

Abstract
Co-assembled peptide amphiphile nanofibers designed to target atherosclerotic plaque and enhance cholesterol efflux are shown to encapsulate and deliver a liver X receptor agonist to increase efflux from murine macrophages in vitro. Fluorescence microscopy reveals that the nanofibers, which display an apolipoprotein-mimetic peptide, localize at plaque sites in LDL receptor knockout mice with or without the encapsulated molecule, while nanofibers displaying a scrambled, non-targeting peptide sequence do not demonstrate comparable binding. These results show that nanofibers functionalized with apolipoprotein-mimetic peptides may be effective vehicles for intravascular targeted drug delivery to treat atherosclerosis.
Keywords: peptides, supramolecular chemistry, self-assembly, atherosclerosis
Table of Contents Entry
Self-assembled peptide amphiphile nanofibers incorporating an apolipoprotein A1-mimetic peptide are designed as vehicles in a targeted drug-delivery platform for atherosclerosis. These nanofibers encapsulate hydrophobic LXR agonist molecules and effectively deliver them to cholesterol-laden mouse macrophages to increase cholesterol efflux. Fluorescence microscopy reveals that these nanofibers successfully localized to plaque sites in atherosclerotic mice.
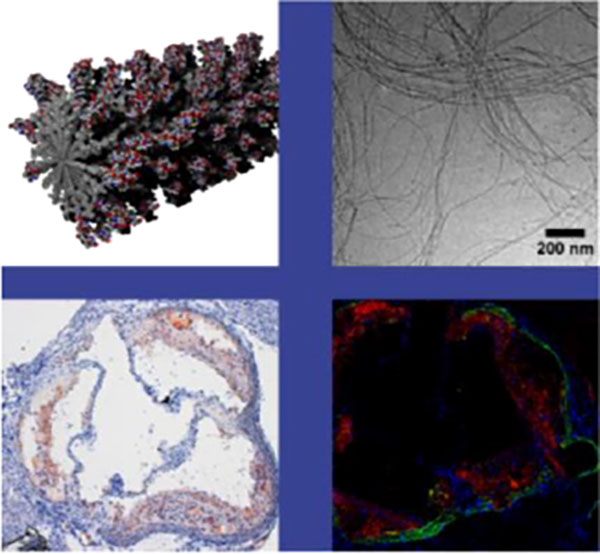
Communication Text
Atherosclerosis is a chronic systemic disease of the vasculature characterized by impaired lipid metabolism and associated inflammatory responses, leading to stenotic or occluded arteries and tissue ischemia.[1] Current interventions include pharmacological treatments to optimize the body’s lipid profile and procedures to restore normal blood flow impeded by arterial stenosis or occlusion. HMG-CoA reductase inhibitors (i.e., statins) are the most commonly prescribed lipid regulators, as they effectively increase LDL catabolism.[2] However, there is still a recurrence rate of over 20% within 30 months of an acute coronary syndrome in patients on statin therapy.[3,4] Balloon angioplasty and stenting, surgical endarterectomy, or bypass grafting may be required when arterial stenosis or occlusion becomes severe, but restenosis or re-occlusion often occurs post-intervention due to neointimal hyperplasia.[5] Hence, there is great need for alternative therapeutic strategies, and targeted nanoparticle-based drug delivery is a promising solution. In particular, our laboratories found that collagen IV-targeted self-assembling peptide amphiphile (PA) nanofibers successfully localize to injured arterial walls, while nanospheres of similar diameter and with the same targeting group do not, suggesting that PA nanofibers are a viable platform for targeting along the vasculature.[6] Nanofibers self-assemble from PA molecules comprising a hydrophobic alkyl tail, a β-sheet region involving amino acids with high propensities to form β-sheets, a charged region composed of acidic or basic amino acids, and an epitope for specific biological activity.[7] PA nanofibers are of particular interest here because of their structural and functional modularity. Furthermore, PAs can be co-assembled with diluent PAs (without a bioactive epitope) as an approach to control the density of biological signals[8] and therefore attain enhanced bioactivity in cases where steric effects play an important role in signaling.[9] This ability to vary the chemical and physical properties of PA supramolecular nanostructures has enabled their design to address diverse medical needs, including treating spinal cord injury,[10,11] inducing angiogenesis,[12–14] drug delivery,[15] regenerating hard or connective tissue,[16–19] inhibiting tumor growth,[20,21] and stopping hemorrhage,[22] amongst other applications.
