

Abstract
Formation of acetaminophen (APAP) protein adducts are a critical feature of APAP hepatotoxicity, and circulating protein adducts have recently been utilized in bioassays for identification of APAP overdose in humans. Despite their clinical significance, mechanisms of adduct release into the circulation are not well understood. Extracellular vesicles (EVs) are discrete membrane bound vesicles, which package cellular cargo and function in extracellular transport. Clarification of their role in transport of APAP adducts is relevant since adduct packaging within these vesicles could shield them from detection by antibody based methods, resulting in under-estimating adduct levels. Hence, this study evaluated EV release after APAP overdose in primary mouse hepatocytes and human HepaRG cells in vitro, in mice and APAP overdose patients in vivo and examined their role in transport of APAP-protein adducts. EVs were characterized by size and protein composition and the levels of APAP-protein adducts were measured. Significant elevations in circulating EV numbers were observed 6h after APAP overdose in vivo and by 4h in primary mouse hepatocytes in culture. EVs were also elevated in media from HepaRG cells by 24h after APAP exposure, an effect recapitulated in APAP overdose patients, where EV numbers were elevated compared to healthy controls. Although APAP-protein adducts were elevated in circulation and media parallel to the increased exosome release, no detectable adducts were observed within EVs. This suggests that although APAP overdose enhances EV release from hepatocytes in mice and humans, it is not a significant mechanism of release of APAP protein adducts into circulation.
Keywords: acetaminophen; drug hepatotoxicity; protein adduct formation, exosomes; HepaRG cells; acetaminophen overdose patients
INTRODUCTION
Acetaminophen toxicity is the leading individual cause of acute liver failure in the United States. Upon ingestion, APAP is rapidly conjugated and eliminated by0020glucuronidation and sulfation reactions. At the same time, a small percent is converted to the toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI), via cytochrome-P450 enzymes, mainly CYP2E1. At therapeutic doses, NAPQI is easily detoxified by glutathione, however, at supra-therapeutic doses, the sulfation pathway is saturated and the glucuronidation reaction becomes overwhelmed, leading to extensive formation of NAPQI and depletion of glutathione (McGill and Jaeschke, 2013). The covalent binding of NAPQI to intracellular proteins, in particular mitochondrial proteins, triggers mitochondrial oxidative stress and ultimately hepatocyte necrosis (Du et al., 2016). While the mitochondrial dysfunction and subsequent signaling responsible for cell death have been well-studied, information regarding the fate of adducted proteins is scarce. NAPQI protein adducts have been identified in circulation even after therapeutic doses of APAP in humans (Heard et al., 2011, 2016). In addition, our earlier studies in mice showed release of APAP-CYS adducts into plasma prior to elevations in ALT (McGill et al., 2013), suggesting that adduct release can occur independent of significant hepatocyte necrosis. This raises the question: in the absence of hepatocyte necrosis, how are these adducts being released in measurable quantities? One very likely possibility is that PAP-CYS adducts are released via extracellular vesicles (EVs), which are discrete membrane bound vesicles. EVs are thought to package cellular cargo and function in extracellular transport (Hessvik and Llorente, 2018). EVs are typically about 150 nm in diameter and are synthesized within the cell from the endosomal system, which ultimately fuses with the plasma membrane for release into the circulation. EVs contain a variety of proteins, lipids and nucleic acids; their ultimate composition in dependent on the cell type (Hessvik and Llorente, 2018).
These issues are clinically relevant, since circulating protein adducts have been suggested to be biomarkers for APAP-induced liver injury (Davern et al., 2006; McGill and Jaeschke, 2018). Consequently, APAP protein adducts have recently been the focus of a rapid bioassay for identification of APAP overdose in humans (Roberts et al., 2017). Clarification of the role of EVs in transport of APAP adducts is relevant, since adduct packaging within these membrane-bound vesicles could shield them from detection by antibody-based methods, resulting in under-estimation of circulating adduct levels. Hence, this study evaluated EV release after APAP overdose in primary mouse hepatocytes and human HepaRG cells in vitro, in mice in vivo, as well as in plasma from APAP overdose patients and examined their potential role in transport of APAP-protein adducts.
MATERIALS AND METHODS
Cell culture experiments.
