells from Rag2−/− OT-II and Rag2−/− OT-II Foxp3Sf mice (henceforth termed Foxp3WT and Foxp3Sf) in vitro under Th9-promoting conditions. Foxp3WT and Foxp3Sf Th9 cells exhibited equivalent production of IL-9, indicating that Th9 differentiation was not globally dysregulated in Foxp3Sf T cells. RA suppressed IL-9 production 3–4-fold in Foxp3Sf T cells despite the absence of functional Foxp3 (Figure 4A–D,S2D). This effect was equivalent to that seen in Foxp3WT T cells, establishing that RA inhibits Th9 differentiation independently of Foxp3.
Figure 4: RA acts independently of Foxp3.

A-D. Flow cytometric analysis of Rag2−/− OT-II (FoxpWT) and Rag2−/− OT-II Foxp3Sf (Foxp3Sf) cells cultured under Th9 conditions in the presence of RA vs. vehicle control. A. Representative plots of IL-9 expression in FoxpWT and Foxp3Sf cells cultured with vehicle control or RA. B. Bar graph summarizing IL-9 expression (n=6). C. Representative plots of Foxp3 expression in cells cultured with vehicle control or RA. D. Bar graph summarizing Foxp3 expression (n=6). (data shown as mean ± SEM; **p<0.01, ***p<0.005, ****p<0.001, paired ttest.) E,F. Scatterplots comparing average gene expression in Foxp3WT and Foxp3Sf cells cultured under Th9 conditions, with vehicle control (E) and RA (F). Black lines indicate fold-change in gene expression (FC) ≥2 or ≤−2 in FoxpWT vs. Foxp3Sf cells. No significant differentially expressed genes were seen (FC≥2 or ≤−2, FDR<0.05) (n=2). RA = 1000 nM for all experiments. See also Figure S2.
We expected Foxp3 would regulate the expression of many targets besides Il9, since Foxp3 was expressed in 20–40% of Foxp3WT cells differentiated under Th9 conditions. Yet comparing Foxp3WT and Foxp3Sf Th9 cells revealed no DEGs, regardless of whether the cells were differentiated in the presence or absence of RA (Figure 4E,F). These results led us to conclude that Foxp3 is dispensable for RA-mediated gene regulation of Th9 cells.
RA disrupts promoter-enhancer interactions of the extended Il9 locus
Because Foxp3 did not mediate RA effects on Th9 cells, we next asked whether RA directly repressed Th9 differentiation. We addressed this question by examining RA’s effects on chromatin accessibility and covalent histone modifications in Th9 cells: active proximalpromoter elements (Histone3 lysine4 trimethylation, H3K4M3), poised enhancers (H3K4 monomethylation, H3K4M1), and active enhancers (H3K27 acetylation, H3K27Ac). As a positive control, RA treatment increased H3K4M1, H3K4M3, and H3K27Ac at the extended locus of the canonical RA target gene Ccr9, including potential cis-regulatory elements (cis-REs) (Figure S3A).
To further dissect the link between RA-mediated chromatin remodeling, changes in gene expression, and Th9 differentiation, we examined the extended Il9 locus. Cell-specific accessibility was seen in the Il9 promoter and in three upstream REs (E1–E3, Figure S3B) (Shih et al., 2016). The most distal upstream element, E3 (previously described as SEc or CNS-25) (Koh et al., 2018; Perumal and Kaplan, 2011; Xiao et al., 2018), was broadly accessible. In contrast, E1 (previously described as CNS0 or SEb) and E2 (previously described as CNS-25 or SEc) were more accessible in Th9 cells relative to other cells. All three REs bore H3K27Ac marks (Figure 5A). RA reduced H3K4M3 at the promoter, reduced accessibility and H3K27Ac at the promoter and E1–E3, but did not change H3K4M1, indicating that the Il9 locus remains poised but inactive. Treatment with RA did not change the accessibility of a downstream element (DS, previously described as CNS2 or SEa) that did not display differential cell-specific accessibility (Figure 5A).
Figure 5. RA interferes with Il9 promoter-enhancer interaction and TF recruitment A. Il9 regulatory elements (REs) identified by histone epigenetic marks and chromatin accessibility, STAT5 and STAT6 binding sites within REs, and RA effect on REs.

Representative Il9 gene tracks of ATAC, H3K4M1, H3K4M3, and H3K27Ac in Th9 cells polarized in the presence of vehicle control or RA. 5 regulatory elements are marked: the Il9 promoter (Il9p), a downstream element (DS), and 3 upstream elements (E1–E3). E1–E3 and DS bear poised (H3K4M1) and active (H3K27Ac) enhancer marks, but not promoter (H3K4M3) marks (n=2). Gene tracks also show STAT5B and STAT6 binding sites in Th9 cells based on public data (GSE41317). B,C. Flow cytometric analysis of IL-9 expression in Th9 cells lacking different Il9 enhancers. B. A 3kb region containing E1 and an 8kb region containing E2–E3 were deleted to generate E1−/− and E2E3−/− mice, respectively. Flow cytometric plots show IL-9 expression in WT, E1−/−, and E2/E3−/− Th9 cells cultured with vehicle control or RA. C. Bar graph summarizing IL-9 expression (n=4). (data shown as mean ± SEM; *p<0.05,**p<0.01,****p<0.0001, paired t-test) D. Activation-dependent looping of extended Il9 locus, as measured by chromatin conformational capture (3C). Line graph depicts binding enrichment of distal regions to Il9 promoter. For Th0 cells, enrichment decreases with increased distance from the promoter. For Th9 cells, enrichment is elevated for E1, decreases for an inaccessible region between E1–E2, increases for E2, and decreases for E3. Treatment with RA reduces enrichment across the Il9 locus. Results are significant for Th9 vs. Th0 and Th9 vs. Th9+RA (n=5). E-G. ChIP-qPCR for STAT5, STAT6, and CTCF at Il9 regulatory elements in Th9 cells treated with vehicle control or RA. Bar graphs summarize binding enrichment for STAT5 (E), STAT6 (F), and CTCF (G) at the five Il9 regulatory elements, in Th9 cells cultured with vehicle control or RA (n=3). Pooled data shown as mean ± SEM; RA = 1000 nM; *p<0.05,**p<0.01,***p<0.005,****p<0.001, unpaired t-test. See also Figure S3.
We next determined whether E1–E3 were functional enhancers by assessing their responsiveness to STAT5. STAT5 directly regulates Il9 expression by binding to consensus sequences near the Il9 promoter (Liao et al., 2014), and STAT5 and STAT6 bound to E1–E3 (Figure 5A). A reporter construct containing the Il9 promoter element was induced 3-fold in the presence of constitutively active Stat5, whereas >2-fold enhancer activity was seen from E1 and E2 and <1.5-fold from E3, establishing that these are STAT5-responsive enhancers (Figure S3C). We next generated mice lacking site E1 or site E2–E3; these mice displayed no spontaneous phenotype or global alterations in T cell development (Figure S3D,E). Th9 cells generated in vitro from either of these lines had reduced IL-9 production, with a larger effect size from deletion of E2–E3 (Figure 5B,C).
To probe the mechanisms by which E1–E3 influence Il9 transcription, we sought to determine whether these regions displayed any long-range physical interactions, or looping, with the Il9 promoter. E1 and E2 interacted significantly with the Il9 promoter in Th9 cells compared to nonpolarized activated T cells (Th0) (Figure 5D). Addition of RA to Th9 cultures significantly reduced E1 and E2 interaction with the Il9 promoter, similar to levels in Th0 cells. CTCF, which contributes to looping between genes and distal REs, bound the Il9 promoter and E1–E3; and RA reduced CTCF and STAT binding across the Il9 locus (Figure 5E–G) (Handoko et al., 2011). Thus, RA interferes with the activation of the extended Il9 locus by reducing accessibility and disrupting normal looping.
RA-RARα directly targets the extended Il9 locus
To further dissect the mechanism by which RA interdicts normal function of the Il9 locus, we next ascertained whether RAR directly regulates Il9. Comparing expression of different RAR isoforms revealed that Th9 cells expressed Rarα and Rarγ but not Rarβ. (Figure S4A–C). Treating Th9 cells with a selective agonist of RARα the most abundant RA receptor, repressed IL-9. Conversely, treatment with a RARα antagonist significantly increased IL-9 production (Figure 6A,B).
Figure 6: RA directly represses IL-9 via RARα.