In this work, we explore the use of a nanofiber-forming PA co-assembly that displays an apolipoprotein A1 (ApoA1) mimetic peptide known as “4F”[23] as the bioactive epitope to target the nanofiber to areas of atherosclerotic plaque. ApoA1 is the primary protein component of high-density lipoprotein (HDL) particles, and its amphipathic α-helices are instrumental in lipid binding, transport, and metabolism.[24] By displaying an ApoA1 mimetic peptide on the nanofiber, we take advantage of the peptide’s lipid-binding capabilities. We then investigate drug encapsulation using co-assembled PA to deliver the LXR agonist GW3965 to mouse macrophages. LXR agonists increase the expression of cholesterol efflux-associated genes; however, in a previous study in which atherosclerosis-prone apolipoprotein E knockout mice were fed GW3965 with a high-fat diet, a reduced lesion area was accompanied by elevated plasma triglyceride levels compared with controls,[25] indicating that GW3965 may benefit from a plaque-targeted delivery vehicle for optimal anti-atherosclerotic activity. Herein, we report on the characterization of PA nanostructures that encapsulate LXR agonist and their effect on cholesterol efflux from macrophages in vitro. We then demonstrate PA targeting of atherosclerotic plaques in an in vivo low density lipoprotein receptor knockout (LDLR KO) mouse model.
The targeting PA (ApoA1 PA) molecule was designed with a C16 aliphatic tail, the β-sheet-forming sequence V2A2, negatively-charged E2 amino acid residues, a single glycine residue as a spacer, and the 4F ApoA1 mimetic sequence (DWFKAFYDKVAEKFKEAF-NH2) displayed as the epitope (Figure 1a). The two glutamic acid residues were incorporated to render overall negative charge to the PA, since negatively charged particles are typically more soluble in physiologic solutions and are more likely to be taken up by macrophages.[26] Furthermore, previous work from the Stupp laboratory found that PA supramolecular nanostructures with high positive charge may be cytotoxic,[27] a phenomenon commonly reported with positively charged nanoparticles.[26] The 4F peptide was developed by the Segrest and Ananthamaraiah research groups as a variant of a parent peptide 18A,[23] a synthetic structural mimetic of apolipoprotein α-helices.[28] These α-helices promote high affinity binding of lipids – especially oxidized lipids – to the 4F peptide sequence.[29] Since oxidative modification of LDL has been shown to correlate with atherosclerotic severity, high-affinity binding to oxidized lipids could offer a valuable targeting strategy.[30–32] Hence, we developed the ApoA1 PA as a platform to use oxidized-lipid binding to selectively target atherosclerotic plaques. The ApoA1 PA alone did not form nanofibers, but instead formed aggregated mesh-like networks (Figure S1). It was therefore co-assembled with a diluent PA (C16V2A2E2-NH2) that on its own self-assembles into long nanofibers (complete experimental details can be found in the Supporting Information). Co-assembly was achieved by mixing solutions of the component PAs in HFIP (1,1,1,3,3,3-hexafluoro-2-propanol), a peptide-disaggregating solvent later removed by evaporation. After co-assembly, nanofibers were observed by cryogenic-transmission electron microscopy (cryo-TEM) (Figure 1b). From a series of co-assembled structures characterized by TEM, the 40 mol% co-assembly with diluent PA incorporated the highest epitope concentration without compromising the fiber morphology. The PA nanofibers’ high aspect ratio makes their exact lengths difficult to determine. However, the presence of fiber ends visible for example in Figure 1b indicates their discrete nature. While the fibers in this image tend to exceed 1 μm in length, other images taken show individual fibers with shorter end-to-end lengths on the order of 300 nm. Circular dichroism (CD) measurements confirmed that the 4F targeting peptide displayed on the ApoA1 PA maintained the expected α-helical conformation (Figure S2) upon nanofiber assembly (Figure 1c).[33]
Figure 1.

Structure and characterization of ApoA1 PA and 40% ApoA1 PA co-assembled with 60% diluent PA. (a) Chemical structures of ApoA1 PA and diluent PA along with a molecular graphics representation of the co-assembly. (b) Cryo-TEM of 40% ApoA1 PA (1 mM in 1X PBS). (c) CD spectra of 40% and 100% ApoA1 PA (500 μM in 1X PBS).