Primary hepatocytes were isolated from mice by means of a 2-step collagenase perfusion technique as described previously (Bajt et al., 2004). Cell viability was generally more than 90% based on trypan blue exclusion, and cell purity of hepatocytes was more than 95%. Cells were plated in Williams E medium (Life Technologies, Grand Island, NY) containing 100 U/mL penicillin/streptomycin, 1×10−7 M insulin, and 10% fetal bovine serum. After 3.5h for attachment, cells were washed with PBS and treated with 5 mM APAP for the indicated time periods. Human HepaRG cell culture was carried out as described previously (McGill et al., 2011). Briefly, cells obtained from Biopredic International (Rennes, France) were seeded at 1 × 105 undifferentiated cells/cm2 in HepaRG growth medium containing HepaRG growth supplement 711 (Biopredic). Undifferentiated cells were cultured for two weeks at 37°C, followed by induction of differentiation using HepaRG differentiation medium (Biopredic) containing HepaRG growth supplement 721 for two weeks. For APAP treatment, cells were washed with PBS and treated with 20 mM APAP dissolved in DMSO-free Williams’ E medium containing 10% FBS (exosome free), insulin and penicillin/streptomycin for indicated time points. Both mouse primary hepatocytes and HepaRG cells and their medium were then harvested at various time points for measurement of lactate dehydrogenase (LDH) activity (Bajt et al., 2004), alanine aminotransferase (ALT) activity (ALT assay kit, Pointe Scientific, MI), APAP-protein adduct measurement (McGill et al., 2011) and EV isolation.
Animal experiments.
Male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) 8–12 weeks of age, housed in an environmentally controlled room with a 12h light/dark cycle, were used for experiments. All experimental protocols followed the criteria of the National Research Council for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center. All animals were fasted overnight and then treated i.p. with 300 mg/kg APAP (Sigma-Aldrich, St. Louis, MO) diluted in warm saline. At the indicated time points, groups of mice were sacrificed via cervical dislocation and exsanguination via the hepatic vena cava under isoflurane anesthesia. Blood was drawn into a heparinized syringe and centrifuged to collect plasma for determination of ALT activities, P protein adducts and isolation of EVs.
Human samples.
Plasma samples from three APAP overdose patients admitted to Banner – University Medical Center Phoenix in Phoenix, AZ, were used for the study. A diagnosis was made by physicians based on standard clinical criteria including reported history of APAP overdose and detectable serum APAP levels. All three patients were female and ranged in age from 41 to 61 years, with samples collected after informed consent. Patient samples were compared to plasma from three healthy volunteers (all female, age range 28–32) who served as controls. Sample collection for all patients was conducted as per approved B protocols.
Extracellular vesicle isolation and nanoparticle tracking.
EVs were isolated from mouse and human plasma as well as from cell culture medium by ultracentrifugation. Specifically, for cell culture media EV isolation, the culture supernatant was centrifuged at 1500 × g for 10 min to remove lifted cells and cellular debris, as well as large microvesicles/microparticles and apoptotic bodies. The resultant supernatant was subjected to centrifugation at 10,000 × g for 60 min, and the supernatant filtered through 0.22-μm pore filters (Corning, Corning USA). After filtration, the filtrate was ultracentrifuged at 100,000 × g for 2h, to pellet EVs, which were then subjected to an additional PBS wash and centrifuged again at 100,000 × g for 60 min. Plasma EVs were isolated in a similar fashion, except for the initial 1500 x g spin, which was omitted. The final pellet of small EVs was either resuspended in 150 μL of PBS for western blotting and nanoparticle tracking analysis or resuspended in 150 μL of sodium acetate buffer for adduct measurement. Size distribution within EV preparations was analyzed by measuring the rate of Brownian motion using a NanoSight LM10 system (NanoSight, Amesbury, U.K.). The system is equipped with a fast video capture and particle-tracking software. Nanoparticle Tracking Analysis (NTA) software post-acquisition settings were kept constant for all samples, and each video was analyzed to give the mean, mode, and median vesicle size, and an estimate of the concentration (Dragovic et al., 2011).
Western blotting.
Protein lysates were resolved on a 4–20% SDS polyacrylamide gel under reducing conditions, followed by transfer onto PVDF membranes. After blocking for an hour in 5% milk in Tris buffered saline, membranes were incubated with primary antibodies at 4°C overnight. The antibodies used were a rabbit anti–Alix polyclonal IgG, rabbit anti-CD63 polyclonal IgG or rabbit anti–β actin polyclonal IgG (Santa Cruz Biotechnology). The membranes were washed and then incubated with anti-rabbit IgG-HRP ( anta Cruz Biotechnology) for 1 h, followed by protein visualization on a LICOR Odyssey imaging system.
Statistics.
Data are expressed as mean ± SE. Differences between two groups were evaluated with Student’s t-test. A two-tailed p < 0.05 was considered significant.
RESULTS
Cytotoxicity and EV release after APAP overdose in primary mouse hepatocytes.
