

Abstract
Transcriptional enhancers play a major role in regulating metazoan gene expression. Recent developments in genomics and next-generation sequencing have accelerated and revitalized the study of this important class of sequence elements. Increased interest and attention, however, has also led to troubling trends in the enhancer literature. In this Perspective, I describe some of these issues and show how they arise from shifting and non-uniform enhancer definitions, and genome-era biases. I discuss how they can lead to interpretative errors and an unduly narrow focus on certain aspects of enhancer biology to the potential exclusion of others.
Keywords: transcription, enhancer, cis-regulatory module, bias, genomic assay
The state of enhancer research
Since their first description almost 40 years ago [1], there has been an increasing recognition of the importance of transcriptional enhancers for all aspects of metazoan biology including development, physiology, evolution, and disease [2–5]. In particular, the era of sequenced genomes has brought with it renewed interest in establishing a more complete annotation of the regulatory genome, and the emergence of genome-scale methods, especially nextgeneration sequencing, has created unprecedented opportunities for enhancer discovery and characterization [6, 7]. However, this resurgent interest in enhancers has also generated increased opportunities for shifting definitions, inconsistent interpretations, and subtle biases. In the following paragraphs I discuss several problems pervading the current enhancer biology literature, providing some recent examples of each. It is worth noting that these are drawn from excellent papers by insightful scientists, and are chosen simply as representative illustrations from the broader literature.
What are enhancers?
Enhancers are cis-regulatory sequences that work in concert with a gene’s core promoter to regulate much of the spatiotemporal control of gene expression in metazoa. Typically on the order of a few hundred basepairs in length, enhancers serve as a scaffold for the recruitment of transcription factors and chromatin modifying enzymes. They can regulate gene expression irrespective of their orientation, position, or distance from the transcription start site. Enhancer biology is the subject of several excellent recent reviews, and the reader is referred to these for details [3, 5, 8, 9].
The traditional definition of an enhancer is a functional one—enhancers were originally characterized by their activity, rather than by a physical property. This makes enhancers rare if not unique among annotated genomic features in that their annotated sequences are not based on objective and unambiguous criteria such as a sequenced transcript, translation product, defined nucleotide sequence, et cetera. (A major exception could be argued to be the gene itself, on whose definition biologists are notoriously in disagreement [10–12]. But in terms of genome annotation there is general consensus that an annotated gene begins at the transcription start site of its most 5’ exon and continues through the sequence of its most 3’ exon.) With the development of high-throughput genomic methods for enhancer discovery (reviewed in [7]) has come a shift from this functional definition of the enhancer to one based on one or more of a variety of chromatin and transcriptional properties. Enhancers are now frequently defined as sequences possessing any of a range of characteristics including binding by specific sets of transcription factors or co-activators; containing certain histone modifications, either alone or in combination; being nucleosome-depleted regions; or transcribing ‘enhancer RNAs’ (eRNAs) [3–5, 8]. Indeed, this definitional shift has become so entrenched that a recent paper erroneously states that enhancers “were first described as nucleosome-depleted regions with a high density of sequence motifs recognized by DNA-binding transcription factors” (ref. [13], emphasis added). However, there is no clear consensus as to which new enhancer definition to use, and all of the current measures lead to overlapping but non-identical sets of “enhancers” when applied to the same cell types and genomes. Significantly, all of these enhancer definitions assume that enhancers function as compact, modular units. Nevertheless, there are several clear and long-recognized exceptions to this [e.g. 14, 15, 16], and from a biochemical standpoint, there “is no inherent reason” that enhancers must be short contiguous segments of DNA, nor that they must contain tight clusters of transcription factor binding sites [17, see also 18, 19].
This fundamental question of how to define an enhancer is not one that is readily solved. In the meantime, however, it creates significant opportunities for ambiguities, contradictions, and interpretive confusion in the literature and even within individual studies. In particular, several common problems bear further scrutiny.
The founder fallacy
The founder fallacy comes into play when multiple experiments have defined overlapping or nested enhancers in a small genomic region, and results from giving primacy of function to an earlier-defined sequence over a more recently-defined one. We can state the founder fallacy as “defining an enhancer sequence based on its historical (earlier) description despite the availability of updated functional information.” Figure 1A illustrates a common scenario in which successive functional assays over a span of years have characterized a regulatory region. Enhancer e2 has identical activity to Enhancer e1, but was described several years later. Had the two been identified contemporaneously, for instance in a single set of deletion experiments, Enhancer e2 would have been named as the enhancer and Enhancer e1 consigned to the dustbin of history as one of a number of sequence fragments tested on the way to defining the enhancer’s boundaries. The founder fallacy comes about when, because for a period of years Enhancer e1 was referred to as “the” enhancer, it continues to be considered so even now that the functional boundaries have shifted substantially narrower. Note that the “true” enhancer does not necessarily mean a shorter sequence; in Figure 1B the newer Enhancer e2 is longer than the original Enhancer e1, but regulates a cleaner expression pattern lacking the ectopic expression observed in tissue C due to inclusion of important repressor sequences.