A,B. Effect of RA, RARα agonist, and RARα antagonist on generation of Th9 cells. A. Representative flow cytometric plots of IL-9 expression in cells cultured under Th9 conditions with vehicle control, 1000 nM RA, 1000 nM RARα agonist (BMS753), or 3000 nM RARα antagonist (ER50891). B. Bar graph summarizing IL-9 expression (n=5). C,D. IL-9 expression in WT vs. RARαΔCD4 Th9 cells. C. Representative flow cytometric plots of IL-9 expression in cultured under Th9 conditions with vehicle control or 1000 nM RA. D. Bar graph summarizing IL-9 expression (n=5). Flow data shown as mean ± SEM; *p<0.05, **p<0.01,***p<0.005,****p<0.001, paired t-test. E. RARα suppresses net expression of Th9high genes. Gene Set Enrichment Analysis (GSEA) plot depicts effect of RARα deletion on Th9-high genes, accounting for the net, or average, effect on all the genes in the geneset. F. ChIP-qPCR for RARα at Il9 regulatory elements in Th9 cells treated with vehicle control or RA. Bar graphs summarize binding enrichment for RARα relative to input at the five Il9 regulatory elements (n=3). G. RA effect on histone modifications and accessibility of Il9 regulatory elements (REs) in WT vs RARαΔCD4 Th9 cells. Representative Il9 gene tracks of ATAC, H3K4M1, H3K4M3, and H3K27Ac in WT or RARαΔCD4 Th9 cells polarized in the presence of vehicle control or RA. (n=2–4). H-J. ChIP-qPCR for STAT5, and STAT6, and CTCF at Il9 regulatory elements in WT or RARαΔCD4 Th9 cells. Bar graphs summarize binding enrichment for STAT5 (H), STAT6 (I), and CTCF (J) at the five Il9 regulatory elements, in WT or RARαΔCD4 Th9 cells (n=3–5); ChIP-qPCR data shown as mean ± SEM; *p<0.05,**p<0.01,***p<0.005,****p<0.001, unpaired t-test. See also Figure S4.
We next sought to confirm our findings using a genetic approach. We crossed mice bearing RARα flanked by two loxP sites with mice expressing CD4-Cre to generate RARαΔCD4 mice. Compared to WT cells cultured under Th9 conditions, Th9 cells from RARαΔCD4 mice produced more IL-9 (Figure 6C,D). Treating RARα-deficient T cells with RA failed to affect IL-9 levels, indicating that RARα was a nonredundant, physiological regulator of Th9 differentiation in vitro.
To determine the effect of Rara deletion on the Th9 transcriptome and epigenome, we analyzed global changes in chromatin accessibility (ATAC-seq) and transcription (RNA-seq) in WT and RARα−deficient Th9 cells. Deletion of RARα increased the motif enrichment and expression of Th9-promoting TFs and increased expression of Th9-high genes, consistent with RA-mediated repression of the Th9 program via RARα (Figure 6E;S4D–F). RA induced Foxp3 expression in RARα-deficient Th9 cells, although less than in WT Th9 cells, indicating that alternative or redundant mechanisms underlie a portion of RA actions in Th9 cells (Figure S4G,H).
Having established that RA antagonized Th9 differentiation via RARα, we next determined whether RARα bound to Il9 REs. As expected, RA increased RARα binding to a known RE associated with Ccr9 (Figure S4I) (Ohoka et al., 2011). RA also induced binding of RARα to the Il9 promoter and E1–E3, but not to DS (Figure 6F). RA failed to reduce Il9 locus accessibility or histone epigenetic marks in RARα-deficient Th9 cells. In addition, binding of STAT5, STAT6 and CTCF increased across the locus in the absence of RARα (Figure 6G–J). RARs act by recruiting ligand-dependent coactivators and corepressors (le Maire and Bourguet, 2014), which in turn promote chromatin remodeling to modulate gene expression (Carroll et al., 2006; Chatagnon et al., 2015). Five ligand-dependent RAR corepressors were expressed in Th9 cells, but RA induced only one of them, Nrip1 (Figure S4J). RA significantly increased NRIP1 binding to the promoter and E1–E3, suggesting that RARα recruited NRIP1 to repress Il9 (Figure S4K). Because RARα would not be expected to recruit corepressors to RA-induced REs, Ccr9 provided a useful negative control (Figure S4L).
Examining the broader impact of RARα on the Th9 epigenome revealed that RA reduced accessibility and permissive histone marks at cis-REs associated with multiple Th9 genes (Figure S4M). Many of these RA-repressed loci contained STAT5 or STAT6 binding sites, consistent with our finding that RA reduced STAT5 or STAT6 binding site accessibility. The effect of RA treatment on locus accessibility and permissive histone marks was diminished in RARαΔCD4 mice. We selected two representative Th9 genes whose accessibility and permissive histone marks were repressed by RA in an RARα-dependent manner (Figure S4N). RA increased RARα occupancy at the promoters of these genes, suggesting that RARα can contribute directly to the repression of genes associated with a Th9 program (Figure S4O). In summary, RARα is not only a nonredundant negative regulator of Il9 architecture and transcription, but also has broader repressive effects on Th9 differentiation.
RA ameliorates disease in a model of allergic lung inflammation
To assess the in vivo relevance of our findings, we employed the chronic papain model of allergic airway inflammation (Richard et al., 2015; Sehra et al., 2015; Wilhelm et al., 2011) (Figure S5A). Mice exposed to intranasal papain developed severe peribronchial leukocyte infiltration, mucus production with airway plugging, goblet cell hyperplasia, and airway resistance. RA treatment decreased goblet cell hyperplasia, reduced peribronchial cellular infiltration, and improved airway resistance (Figure 7A–C). In lung-resident CD4+ cells, treatment with RA diminished IL-9 and IL-13, but not IL-2 production (Figure 7D, S5B). RAtreated mice had fewer pulmonary Treg cells, consistent with lower overall cellular infiltration (Figure S5B). These findings confirm the in vivo suppressive role of RA on the Type 2 allergic response including IL-9 production and suggest that RA-mediated suppression of Type 2 immunity is independent of Tregs.
Figure 7. RA is essential to control pathology in allergic lung disease.

A-C. RA effect on lung inflammation and airway resistance. A. Representative images of Periodic Acid Schiff (PAS) stained lung tissue demonstrate reduced mucus production and lymphocytic infiltration in RA-treated vs. vehicle-treated mice with papain-induced asthma. B. Pulmonary histology scores in mice treated with RA and vehicle (3 replicates, n=3–6 per replicate). C. Airway resistance in mice exposed to escalating doses of intratracheal methacholine to induce bronchospasm. (2 replicates, n=1–3 per replicate) D. Flow cytometric analysis of IL-9 producing T cells in lung tissue at d14 of papain-induced asthma. Graphs show total numbers of Lin−TCRβ+CD4+CD44hi cells and of IL-9+ Lin−TCRβ+CD4+CD44hi T cells extracted from lung tissue of mice treated with vehicle control or RA. (4 replicates, n=3–5 per replicate) E-G. Lung inflammation and airway resistance in WT and RARα−/− mice at d14 of papain-induced asthma. E. Representative images of PAS stained slides demonstrate increased lymphocytic infiltration in RARα−/− vs. WT mice. F. Pulmonary histology scores in WT and RARα−/− mice. (3 replicates, n=3–5 per replicate) G. Airway resistance in mice exposed to escalating doses of intratracheal methacholine to induce bronchospasm. (3 replicates, n=1–3 per experiment) H. Flow cytometric analysis of IL-9 producing T cells in lung tissue at d14 of papain-induced asthma. Graphs show total numbers of Lin−TCRβ+CD4+CD44hi cells and of IL9+ Lin−TCRβ+CD4+CD44hi T cells extracted from lung tissue of WT and RARα−/− mice (2 replicates, n=3–5 per replicate). *p<0.05,**p<0.01,***p<0.005, Mann-Whitney; AW, Airway; BV, blood vessel. See also Figure S5–S7.
To specifically address the contribution of RA-mediated Th9 suppression, we employed an ovalbumin (Ova)-induced model of airway inflammation in which antigen-specific Th9 cells were differentiated in the presence or absence of RA and transferred into congenic hosts. To eliminate the potential contribution of Foxp3, we used Ova-specific Th9 cells from Foxp3Sf mice (Figure S5C). RA inhibited IL-9 production in donor OT-II Th9 cells, before and after transfer (Figure S5C,D) and pathology was significantly reduced in recipients of RA-treated Th9 cells (Figure S5E). These results indicate that treatment with RA reduces Th9 cell pathogenicity, and that this effect is independent of Foxp3 induction.
To determine whether endogenous RA-RARα regulate Th9 generation and immunopathology in vivo, we next sensitized RARαΔCD4 mice and littermate controls to intranasal papain. RARαΔCD4 mice displayed more severe airway pathology and increased numbers of IL-9-producing pulmonary CD4+ T cells compared with WT controls, but no difference in Foxp3 induction (Figure 7E,F,S5G). Using a papain dose titrated down to avoid mucus plugging in WT mice, RARαΔCD4 mice displayed heightened airway reactivity (Figure 7G,H). Thus, physiologic RARα signaling in CD4+ T was highly relevant for in vivo Th9 specification and allergic pathology.