Initial experiments evaluating EV release after APAP exposure were carried out in vitro, with primary mouse hepatocytes exposed to 5 mM APAP. No significant cell death was evident in cells 2 h after APAP treatment (Figure 1A). There was a significant increase in cell death by 4 h post-APAP, however, it was still only around 20%. EVs were isolated from the cell culture media at both 2 and 4 h after APAP, and the EV marker protein Alix was used to confirm the identity of the isolated vesicles by western blotting (Figure 1B). Alix was only detectable in EVs isolated from the cell culture media, not in the cell lysate (Figure 1B). An examination of the size of the isolated vesicles revealed that the majority were around 50–150 nm, further corroborating their identity as exosomal vesicles (Figure 1C). The total number of EVs was increased by 2 h compared to controls and further increased by 4 h after APAP (Figure 1C,D). Since cell death was negligible at the 2 h time point, this indicates that EV release is an active process occurring prior to significant cell death of hepatocytes. Measurement of APAP protein adducts were then carried out in cell lysates, media, as well as in the EV fraction to determine if they could be vehicles of APAP adduct release from the cell. Interestingly, while significant levels of APAP adducts could be measured in both cell lysates and media, no detectable levels were found in the EV fraction (Figure 1E), indicating that exosomal transport was not a predominant mechanism of adduct release from mouse hepatocytes in vitro.
Figure 1: Enhanced EV release from primary mouse hepatocytes after APAP overdose.
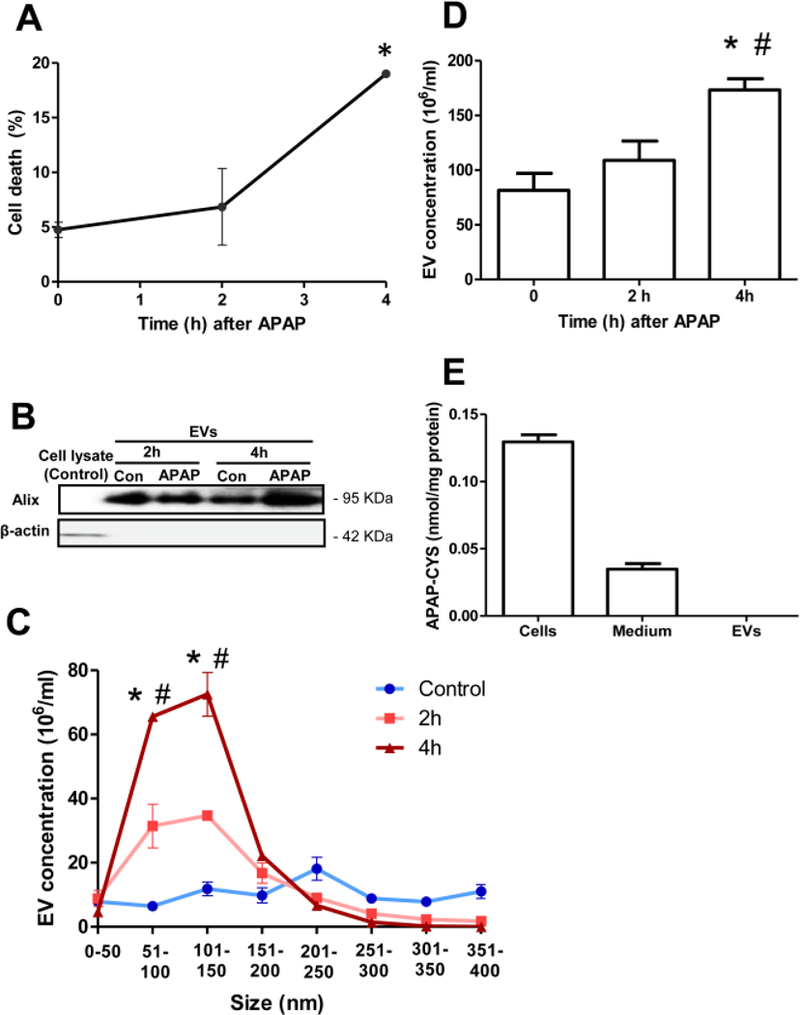
Hepatocytes isolated from maleC57BL/6JD mice were treated with 5 mM APAP, followed by collection of media and cell lysates at the indicated time points (A) Hepatocyte viability as measured by LDH release at 2 and 4 h after treatment with APAP. (B) Enrichment of the EV marker Alix in isolated EVs at both 2 and 4 h after APAP. (C) Size range of isolated EVs. (D) Total EV numbers before and after APAP exposure of hepatocytes in culture. (E) Measurement of APAP- Cysteine adducts in cells, media and the EV fraction from hepatocytes 4 h after APAP exposure. Values are mean ± SEM of n=3 hepatocyte isolation preparations. *P<0.05 (compared to untreated controls), # < 0.05 (compared to 2 h time point).
EV release and viability in human HepaRG cells in culture after APAP exposure.
