

Abstract
Background:
No approved systemic therapy exists for von Hippel-Lindau disease, an autosomal dominant disorder with pleiotropic organ manifestations, including clear cell renal cell carcinomas, retinal, cerebellar and spinal hemangioblastomas, pheochromocytomas, pancreatic serous cystadenomas, and pancreatic neuroendocrine tumors. This prospective phase II study was designed to assess the efficacy and safety of pazopanib in patients with von Hippel-Lindau disease.
Methods:
Adult patients with clinical manifestations of von Hippel-Lindau disease were treated with pazopanib 800mg PO daily for 24 weeks, with an option to continue treatment if desired by patient and treating physician. Primary endpoints included objective response rate (ORR) and safety by intention to treat. ORR was measured both per patient and per lesion type. Radiographic assessments were performed at baseline and every 12 weeks using appropriate imaging modalities. Efficacy and safety were assessed using continuous monitoring and a Bayesian design. Study was registered as NCT01436227 in http://ClinicalTrials.gov. Trial accrual was complete at time of analysis.
Findings:
Thirty-one eligible patients were treated with pazopanib. The ORR was 42 percent. Organ specific response rate was 52 percent in renal cell carcinoma, 53 percent in pancreatic lesions, and 4 percent in central nervous system hemangioblastomas. The majority of patients chose to stay on treatment after 24 weeks. Four patients came off study because of grade-3/4 transaminitis, and three additional patients discontinued because of general treatment intolerance due to multiple intercurrent grade 1-2 toxicities. Treatment related serious adverse events included one case each of appendicitis and gastritis. One patient experienced a fatal central nervous system bleed.
Interpretation:
Pazopanib resulted in significant response rates in von HippelLindau disease, with an acceptable side effect profile. Pazopanib should be considered a treatment choice in patients with von Hippel-Lindau disease and growing lesions, or in patients with unresectable lesions where decrease in tumor size is desired.
Introduction:
Von Hippel-Lindau disease is an autosomal dominant hereditary cancer syndrome that affects approximately one in 35,000 live births per year(1). The primary manifestations of von Hippel-Lindau disease include retinal, cerebellar and spinal hemangioblastomas, renal cysts and clear cell renal cell carcinomas, pancreatic cysts and pancreatic neuroendocrine tumors, pheochromocytomas, endolymphatic sac tumors of the middle ear, and epididymal or round ligament cysts(1). von HippelLindau disease arises due to germline mutations in the eponymous VHL gene(2), which encodes a 213 amino acid protein(3). One of the main functions of the VHL protein is to act as an E3 ubiquitin ligase for hypoxia inducible factor 1α (HIF1α) and HIF2α(4). Loss of HIF regulation results in highly angiogenic lesions, exemplified by hemangioblastomas and renal cell carcinomas(5).
Treatment for patients with von Hippel-Lindau disease includes lifelong surveillance, with surgical intervention when necessary(5). If renal cell carcinomas or neuroendocrine tumors increase beyond 3 centimeters (cm) in size they can develop metastatic potential(6). If hemangioblastomas become symptomatic, they need to be surgically removed to reduce the possibility of patients developing permanent neurological sequelae. Retinal hemangioblastomas require ablation to prevent irreversible retinal damage. The overall impact these multiple tests and interventions have on the patient is significant. Currently there are no established systemic therapies for von Hippel-Lindau disease(1). Prior experience with targeted therapy administered to patients with von Hippel-Lindau disease is largely anecdotal, and the results are mixed. A number of agents have been used to treat retinal hemangioblastomas. Intravitreal treatment with bevacizumab resulted in stabilization in some patients (7-9)(10). Intravitreal ranibizumab did not provide consistent benefit in a case series of five patients(11). Treatment with systemic bevacizumab provided marginal ocular benefit in individual case reports(12). Systemic treatment with sunitinib resulted in possible visual stabilization in three patients but with significant concomitant toxicity(13). Systemic therapy has been more broadly tested in patients with von Hippel-Lindau disease. Multiple studies reported outcomes in individuals treated with sunitinib(14-16). A 2011 study prospectively evaluated the safety and efficacy of sunitinib in 15 patients with von Hippel-Lindau disease(17). Individuals on the study received 50 mg of sunitinib daily for 28 days, followed by 14 days off for up to four cycles, with a primary endpoint of toxicity. Grade 3 toxicity included fatigue in five patients (33%); dose reductions were made in ten patients (75%). Eighteen renal cell carcinomas and 21 hemangioblastomas were evaluable. Of these, six renal cell carcinomas (33%) exhibited PR, and no hemangioblastomas showed response. A retrospective study of 14 von Hippel-Lindau disease patients with renal cell carcinoma, 10 of whom had metastatic disease, demonstrated significant response in metastatic and primary renal cell carcinoma lesions. Eleven patients had cerebellar hemangioblastomas, and eight had spinal hemangioblastomas. No response was seen in hemangioblastomas(18).
A retrospective assessment of angiogenic receptor expression in renal cell carcinomas and hemangioblastomas was reported in the 2011 sunitinib study(17). Laser scanning cytometry was used to assess the expression levels of vascular endothelial growth factor receptor, platelet derived growth factor receptor, Tie2, fibroblast growth factor receptor 3, and phospho-fibroblast growth factor receptor substrate 2 (pFRS2). The fibroblast-growth factor receptor ligand pFRS2 was higher in hemangioblastomas than in renal cell carcinomas. These data suggested that FGFR blockade may be necessary to impact hemangioblastoma endothelium and shrink hemangioblastomas. Around that time a patient with von Hippel-Lindau disease who had previously been treated on the prospective sunitinib study received pazopanib, and demonstrated dramatic response in several hemangioblastomas that had previously not responded to sunitinib(19). These data, coupled with the capacity of pazopanib to induce regression in metastatic renal cell carcinoma(20-23) as well as providing primary tumor size reduction in a neoadjuvant or presurgical setting(24, 25) provided a mechanistic and clinical rationale for testing pazopanib in the von Hippel-Lindau disease patient population, with the expectation that renal cell carcinomas would exhibit response, and the hope that hemangioblastomas would demonstrate treatment effect as well.