HFIP was also used to dissolve and encapsulate the LXR agonist GW3965. Since HFIP is highly volatile and each sample was placed under vacuum twice, any residual HFIP would have been negligible in the final PA or PA samples containing encapsulated drug. Addition of base was required for solubilizing the PA co-assembly itself, and since the LXR agonist was supplied as a hydrochloride salt, excess sodium hydroxide was added to neutralize the acid. Upon addition of this excess base to pH 7.5, 40% ApoA1 PA encapsulated LXR agonist in a 1:1 weight ratio (the encapsulation will be referred to as LXR-40% ApoA1 PA). Encapsulation did not impact the ability of the PA to form nanofibers (Figure 2a). CD measurements on the encapsulation showed that the presence of LXR agonist diminished the overall intensity of the CD signal compared with that of PA alone and somewhat reduced the α-helicity of the PA (Figure 2b), suggesting a slight conformational change upon binding. As discussed below, the drug-free and drug-containing PA nanostructures showed similar plaque-binding efficacy in vivo. However, in this context one cannot discount the possibility that conformational changes occur upon lipid binding in vivo, something which is at this time very difficult to establish experimentally.
Figure 2.
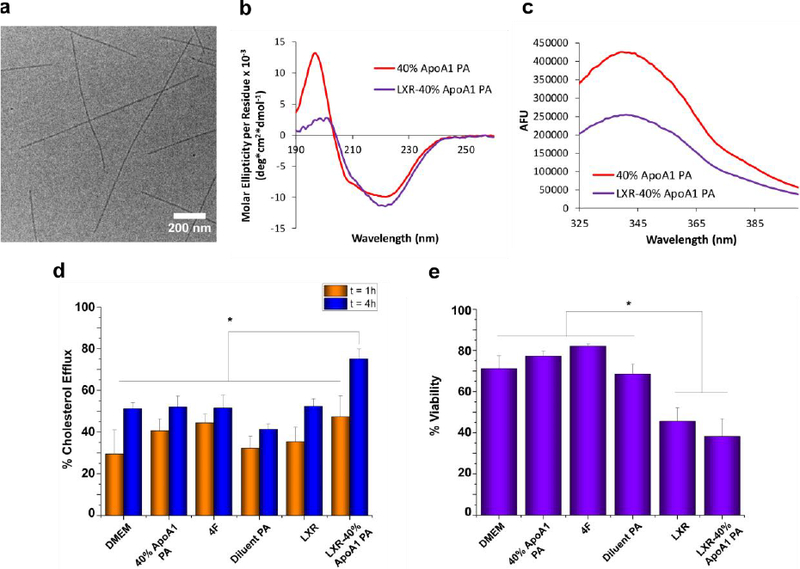
Characterization of LXR-40% ApoA1 PA encapsulation. (a) Cryo-TEM shows formation of nanofibers similar to those of PA without encapsulated LXR agonist. (b) LXR-encapsulation by 40% ApoA1 PA resulted in a more β-sheet-like CD spectrum. (c) Tryptophan fluorescence quenching of PA occurred upon encapsulation of LXR agonist. (d) Neither the PA, 4F, LXR, nor the LXR-PA treatments increased percent cholesterol efflux above the control (DMEM) at t = 1 h, but LXR-40% ApoA1 PA did induce significant (*p < 0.05) efflux above the control, 4F, 40% ApoA1 PA, Diluent PA, and LXR at t = 4 h. (e) Effect of PA, peptide, and LXR treatments upon macrophage cell viability in vitro. * indicates p<0.05 vs. LXR-40% ApoA1 PA and LXR.