To confirm that the findings from mouse hepatocytes were relevant to human cells, experiments were then repeated in the human HepaRG cell line. Hepatocytes were treated with 20 mM APAP, followed by analysis of culture medium and cell lysates at 6, 12 and 24 h because metabolism and cell death in human cells (McGill et al., 2011; Xie et al., 2014) are more delayed compared to the mouse (McGill et al., 2012). While a slight increase in cell death was evident by 12 h after APAP, only by 24 h was cell death prominent in HepaRG cells, with around 20% loss of cell viability (Figure 2A). EV isolation was then carried out; only the EV fraction was enriched in markers Alix and CD63, with no detectable levels in the cell lysates (Figure 2B). Analysis of EV size indicates a slightly broader size range (50–200 nm) than that in mice (Figure 2C). Interestingly however, a significant increase in EV numbers in this size range only occurred at 24 h after APAP, unlike in mouse hepatocytes. This increase was also seen in total exosome numbers, (Figure 2D), which also indicates no significant change in EV number at 6 and 12 h after APAP, with a doubling of EV numbers only by 24 h after APAP. This indicates that human HepaRG cells delay release of EVs in response to APAP until appreciable cell death has been initiated. APAP protein adducts in cell lysates, culture medium and EVs at 24 h after APAP were measured. APAP protein adducts could not be detected in EVs, while appreciable levels were found in both cell lysates and culture medium (Figure 2E). This indicates that human HepaRG cells, like mice, use an alternate mechanism for release of the clear majority of APAP adducts into the surrounding milieu.
Figure 2: Elevations in EV release from human HepaRG cells after APAP overdose.
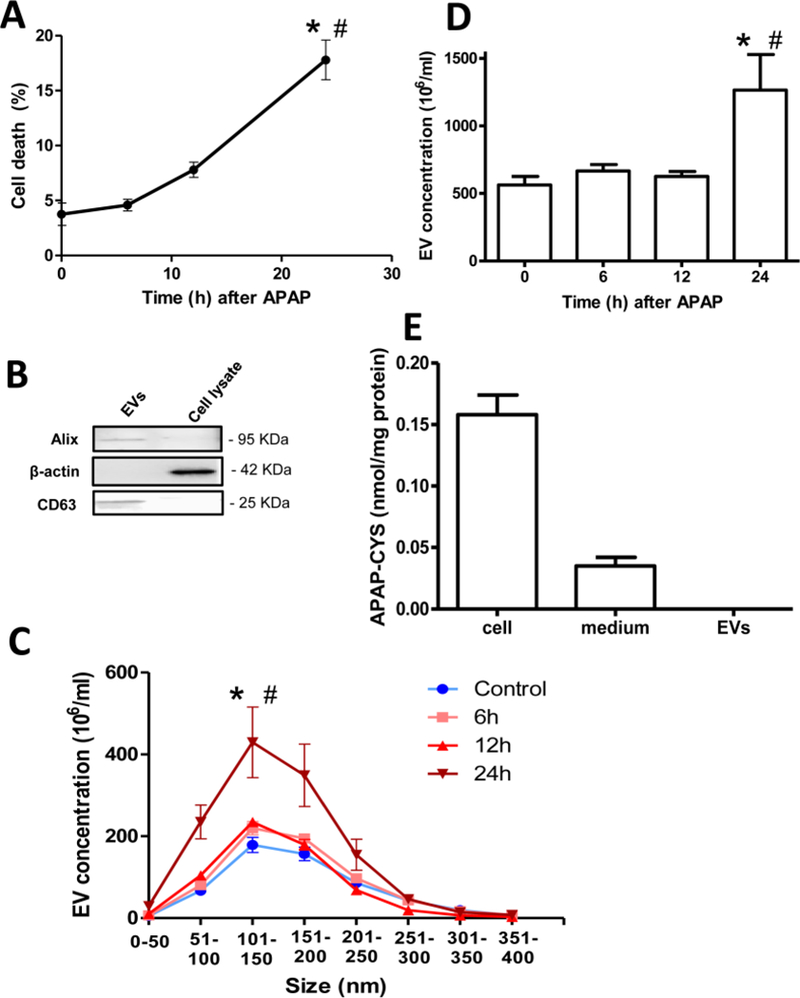
HepaRG cells were treated with 20 mM APAP and media and cell lysate harvested at the indicated time points. (A) Temporal profile of HepaRG cell viability based on ALT release after treatment with 20 mM APAP. (B) Western blotting of EVs and cell lysates using antibodies against the EV marker Alix and CD63 and the cytosolic marker β-actin. (C) Characterization of HepaRG cell derived EV size at various time points after APAP exposure (D) Total EV numbers in HepaRG cell culture supernatant 24 h after APAP exposure. (E) APAP-Cysteine adduct measurement in cells, medium and EV fraction from HepaRG cells 24 h after APAP exposure. Values are mean ± SEM of n=3 separate experiments. *P<0.05 (compared to untreated controls), #P< 0.05 (compared to 6 or 12 h time points).
Early elevations of EV release into plasma of mice after APAP-induced liver injury.