This study set out to determine the efficacy of pazopanib in the treatment of patients with clinical features of or genetically confirmed von Hippel-Lindau disease.
Methods:
Study design and participants
Institutional review board (IRB) approval was obtained for this prospective phase II, single-center, single-arm open-label study and written informed consent was obtained from patients with genetically confirmed von Hippel-Lindau disease or clinical features consistent with von Hippel-Lindau disease who were undergoing surveillance for at least one measurable von Hippel-Lindau disease-related lesion. Patients were enrolled from the USA and from Canada. Patients with negative VHL genetic testing were permitted provided they had lesions that were consistent with a von Hippel-Lindau disease-like syndrome, including multiple retinal or central nervous system hemangioblastomas. Prior systemic treatment for von HippelLindau disease was permitted, provided at least 14 days or 5 half-lives of prior drug had elapsed prior to first dose of pazopanib.
Those with the following were eligible: Performance status of 2 or better, retinal hemangioblastomas discernible with ophthalmoscopy; cerebellar and spinal hemangioblastomas at least 5 mm in longest diameter; renal cell carcinomas and renal cysts with solid nodules at least 1 cm in longest diameter; and pancreatic cysts and neuroendocrine tumors at least 1 cm in longest diameter. Patients were required to have normal organ and marrow function, as defined by serum aspartate and alanine transaminase concentrations μ2.0 times the local laboratory’s upper limit of normal (ULN), total serum bilirubin concentrations μ1.5 times the ULN, absolute neutrophil count >1500 cells/mm3, platelet count >100 000 cells/microliter, hemoglobin concentration >9.0 g/dL, and serum creatinine ≤2.0 mg/dL. There was no lower age limit for eligibility. Patients were eligible if they had no immediate need for intervention in any of their von Hippel-Lindau disease related lesions, including lesions that were deemed not to be in imminent danger of causing neurological complications, and were not deemed to be a significant metastatic threat. Patients were excluded if they had a myocardial infarction or cerebrovascular accident within the past six months, or if they had active bleeding or a bleeding diathesis.
Procedures
Pazopanib was initiated at 800mg PO daily, with dose reduction in 200mg increments permitted if patients developed grade 3 or greater toxicity. A cycle was defined as 28 days of treatment. Six cycles of treatment were planned. Imaging was performed after 3 and six cycles. After six cycles, if patient and the treating team determined that patient continued to experience clinical benefit from pazopanib (due to stabilization or decrease in lesion size, decrease in von Hippel-Lindau disease related symptoms or other factors deemed to confer benefit to the patient, and benefit outweighed any intercurrent toxicities), and if the patient wished to continue therapy, then treatment was continued until treatment intolerance or disease progression occurred.
Baseline and follow-up evaluations of target lesions were carried out by using computed tomography (CT) scanning or magnetic resonance imaging (MRI), as appropriate. Direct ophthalmoscopy, using fluorescein angiography with photographs, color testing, and visual field testing, was used to follow retinal lesions. Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 was used to evaluate response, and was also modified to uncouple target organ systems, to allow each organ system to be evaluated separately. Summation of size was carried out separately for lesions in the kidney, pancreas, central nervous system (brain + spine), and retina, and these measurements were used to compare changes from baseline size. Laboratory testing included assessment of transaminase levels every two weeks for the first eight weeks and then monthly, as well as monthly complete blood counts, serum chemistries and kidney function tests. Adverse event monitoring included assessment of patient with history and physical exam every 12 weeks, as well as through direct communication with study team at any point during study if an adverse event occurred. Dose reductions to 600, 400 and 200mg by mouth per day were permitted if intolerance to 800mg was noted. Dose reescalation was permitted if considered appropriate by treating physician. If dose interruptions of greater than 21 days were required, review of patient’s condition was required prior to re-initiation of treatment.
Outcomes
The primary study objective was to evaluate the efficacy and safety of pazopanib, with efficacy measured as objective response rate (ORR), which is the sum of complete responses (CRs) + partial responses (PRs). Secondary objectives included assessing growth rate over time in target lesions before and after pazopanib treatment, evaluating the need for surgical intervention in individuals who received pazopanib in comparison to rate prior to receipt of drug, and creating an annotated tissue resource from patients on study. All endpoints, including safety, were assessed by intent-to-treat.