Since the ApoA1 mimetic peptide contains a tryptophan residue near its N-terminus, and tryptophan is a highly sensitive solvatochromic fluorophore, we compared the fluorescence emission spectra of 40% ApoA1 PA in the absence and presence of the LXR agonist to determine if its interaction with the PA would result in a change to the emission profile. LXR agonist encapsulation resulted in tryptophan fluorescence quenching (Figure 2c), but did not shift the λmax of emission (338 nm) as typically occurs when tryptophan solvent exposure changes.[34] Tryptophan quenching is commonly used to study ligand–protein interactions, with quenching occurring due to the inner filter effect (significant absorption at the excitation wavelength), collisional quenching, or ligand binding.[35] Absorption values at the excitation wavelength used in this work (295 nm for exclusive excitation of tryptophan residues over tyrosine residues[36]) were almost identical between 40% ApoA1 PA and LXR-40% ApoA1 PA (Figure S3), which means inner filter effects were not contributing significantly to the observed quenching. Hence, collisional quenching or binding-associated interactions, or both, were likely occurring, indicating close interactions between the PA and LXR agonist. Together the CD and fluorescence results suggest that the LXR agonist interacts closely with the α-helices presented on the PA nanofibers. The peptide epitopes are amphiphilic and each has a nonpolar face consisting of most of the peptide’s hydrophobic residues;[37] it is therefore likely that the encapsulation of LXR agonist, a molecule with very low water-solubility, is stabilized by hydrophobic interactions between the agonist and peptide nonpolar residues. Another possible stabilizing interaction is that of pi-pi stacking between the agonist’s aromatic rings and the aromatic side chains on the PA molecules. Both hydrophobic interactions and pi-pi stacking have been used successfully in designing self-assembling nanomaterials with enhanced encapsulation efficiency, drug retention, and controlled release of hydrophobic drugs.[38–41]
Since cholesterol efflux is a key step in reverse cholesterol transport, we investigated the in vitro efflux activity of PA-encapsulated LXR agonist. J774.2 mouse macrophages were loaded with the fluorescent cholesterol analogue NBD-cholesterol for 24 hours, equilibrated for 18 hours with 0.2% bovine serum albumin, and then treated with each condition in triplicate for t = 1 or 4 hours. This was performed in serum-free DMEM culture medium to avoid confounding affects with the lipoproteins present in serum, which could also enhance efflux. After 1 hour, none of the treatment conditions resulted in significantly different cholesterol efflux compared with the DMEM culture medium control (Figure 2d). However, after 4 hours cholesterol efflux was significantly higher in macrophages treated with LXR-40% ApoA1 PA in comparison to DMEM (p=0.0127), 4F (p=0.0142), 40% ApoA1 PA (p=0.0154), diluent PA (p=0.0007), and LXR at 20 μg/mL (p=0.0171). We also examined the effects of two- and three-fold higher concentrations of LXR, but did not see any significant increases in LXR effects upon macrophage efflux (data not shown). These results may be due to limits in LXR solubility within the culture media as the LXR-40% ApoA1 PA at similar concentrations did not show solubility issues and demonstrated a therapeutic effect. In order to analyze the effect of each treatment on cell viability, we carried out a MUSETM Count and Viability experiment (Figure 2e). Although treatment with the LXR-40% ApoA1 PA co-assembly significantly decreased cell viability in comparison to DMEM (p=0.0010), 4F (p<0.0001), ApoA1 PA (p=0.0002), and diluent PA (p=0.0019), there was not a significant difference in comparison to treatment with LXR agonist alone (p=0.3785). It has been found that LXR agonists reduce cell viability in some cell lines, particularly at higher concentrations (e.g. 20 μM; we used a concentration of 20 μg/ml or ~ 32 μM)[42,43] whereas when used at lower concentrations (e.g. 1 μM)[44,45] viability is not affected, suggesting that the observed toxicity may be due to the higher concentration of LXR agonist used in the treatments.
Dialysis experiments with LXR-40% ApoA1 in PBS at 37°C yielded no release of drug from the dialysis chamber over 24 hours (Figure S4). No release was observed for up to one week in PBS (pH 7.4) at room temperature either. However, dialysis with non-encapsulated LXR agonist in PBS with excess base (final pH 8–8.5; necessary for dissolution) showed rapid release over first hour, confirming that GW3965 alone was capable of crossing the dialysis membrane (Figure S4). Cholesterol efflux suggested that macrophages were able to access LXR agonist. The drug molecule may be too hydrophobic to diffuse passively out of the PA nanofiber in PBS, but in the presence of lipid-binding proteins in the cell under physiologic conditions, it may be induced to leave the nanofiber. To investigate this possibility, dialysis with non-encapsulated LXR agonist was repeated in serum-containing culture medium, as serum contains lipoproteins, albumin, and other potential lipid-carriers, and binding between the drug molecule and proteins would prevent LXR agonist from crossing the dialysis membrane. Compared with dialysis in PBS, dialysis in culture medium resulted in both slower initial release and greatly reduced overall release over 24 hours (Figure S4), suggesting binding between LXR agonist and culture medium components. Since “release” of non-encapsulated LXR agonist across the membrane appears to plateau at around 25%, this reduced amount of drug may be enough to enhance efflux. It is also possible that other yet-to-be-discovered mechanisms are responsible for the efficacy of LXR-40% ApoA1 PA in macrophage cholesterol efflux. It is true that the dialysis set-up with PA-containing samples does not mimic the in vivo situation in which proteins and protein-lipid complexes are present. However, due to the nature of the measurements (i.e. analysis of remaining sample post-dialysis via LC-MS), including such complexes would confound our ability to measure the remaining quantity of PA or drug, as their associated m/z peaks could overlap. Regardless, the lack of LXR agonist crossing the dialysis membrane in the LXR-40% ApoA1 PA experiment also provides further confirmation of encapsulation of the drug molecule, since the experiments performed with LXR agonist alone show that any non-encapsulated drug crosses the dialysis membrane readily.