RA-RARα signaling suppresses a shared in vitro and in vivo Th9 transcriptional program
The finding that RA ameliorated Th9 pathology in vivo led us to hypothesize that RARARα suppressed transcriptional programs of in vivo IL-9-producing cells. To this end, we identified genes expressed by IL-9-producing T cells using two different IL-9 reporter mice (GFP+ and eYFP+) challenged with papain (Licona-Limon et al., 2013; Richard et al., 2015; Sehra et al., 2015) (Figure S6A,B). We noted substantial differences between the genes expressed in IL-9-producing cells identified by the two different reporters and so compiled DEGs in at least one reporter model (Figure S6C,D). In addition to Il9 and other genes associated with Th9 differentiation (Spi1, Il17rb, Ccl17), this cassette also included a number of Th2 (Il4, Il13, Gata3) and Th17 (Ccl3, Lag3, Csf2) signature genes (Hu et al., 2017; Humblin et al., 2017; Li et al., 2013; Perumal and Kaplan, 2011). We therefore excluded genes expressed by alternative fates by determining the intersection with genes preferentially expressed by in vitro differentiated Th9 cells relative to other subsets (average FC≥1.5, FDR<0.05, ANOVA). Using these criteria, a cassette of 62 genes was designated as “shared Th9” (Figure S6E). Treatment with RA reduced expression of shared Th9 genes, whereas deletion of RARα increased expression of these genes (Figure S6F–I). Taken together, these findings indicate that RA and RARα broadly repress a transcriptomic program in Th9 cells, recognizing that this is a heterogeneous group.
RA-RAR signaling is aberrant in human Th9-associated disease
The finding that RA-RAR signaling repressed in vivo Th9 genes and improved Th9-associated immunopathology led us to question whether RA signaling had a role in human Th9associated inflammation. RA inhibited human Th9 differentiation, similar to its effects in mice (Figure S7A,B). RA target genes (Figure 1B) were also dysregulated in skin from nickelallergic patients, and in CD4+ T cells from atopic individuals stimulated in vitro with house dust mite extract (Pedersen et al., 2007; Troy et al., 2016). House dust mite stimulation was associated with decreased expression of RA-induced genes and increased expression of RArepressed genes, including increased IL9 and IL13 expression (Figure S7C–E). Nickel exposure was similarly associated with increased expression of RA-repressed genes, including IL9 (Figure S7F–H). Expression of NRIP1 and RA-metabolizing enzymes was lower in nickel-allergic skin than in healthy skin. Conversely, expression of the retinaldehyde-reducing enzyme DHRS3 was higher in allergic skin (Figure S7I,J). These results suggest that allergic inflammation in humans is associated with reduced RA signaling, indicating potential translational relevance.
Discussion
Vitamin A and its metabolite RA have long been identified as key immunomodulators, yet a number of issues remain unclear including the comprehensive effect of RA across T effector subsets, the role of Foxp3 in mediating RA signaling, and the direct effects of RAR in Th9 cells that reside in RA-rich mucosal environments. Herein, we addressed these questions using genomic and functional approaches, revealing molecular mechanisms underlying RA actions in Th9 cell specification and function in vitro and in vivo.
RA antagonized Th9-promoting TFs and repressed a broader Th9 transcriptional program indicating a major role for RA signaling in this Th subset. Vitamin A deficiency is associated with increased prevalence and severity of asthma (Arora et al., 2002; Marquez and Cardoso, 2016), and our analysis suggests that RA signaling is reduced in the setting of acute allergic inflammation. Moreover, RA triggered chromatin remodeling across the Il9 locus and at the loci of other Th9 genes via RARα. While direct RARα binding was shown at selected Th9 genes, we were unable to obtain reliable RARα ChIP-seq data. Improved reagents with more specificity could conclusively document genome wide RARα binding and establish a direct mechanism for the broader Th9 program. Critically, Th9 differentiation was repressed across a range of doses and under physiologic conditions, as RARα-deficient Th9 cells differentiated more efficiently and mediated more severe lung disease. This distinguishes our work from previous studies, where low concentrations of RA have been found essential for a productive immune response (Brown et al., 2015; Hall et al., 2011a; Iwata et al., 2003).
Our findings also advance the understanding of how the Il9 locus is dynamically regulated, which is important because the factors governing Th9 differentiation and function are incompletely characterized. (Kaplan et al., 2015). Th9 cells are regulated by a network of TF modules including cytokine-dependent TFs (STATs, Smad/RBP-Jκ/Notch, s, TAK1), antigen receptor dependent TFs (BATF/IRF4, NFAT/NFκB), and lineage-determining TFs (ETV5/PU.1) (Kaplan, 2017). RARα can now be added to this network as a metabolite-dependent TF, distinguishing it from other Th9-regulating signals. Moreover, the characterization of enhancers within the extended Il9 locus may provide insight into the actions of other Th9-promoting TFs. For example, enhancer DS is more relevant to the activity of OX40 and RelB than E1–E3, whereas E1–E3 were important for STAT5-dependent IL-9 production (Koh et al., 2018; Xiao et al., 2018).
Aside from its effect on the Il9 locus, RA had a broad impact on chromatin status and transcriptomes of IL-9-producing cells. Analysis of in vivo Th9 cells revealed a core set of genes highly expressed in both in vitro differentiated and in vivo Th9 cells. Nonetheless, substantial differences existed between in vitro and in vivo Th9 cells, and between the two different reporter models of IL-9 in vivo. The reasons for these differences are unclear, but IL-9 producing cells apparently represent a heterogeneous collection, and there is much to learn about exactly what a “Th9 cell” is (Malik and Awasthi, 2018). A comprehensive and dynamic understanding of IL-9 producing Th cells and their relationship to other subsets will require further investigation and methodologies, including multi-cytokine reporter mice and dual reporter/fate reporter models. These insights will clarify the identity and functionality of IL-9 producing cells in the context of not only Type 2 immunity but also inflammatory bowel disease and antitumor immunity, where Th9 cells are implicated in disease pathogenesis.
Although RA strongly induced Foxp3, which profoundly limits T effector function, Foxp3 was unexpectedly dispensable for RA-mediated regulation of the Th9 transcriptome. Instead, RARα directly targeted the Il9 locus by recruiting the corepressor Nrip1 and reducing chromatin accessibility. RA-mediated chromatin remodeling interfered with TFs that promoted Il9 transcription and prevented binding of CTCF, blocking promoter-enhancer looping. These results address a major gap in retinoid biology, as the regulation and function of RA-dependent coregulators in CD4+ T cells has not been described (Larange and Cheroutre, 2016).
Although Vitamin A can also enhance Type 2 immunity (Schuster et al., 2008), the discrepancy with our findings could be due to the existence of several mechanistically distinct Vitamin A-driven regulatory networks. RA-RARα signaling repressed Th9 differentiation, but retinoid X receptor (RXR) ligands may enhance the production of cytokines like IL-5, which are more important for some models of airway disease (Stephensen et al., 2002). Retinoids also regulate respiratory epithelial cells and can indirectly regulate Th-driven immune responses by priming dendritic cells, which activate Th cells (Maret et al., 2007; Rampal et al., 2016). It will be critical to consider these and other potentially competing mechanisms, including the effects of RA on other cell types, in elucidating the immunomodulatory effects of Vitamin A and its metabolites. Future investigations exploring the specific role of RARα using genetic models should address these questions.
By systematically characterizing RA-regulated genes in all major in vitro generated Th subsets, our findings can also be used to explain some of the paradoxical effects of RA. For example, RA repressed a cassette of genes in iTreg and Th9 cells but induced the same cassette in Th17 cells. This could be because RARs work cooperatively with cofactors; the cell-specific expression of a particular cofactor could determine the directionality of RA’s effect on a specific gene in that cell type. Dissecting the mechanisms by which divergent regulation occurs in different subsets will be an interesting area for future investigation. RA also had divergent impacts on various Th17-promoting genes, repressing Il6ra while inducing Runx1. Such findings help explain the ostensibly contradictory effects of RA in Th17 specification, although confirmation will require in vivo models and genetic tools (Takahashi et al., 2012).
In summary, RA signaling had a major effect in Th9 cells that involved suppression of the Th9 transcriptional program, independently of Foxp3 induction. RARα suppressed the global Th9 epigenome and targeted three functional enhancers within the extended Il9 locus. Finally, RA signaling was aberrant in the setting of Th9-associated human inflammation. These results advance the field of Th9 biology, which is becoming increasingly relevant as new roles emerge for Th9 cells in the pathogenesis of human disease.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Daniella M. Schwartz (Daniella.Schwartz@nih.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were performed in the AAALAC-accredited animal housing facilities at NIH. All animal experiments were conducted in accordance with NIH guidelines for the use and care of live animals and were approved by the Institutional Animal Care and Use Committee of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). Mice were bred and maintained at the National Institutes of Health specific pathogen free animal facility, in conventional open cages. Food and water were continuously available, mice were maintained on a 12-hour light/dark cycle, and mice were checked periodically to ensure normal health, and checked daily if adverse effects were anticipated.
All mice were on a C57Bl/6 background. Foxp3Sf, eYFP IL-9 fate reporter, and B6 CD45.1 mice were purchased from the Jackson Laboratory. Rag2−/− OT-II mice were purchased from Taconic Biosciences. Rara−/− mice were graciously provided by Yasmine Belkaid, INFER mice were graciously provided by Richard Flavell. Enhancer knockout mice were generated by the NHLBI Crispr Core Facility (see below). Mice were genotyped by standard PCR protocols.