Statistical analyses
The sample size of 40 patients was chosen to estimate the response rate at 24 weeks with a standard error no larger than 0.08 so the half length of the 95% confidence interval would not exceed 15%. The trial was not designed to perform formal hypothesis testing, but was designed to provide precise estimates of response and toxicity. There was no pre-defined stopping rule for slow accrual. Patients not completing 1 month of therapy due to a reason other than drug toxicity or progressive disease were considered inevaluable, and all other patients were considered assessable for toxicity and efficacy. With all 40 patients accrued, the response rate, early progression rate, toxicity rate and their corresponding 95% posterior credible intervals would be assessed. Biomarker endpoints and genetic mutations were to be measured at baseline and/or over time. Summary statistics, including frequency tabulation, means, standard deviations, median, and range, were to be used to describe subject characteristics and marker data. The chisquared (χ2) test or Fisher’s exact test were to be used to test the association between two categorical variables, such as response and prognostic factors and discrete markers. The association among various continuous and discrete markers and response were assessed first by the exploratory data analysis graphically and tested using Wilcoxon rank sum test. Correlation among continuous biomarkers were to be examined by Pearson or Spearman rank correlation coefficients. Both univariate and multivariate logistic regressions were to be performed to model the baseline prognostic factors on response, when appropriate. The generalized estimating equations to model were to be used to correlate discrete variables. Repeated measures analysis including mixed effects model were to be performed to analyze continuous biomarkers change over time. Time to progression (TTP) were estimated using the Kaplan-Meier method. Log-rank test was to be performed to test the difference in survival between prognostic groups. Regression analyses of survival data based on the Cox proportional hazards model was to be conducted on TTP. The proportional hazards assumptions were to be evaluated graphically and analytically, and regression diagnostics (e.g., martingale and Shoenfeld residuals) were examined to ensure that the models are appropriate. There was no plan for sensitivity analysis. Continuous monitoring for lesion progression (PD) and drug discontinuation due to toxicity (DD) occurred during the entire treatment period. If there was a high probability that the PD rate exceeded 20% or the DD rate exceeded 10%, the trial would be stopped early. Formally, the study would be terminated if Pr(PD Rate > 0.20 | Data) > 0.90 or Pr(DD Rate > 0.10 | Data) > 0.90. The assumed prior distributions for PD rate and DD rate were Beta (0.2, 0.8) and Beta (0.1, 0.9), respectively, with one patient’s worth of information. The study was monitored in cohorts of 10 patients. SAS 9.4 and RStudio Version 1.0.136 were used for the analysis. The study was registered with http://ClinicalTrials.gov under number NCT01436227.
Role of funding source
The Sponsor approved the study design. The corresponding author and coinvestigators collected, analyzed and interpreted the data, and wrote the manuscript with no input from the sponsor. The corresponding author (EJ), LD, CCR, MFV, and JAW had access to the raw data. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Results:
Patient demographics:
Thirty-two patients were enrolled on the trial from 18/01/2012 to 10/08/2016 (Figure 1). Thirty-one patients were evaluable for efficacy, and one patient registered but did not initiate therapy. Table 1 summarizes the patient demographics. There was a slightly higher percentage of female participants. Median age at time of enrollment was 38 years (IQR 32-42 years). Twenty-eight (87.5%) of 32 patients had either genetically confirmed von Hippel-Lindau disease or a strong family history and a clinical diagnosis of von Hippel-Lindau disease. Four (12.5%) of 32 patients with negative germline VHL gene testing and with multiple central nervous system or retinal hemangioblastomas were included in the trial. Four (12.5%) of 32 patients had been previously treated on a trial testing sunitinib in von Hippel-Lindau disease patients (17).
Figure 1:
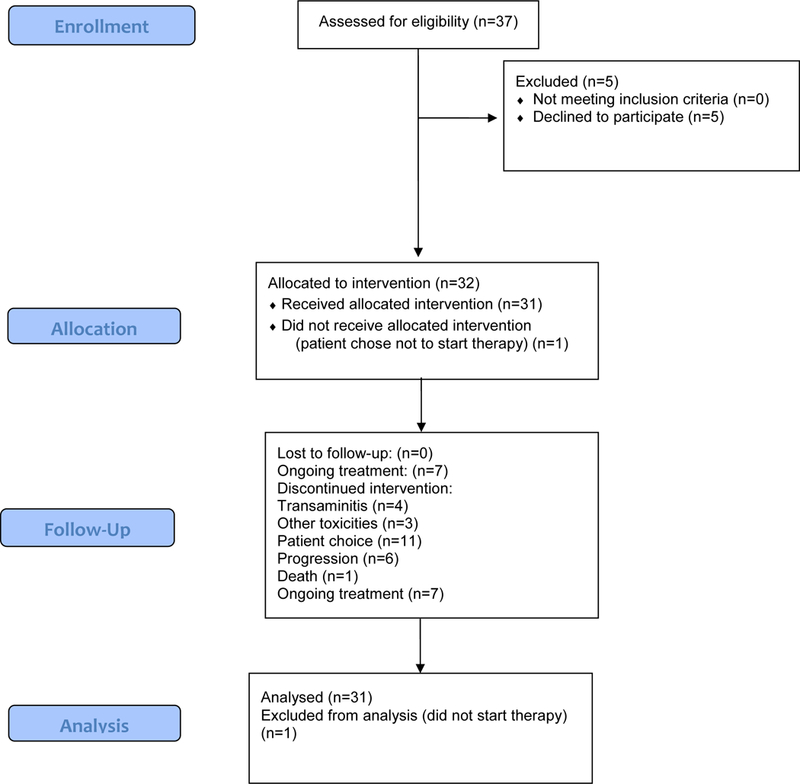
CONSORT diagram
Table 1:
Patient Demographics
| Characteristic | N (% unless specified) |
|---|---|
| Total Patient Number | 32 |
| Gender | |
| Female | 18 (56) |
| Male | 14 (44) |
| Median age | 38 (IQR 32-42) |
| VHL Status | |
| Confirmed VHL mutation | 24 (75) |
| Patient not tested but positive family history | 4 (13) |
| Tested negative for VHL mutation but had clinical features of von Hippel-Lindau disease | 4 (13) |
| Lesions (Any Per Patient) | |
| Central nervous system | 23 (72) |
| Kidney | 22 (69) |
| Pancreas | 9 (28) |
| Eye | 3 (9) |
| Adrenal | 1 (3) |
| Number of Organ Sites Involved (Per Patient) | |
| 1 | 11 (34) |
| 2 | 16 (50) |
| 3 | 5 (16) |
Efficacy:
The 31 evaluable patients received a median of six cycles of therapy (range 1-36). Median follow up was 12 months (IQR 7, 32 months). Median time to RECIST response was 3 months (IQR 3, 7 months) for renal cell carcinoma, six months (IQR 4.5, 13 months) for hemangioblastomas and six months (IQR 4, 9 months) for pancreatic lesions. Thirteen (42%) of 31 patients demonstrated overall PR to therapy, and the remaining 18 (58%) of 31 patients exhibited SD as best response (Table 2). Assessment by organ site revealed 31 (52%) of 59 target renal cell carcinomas responded, including a 2 (3%) out of 59 complete responses (CRs). Nine (53%) of 17 pancreatic lesions, which were predominantly serous cystadenomas, responded, with no CRs. Two (4%) out of 49 hemangioblastomas responded, with no CRs. The median percentage shrinkage in the renal lesions was 40.5 percent (IQR 21, 53 percent), in pancreatic lesions 30.5 percent (IQR 18, 36 percent) and in hemangioblastomas 13 percent (IQR 7, 23 percent). One patient with retinal hemangioblastomas and deteriorating vision continued on treatment for over two years with stabilization of visual acuity. Six (19%) of 32 patients eventually came off study for progressive disease (Table 4). A swimmer’s plot (Figure 2) summarizes overall duration of therapy for each patient and response status by organ site. Response by lesion is summarized in waterfall plots (Figure 3). No patient developed new lesions in any organ site while on therapy, and no patients developed metastatic disease from renal cell carcinoma. Secondary endpoints of the study, including rate of growth over time in target lesions before and after pazopanib treatment; the need for surgical intervention over time in patients who receive pazopanib and compared with the rate before receipt of the drug; and the creation of the annotated tissue resource from patients with von Hippel-Lindau disease will be reported on in future manuscripts.