After in vitro characterization of the PA nanofiber, we completed in vivo proof of concept studies in a murine model. The LDLR KO mouse was used for experiments rather than the ApoE KO one because the former model more closely resembles a known human condition (familial hypercholesterolemia)[46] compared to the latter which does not emulate a human disease. Wild type C57/Bl6 mice do not develop atherosclerosis on regular chow and LDLR KO mice fed a high fat diet develop atherosclerosis in the aortic root (Figures 3a-b). Quantification of Oil Red O staining shows that approximately 45% of the aortic root area in the high fat diet-LDLR KO model contained lipid droplets, while less than 1% did in the C57/Bl6 fed regular chow (Figure 3c). LDLR KO mice fed the high fat diet for 14 weeks were administered ApoA1 PA and LXR-40% ApoA1 PA intravenously. Nanofiber solutions were prepared under sterile conditions and injected using surgical techniques. Fluorescent microscopy revealed localization of the nanofibers to areas of atherosclerotic plaque in the aortic root 24 hours after injection. Therefore, 40% ApoA1 PA may be a viable delivery vehicle for in vivo targeted therapy. Furthermore, addition of the LXR agonist GW3965 does not compromise the delivery vehicle’s ability to target atherosclerosis (Figures 3e-f). As a non-targeting control, we designed scrambled ApoA1 PA with identical composition to ApoA1 PA, but displaying a non-helical scrambled 4F sequence.[47] Intravenous injection of scrambled ApoA1 PA does not reveal evidence of nanofiber localization at areas of atherosclerosis 24 hours after intravenous injection in atherosclerotic LDLR KO mice fed the high fat diet for 14 weeks (Figure 3d; quantification of PA binding in Figures 3d-f shown in Figure 3g). Due to the marginalization to the periphery of the vascular lumen conferred by the targeting peptide sequence and nanofiber shape, this serves as a promising platform for in vivo drug delivery and therapy for atherosclerosis in humans. There has been work suggesting that vessel margination occurs for microparticles but not nanoparticles.[48] However, these results are difficult to compare to ours since the polystyrene particles used are very different from PA nanofibers in terms of shape, mechanical properties, and chemistry. In fact, while Eniola et al. found that elongated micron scale rods do not have improved margination compared with spheres of similar volume, in our previous work targeted PA nanofibers localized to the vessel wall whereas targeted PA nanospheres did not as mentioned previously.[6] Mechanistic binding studies and in vivo testing are beyond the scope of this paper but will be required to determine binding constants, pharmacokinetics, extravascular effects, and optimum dose/concentration for safety and efficacy.
Figure 3.
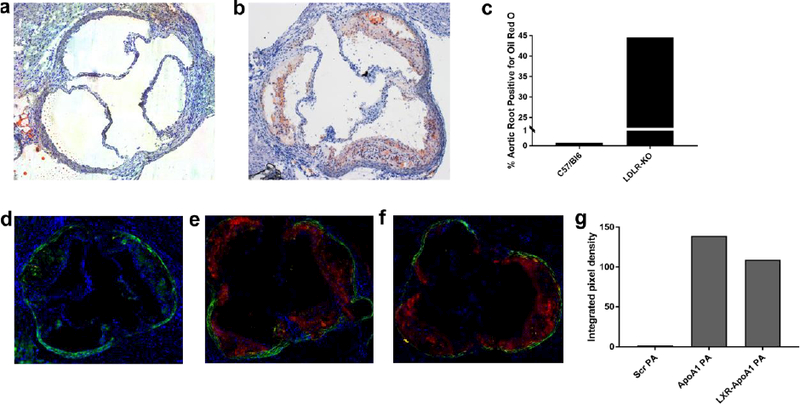
Bright field images of aortic roots of (a) a non-atherogenic wild type C57/Bl6 mice on regular chow stained with H&E and (b) an atherosclerotic LDLR-KO mice fed a high fat diet for 14 weeks stained with H&E and oil-red-O, note lipids/atherosclerosis stained in red. (c) Quantification of Oil Red O staining of aortic root regions in (a) and (b). Fluorescent microscopy of aortic roots of LDLR-KO mouse fed a high fat diet for 14 weeks, injected with (d) non-targeted PA (e) targeted PA (ApoA1 PA), and (f) therapeutic targeted PA (LXR-40% ApoA1 PA); all mice in C-E sacrificed 24 hours after injection. Red fluorescence represents Alexa Fluor 546 and indicates presence of nanofibers. The elastic laminae of the vessel walls are auto fluorescent and were visualized with a green filter. (g) Quantification of PA binding to the aortic root in (d)-(f). Injections were given at a dose of 6 mg/kg PA or 6 mg/kg PA + 6 mg/kg LXR. All images were taken with the 5× objective.