3–6 replicates were used for all in vitro experiments except for sequencing experiments (RNAseq, ChIP-seq, ATAC-seq), where 2–3 replicates were used. For all papain experiments where Th9 cells were isolated from lung, 2–4 separate experiments were performed, with 3–5 subjects per experimental group (18–20 subjects per experiment). For flexivent experiments, 2–3 replicates with 1–3 subjects per experimental group (9–10 subjects per experiment) were performed. For Th9 transfer experiments, two separate experiments with 1–3 subjects per experimental group (6–10 subjects per experiment) were performed.
The majority of experiments were performed on 8–14-week-old mice, with the exception of the flexivent experiments, which were performed on 12–18-week-old mice. Experiments on Foxp3Sf mice and FoxpWT littermate controls were done in male mice because the Foxp3Sf allele is Xlinked and the experiments could not be done in female mice. Th9 transfer experiments were all done in male mice because Foxp3Sf donor Th9 cells were obtained from male mice and could not be transferred to female recipients due to risk of rejection. All remaining experiments were done in both male and female mice and were age and sex-matched within experiments.
METHOD DETAILS
Generation of enhancer knockout mice
The Il9.E1−/− and Il9.E2E3−/− mice were generated using the CRISPR/Cas9 method as reported previously (Wang et al., 2013). Briefly, for each line, an upstream sgRNA (AGAGCTCCTGGGGCCGAGAC) and a downstream sgRNA (TCCTATCCAGACATTGAGGC) were made using T7 promoter-driven in vitro transcription. These two sgRNAs (5 ng/ul each) were co-microinjected with Cas9 mRNA (10 ng/ul, purchased from Trilink Biotechnologies) into the cytoplasm of fertilized eggs collected from C57BL/6N mice (Charles River). The injected zygotes were cultured overnight in M16 medium at 37°C in 5% CO2. The next morning, embryos that had reached the 2-cell stage of development were implanted into the oviducts of pseudopregnant foster mothers (Swiss Webster, Taconic Farm). The mice born to the foster mothers were genotyped using PCR, and then confirmed by DNA sequencing. Founder mice with the desired mutations, resulting in deletion of either entire enhancer E1 (3kb deletion) or the contiguous enhancer E2–E3 region (8kb deletion), were bred with C57BL/6N mice for propagating the line, as well as for eliminating possible mosaicism and diluting out off-target effects, if any. Germline transmission of the E1 deletion (Il9.E1−/− mice) and the E2–E3 region (Il9.E2E3−/−) mice was confirmed by DNA sequencing after 4 generations of breeding.
Media
All cell cultures were performed in RPMI medium with 10% (vol/vol) FCS (Invitrogen), 2 mM glutamine (Invitrogen), 100 IU/mL penicillin (Invitrogen), 0.1 mg/mL streptomycin (Invitrogen), 20 mM HEPES buffer (pH 7.2–7.5) (Invitrogen), and 2 mM β-mercaptoethanol (SigmaAldrich).
Reagents
RA was purchased from Sigma (cat#R2625) and reconstituted in DMSO at a stock concentration of 100mM (in vitro experiments) or 40mg/mL (in vivo experiments). BMS753 (Tocris cat#3505) and ER50891 Tocris (cat#3823) were reconstituted in DMSO to stock concentrations of 100mM. Aliquots of RA and agonists/antagonists were serially diluted in PBS immediately prior to in vitro treatment of cells. Stocks were replaced every 3–6 months.
Cell culture
Naïve CD4+/CD25−/CD44lo/CD62Lhi mouse T cells were isolated by cell sorting from spleen and lymph nodes to >95% purity using a FACSAria III Cell Sorter, after magnetic enrichment (StemCell Technologies or Miltenyi Biotec). T cells were plated at a density of 0.5×106/mL and activated with 10μg/mL plate bound anti-CD3/anti-CD28 for 72h in the presence of polarizing cytokines and antibodies to promote the differentiation of Th0 (none), Th1 (10ng/mL IL-12, 10 μg/mL anti-IL-4), Th2 (20 ng/mL IL-4, 10 μg/mL anti-IFN-gamma, Th9 (20ng/mL IL-4, 10 μg/mL anti-IFN-gamma, 2.5 ng/mL TGF-beta, 100 units/mL hIL-2), Th17 (10ng/mL IL-6, 10μg/mL anti-IFN-gamma 10μg/mL anti-IL-4, 2.5ng/mL TGF-beta), iTreg (10ng/mL TGFbeta, 100 units/mL hIL-2).
For experiments examining the effects of RA on established Th9 cells, naïve T cells were purified and cultured to promote Th9 differentiation, adding either DMSO or RA (1 μM) on d0 of culture. After 72 hours, cells were harvested, washed, and resuspended in Th9-promoting cytokines and antibodies. Cells were split to two aliquots of 0.5×106 cells/mL on 10 μg/mL plate bound anti-CD3/anti-CD28. One aliquot was maintained under the same culture conditions (control → control or RA → RA) and one aliquot was switched to different culture conditions (control → RA or RA → control). Cytokine production was assessed by intracellular staining on d6 of culture.
For transfer experiments, antigen presenting cells (APCs) from C57Bl/6 mice were depleted of T-cells by magnetic separation (Miltenyi) using biotinylated anti-Thy1.2 (53–2.1, Biolegend). Tcell depleted APCs (> 95% purity) were resuspended to a density of 2 million/mL in complete RPMI and irradiated at 3000 Gray. Rag2−/− OT-II Foxp3Sf CD45.2+ CD4+ T cells were isolated in vitro using magnetic enrichment and cultured in complete IMDM under the following conditions: 1:5 CD4+ T cells:T-cell depleted irradiated APCs, 10ng/mL IL-4, 2ng/mL human TGF-β, 0.5μg/mL soluble anti-CD28, 10μg/mL anti-IFN-γ, 1μM ovalbumin peptide (S7951, Sigma).
For human T cell cultures, peripheral blood mononuclear cells (PBMCs) from de-identified healthy donors were obtained from the NIH Blood Bank, isolated using Ficoll, and frozen in liquid nitrogen for up to 6 months. PBMCs were thawed, and CD45RA+ CD45RO− CD4+ T cells were isolated using magnetic columns (Miltenyi) to >90% purity. T cells were plated at a density of 1×106 cells/mL and activated with 1μg/mL plate bound anti-CD3 (ebioscience clone OKT3) and 0.5 μg/mL soluble anti-CD28 (BD clone L293) for 5 days in the presence of polarizing cytokines and antibodies to promote Th9 differentiation (30 ng/mL IL-4, 5 μg/mL anti-IFNgamma (BD clone B27), 5 ng/mL TGF-beta, 10 ng/mL IL1β, 10 units/mL IL-2).
Anti-CD3, anti-CD28, anti-IFN-gamma and anti-IL-4 were purchased from BioXcell, human IL2 was purchased from the NIH/NCI BRB Preclinical Repository, and all other mouse antibodies and cytokines were purchased from R&D Systems. Human cytokines were purchased from Peprotech, and antibodies were purchased from BD unless otherwise specified. Where specified, RA (0.1nM-1μM as specified), DMSO (vehicle control), BMS753 (RARα agonist), or ER50891 (RARα antagonist) were added on d0 of culture. Concentrations of RARα selective agonists and antagonists were chosen based on IC50 data provided by the manufacturer.
Flow Cytometry
For analysis of cytokine production, cells were stimulated in 96-well plates for 4–6 hours with phorbol 12-myristate 13-acetate (PMA), ionomycin (50ng/mL and 500ng/mL, respectively), treated with brefeldin A (10 μg/mL; Sigma Aldrich) Cells were stained with LIVE/DEAD Cell Stain (Invitrogen), followed by staining for cell surface markers, and then fixed and permeabilized with the Cytofix/Cytoperm kit (BD Biosciences, Cat# 554714) for intracellular staining.
Viability was assessed with LIVE/DEAD Fixable Aqua Dead Cell Stain or LIVE/DEAD Fixable Near-IR Dead Cell Stain (ThermoFisher).
For intracellular staining of in vitro cultured mouse cells, the following antibodies were used: anti-IL-9 PE or APC (RM9A4 and D9302C12); anti-IL-13 PE-Cy7 (eBio13A); anti-IL-17 FITC (eBio17B7, eBiosciences); anti-IFN-gamma PE (XMG1.2); anti-Foxp3 eFluor 450 (FJK-16s), anti-CD4 PerCp-Cy5.5 (RM4–5). For intracellular staining of in vitro cultured human cells, the following antibodies were used: anti-IL-9 PE or PerCP-Cy5.5 (MH9A4 or MH9A3, BD or Biolegend), anti-CD3 Alexa Fluor 700 (SP34–2, BD), anti-Foxp3 eFluor 450 (236A/E7, eBioscience), anti-CD45RO PE-Texas Red (UCHL1, Beckman Coulter).