Table 2:
Efficacy
| Overall Response Rate | Number of patients (%) |
|---|---|
| Total Evaluable | 31 (100) |
| CR | 0 |
| PR | 13 (42) |
| SD | 18 (58) |
| PD | 0 |
| Inevaluable | 1 |
| Response by Organ Site | Number of sites (%) | |||
| Renal | Central System | Nervous | Pancreas | |
| #Target Lesions | 59(100) | 49(100) | 17(100) | |
| CR (%) | 2 (3) | 0 | 0 | |
| PR (%) | 29 (49) | 2 (4) | 9 (53) | |
| SD (%) | 28 (47) | 47 (96) | 8 (47) | |
| PD (%) | 0 | 0 | 0 | |
Table 4:
Dosing and Reasons for Treatment Discontinuation
| Number of Patients (%) | |
|---|---|
| Total Evaluable | 31 (100) |
| 800mg | 13 (42) |
| 600mg | 12 (39) |
| 400mg | 6 (19) |
| Reason off therapy | 32 (100) |
| Transaminitis | 4 (13) |
| Other toxicities | 3 (10) |
| Progression | 6 (20) |
| Patient choice | 11 (35) |
| Ongoing treatment | 7 (32) |
| Death | 1 (3) |
Figure 2: Swimmer’s plot broken down by major organ site.
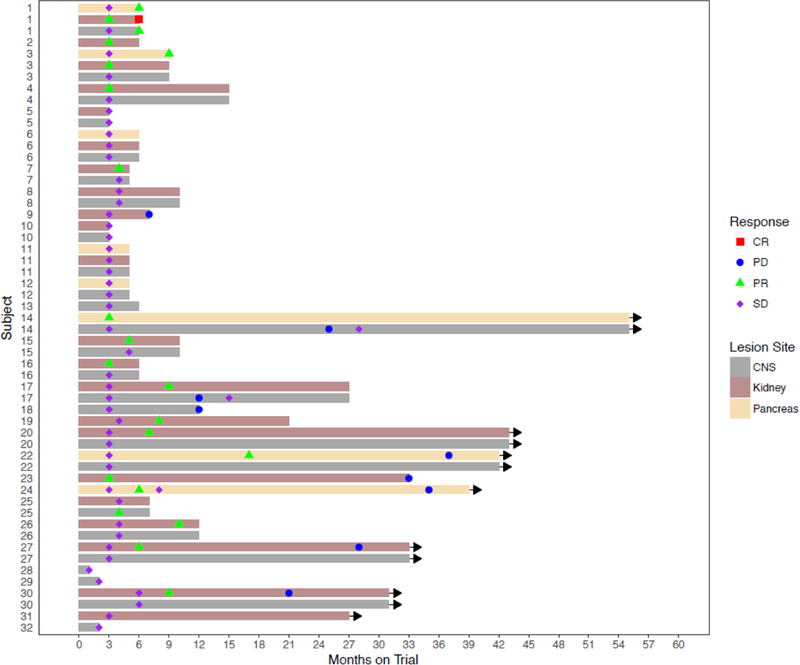
Plot showing duration of treatment by patient, type of lesion per patient, and time response occurred in lesions. CR=complete response; PD=progressive disease; PR=partial response, SD=stable disease, CNS=central nervous system.
Figure 3: Waterfall plots of response by lesion type.
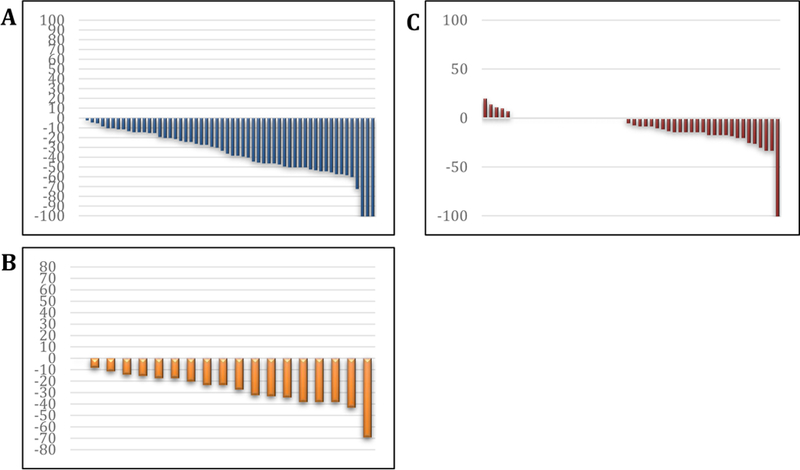
Waterfall plots demonstrating response in A. renal cell carcinomas, B. pancreatic lesions and C. hemangioblastomas. Lines in X-axis represent individual lesions, and Y-axis shows percentage growth (positive numbers) or decrease (negative numbers) in percent.