In conclusion, we have synthesized and characterized a targeting PA nanofiber-based drug delivery vehicle that is injected intravenously and reaches atherosclerotic sites with high specificity. Future studies will focus on the pharmacokinetics and therapeutic effect of both the LXR-40% ApoA1 PA and the 40% ApoA1 PA without LXR agonist in vivo. The nanofiber morphology and multivalent presentation of ApoA1-mimetic peptides combine to offer a robust plaque-targeting strategy. Hence, results from our in vitro structural characterization and study on cholesterol efflux from macrophages, together with in vivo targeting of atherosclerotic plaques, all support this PA nanofiber as a promising vehicle for drug delivery and targeted therapy for atherosclerosis.
Supplementary Material
Acknowledgments
This research was supported by funding from the National Institutes of Health (NIH)/NHLBI Bioengineering Research Partnership grant (5R01HL116577), the Northwestern Memorial Foundation Dixon Translational Research Innovation Award and the NUCATS Institute funded, in part, by the National Center for Advancing Translational Sciences (NCATS) of the NIH research grant UL1TR001422 NUCATS. MS was supported by a National Defense Science and Engineering Graduate Fellowship (NDSEG), and NM partially supported by a National Institutes of Health/National Heart, Lung and Blood Institute (NHLBI) Training Grant (2T32HL094293) and the American Medical Association Foundation 2016 Seed Grant. Peptide amphiphile synthesis was performed in the Peptide Synthesis Core Facility of the Simpson Querrey Institute at Northwestern University. The U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University provided funding to develop this facility and ongoing support is being received from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205).
Electron microscopy was performed at the Northwestern University Biological Imaging Facility generously supported by the NU Office for Research.
Circular dichroism and fluorescence experiments were performed in the Keck Biophysics Facility at Northwestern University. Circular dichroism was also performed at the UNC Macromolecular Interactions Facility, supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086.
The authors would like to thank L. Palmer for assistance with the manuscript and M. Seniw for preparation of molecular graphics.
Footnotes
** This is the accepted version of the following article: So, M. M.; Mansukhani, N. A.; Peters, E. B.; Albaghdadi, M. S.; Wang, Z.; Rubert Perez, C. M.; Kibbe, M. R.; Stupp, S. I. “Peptide Amphiphile Nanostructures for Targeting of Atherosclerotic Plaque and Drug Delivery” Advanced Biosystems 2018, 2(3), 1700123 which has been published in final form at [https://onlinelibrary.wiley.com/doi/abs/10.1002/adbi.201700123]. This article may be used for non-commercial purposes in accordance with the Wiley Self-Archiving Policy [https://authorservices.wiley.com/author-resources/Journal-Authors/licensing-open-access/open-access/self-a]
Supporting Information
Supporting Information is available from the Wiley Online Library or from the authors.
Authors MS and NM contributed equally to this work and share first authorship.
Authors MK and SS contributed equally to this work and share senior authorship.
Contributor Information
Miranda M. So, Simpson Querrey Institute for BioNanotechnology, Northwestern University, Chicago, IL 60611, USA Department of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA.