The next series of experiments were carried out in vivo, initially in the mouse model of APAP overdose. Male C57BL/6J mice were treated with 300 mg/kg APAP and sacrificed at 2, 4 and 6 h after APAP. Liver damage were evaluated by measuring ALT levels (Figure 3A). No significant change in plasma ALT activities was evident at 2 h after APAP, indicating no appreciable necrotic cell death at that early point. Some liver injury was evident by 4 h, which further increased significantly by 6 h, indicating robust necrotic cell death at that point (Figure 3A). EVs were isolated from mouse plasma at the indicated time points, and vesicular identity was confirmed by measurement of EV protein markers Alix and CD63, which were detectable in the EV fraction and not in plasma (Figure 3B) indicating enrichment of isolated EVs. Quantitation of EVs in mouse plasma revealed significant elevation in the total EV number within 2 h after APAP (Figure 3C), when appreciable necrotic death was not evident (Figure 3A). EV numbers then dropped by 4 and 6 h, though still significantly higher than controls (Figure 3C). Characterization of EV size indicated that the elevation at 2 and 4 h were of vesicles around 50–150 nm in size (Figure 3D), which are characteristic of circulating EVs. This again reiterates the early release of these vesicles prior to significant necrosis in vivo. An evaluation of APAP-CYS adducts in plasma and the exosome fractions at the various time points indicated that adduct levels were elevated within 2 h and further increased by 4 h post APAP in plasma and decreased subsequently at 6 h (Figure 3E). However, no APAP-CYS adducts were detectable in the EV fraction at any of the time points tested (Figure 3E).
Figure 3: EV release into circulation after APAP overdose in mice in vivo.
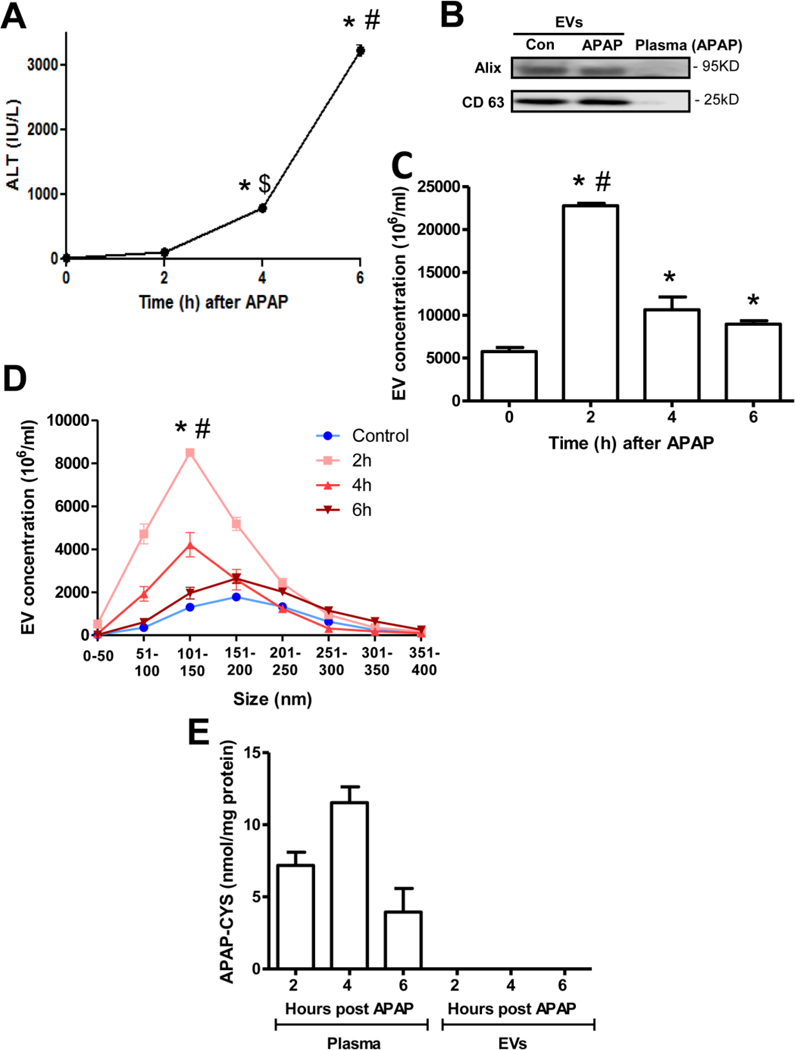
Mice were treated with 300 mg/kg APAP and sacrificed at the indicated time points. (A) Plasma ALT levels as an indicator of liver injury after APAP overdose. (B) Western blotting showing enrichment of the EV markers Alix and CD63 in isolated plasma EVs from both control and APAP-treated mice at 2 h after APAP. (C) Total EV numbers before and after APAP treatment in vivo. (D) Size range of EVs isolated at various time points. (E) APAP-Cysteine adduct measurement in plasma and EV fractions at various time points after APAP. Values are mean ± SEM of n=3 animals per time point. *P<0.05 (compared to untreated controls), #P< 0.05 (compared to 4 or 6h time points), $P< 0.05 (compared to 2 h time point).