Toxicity and dose adjustments:
Side effects experienced in at least 10 percent of patients are summarized in Table 3. Patients experienced fatigue, diarrhea or transaminitis at rates comparable to prior studies with pazopanib. Four (13%) of 31 patients discontinued treatment due to grade 3 or greater transaminitis (Table 2). Three (10%) of 31 additional patients discontinued treatment due to general intolerance to pazopanib, with a collection of grade 2 toxicities (Grade 2 back pain; Grade 2 diarrhea and fatigue; grade 2 abdominal pain, diarrhea, and fatigue) that were considered unacceptable to the patients, resulting in study withdrawal. Treatment related serious adverse events included one case each of appendicitis and gastritis. A number of patients who responded came off study at the six-month mark, as illustrated in Figure 2. They stopped for a variety of reasons, including logistics (needing to come from a distance, cost of travel, etc.), a lack of imperative to remain on study (duration of treatment was not an endpoint and patients were free to stop after six months, and patients were generally not in an immediate need of surgical intervention), and a decrement in quality of life (even if grade 3 toxicities were not experienced, patients did experience toxicities typical of pazopanib which affected quality of life). One patient with advanced, inoperable hemangioblastomas sustained a fall and head trauma while on treatment, with hemorrhage of a central nervous system hemangioblastoma, ultimately resulting in patient death. An additional patient who demonstrated progression in a central nervous system hemangioblastomas after two years of treatment also developed a central nervous system bleed after treatment discontinuation while awaiting surgical intervention. The patient underwent resection, followed by full recovery. No additional major toxicities were observed in this patient population. With 31 patients accrued, seven treatment discontinuations for side effects triggered the predetermined toxicity stopping rule. Due to the slowing accrual rate, and as per recommendations by the statistician and by the departmental and institutional protocol offices, we did not pursue a protocol modification for the toxicity stopping rule, and closed the study to permit efficacy analysis.
Table 3:
Side effects observed in at least 10 percent of patients and all grade 3-5 events while on study (N=31)
| Adverse Event | Grade 1-2 | Grade 3 | Grade 4 | Grade 5 |
|---|---|---|---|---|
| Diarrhea | 23 | |||
| Fatigue | 21 | 1 | ||
| Aspartate aminotransferase increased | 17 | 3 | ||
| Alanine aminotransferase increased | 15 | 3 | 1 | |
| Skin hyperpigmentation | 18 | |||
| Hypertension | 14 | |||
| Nausea | 12 | |||
| Dysgeusia | 10 | |||
| Proteinuria | 8 | 1 | ||
| Mucositis | 8 | |||
| Leukopenia | 8 | |||
| Alopecia | 8 | |||
| Vomiting | 7 | |||
| Platelet count decreased | 6 | |||
| Hair pigment change | 6 | |||
| Rash | 6 | |||
| Anemia | 5 | |||
| Alkaline phosphatase increased | 4 | |||
| Creatinine increased | 4 | |||
| Depression | 4 | |||
| Epistaxis | 4 | |||
| Hyperglycemia | 4 | |||
| Hyperthyroidism | 4 | |||
| Hypoalbuminemia | 4 | |||
| Hypernatremia | 3 | |||
| Hypocalcemia | 3 | |||
| Hypokalemia | 3 | |||
| Hyponatremia | 2 | 1 | ||
| Appendicitis | 1 | |||
| Gastritis | 1 | |||
| GGT increased | 1 | |||
| Peripheral Motor Neuropathy | 1 | |||
| Nervous system disorders - Other | 1 |
Ten (32%) of 31 patients remained on 800mg pazopanib. Twelve (39%) of 31 patients dose reduced to 600mg PO daily and six (19%) of 31 patients to 400mg PO daily (Figure 3). The remaining patients came off study early without dose reduction due to patient preference/therapy intolerance.
Association between genomic status and response:
Four (13%) of 31 evaluable patients did not have germline VHL mutations but showed clinical features of von Hippel-Lindau disease including multiple central nervous system and/or retinal hemangioblastomas. All demonstrated SD as best response, with no PRs. There was no association between response and the presence or absence of a positive VHL mutation (p=0.37) in central nervous system lesions, likely due to the small sample size. In addition, there was no association between mutational type (point mutation vs deletion) and response in renal cell carcinomas, pancreatic lesions or hemangioblastomas in individuals with known VHL mutations (p=1.0) (See Web Appendix, Supplemental Table 1 for data on mutational status). In a post-hoc exploratory analysis, when patients who tested negative for von Hippel-Lindau disease were excluded from analysis, the ORR for central nervous system lesions in the 27 remaining patients increased to 5 percent, and ORR remained the same for renal cell carcinomas and pancreatic lesions. (See Web appendix, Supplemental Table 2).