Dr. Neel A. Mansukhani, Simpson Querrey Institute for BioNanotechnology, Northwestern University, Chicago, IL 60611, USA Division of Vascular Surgery, Department of Surgery, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
Dr. Erica B. Peters, Department of Surgery, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Dr. Mazen S. Albaghdadi, Simpson Querrey Institute for BioNanotechnology, Northwestern University, Chicago, IL 60611, USA
Zheng Wang, Division of Vascular Surgery, Department of Surgery, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
Dr. Charles M. Rubert Pérez, Simpson Querrey Institute for BioNanotechnology, Northwestern University, Chicago, IL 60611, USA
Prof. Melina R. Kibbe, Simpson Querrey Institute for BioNanotechnology, Northwestern University, Chicago, IL 60611, USA Division of Vascular Surgery, Department of Surgery, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA; Department of Surgery, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Prof. Samuel I. Stupp, Simpson Querrey Institute for BioNanotechnology, Northwestern University, Chicago, IL 60611, USA Department of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA; Department of Chemistry and Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA; Department of Medicine, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
References
- [1].Weber C, Noels H, Nat. Med 2011, 17, 1410. [DOI] [PubMed] [Google Scholar]
- [2].Maron DJ, Fazio S, Linton MF, Circulation 2000, 101, 207. [DOI] [PubMed] [Google Scholar]
- [3].Libby P, Ridker PM, Hansson GK, Nature 2011, 473, 317. [DOI] [PubMed] [Google Scholar]
- [4].Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM, N. Engl. J. Med 2004, 350, 1495. [DOI] [PubMed] [Google Scholar]
- [5].Jukema JW, Verschuren JJW, Ahmed TAN, Quax PHA, Nat. Rev. Cardiol. 2012, 9, 53. [DOI] [PubMed] [Google Scholar]
- [6].Moyer TJ, Kassam HA, Bahnson ESM, Morgan CE, Tantakitti F, Chew TL, Kibbe MR, Stupp SI, Small 2015, 11, 2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cui H, Webber MJ, Stupp SI, Biopolymers 2010, 94, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI, Biomaterials 2007, 28, 4608. [DOI] [PubMed] [Google Scholar]
- [9].Boekhoven J, Stupp SI, Adv. Mater 2014, 26, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, Stupp SI, Kessler JA, J. Neurosci 2008, 28, 3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tysseling VM, Sahni V, Pashuck ET, Birch D, Hebert A, Czeisler C, Stupp SI, Kessler JA, J. Neurosci. Res 2010, 88, 3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rajangam K, Behanna HA, Hui MJ, Han X, Hulvat JF, Lomasney JW, Stupp SI, Nano Lett. 2006, 6, 2086. [DOI] [PubMed] [Google Scholar]
- [13].Rajangam K, Arnold MS, Rocco MA, Stupp SI, Biomaterials 2008, 29, 3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chow LW, Bitton R, Webber MJ, Carvajal D, Shull KR, Sharma AK, Stupp SI, Biomaterials 2011, 32, 1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Webber MJ, Matson JB, Tamboli VK, Stupp SI, Biomaterials 2012, 33, 6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang Z, Sargeant TD, Hulvat JF, Mata A, Bringas P Jr., Koh CY, Stupp SI, Snead ML, J. Bone Miner. Res 2008, 23, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mata A, Geng Y, Henrikson KJ, Aparicio C, Stock SR, Satcher RL, Stupp SI, Biomaterials 2010, 31, 6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shah RN, Shah NA, Del Rosario Lim MM, Hsieh C, Nuber G, Stupp SI, Proc. Natl. Acad. Sci 2010, 3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee SS, Huang BJ, Kaltz SR, Sur S, Newcomb CJ, Stock SR, Shah RN, Stupp SI, Biomaterials 2013, 34, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Soukasene S, Toft DJ, Moyer TJ, Lu H, Lee H-K, Standley SM, Cryns VL, Stupp SI, ACS Nano 2011, 5, 9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Toft DJ, Moyer TJ, Standley SM, Ruff Y, Ugolkov A, Stupp SI, Cryns VL, ACS Nano 2012, 6, 7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Morgan CE, Dombrowski AW, Rubert Perez CM, Bahnson ES, Tsihlis ND, Jiang W, Jiang Q, Vercammen JM, Prakash VS, Pritts TA, Stupp SI, Kibbe MR, ACS Nano 2016, 10, 899. [DOI] [PubMed] [Google Scholar]