Cells extracted from lung tissue were stained with LIVE/DEAD Fixable Blue Dead Cell Stain (Life Technologies), and with antibodies against the following cell surface markers for lineage negative selection: TCR-β, CD8, NK1.1, GR1, CD11b, CD11c, and CD19 (PerCp-Cy5.5, eBioscience). Cells were also stained with the following antibodies TCRβ APC-eFluor 780 (eBioscience), CD45.2 PeCy7 (BioLegend), Thy1.2 eFluor 450 (eBiosciences), CD44 AlexaFluor 700 (eBioscience), CD4 V500 (BD), IL-9 PE (BioLegend) or IL-9 APC (eBioscience), IL-13 PE or IL-13 eFluor 660 (eBioscience), IL-17 FITC (eBioscience) or IL-2 FITC (eBioscience), Foxp3 v450 (eBioscience). For Th9 transfer experiments, the following panel was used: CD45.1 FITC (eBioscience), CD45.2 APC-eFluor780 (eBioscience), TCRβ APC (Biolegend), CD4 PerCp-Cy5.5 (Biolegend), IL-9 PE (BioLegend), and Foxp3 v450 (eBioscience). For isolation and sorting of ex vivo Th9 cells and other Th subsets, antibodies against the following antigens were used on unstimulated cells: CD45.2 PeCy7 (eBioscience), TCRβ APC-eFluor 780 (Biolegend), CD44 AlexaFluor 700 (eBioscience), CD4 PerCp-Cy5.5 (Biolegend), GFP/YFP AlexaFluor 488 (Biolegend), CXCR3 PE (Biolegend), CCR4 APC (Biolegend), CCR6 Brilliant Violet 421 (BD).
Data were collected on a FACS Verse or LSR Fortessa (BD Biosciences) and analyzed using Flowjo software (Tree Star).
RNA-seq
For in vitro polarized cells, total RNA was prepared from approximately 500,000 cells using an Ambion mirVana miRNA isolation kit following the manufacturer’s protocol (ThermoFisher AM1560). For in vivo IL-9 producing cells and other subsets, total RNA was prepared from approximately 2,000–10,000 cells using a Trizol-based extraction technique following the manufacturer’s protocol (ThermoFisher 15596026). For all samples, mRNA was processed to generate single end mRNA-seq libraries using NEBNext Poly(A) mRNA Magnetic Isolation and Ultra II Directional RNA Library Prep kit for Illumina (NEB E7490, E7760, E7335). After recovering purified DNA, libraries were generated according to the vendor’s manual for the Illumina platform. Illumina HiSeq 2500 was used for 50-cycle single-end read sequencing. Raw sequencing data were processed with bcl2fastq 2.17.1 to generate FastQ files.
ATAC-seq
ATAC-seq samples were prepared as previously described (Shih et al., 2016). Briefly, cells were harvested after 72 hours of culture, and dead cells were removed by flow cytometric sorting. Approximately 50,000 cells were isolated, washed with cold PBS, and lysed for 10 minutes in cold lysis buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). After pelleting the nuclei by centrifugation (500×g for 10 min), pellets were resuspended in a 40-μl transposition reaction with 2 μl Tn5 transposase (FC-121–1030; Illumina) to tag and fragmentalize accessible chromatin. The reaction was incubated at 37°C, 400 rpm for 30 min; DNA was then purified using a MinElute kit (QIAGEN) and amplified with 8–12 cycles of PCR based on the amplification curve. After purification using a QIAquick PCR cleanup kit (QIAGEN) and Ampure XP beads (Beckman Coulter), samples were sequenced for 75 cycles (paired-end reads) on an Illumina HiSeq 2500.
Histone Chromatin Immunoprecipitation and ChIP-seq
ChIP-seq and ChIP-qPCR were performed using in vitro differentiated Th9 cells treated with RA 1μM or DMSO control. At least 20 million cells were used for transcription factor ChIP, and at least 1 million cells were used for histone mark ChIP. After chemical chromatin cross-linking (1% formaldehyde), cells were washed and frozen at −80C for ≤6 months.
Sonication: Histone Chromatin Immunoprecipitation
Cells were resuspended in high SDS shearing buffer (50mM Tris-HCl, 10mM EDTA, 1% SDS, protease inhibitors) at a concentration of 10 million/mL, and DNA fragmentation was performed on a Diagenode Bioruptor at high amplitude (40 cycles, 30s on/30s off) to an average length of 200–500 bp. After sonication, lysates were diluted to 0.1% SDS for immunoprecipitation.
Transcription Factor Chromatin Immunoprecipitation and ChIP-qPCR
20 million cells were suspended in 10 mL cell lysis buffer (50mM HEPES-KOH, 140mM NaCl, 1mM EDTA, 10% Glycerol, 0.5% Triton-X-100, Roche Complete mini EDTA free tablet 10X #11836170001) for 10 minutes at 4oC, pelleted, resuspended in 5 mL protein extraction buffer (200mM NaCl, 1mM EDTA, 0.5mM EGTA, 10mM Tris pH8) for 10 minutes at room temperature. Cells were pelleted and resuspended in 0.5 mL chromatin extraction buffer (protein extraction buffer with 0.1% Na-Deoxycholate, 0.5% N-laurylsarcosine, 0.1% SDS) and sonicated using a Bioruptor sonicator (high frequency, 30s on/30s off, 40 cycles) to a major band size of 300–500bp.
Immunoprecipitation, Library preparation, and ChIP-qPCR
After sonication, cells were immunoprecipitated with anti-H3K27Ac (ab4729; Abcam), anti-H3K4m1 (ab8895; Abcam), anti-H3K4m3 (ab8580; Abcam), anti-RARα (c15310155; Diagenode), anti-NRIP1 (ab42126; Abcam), a 1:1 mix of anti-STAT5A(PA-ST5A; R&D Systems)/anti-STAT5B (AF1584, R&D Systems), anti-STAT6 (ab32520; Abcam), or anti-CTCF (07–729; Millipore). Aliquots of genomic DNA (input) and immunoprecipitated samples were treated with proteinase K, heated to induce de-cross linking, and purified using columns (D4014, Zymo). After recovering purified DNA, 5ng or more of DNA was used to generate libraries according to the vendor’s manual for the Illumina platform (Cat#0344; NuGEN). Illumina HiSeq 2500 was used for 50-cycle single-read sequencing.
For ChIP-qPCR, quantitative PCR reactions were performed in triplicate on specific genomic regions using SYBR Green supermix (Bio-Rad). See STAR Methods Table for primer details. Data was normalized for primer efficiency by carrying out reactions on input DNA and normalizing relative to input signal. For all reactions, an inaccessible region within the Il9 locus (gene desert) was used as a negative control for normalization.
Cell lines and transfection
HEK293T cells were obtained from ATCC and maintained in DMEM supplemented with 10% FBS and antibiotics. Cells were transfected using lipofectamine 2000 (cat#11668019; Thermofisher) per the manufacturer’s protocol.
Luciferase detection
The Il9 promoter was cloned either alone or in tandem with E1, E2, or E3 into pGL4.23, a luciferase reporter vector with a minimal promoter. Sequences of specific constructs are provided in the Supplementary Data. We transfected HEK293T cells with each construct, and co-transfected cells with constitutively active Stat5A. Cells were also co-transfected with pRLTK (Renilla luciferase vector) for background normalization. After 48 hours, cells were lysed, and luciferase activity was detected using the Genecopoiea Luc-Pair Duo-Luciferase Assay Kit (Cat# LPFR-P030).
Chromatin Conformational Capture (3C)
3C was performed using in vitro differentiated Th0 cells, Th9 cells treated with RA 1μM, or Th9 cells treated with DMSO control. At least 10 million cells were used for each condition. After chemical chromatin cross-linking (1% formaldehyde), cells were washed and lysed (10mM Tris pH8, 10mM NaCl, 0.2% NP-40). Crosslinked DNA digested overnight at 37oC with 200 units of restriction enzyme (BglII, R0144L), then digested for another 24 hours with 200 additional units. Digestion efficiency was determined to be >95%. Restriction enzyme was heat-inactivated at 68oC for 10 minutes, and fragments were religated (T4 ligase, 100 weiss units) at 16oC overnight, after which an additional 100 units of T4 ligase was added for 4 hours. Crosslinking was reversed, after which DNA was isolated and purified for analysis using RT-qPCR. A bacterial artificial chromosome (BAC) containing the Il9 locus and surrounding gene loci was used for quantification. Primers and probes for the Fbxl21 locus, which is adjacent locus to Il9 and which was not affected by RA, were used for normalization. Primer and probe information are provided in STAR Methods Table.
Papain induced asthma
For papain-induced lung inflammation, mice were anaesthetized with isoflurane and exposed intranasally to 25 μg papain (Calbiochem) in 30 μL PBS on day 0, 3, 6 and 14. 12–16 hours after the last challenge, lung-isolated cell analyses, and/or measurements of airway reactivity were performed.
Lung tissues were fixed in 4% neutral buffered formaldehyde, embedded in paraffin, sectioned, and stained with hematoxilin and eosin (H&E) or periodic acid-Schiff (PAS) stain. Cells were isolated from lungs by incubating lung fragments with 100U collagenase for 1 hour and 10 min. Lung cells were stained for surface antigens and intracellular cytokines after stimulation with PMA/ionomycin for 4–5 hours, as detailed above.