Discussion:
This study demonstrates that pazopanib can induce a significant reduction of disease burden in von Hippel-Lindau disease patients. Efficacy data from this study indicate a clear clinical benefit in individuals with renal cell carcinomas and pancreatic lesions, and some potential efficacy signals in hemangioblastomas as well. Renal cell carcinoma specific response exceeded 50 percent, and response appeared to be durable in patients who chose to remain on study. We did not formally categorize lesions as being resectable versus nonresectable, but response in either situation would be potentially beneficial. Although duration of therapy was not one of the formal endpoints of the study, it is of inherent interest to patients and treating physicians whether treatment effect is longstanding. A number of individuals remain on study with no evidence of tumor regrowth in renal cell carcinomas (Figure 2). Others have shown slow regrowth after initial shrinkage, and ultimately underwent surgical resection. Still others chose to go on observation after a six month treatment period, due to the modest but persistent decrement in quality of life they experienced. Patient 1 on the swimmer’s plot is a case in point. Despite showing a CR in a renal cell carcinoma, the patient experienced grade 1 and 2 side effects from pazopanib they did not want to deal with on an ongoing basis. The patient discontinued therapy at the predetermined six-month mark and was observed. They experienced slow regrowth of a renal mass, and underwent a partial nephrectomy three years later. This patient is a good example of a person who could have benefitted from studies permitting intermittent treatment, which was not possible with the study as it was written. Future studies are needed to measure duration of response (with provisions to allow breaks in therapy and re-initiation of therapy if needed).
Similar benefit was seen in pancreatic serous cystadenomas. While these lesions are not cancerous per se, involvement of the pancreas can be massive, and ultimately can impact both exocrine and endocrine pancreatic function. No surgical solution exists for this disease manifestation beyond pancreatic resection. Several individuals experienced dramatic reduction of their pancreatic serous cystadenomas with pazopanib, with continued benefit after several years of treatment. The PR rate for hemangioblastomas was low, but was higher than that observed in the previously published sunitinib study (17). Although our data suggest activity of pazopanib and stabilization of central nervous system lesions, it is difficult to draw firm conclusions as these lesions can be stable for long periods of time without therapy, and the duration of follow-up is not long enough to be certain that the treatment is changing the course of the lesions. Longitudinal assessment of lesion growth prior to, and after participation in the clinical trial is part of an ongoing analysis. The waterfall plot in Figure 3 suggest that a salutary effect was achieved in hemangioblastomas. Due to the small size of some of these lesions, the observed responses may be due varying contrast timing and technique. However, directionality of response was fairly consistent within individual lesions, and it is unlikely that contrast variability influenced measurement of tumor diameter as the outer perimeters of renal cell carcinomas and hemangioblastomas are relatively discrete on CTs and MRIs due to their inherent vascularity. Several individuals with extensive hemangioblastomas have remained on treatment for years, with no evidence of tumor regrowth. One individual with declining vision from retinal hemangioblastomas experienced stabilization of visual acuity on pazopanib, and remained on treatment for several years. A second individual with retinal hemangioblastomas also reported visual stabilization while on study. Overall the impact of pazopanib on the ophthalmological manifestations of von Hippel-Lindau disease are hard to assess due to the small number of patients with retinal lesions, but a potential efficacy signal exists.
One of the concerns that arose with the treatment of hemangioblastomas and other vascular lesions with agents that may disrupt vascular integrity was the potential for catastrophic intracranial or intraspinal hemorrhage. Two individuals with hemangioblastomas did experience hemorrhage while on or immediately after discontinuing pazopanib. The first individual had extensive, refractory hemangioblastomas, which were not deemed to be surgically resectable and experienced trauma while on treatment, leading to a bleed. Nonetheless we have to consider pazopanib as a potential contributor to the hemorrhagic event. The second individual experienced an initial non- RECIST shrinkage of hemangioblastomas, followed by slow regrowth. He then experienced bleeding in a hemangioblastoma immediately prior to undergoing surgery. The patient recovered well after surgery. It is possible that pazopanib contributed to vascular friability in this case. The remaining individuals have not experienced bleeding, nor are there reports in the literature of similar events, despite the reported use of pazopanib in patients with hemangioblastomas (19, 26, 27). Additional pazopanib related toxicities were observed in this study, with grade 3-4 transaminitis requiring treatment discontinuation in four patients (Table 3). Despite not seeing a high rate grade 3 hypertension in this group of individuals, the overall pattern of toxicity suggested that dosing was appropriate, and that dose escalation would not have been feasible. Some patients also reported general intolerance to pazopanib, and several chose to discontinue study drug after a short treatment interval without attempting dose reduction. The rate of treatment discontinuation was similar to that seen in large phase III studies (21). These toxicity readouts provide a cautionary note and need to be weighed against the potential benefits of pazopanib therapy, as well as the risks and benefits of alternate treatments, including surgery, thermal ablation, and radiation therapy. Since many patients enrolled on the study had the option to revert to observation, and did not have the immediate threat of shortened survival as would be the case in metastatic disease, the incentive to remain on an agent that caused side effects was lower than in other settings. Despite that, a number of patients chose to remain on treatment for prolonged periods, suggesting that a reasonable balance between toxicity and efficacy can be achieved with this agent in some patients.
The ultimate goal of systemic therapy is to decrease the rate of surgical intervention in patients with von Hippel-Lindau disease by manipulating the underlying molecular mechanism that affects multi-organ manifestations. One of the major challenges in assessing the efficacy of systemic therapy in this patient population is precisely defining rates of von Hippel-Lindau disease-related lesion growth and the baseline frequency of intervention required in patients with this disease. Some data are available to help provide context. A large, prospective study followed the natural history of hemangioblastomas in 225 patients with von Hippel-Lindau disease(28). After a mean follow-up of 7 years, 72% developed new lesions. Forty nine percent of existing hemangioblastomas demonstrated heterogeneous growth rates, with cerebellar and brainstem lesions growing faster than those in the spinal cord or cauda equina. Increased tumor burden or total tumor number detected was associated with male sex, longer follow-up, and genotype. Similar natural history data are available for renal cell carcinomas (6, 29). Future trials can be designed using either ORR or intervention rate as compared to historical or intra-patient controls as primary efficacy criteria.