- [23].Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM, J. Lipid Res 2001, 42, 1096. [PubMed] [Google Scholar]
- [24].Gursky O, Atkinson D, Proc. Natl. Acad. Sci 1996, 93, 2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P, Proc. Natl. Acad. Sci 2002, 99, 7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fröhlich E, Int. J. Nanomedicine 2012, 7, 5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Newcomb CJ, Sur S, Ortony JH, Lee O-S, Matson JB, Boekhoven J, Yu JM, Schatz GC, Stupp SI, Nat. Commun 2014, 5, 3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Anantharamaiah GM, Jones JL, Brouillette CG, Schmidt CF, Chung BH, Hughes TA, Bhown AS, Segrest JP, J. Biol. Chem 1985, 260, 10248. [PubMed] [Google Scholar]
- [29].Van Lenten BJ, Wagner AC, Jung C-L, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM, J. Lipid Res 2008, 49, 2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Briley-Saebo KC, Cho YS, Shaw PX, Ryu SK, Mani V, Dickson S, Izadmehr E, Green S, Fayad ZA, Tsimikas S, J. Am. Coll. Cardiol 2011, 57, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Parthasarathy S, Raghavamenon A, Garelnabi MO, Santanam N, Methods Mol. Biol 2010, 610, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ito T, Fujita H, Tani T, Ohte N, Atherosclerosis 2015, 239, 311. [DOI] [PubMed] [Google Scholar]
- [33].Epand RM, Epand RF, Sayer BG, Melacini G, Palgulachari MN, Segrest JP, Anantharamaiah GM, Biochemistry 2004, 43, 5073. [DOI] [PubMed] [Google Scholar]
- [34].Vivian JT, Callis PR, Biophys. J 2001, 80, 2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].van de Weert M, Stella L, J. Mol. Struct 2011, 998, 144. [Google Scholar]
- [36].Epps DE, Raub TJ, Caiolfa V, Chiari A, Zamai M, J. Pharm. Pharmacol 1999, 51, 41. [DOI] [PubMed] [Google Scholar]
- [37].Datta G, Epand RF, Epand RM, Chaddha M, Kirksey MA, Garber DW, Lund-Katz S, Phillips MC, Hama S, Navab M, Fogelman AM, Palgunachari MN, Segrest JP, Anantharamaiah GM, J. Biol. Chem 2004, 279, 26509. [DOI] [PubMed] [Google Scholar]
- [38].Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ, Biomaterials 2011, 32, 5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shi Y, van Steenbergen MJ, Teunissen EA, Novo L. s., Gradmann S, Baldus M, van Nostrum CF, Hennink WE, Biomacromolecules 2013, 14, 1826. [DOI] [PubMed] [Google Scholar]
- [40].Liang Y, Deng X, Zhang L, Peng X, Gao W, Cao J, Gu Z, He B, Biomaterials 2015, 71, 1. [DOI] [PubMed] [Google Scholar]
- [41].Shi C, Guo D, Xiao K, Wang X, Wang L, Luo J, Nat. Commun 2015, 6, 7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Meng ZX, Nie J, Ling JJ, Sun JX, Zhu YX, Gao L, Lv JH, Zhu DY, Sun YJ, Han X, Diabetologia 2008, 52, 125. [DOI] [PubMed] [Google Scholar]
- [43].El Roz A, Bard J-M, Huvelin J-M, Nazih H, Anticancer Res. 2012, 32, 3007. [PubMed] [Google Scholar]
- [44].Cao F, Castrillo A, Tontonoz P, Re F, Byrne GI, Infect. Immun 2007, 75, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yoon C-H, Kwon Y-J, Lee S-W, Park Y-B, Lee S-K, Park M-C, J. Clin. Immunol 2013, 33, 190. [DOI] [PubMed] [Google Scholar]
- [46].Moghadasian MH, Frohlich JJ, McManus BM, Lab. Investig 2001, 81, 1173. [DOI] [PubMed] [Google Scholar]
- [47].Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Wagner AC, Frank JS, Datta G, Garber D, Fogelman AM, Circulation 2004, 109, 3215. [DOI] [PubMed] [Google Scholar]
- [48].Thompson AJ, Mastria EM, Eniola-Adefeso O, Biomaterials 2013, 34, 5863. [DOI] [PubMed] [Google Scholar]
- [49].Sankaranarayanan S, de la Llera-Moya M, Drazul-Schrader D, Phillips MC, Kellner-Weibel G, Rothblat GH, J. Lipid Res 2013, 54, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.[] Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, Wang Y, PLOS ONE 2012, 7, e35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.[] Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J, J. Clin. Invest 1993, 92, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.[] Baglione J, Smith JD, Methods Mol. Med 2006, 129, 83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.