EV release into circulation after APAP overdose in humans.
Finally, to confirm relevance of these findings in humans, plasma samples from APAP overdose patients were examined. Peak plasma ALT values for each APAP overdose patient was higher (138, 2299, 8614 U/L) compared to controls (31, 25, 37 U/L). EV isolation from their plasma and assessment of exosome markers such as Alix and CD63 show enrichment in the EV fraction (Figure 4A), confirming the identity of the isolated particles. This was further corroborated by size analysis of isolated vesicles, which indicated that healthy volunteers had the majority of vesicles between 150–200 nm in size, while APAP overdose patients showed vesicles ranging from 50–200 nm, with predominant elevation in vesicles between 100–150 nm (Figure 4B). dramatic elevation was also seen in the total number of EVs in APAP overdose patients (Figure 4C), indicating that like in the mouse, APAP overdose induces significant EV release in humans. Interestingly, EV numbers seem to be independent of circulating ALT levels, as seen in Figure 4D, where the patient with low ALT also had elevations in EV numbers, similar to the patient with much higher ALT activities. Measurement of APAP-CYS adduct levels in plasma and the EV fraction from the overdose patients indicated that while adducts could be readily measured in plasma, they were not detectable in the EV fraction (Figure 4E), again indicating that in humans too, alternate mechanisms may be responsible for most of the APAP-CYS adduct release into circulation.
Figure 4: EV release into plasma in humans after APAP overdose.
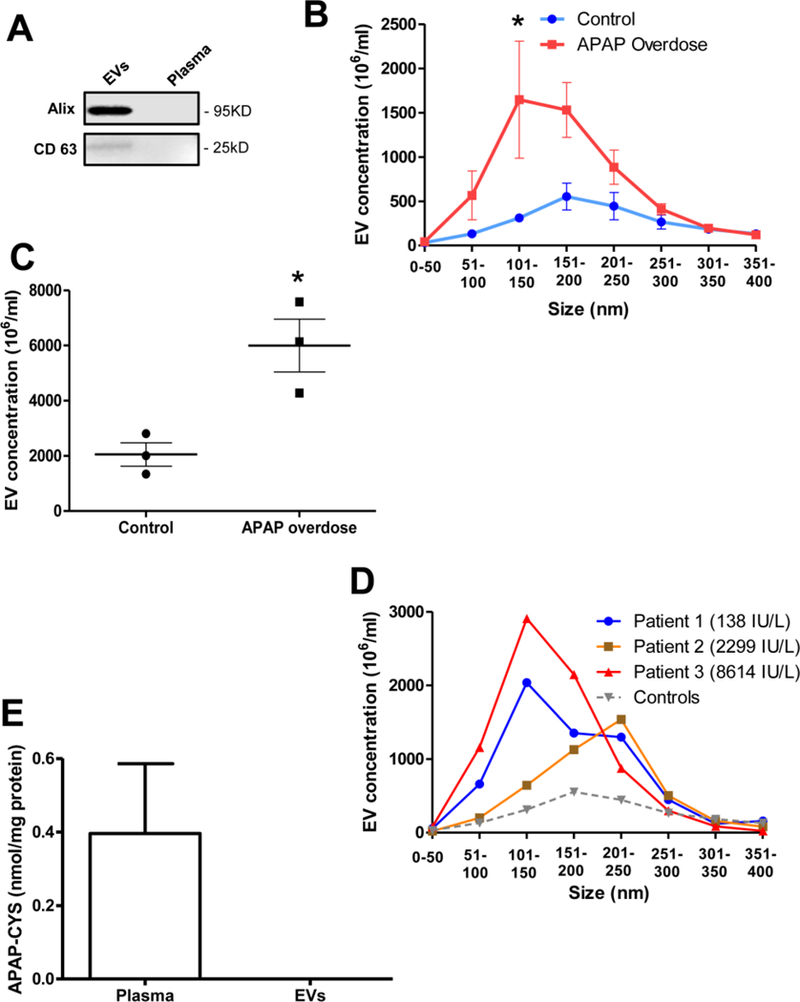
EVs were isolated from plasma of APAP overdose patients and healthy volunteers. (A) Western blot indicating enrichment of markers such as Alix and CD63 in EVs compared to EV-free plasma (B) Size distribution of EVs isolated from human plasma of controls and APAP overdose patients. (C) Total circulating EV numbers in controls and after APAP overdose in humans. (D) Size distribution and elevations in EV numbers from individual APAP overdose patients (ALT levels in parenthesis) compared to the average of all controls (E) APAP-Cysteine adduct measurement in plasma and EVs fractions from APAP overdose patients. Values are mean ± SEM of n=3 patients per group. *P<0.05 (compared to controls)
DISCUSSION
Formation of APAP-CYS adducts on cellular proteins, especially mitochondrial proteins, is a critical feature of APAP-induced cellular necrosis (Ramachandran and Jaeschke, 2017, 2018), and their removal by autophagy is an important adaptive response in hepatocytes (Ni et al., 2016). APAP protein adducts in circulation are specific biomarkers of APAP-induced hepatotoxicity (Davern et al., 2006; McGill and Jaeschke, 2018), and this has been leveraged recently by development of a rapid bioassay for identification of APAP overdose in humans (Roberts et al., 2017). However, adducts can appear in circulation without significant hepatic necrosis in both humans and mice (Heard et al., 2011; McGill et al., 2013). better understanding of the mechanisms by which this occurs would be pertinent in the light of development of antibody-based point-of-care assays (Roberts et al., 2017), which are based on the assumption that adducts in circulation are freely available to interact with immobilized antibodies on the test strip. However, if a substantial amount of the protein adducts in plasma are packaged in EVs, the assay could significantly underestimate the actual adduct levels in plasma. Since even therapeutic doses of APAP can result in detectable APAP protein adduct levels in plasma of <0.5 nmol/ml (Heard et al., 2011) and hepatotoxic doses of APAP are generally associated with adduct levels >1.1 nmol/ml (James et al., 2009), accurate quantitation of these adducts in plasma is necessary to be useful for the diagnosis.