This study was stopped before the originally planned 40 patient accrual was reached. Two factors led to this decision. The first was that accrual was slowing considerably, and the trial had been open for three years. The second was that the trial met a prespecified toxicity stopping rule after 30 patients were accrued. The assessment of other trials using pazopanib indicated this rule was overly stringent as treatment discontinuation rates of 24 percent are reported(21), and would have been revised upward to keep the study open for accrual. To ensure timely dissemination of data, the decision was made to close the trial after 31 evaluable patients were accrued. Additional study limitations include a non-randomized study design and a small sample size.
In conclusion, pazopanib resulted in significant response rates in renal cell carcinomas and pancreatic serous cystadenomas, and showed potential signs of efficacy in central nervous system and retinal hemangioblastomas, with a side effect profile consistent with that seen in other treatment settings. Pazopanib could be considered in patients with von Hippel-Lindau disease and growing lesions where surgical resection may be required in the relatively near future, or in patients with unresectable lesions where decrease in tumor size is desired.
Supplementary Material
Acknowledgements:
The authors would like thank Muammer Altok, Marcia Holloway, Jacqueline Rivas, Savannah Lothringer and Tanisa Johnson for patient management and/or technical assistance with this manuscript. Funding was provided by Novartis Inc. and the NIH/NCI Core Grant under award number P30CA016672-39
Funding: Novartis Inc; NIH/NCI Core Grant under award number P30CA016672-39
Evidence before this study
A PubMed search was performed on March 11, 2018 between January 1980 and March 2018 to identify studies with search terms VHL, von Hippel-Lindau disease, therapy, antiangiogenic therapy, prospective trial, treatment, and surgery. Only one prospective, 15 patient study using systemic therapy in von Hippel-Lindau disease patients was identified. Multiple larger retrospective studies on the natural history and/or resection of von Hippel-Lindau related lesions had been published. We concluded that a major unmet need existed for larger prospective therapeutic studies in von Hippel-Lindau disease using systemic therapy to control von HippelLindau disease related lesions.
Added value of this study
The current study is the largest therapeutic trial to date testing a systemic therapy in patients with von Hippel-Lindau disease, and demonstrates clear efficacy of this agent in multiple von Hippel-Lindau disease manifestations. Pazopanib treatment resulted in significant shrinkage of renal cell carcinoma and pancreatic lesions, as well as stabilization of hemangioblastomas. Side effects of pazopanib in von HippelLindau disease patients were largely consistent with those seen in phase III clinical trials. These data show that pazopanib therapy is a realistic option for patients with von Hippel Lindau disease.
Implications of all the available evidence
The data from this study are potentially practice changing for patients with von Hippel-Lindau disease as they provide an alternative approach to surgical intervention for individuals with growing von Hippel-Lindau disease related lesions.
Declaration of Interest:
Eric Jonasch has competing interest: Research Support and Honoraria from Novartis
All other authors of this manuscript declare no competing interest
Footnotes
Research in context
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Eric Jonasch, Departments of Genitourinary Medical Oncology, UT MD Anderson Cancer Center, Houston, TX, USA.
Ian E. McCutcheon, Neurosurgery, UT MD Anderson Cancer Center, Houston, TX, USA
Dan S. Gombos, Ophthalmology, UT MD Anderson Cancer Center, Houston, TX, USA
Kamran Ahrar, Interventional Radiology, UT MD Anderson Cancer Center, Houston, TX, USA.
Nancy D. Perrier, Surgical Oncology, UT MD Anderson Cancer Center, Houston, TX, USA
Diane Liu, Biostatistics, UT MD Anderson Cancer Center, Houston, TX, USA.
Christine C. Robichaux, Departments of Genitourinary Medical Oncology, UT MD Anderson Cancer Center, Houston, TX, USA
Mercedes F. Villarreal, Departments of Genitourinary Medical Oncology, UT MD Anderson Cancer Center, Houston, TX, USA
Justin A. Weldon, Departments of Genitourinary Medical Oncology, UT MD Anderson Cancer Center, Houston, TX, USA
Ashley H. Woodson, 6, Clinical Cancer Genetics, UT MD Anderson Cancer Center, Houston, TX, USA
Patrick G. Pilie, Departments of Genitourinary Medical Oncology, UT MD Anderson Cancer Center, Houston, TX, USA
Gregory N. Fuller, Pathology, UT MD Anderson Cancer Center, Houston, TX, USA
Steven G. Waguespack, Endocrinology, UT MD Anderson Cancer Center, Houston, TX, USA
Surena F. Matin, Urology, UT MD Anderson Cancer Center, Houston, TX, USA
References:
- 1.Ho TH, Jonasch E. Genetic kidney cancer syndromes. Journal of the National Comprehensive Cancer Network : JNCCN. 2014;12(9):1347–55. [DOI] [PubMed] [Google Scholar]
- 2.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):131720. [DOI] [PubMed] [Google Scholar]
- 3.Iliopoulos O, Ohh M, Kaelin WG, Jr. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A. 1998;95(20):11661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–5. [DOI] [PubMed] [Google Scholar]
- 5.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet. 2003;361(9374):2059–67. [DOI] [PubMed] [Google Scholar]