Figure 1: The founder fallacy.
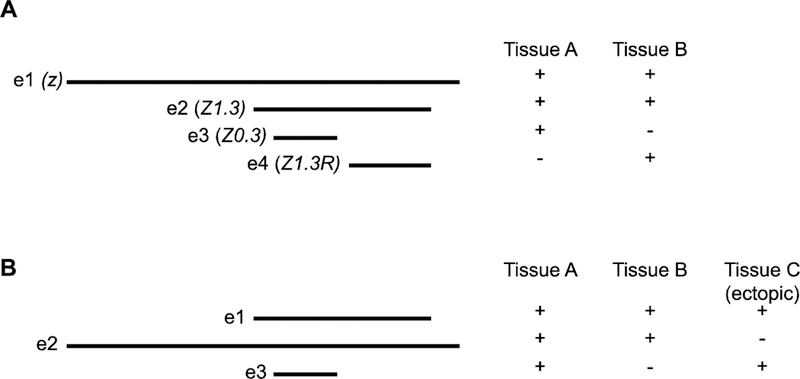
(A) A set of sequence fragments (e1–e4), each of which functions as an enhancer in a reporter gene assay. Fragment e2 is active in both Tissue A and Tissue B, identical to e1, but was identified more recently. e3 and e4 each have a subset of the function of e1 and e2, with distinct activity. (B) In this second set of sequence fragments, the more recently-identified e2 is longer than the original e1 enhancer, but lacks ectopic regulatory activity in Tissue C. Thus e2 is more properly the “true” enhancer than the shorter e1.
Reasons for the founder fallacy can be many and are often quite innocuous, such as a lag in updating the genome annotation or the well-known preference for authors to continue to use their own original designations for features. However, failure to consider critically the implications for the regulatory architecture of the locus can lead to important interpretive consequences. (See Box 1 for a discussion of how this issue is dealt with in a regulatory genome annotation project.)
Box 1: Enhancers and Genome Annotation.
How to properly reflect enhancer sequences in a genome annotation is a significant challenge. Genome features must be annotated with specific nucleotide coordinates, but the indefinite ways used to define enhancers makes this difficult. Nested and overlapping sequences must be resolved, and the annotation must be kept current as older enhancer boundaries are refined through subsequent experiments.
An example of a highly curated metazoan regulatory annotation is that of Drosophila melanogaster. While the Drosophila annotation is maintained by FlyBase [53], enhancer annotations are primarily based on data from the REDfly database [54]. REDfly annotates regulatory data based on a variety of considerations, but applies the label of “cis-regulatory module (CRM),” essentially used synonymously with “enhancer,” only in specific circumstances. To qualify, a REDfly regulatory sequence must meet both of the following criteria:
-
(1)
The sequence must have demonstrated regulatory activity, typically based on sufficiency to regulate reporter gene activity in either transgenic flies or cultured cells
-
(2)
the sequence must be the minimal-length sequence in a set of one or more nested sequences that regulate the same expression pattern
Sequences that fail to meet these requirements are still annotated by REDfly, but are not labeled as CRMs.
The CRM designation is re-evaluated each time new data are added to the database, so that if a new, more minimal sequence with identical activity is added within the boundaries of a current CRM, the new sequence is awarded the CRM label and the older sequence reverts to a non-CRM classification. FlyBase includes only currently-designated CRMs in the Drosophila genome annotation, so that in any given version the two enhancer criteria listed above are maintained.
The choice to use the shortest of a set of nested sequences to represent the enhancer can be debated, as it can be difficult to determine whether a particular reporter construct captures all of the subtleties of endogenous expression as well as another, slightly larger sequence; also, this definition enforces the conceptualization of enhancers as strictly modular units, for instance disallowing the possibility that in the context of the genome aspects of enhancer function might be distributed over large distances [3, 18]. Nevertheless, this is the typical accepted practice across the enhancer field and seems necessary to avoid considering everlonger genomic sequences as potential contributors to enhancer function until the reductio ad absurdum of including entire chromosomes is reached.
REDfly is also careful to distinguish functionally-demonstrated regulatory sequences from those predicted by genomic or computational methods (“predicted,” or “pCRMs”), or from those inferred by the overlapping portions of two enhancers with similar function (“inferred,” or “iCRMs”). By segregating out these sequences, a function-based enhancer definition is maintained in the genome annotation.