Circulating EVs with various cellular cargo, including proteins and nucleic acids, are being intensely studied as mediators of inter-cellular communication within the circulation. EVs were initially considered to be involved in expulsion of cellular waste (Hessvik and Llorente, 2018; Johnstone et al., 1987) and have been considered to act in concert with autophagy to maintain cellular homeostasis (Baixauli et al., 2014). In addition, these vesicles are now associated with numerous physiological and pathological functions (Hessvik and Llorente, 2018). Circulating microRNAs in EVs have been suggested to indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases (Bala et al., 2012). Studies in rats in vivo as well as in primary rat and human hepatocytes demonstrated no significant change in the number of EVs after exposure to subtoxic concentrations of APAP, though alterations in EV cargo composition were evident (Holman et al., 2016). However, rats are not a good model for APAP hepatotoxicity (McGill et al., 2012), and our data clearly indicate that the numbers of EVs are significantly elevated after toxic doses of APAP in mice and in humans. The relationship of EV release with cytotoxicity is influenced by the experimental model, since in both primary mouse hepatocytes as well as in human HepaRG cells, elevation in the number of EVs was appreciable only when some cell death was evident. In contrast, in mice exposed to an APAP overdose in vivo, significant elevations in EV numbers were obvious prior to significant liver cell necrosis. This indicates that mechanisms of early EV release functioning in the mouse in vivo are probably compromised by in vitro culture conditions, resulting in delayed release only with cell death. The data from humans also corroborate the mouse in vivo data, with elevations in EV numbers independent of circulating ALT levels in plasma. These elevations in EVs do not seem to be unique to APAP overdose, since it has been shown that EV numbers were also significantly elevated in human subjects with alcoholic hepatitis (Momen-Heravi et al., 2015).
Interestingly, despite consistent elevations in APAP-CYS adducts in media after APAP in cells in culture, as well as in circulation after in vivo APAP overdose in mice and humans, none of these adducts were detected in the EV fractions. Maximal EV release also seems to occur prior to maximal adduct release in mice in vivo. One possibility for lack of adducts within EVs could be the origin of the exosomal vesicles, which arise from the endosomal compartment and are derived from internal vesicles of multi-vesicular bodies (Hessvik and Llorente, 2018; Masyuk et al., 2013). APAP-CYS adducts inducing cellular necrosis involve, to a substantial degree, those in mitochondria (Ramachandran and Jaeschke, 2018). However, a proteomic analysis of melanoma-derived EVs indicated the absence of mitochondrial proteins (Mears et al., 2004). This may be one possible reason for absence of adducts within EVs. These findings have precedent, since although microRNAs have been shown to be packaged within hepatocyte-derived EVs (Bala et al., 2012; Fang et al., 2018), a porcine model of acute liver failure showed increases in global microRNA concentrations 4 h prior to acute liver failure in plasma but not in isolated EVs (Baker et al., 2015).
In conclusion, our studies of mouse primary hepatocytes and human HepaRG cells in vitro, as well as mice and humans in vivo, showed significant elevations in numbers of EVs released into the medium and circulation, respectively, after an APAP overdose, which occurs prior to significant liver injury in the mouse in vivo and seems to be independent of liver injury in humans, as well. However, release of APAP-CYS adducts into the circulation after an APAP overdose does not seem to involve EVs, and the mechanisms behind the early EV elevations, as well as independent APAP-CYS adduct release, need further investigation. The fact that no relevant fraction of APAP-protein adducts was detected within EVs in both mice and humans suggests there is no relevant interference in detecting these adducts in plasma with the new antibody-based point of care assay (Roberts et al., 2017).