- 6.Choyke PL, Glenn GM, Walther MM, Zbar B, Weiss GH, Alexander RB, et al. The natural history of renal lesions in von Hippel-Lindau disease: a serial CT study in 28 patients. AJR Am J Roentgenol. 1992;159(6):1229–34. [DOI] [PubMed] [Google Scholar]
- 7.Ach T, Hoeh AE, Ruppenstein M, Kretz FT, Dithmar S. Intravitreal bevacizumab in vascular pigment epithelium detachment as a result of subfoveal occult choroidal neovascularization in age-related macular degeneration. Retina. 2010;30(9):1420–5. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal A, Kumari N, Singh R. Intravitreal bevacizumab and feeder vessel laser treatment for a posteriorly located retinal capillary hemangioma. Int Ophthalmol. 2016;36(5):747–50. [DOI] [PubMed] [Google Scholar]
- 9.Hrisomalos FN, Maturi RK, Pata V. Long-term use of intravitreal bevacizumab (avastin) for the treatment of von hippel-lindau associated retinal hemangioblastomas. The open ophthalmology journal. 2010;4:66–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slim E, Antoun J, Kourie HR, Schakkal A, Cherfan G. Intravitreal bevacizumab for retinal capillary hemangioblastoma: A case series and literature review. Can J Ophthalmol. 2014;49(5):450–7. [DOI] [PubMed] [Google Scholar]
- 11.Wong WT, Liang KJ, Hammel K, Coleman HR, Chew EY. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von HippelLindau disease. Ophthalmology. 2008;115(11):1957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wackernagel W, Lackner EM, Pilz S, Mayer C, Stepan V. von Hippel-Lindau disease: treatment of retinal haemangioblastomas by targeted therapy with systemic bevacizumab. Acta Ophthalmol. 2010;88(7):e271–2. [DOI] [PubMed] [Google Scholar]
- 13.Knickelbein JE, Jacobs-El N, Wong WT, Wiley HE, Cukras CA, Meyerle CB, et al. Systemic Sunitinib Malate Treatment for Advanced Juxtapapillary Retinal Hemangioblastomas Associated with von Hippel-Lindau Disease. Ophthalmol Retina. 2017;1(3):181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oudard S, Elaidi R, Brizard M, Le Rest C, Caillet V, Deveaux S, et al. Sunitinib for the treatment of benign and malignant neoplasms from von Hippel-Lindau disease: A single-arm, prospective phase II clinical study from the PREDIR group. Oncotarget. 2016;7(51):85306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi A, Takahashi M, Imai H, Akiyama S, Sugiyama S, Komine K, et al. Attainment of a Long-term Favorable Outcome by Sunitinib Treatment for Pancreatic Neuroendocrine Tumor and Renal Cell Carcinoma Associated with von Hippel-Lindau Disease. Intern Med. 2016;55(6):629–34. [DOI] [PubMed] [Google Scholar]
- 16.Jimenez C, Cabanillas ME, Santarpia L, Jonasch E, Kyle KL, Lano EA, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von HippelLindau disease-related tumors. J Clin Endocrinol Metab. 2009;94(2):386–91. [DOI] [PubMed] [Google Scholar]
- 17.Jonasch E, McCutcheon IE, Waguespack SG, Wen S, Davis DW, Smith LA, et al. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann Oncol. 2011;22(12):2661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roma A, Maruzzo M, Basso U, Brunello A, Zamarchi R, Bezzon E, et al. FirstLine sunitinib in patients with renal cell carcinoma (RCC) in von Hippel-Lindau (VHL) disease: clinical outcome and patterns of radiological response. Fam Cancer. 2015;14(2):309–16. [DOI] [PubMed] [Google Scholar]
- 19.Kim BY, Jonasch E, McCutcheon IE. Pazopanib therapy for cerebellar hemangioblastomas in von Hippel-Lindau disease: case report. Target Oncol. 2012;7(2):145–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28(6):1061–8. [DOI] [PubMed] [Google Scholar]
- 21.Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369(8):72231. [DOI] [PubMed] [Google Scholar]
- 22.Hutson TE, Davis ID, Machiels JP, De Souza PL, Rottey S, Hong BF, et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2010;28(3):475–80. [DOI] [PubMed] [Google Scholar]
- 23.Choueiri TK, Fay AP, Gagnon R, Lin Y, Bahamon B, Brown V, et al. The role of aberrant VHL/HIF pathway elements in predicting clinical outcome to pazopanib therapy in patients with metastatic clear-cell renal cell carcinoma. Clin Cancer Res. 2013;19(18):5218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powles T, Sarwar N, Stockdale A, Sarker SJ, Boleti E, Protheroe A, et al. Safety and Efficacy of Pazopanib Therapy Prior to Planned Nephrectomy in Metastatic Clear Cell Renal Cancer. JAMA Oncol. 2016;2(10):1303–9. [DOI] [PubMed] [Google Scholar]
- 25.Rini BI, Plimack ER, Takagi T, Elson P, Wood LS, Dreicer R, et al. A Phase II Study of Pazopanib in Patients with Localized Renal Cell Carcinoma to Optimize Preservation of Renal Parenchyma. J Urol. 2015;194(2):297–303. [DOI] [PubMed] [Google Scholar]
- 26.Taylor DG, Ilyas A, Mehta GU, Chen CJ, Schiff D, Oldfield EH, et al. Variable response of CNS hemangioblastomas to Pazopanib in a single patient with von Hippel-Lindau disease: Case report. J Clin Neurosci. 2018. [DOI] [PubMed] [Google Scholar]
- 27.Migliorini D, Haller S, Merkler D, Pugliesi-Rinaldi A, Koka A, Schaller K, et al. Recurrent multiple CNS hemangioblastomas with VHL disease treated with pazopanib: a case report and literature review. CNS Oncol. 2015;4(6):387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huntoon K, Wu T, Elder JB, Butman JA, Chew EY, Linehan WM, et al. Biological and clinical impact of hemangioblastoma-associated peritumoral cysts in von Hippel-Lindau disease. J Neurosurg. 2016;124(4):971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walther MM, Choyke PL, Glenn G, Lyne JC, Rayford W, Venzon D, et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999;161(5):1475–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.