Th9 transfer experiment
For Th9 transfer experiments, Rag2−/− OT-II Foxp3Sf (ovalbumin specific) Th9 cells were cultured in vitro in the presence of either vehicle control or RA 1μM as detailed above. After 72 hours, Th9 cells were harvested and washed three times with PBS, and then 0.9 million Th9 cells were injected into the tail veins of B6 CD45.1 mice. On day 1 after Th9 transfer, mice were anesthetized by i.p. administration of ketamine/xylazine mixture (1 mL ketamine [100 mg/mL], 0.5 mL xylazine [20 mg/mL], and 8.5 mL PBS) and challenged intratracheally with 100μg of ovalbumin (Sigma-Aldrich A5503–50G). On day 2, were anaesthetized with isoflurane and challenged intranasally with 100μg of ovalbumin; 12–20 hours after the last challenge, lungisolated cell analyses were performed as detailed above.
Measurement of allergic airway reactivity
For RA treatment experiments, mice were anaesthetized with isoflurane and exposed intranasally to 25 μg papain as detailed above. For experiments in WT vs. RARα−/− mice, 12.5μg papain was used but the same volume/schedule was maintained, due to high amounts of mucus plugging noted in histology slides with full-dose (25μg) papain, which limits the sensitivity of airway reactivity measurement. Bronchial reactivity was determined 12–18h after the last challenge of papain. Mice were anesthetized by i.p. administration of ketamine/xylazine mixture (1 mL ketamine [100 mg/mL], 0.5 mL xylazine [20 mg/mL], and 8.5 mL PBS). A 19-gauge blunt-end needle was inserted into the trachea, and the animals then were ventilated mechanically. Baseline measurements were recorded after the aerosol administration of saline, followed by doubling doses of methacholine (6.25–100 mg/mL) using flexiVent (Scireq Scientific Respiratory Equipment).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq analysis
Reads of 50 bases were mapped to the mouse transcriptome and genome mm9 using TopHat 2.0.8 with Bowtie2–2.1.0. Gene expression values (RPKM: Reads Per Kilobase exon per Million mapped reads) were calculated with Partek Genomics Suite 6.6. 2–3 replicate RNA-seq experiments were performed for each condition.
Datasets were normalized based on RPKM and purged of micro-RNAs, sno-RNAs and scaRNAs. To minimize fold-change artifact from low abundance transcripts, a small offset was added to all RPKM values (equal to the averaged second quartiles of each dataset). When multiple transcripts were detected for a single gene, only the most abundant (highest average RPKM across all replicates) was analyzed. Transcripts with RPKM < 1 were excluded. Heatmaps were created using Morpheus (Broad Institute). Other downstream analyses and figures were created with R 3.0.1 and custom R programs, unless otherwise specified.
Cell-specific and shared RA-regulated modules
Individual lists of RA-regulated genes were generated separately for each subset (Th0, Th1, Th2, Th9, Th17, and iTreg). Differential gene expression was determined by ANOVA using FDR <0.05 and 2-fold change relative to vehicle-treated controls (Partek Genomics Suite). The six gene lists were combined, and duplicates were removed, to generate a master set of 1025 RA-regulated targets genes. K-means clustering (k=15) was performed based on fold-change in gene expression (RA vs. vehicle control). K-means clusters (Supplementary Table 1) were defined as regulated in multiple subsets if ≥50% of the genes were RA-regulated (FDR<0.05) in ≥3 subsets and regulated in a subset-specific fashion if ≥50% of the genes were RA-regulated in ≤2 subsets.
Evaluation of functional enrichment
All we identified Kegg Pathway and Gene Ontology terms enriched in sets of differentially expressed genes using metascape (http://metascape.org). Complete lists of enriched pathways are included in Supplementary Table 1.
Gene Sensitivity Enrichment Analysis
GSEA analysis was performed as described (Subramanian et al., 2005). Enrichment score curves and member ranks were generated by the GSEA software (Broad Institute). RNA-seq datasets were used in conjunction with the following user-generated Gene Sets (1) Th9-high genes based on the overlap of in vitro and in vivo data. For in vitro data, Th9-high genes were defined as FDR<0.05 and average gene expression ≥ 1.5 fold higher in Th9 cells using a 1-way ANOVA of Th9 cells vs. (Th1 or Th2 or Th17 or iTreg cells). For INFER (GFP IL-9 reporter) mice, Th9high genes were defined as FDR<0.05 and average gene expression ≥ 1.5 fold higher in GFP+ cells compared with GFP− cells. For eYFP IL-9 fate reporter mice, Th9-high genes were defined as FDR<0.05 and average gene expression ≥ 1.5 fold higher in Il9-expressing subsets (eYFP+ or Th2) cells compared non-Il9-expressing (Th1 and Th17) subsets. Genes that were identified as Th9-high in the in vitro dataset and in at least one in vivo dataset were included in the Th9-high gene cassette (2) Th9 high genes based on a more stringent analysis of in vitro data, defined as FDR<0.05 for Th9 vs. Th1, Th2, Th17, and iTreg subsets by 1-way ANOVA; average gene expression ≥ 1.5 fold higher in Th9 cells using pairwise comparisons of Th9 vs. Th1, Th9 vs. Th2, Th9 vs. Th17, and Th9 vs. iTreg subsets; rpkm>5 in Th9 cells (30 members) (3) Th9 genes as defined by a separate dataset (GSE44937). Because this dataset did not test relevant comparators (Th1, Th17 subsets), genes associated with these subsets were manually removed prior to analysis. For Figure S7, gene lists from Th9/iTreg-selective and common RA-regulated modules were converted to human orthologs using bioDBnet and combined to generate a list of 335 human orthologs of murine RA-induced genes and 353 human orthologs of murine RArepressed genes.
For Figure 2, lists of TF-induced and TF-repressed genes were obtained by analyzing public datasets: (1) IL-2 regulated Stat5 target genes (GSE77656, 420 genes) (2) IL-4 regulated Stat6 target genes (GSE22801, 251 genes) (3) IL-4 regulated Gata3 target genes (GSE20898, 623 genes) (4) TGF-b regulated Smad3 target genes (GSE19601, 323 genes), (5) Runx1 target genes (GSE6939, 715 genes), (6) Irf4 target genes (GSE39756, 862 genes) (7) canonical NF-kB target genes ((Pahl, 1999), 177 genes), (8) Fosl2 target genes (GSE40918, 95 genes), (9) Atf3 (GSE61055, 46 genes), (10) Vdr target genes in macrophages (GSE2421, 2373 genes), (11) Foxp1 (GSE50725, 60 genes), (12) Nfat target genes in CD8 T cells (GSE64409, 209 genes), (13) Batf target genes in Th17 cells (GSE40918, 156 genes), (14) Junb (GSE98413, 233 genes), (15) Rorc target genes in Th17 cells (GSE40918, 78 genes). Normalized Enrichment Scores for TF-induced and TF-repressed genes were combined to generate a consensus enrichment score. Significance was determined by combining FDR for the two GSEA analyses using Fisher’s method for combining independent tests.
Selection and analysis of human datasets
The Gene Expression Omnibus was searched for datasets or series examining time-course gene expression in atopic or allergic inflammation, which displayed significantly increased IL9 expression during allergic response. Ratios of mean gene expression values at two timepoints were calculated in Partek. To determine the effect of allergen on RA-regulated genes, gene lists from Th9/iTreg-selective and common RA-regulated modules were converted to human orthologs using bioDBnet, to obtain a list of 688 human orthologs of murine RA-regulated genes. Human gene expression arrays were filtered for RA-regulated genes using RStudio: 645 RAregulated genes were expressed in GSE73482 whereas 665 RA-regulated genes were expressed in GDS2935. Lack of expression was confirmed manually for each of the undetected genes in both datasets. Ecdf (empirical cumulative distribution function) plots were generated in RStudio.
ChIP-seq alignment and peak calling
Reads of 50 bases were aligned to the mouse genome build mm9 with Bowtie 0.12.8, allowing two mismatches. Uniquely mapped and non-redundant reads were used for downstream analysis. The aligned file was converted to bam format using samtools (http://samtools.sourceforge.net). Peaks were called using MACS v1.4.3 using a p<1e-5 and with the input sample for background correction. In the case of H3K4me1, H3K4me3, and H3K27Ac samples, “—nomodel” setting was used. BigWig tracks were generated from Bam files and converted into bedGraph format using bedtools. These were further reformatted with the UCSC tool bedGraphToBigWig. Genome browser files are displayed with IGV.
For STAT5 and STAT6 ChIP-seq data in Th9 cells, raw data were downloaded from the Gene Expression Omnibus (GSE41317). Annotation was converted to mm9 (crossmap), files were processed as above, and tdf files were generated from bedGraph files using igvtools.
ATAC-seq alignment and peak calling
ATAC-seq reads from two biological replicates were used for each sample. Redundant pairedend (PE) reads were removed using fastquniq. PE reads of 50 bases were aligned to the mouse genome build mm9 with Bowtie 0.12.8, following the guidelines presented by Buenrostro et al (Buenrostro et al., 2013). Customized python scripts were used to calculate fragment length of each pair of uniquely mapped PE reads for size distribution analysis, and to group uniquely mapped reads into bins of 0 to 175 bases and 180 to 250 bases, respectively. UCSC Genome Browser viewable and normalized BigWig files were generated with the Hypergeometric Optimization of Motif EnRichment program (HOMER) version 4.8.