HIGHLIGHTS.
Acetaminophen protein adducts are biomarkers for reactive metabolite formation
Extracellular vesicles are released by hepatocytes in response to APAP overdose
APAP protein adducts were detected in plasma and cell culture supernatant
No APAP protein adducts were detected in EVs in vivo or in vitro
No relevant elimination of APAP protein adducts from the liver occurs through EVs
ACKNOWLEDGEMENTS
This investigation was supported in part by the National Institutes of Health grant R01 DK102142 and by grants from the National Institute of General Medical Sciences (P20 GM103549 and P30 GM118247) from the National Institutes of Health. J.Y.A. was supported by a NIH Predoctoral Fellowship F31 DK120194-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURE
The authors declare that they have no conflict of interest.
REFERENCES
- Baixauli F, Lopez-Otin C, Mittelbrunn M, 2014. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol 5, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajt ML,Knight TR,Lemasters JJ,Jaeschke H, 2004. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci 80, 343–349. [DOI] [PubMed] [Google Scholar]
- Baker LA, Lee KC, Palacios Jimenez C, Alibhai H, Chang YM, Leckie PJ, Mookerjee RP, Davies NA, Andreola F, Jalan R, 2015. Circulating microRNAs Reveal Time Course of Organ Injury in a Porcine Model of Acetaminophen-Induced Acute Liver Failure. PLoS One 10, e0128076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, Alao H, Kodys K, Szabo G, 2012. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 56, 1946–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davern TJ 2nd, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, Lalani E, Munoz S, Shakil AO, Lee WM; Acute Liver Failure Study Group., 2006. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 130, 687–94. [DOI] [PubMed] [Google Scholar]
- Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, Carr B, Redman CW, Harris AL, Dobson PJ, Harrison P, Sargent IL, 2011. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine 7, 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Ramachandran A, Jaeschke H., 2016. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol 10, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JH, Zhang ZJ, Shang LR, Luo YW, Lin YF, Yuan Y, Zhuang SM, 2018. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 68, 1459–1475. [DOI] [PubMed] [Google Scholar]
- Heard K, Green JL, Anderson V, Bucher-Bartelson B, Dart RC, 2016. Paracetamol (acetaminophen) protein adduct concentrations during therapeutic dosing. Br J Clin Pharmacol 81, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard KJ, Green JL, James LP, Judge BS, Zolot L, Rhyee S, Dart RC, 2011. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol 14, 11:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessvik NP, Llorente A, 2018. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 75, 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman NS, Mosedale M, Wolf KK, LeCluyse EL, Watkins PB, 2016. Subtoxic Alterations in Hepatocyte-Derived Exosomes: An Early Step in Drug-Induced Liver Injury? Toxicol Sci 151, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LP, Letzig L, Simpson PM, Capparelli E, Roberts DW, Hinson JA, Davern TJ, Lee WM., 2009. Pharmacokinetics of acetaminophen-protein adducts in adults with acetaminophen overdose and acute liver failure. Drug Metab Dispos 37, 1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C, 1987. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem 262, 9412–9420. [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, Larusso NF, 2013. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol 59, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H., 2013. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. PharmRes 30,2174–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H, 2018. Biomarkers of drug-induced liver injury: progress and utility in research, medicine, and regulation. Expert Rev Mol Diagn 18, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, Williams CD, Wilkins DG, Rollins DE, Jaeschke H, 2013. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol 269, 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H, 2012. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 264, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H, 2011. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 53, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears R, Craven RA, Hanrahan S, Totty N, Upton C, Young SL, Patel P, Selby PJ, Banks RE, 2004. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 4, 4019–4031. [DOI] [PubMed] [Google Scholar]
- Momen-Heravi F, Saha B, Kodys K, Catalano D, Satishchandran A,Szabo G,2015. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J Transl Med 13, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y, Jaeschke H, Ding WX, 2016. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol 65, 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H,2017Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res 3, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H, 2018. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene Expr 18, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DW, Lee WM, Hinson JA, Bai S, Swearingen CJ, Stravitz RT, Reuben A, Letzig L, Simpson PM, Rule J, Fontana RJ, Ganger D, Reddy KR, Liou I, Fix O, James LP, 2017. An Immunoassay to Rapidly Measure Acetaminophen Protein Adducts Accurately Identifies Patients With Acute Liver Injury or Failure. Clin Gastroenterol Hepatol 15, 555–562 e553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H, 2014. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol 279, 266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]