Only one mapped read to each unique region of the genome that was less than 175 bp was kept and used in peak calling. Regions of open chromatin were identified by MACS (version 1.4.3) using a p-value threshold of 1 × 10−5. Only regions called in both replicates were used in downstream analysis. Peak intensities (“tags” column) were normalized as tags per 10 million reads (RP10M) in the original library.
Analysis of ATAC-seq and ChIP-seq peaks
Signal across all sites (i.e., all annotated genes and all accessible chromatin regions) was analyzed to eliminate potential bias by pre-selection. After calling ATAC-seq peaks with MACS, the union peaks of replicate samples were created using mergePeaks module in HOMER and divided into shared and cell-specific peaks using the same utility. For analysis of chromatin accessibility and epigenetic marks associated with Th9-high genes, peaks were annotated based on proximity to the nearest gene, and tag densities (normalized reads) were calculated using HOMER. For ATAC-seq data, fold-changes in tag density were calculated for RA-treated vs. control-treated WT Th9 cells and for RARαΔCD4 Th9 cells. For ChIP-seq data, tag densities for immunoprecipitated samples were normalized to paired input controls; then fold-changes in normalized tag density were calculated for RA-treated vs. control-treated WT Th9 cells and for RARαΔCD4 Th9 cells. When multiple peaks were annotated to a single gene, the peak with the highest absolute fold change (RA vs. control) in WT Th9 cells was selected for further analysis. Motif analysis of cell-specific peaks was done using HOMER. Other downstream analysis and heatmap generation were performed with R 3.0.1 and morpheus. For downstream analysis, regulatory elements were designated as promoters if they were located <2kb from the transcription start site (TSS) and as enhancers if located >2kb from the TSS.
Statistical analysis of RNA-seq, qPCR, flow cytometry, and in vivo experiments
For 3C, ChIP-qPCR and luciferase assay, statistical significance was calculated by unpaired twotailed Student’s t-test with Graphpad Prism software. For T-cell intracellular cytokine production and dose-response curves, statistical significance was calculated using a paired two-tailed Student’s t-test with Graphpad Prism software. For in vivo experiments, statistical significance was calculated using Mann-Whitney analysis with Graphpad Prism Software. For determination of differentially expressed genes from RNA-seq data, all statistical analysis was performed in Partek using Analysis of Variation (ANOVA), with multiple comparison adjustment to calculate false discovery rate (FDR). For ecdf plots, statistical analysis was done using Kolmogorovsmirnov testing in RStudio. For analysis of GSE73482, paired t-test was performed in Partek, with multiple comparison adjustment to calculate FDR.
Scoring: Papain- and Ova-induced asthma
Lung histology was scored on H&E and Periodic Acid Schiff (PAS) stained sections by a reader with experimental conditions masked, Perivascular and peri-bronchiolar cuffing (PVC and PBC) were each scored as follows: 0: No visible infiltrate. 1: Patchy infiltrate in <25% of bronchioles or vessels, 2: Patchy infiltrate in <50% of bronchioles or vessels, 3: Widespread infiltrate >50% of bronchioles or vessels with circumferential infiltrates in most bronchioles or vessels. 4.
Criteria for score of 3 plus vascular obliteration (for PVC) or bronchiolar plugging (for PBC). Interstitial inflammation was graded from 0–3 depending on the extent of cellular infiltrate into alveoli. Goblet cell hyperplasia was scored for small airways as follows: 0: No visible hyperplasia or mucous production, 1: patchy hyperplasia and/or PAS staining in <25% of bronchioles or vessels, not circumferential, 2: patchy hyperplasia and/or PAS staining of <50% of bronchioles, 3: widespread hyperplasia and >50% PAS staining in most bronchioles, 4: criteria for 3 plus bronchiolar plugging or obliteration. Scores reported were the total score for each lung (0–15).
DATA AND SOFTWARE AVAILABILITY
Raw and analyzed data reported in this paper have been submitted to GEO, accession number GSE123501.
Supplementary Material
Table S1, related to Figure 1. Common and subset-selective retinoic acid (RA)-regulated genes and pathways.The first panel (Lists) displays lists of common (coordinately RAregulated in ≥3 subsets; grey) or subset selective (RA-induced in ≤2 subsets or RA-repressed in ≤2 subsets; green, Th9 and iTreg; red, Th1 and/or Th17; orange, Th2) differentially expressed genes (DEGs, FDR<0.05, fold-change in gene expression >2 or <–2, Partek). The remaining panels display significantly enriched pathways (Metascape) for each group of RA-regulated DEGs. If more than 20 pathways were enriched, then the 20 most significantly enriched pathways are displayed.
Highlights.
Global effects of retinoic acid (RA) were evaluated by RNA-seq in major Th subsets
RA impacts the transcriptome of Th9 cells more so than that of other effector subsets
RA-RARα represses the Il9 locus and Th9 program independently of Foxp3
RA-RARα activity controls pathology in Th9-associated allergic lung disease
Acknowledgments
We thank J. Simone, J. Lay, and K. Tinsley (NIAMS) for expert assistance in flow cytometry and the NIAMS LACU staff for technical support. We thank C. Liu and F. Zhang (NHLBI) for generating knockout mouse lines. We thank G. Gutierrez-Cruz and S Dell’Orso (NIAMS) for deep-sequencing support. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster (NIH). This work was supported by the Intramural Research Programs of NIAMS and NIAID, and by the Office of Dietary Supplements (ODS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: None
References
- Abbas AK, Murphy KM, and Sher A (1996). Functional diversity of helper T lymphocytes. Nature 383, 787–793. [DOI] [PubMed] [Google Scholar]
- Arora P, Kumar V, and Batra S (2002). Vitamin A status in children with asthma. Pediatric Allergy and Immunology 13, 223–226. [DOI] [PubMed] [Google Scholar]
- Bai XF, Li O, Zhou Q, Zhang H, Joshi PS, Zheng X, Liu Y, Wang Y, Zheng P, and Liu Y (2004). CD24 controls expansion and persistence of autoreactive T cells in the central nervous system during experimental autoimmune encephalomyelitis. The Journal of experimental medicine 200, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Pino-Lagos K, Rosemblatt M, and Noelle RJ (2007). All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. The Journal of experimental medicine 204, 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma-Garcia I, Al-Bader R, Ortiz C, Elgueta R, Arno M, et al. (2015). Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 42, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. (2006). Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Chatagnon A, Veber P, Morin V, Bedo J, Triqueneaux G, Semon M, Laudet V, d’AlcheBuc F, and Benoit G (2015). RAR/RXR binding dynamics distinguish pluripotency from differentiation associated cis-regulatory elements. Nucleic Acids Res 43, 4833–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, and Ramsdell F (1999). Cellular and molecular characterization of the scurfy mouse mutant. Journal of immunology (Baltimore, Md. : 1950) 162, 2546–2554. [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, and Powrie F (2007). A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. The Journal of experimental medicine 204, 1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, and Flavell RA (2000). Control of T helper cell differentiation--in search of master genes. Sci STKE 2000, pe1. [DOI] [PubMed] [Google Scholar]
- Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, and Flavell RA (2001). ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 409, 97–101. [DOI] [PubMed] [Google Scholar]
- Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, and O’Shea JJ (2008). Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, and Rudensky AY (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology 4, 330–336. [DOI] [PubMed] [Google Scholar]
- Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, et al. (2011a). Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Grainger JR, Spencer SP, and Belkaid Y (2011b). The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. (2011). CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43, 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Notarbartolo S, Croonenborghs T, Patel B, Cialic R, Yang TH, Aschenbrenner D, Andersson KM, Gattorno M, Pham M, et al. (2017). Transcriptional signature of human pro-inflammatory TH17 cells identifies reduced IL10 gene expression in multiple sclerosis. Nature communications 8, 1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humblin E, Thibaudin M, Chalmin F, Derangere V, Limagne E, Richard C, Flavell RA, Chevrier S, Ladoire S, Berger H, et al. (2017). IRF8-dependent molecular complexes control the Th9 transcriptional program. Nature communications 8, 2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Eshima Y, and Kagechika H (2003). Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. International immunology 15, 1017–1025. [DOI] [PubMed] [Google Scholar]
- Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, and Song SY (2004). Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538. [DOI] [PubMed] [Google Scholar]
- Jabeen R, Goswami R, Awe O, Kulkarni A, Nguyen ET, Attenasio A, Walsh D, Olson MR, Kim MH, Tepper RS, et al. (2013). Th9 cell development requires a BATF-regulated transcriptional network. J Clin Invest 123, 4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, and Rudensky A (2009). Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH (2017). The transcription factor network in Th9 cells. Semin Immunopathol 39, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Hufford MM, and Olson MR (2015). The development and in vivo function of T helper 9 cells. Nat Rev Immunol 15, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh B, Abdul Qayum A, Srivastava R, Fu Y, Ulrich BJ, Janga SC, and Kaplan MH (2018). A conserved enhancer regulates Il9 expression in multiple lineages. Nature communications 9, 4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok SK, Park MK, Cho ML, Oh HJ, Park EM, Lee DG, Lee J, Kim HY, and Park SH (2012). Retinoic acid attenuates rheumatoid inflammation in mice. Journal of immunology (Baltimore, Md. : 1950) 189, 1062–1071. [DOI] [PubMed] [Google Scholar]
- Larange A, and Cheroutre H (2016). Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annu Rev Immunol 34, 369–394. [DOI] [PubMed] [Google Scholar]
- le Maire A, and Bourguet W (2014). Retinoic acid receptors: structural basis for coregulator interaction and exchange. Subcell Biochem 70, 37–54. [DOI] [PubMed] [Google Scholar]
- Lee W, Su Kim H, and Lee GR (2015). Leukotrienes induce the migration of Th17 cells. Immunology and cell biology 93, 472–479. [DOI] [PubMed] [Google Scholar]
- Li H, Edin ML, Bradbury JA, Graves JP, DeGraff LM, Gruzdev A, Cheng J, Dackor RT, Wang PM, Bortner CD, et al. (2013). Cyclooxygenase-2 inhibits T helper cell type 9 differentiation during allergic lung inflammation via down-regulation of IL-17RB. Am J Respir Crit Care Med 187, 812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Spolski R, Li P, Du N, West EE, Ren M, Mitra S, and Leonard WJ (2014). Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proceedings of the National Academy of Sciences of the United States of America 111, 3508–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licona-Limon P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limon I, Ishigame H, Hao L, Herbert DR, and Flavell RA (2013). Th9 Cells Drive Host Immunity against Gastrointestinal Worm Infection. Immunity 39, 744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, and Awasthi A (2018). Transcriptional Control of Th9 Cells: Role of Foxo1 in Interleukin-9 Induction. Front Immunol 9, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, Kasturi SP, Rosenthal KM, Evavold BD, and Pulendran B (2009). Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med 15, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret M, Ruffie C, Periquet B, Campo AM, Menevret M, Phelep A, Dziewiszek K, Druilhe A, and Pretolani M (2007). Liposomal retinoic acids modulate asthma manifestations in mice. The Journal of nutrition 137, 2730–2736. [DOI] [PubMed] [Google Scholar]
- Marquez HA, and Cardoso WV (2016). Vitamin A-retinoid signaling in pulmonary development and disease. Mol Cell Pediatr 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard CL, Hatton RD, Helms WS, Oliver JR, Stephensen CB, and Weaver CT (2009). Contrasting roles for all-trans retinoic acid in TGF-beta-mediated induction of Foxp3 and Il10 genes in developing regulatory T cells. The Journal of experimental medicine 206, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Zu Horste G, Wu C, Wang C, Cong L, Pawlak M, Lee Y, Elyaman W, Xiao S, Regev A, and Kuchroo VK (2016). RBPJ Controls Development of Pathogenic Th17 Cells by Regulating IL-23 Receptor Expression. Cell Rep 16, 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P, Korn T, and Kuchroo VK (2009). Interleukin-17 and type 17 helper T cells. N Engl J Med 361, 888–898. [DOI] [PubMed] [Google Scholar]
- Mizutani N, Nabe T, and Yoshino S (2015). Semaphorin 7A plays a critical role in IgEmediated airway inflammation in mice. Eur J Pharmacol 764, 149–156. [DOI] [PubMed] [Google Scholar]
- Moisan J, Grenningloh R, Bettelli E, Oukka M, and Ho IC (2007). Ets-1 is a negative regulator of Th17 differentiation. The Journal of experimental medicine 204, 2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, and Cheroutre H (2007). Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (New York, N.Y.) 317, 256–260. [DOI] [PubMed] [Google Scholar]
- Naus S, Blanchet MR, Gossens K, Zaph C, Bartsch JW, McNagny KM, and Ziltener HJ (2010). The metalloprotease-disintegrin ADAM8 is essential for the development of experimental asthma. Am J Respir Crit Care Med 181, 1318–1328. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, and Paul WE (2010). Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (New York, N.Y.) 327, 1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka Y, Yokota A, Takeuchi H, Maeda N, and Iwata M (2011). Retinoic acid-induced CCR9 expression requires transient TCR stimulation and cooperativity between NFATc2 and the retinoic acid receptor/retinoid X receptor complex. Journal of immunology (Baltimore, Md. : 1950) 186, 733–744. [DOI] [PubMed] [Google Scholar]
- Pahl HL (1999). Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866. [DOI] [PubMed] [Google Scholar]
- Pedersen MB, Skov L, Menne T, Johansen JD, and Olsen J (2007). Gene expression time course in the human skin during elicitation of allergic contact dermatitis. The Journal of investigative dermatology 127, 2585–2595. [DOI] [PubMed] [Google Scholar]
- Perumal NB, and Kaplan MH (2011). Regulating Il9 transcription in T helper cells. Trends Immunol 32, 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan AT, Goldrath AW, and Glass CK (2017). Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity 46, 714–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino-Lagos K, Guo Y, Brown C, Alexander MP, Elgueta R, Bennett KA, De Vries V, Nowak E, Blomhoff R, Sockanathan S, et al. (2011). A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. The Journal of experimental medicine 208, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal R, Awasthi A, and Ahuja V (2016). Retinoic acid-primed human dendritic cells inhibit Th9 cells and induce Th1/Th17 cell differentiation. Journal of leukocyte biology 100, 111–120. [DOI] [PubMed] [Google Scholar]
- Raverdeau M, and Mills KH (2014). Modulation of T cell and innate immune responses by retinoic Acid. Journal of immunology (Baltimore, Md. : 1950) 192, 2953–2958. [DOI] [PubMed] [Google Scholar]
- Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, and Mucida D (2013). Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nature immunology 14, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AC, Tan C, Hawley ET, Gomez-Rodriguez J, Goswami R, Yang XP, Cruz AC, Penumetcha P, Hayes ET, Pelletier M, et al. (2015). The TNF-family ligand TL1A and its receptor DR3 promote T cell-mediated allergic immunopathology by enhancing differentiation and pathogenicity of IL-9-producing T cells. Journal of immunology (Baltimore, Md. : 1950) 194, 3567–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster GU, Kenyon NJ, and Stephensen CB (2008). Vitamin A Deficiency Decreases and High Dietary Vitamin A Increases Disease Severity in the Mouse Model of Asthma. The Journal of Immunology 180, 1834–1842. [DOI] [PubMed] [Google Scholar]
- Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, and Kaplan MH (2015). TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J Allergy Clin Immunol 136, 433–440 e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR, Urban JF Jr., Davis FP, Kanno Y, and O’Shea JJ (2016). Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 165, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephensen CB, Rasooly R, Jiang X, Ceddia MA, Weaver CT, Chandraratna RAS, and Bucy RP (2002). Vitamin A Enhances in Vitro Th2 Development Via Retinoid X Receptor Pathway. The Journal of Immunology 168, 4495–4503. [DOI] [PubMed] [Google Scholar]
- Su P, Chen S, Zheng YH, Zhou HY, Yan CH, Yu F, Zhang YG, He L, Zhang Y, Wang Y, et al. (2016). Novel Function of Extracellular Matrix Protein 1 in Suppressing Th17 Cell Development in Experimental Autoimmune Encephalomyelitis. Journal of immunology (Baltimore, Md. : 1950) 197, 1054–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciume G, Muljo SA, Kuchen S, Casellas R, Wei L, Kanno Y, and O’Shea JJ (2012). TGF-beta and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nature immunology 13, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy NM, Hollams EM, Holt PG, and Bosco A (2016). Differential gene network analysis for the identification of asthma-associated therapeutic targets in allergen-specific T-helper memory responses. BMC Med Genomics 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Casmediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, Narlikar L, Northrup DL, Tang Q, Paul WE, et al. (2011). Genome-wide analyses of transcription factor GATA3mediated gene regulation in distinct T cell types. Immunity 35, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, and Stockinger B (2011). An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nature immunology 12, 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, and Kuchroo VK (2008). Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. Journal of immunology (Baltimore, Md. : 1950) 181, 2277–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Fan Y, Li J, Zhang X, Lou X, Dou Y, Shi X, Lan P, Xiao Y, Minze L, and Li XC (2018). Guidance of super-enhancers in regulation of IL-9 induction and airway inflammation. The Journal of experimental medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 1. Common and subset-selective retinoic acid (RA)-regulated genes and pathways.The first panel (Lists) displays lists of common (coordinately RAregulated in ≥3 subsets; grey) or subset selective (RA-induced in ≤2 subsets or RA-repressed in ≤2 subsets; green, Th9 and iTreg; red, Th1 and/or Th17; orange, Th2) differentially expressed genes (DEGs, FDR<0.05, fold-change in gene expression >2 or <–2, Partek). The remaining panels display significantly enriched pathways (Metascape) for each group of RA-regulated DEGs. If more than 20 pathways were enriched, then the 20 most significantly enriched pathways are displayed.
Data Availability Statement
Raw and analyzed data reported in this paper have been submitted to GEO, accession number GSE123501.