An example can be found in a recent paper exploring enhancer pleiotropy [20]. Enhancer pleiotropy refers to the situation where a single enhancer regulates gene expression in more than one spatiotemporal domain [21]. This is a more common situation than may be generally realized, but one that has not been well studied. Preger-Ben Noon et al. [20] ask the important question of whether pleiotropic enhancers make use of the same or different transcription factor binding sites to regulate multiple domains of gene expression. Using a previously-described set of enhancers in the Drosophila shavenbaby (svb) locus, the authors claim that seven out of seven enhancers demonstrate pleiotropy by driving expression in both the embryo and the pupa. Closer examination of two of these enhancers, “E6” and “Z1.3,” is said to reveal “two fundamentally distinct models” of enhancer pleiotropy: E6 utilizes common binding sites to execute its function in both tissues (“site pleiotropy”; Figure 2A), whereas Z1.3 does not—the transcription factor binding sites that mediate embryonic versus pupal function “act independently” (Figure 2B, C). The analysis of the E6 enhancer is masterfully carried out and provides a beautiful example of site pleiotropy, leaving no question that this is a key mechanism with important implications for enhancer function and evolution. However, the analysis of the Z1.3 enhancer, and by extension that of the other five less-studied putatively pleiotropic enhancers, falls victim to the founder fallacy. Figure 1A is not a purely hypothetical example but rather a simplified diagram of the svb locus, adapted from [20]. Enhancer e2 corresponds to Z1.3 and is active in both embryos and pupae. Enhancer e3 is an embryo-specific enhancer “Z0.3” and enhancer e4 is the pupa-specific enhancer “Z1.3R.” Viewed without bias toward the originally-identified Z1.3 fragment, the most straightforward interpretation of the results is that instead of Z1.3 being a pleiotropic enhancer with independently-acting binding sites for two tissues, it simply represents two different enhancers, the Z0.3 embryonic enhancer and the Z1.3R pupal one (Figure 2C). A simple thought experiment makes the founder fallacy clear: if the order of discovery had been reversed and the Z0.3 and Z1.3R enhancers defined first, without the larger Z1.3 enhancer having been described, would there be any rationale for combining these into a single pleiotropic enhancer? In this light, the authors’ claim that the remaining five enhancers, which have not been further dissected, are similarly pleiotropic must also be called into question: would further dissection reveal that some or all of these too can simply be separated into functionally distinct sequences?
Figure 2: Enhancer pleiotropy and site pleiotropy.
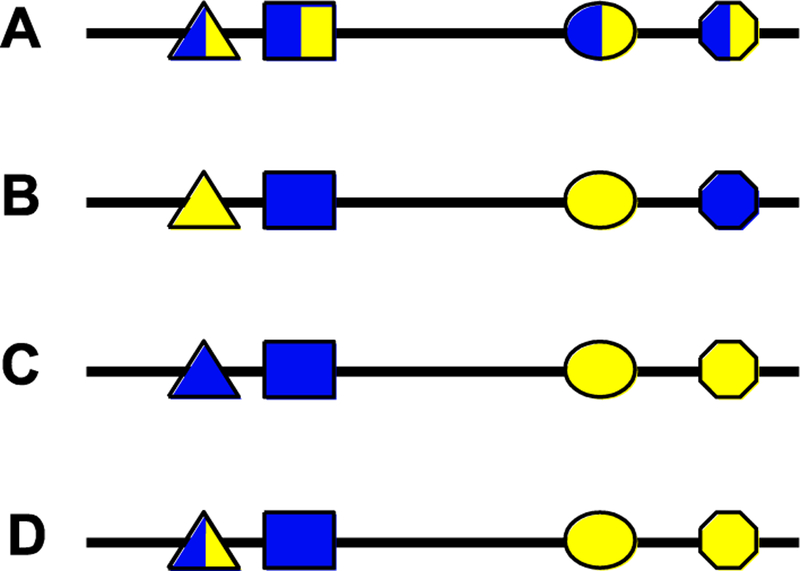
Four examples of potentially pleiotropic enhancers. Each polygon represents a transcription factor binding site (TFBS), with blue fill indicating a required role in regulating gene expression in one tissue and yellow fill indicating a required role in a second tissue. (A) An enhancer with complete “site pleiotropy”: each TFBS is used in both regulatory contexts. This is similar to the “E6” enhancer in [20]. (B) A pleiotropic enhancer lacking site pleiotropy such that each TFBS contributes to only one of the two activities of the enhancer. The sites are integrated in the enhancer sequence in such a way that the two activities cannot be separated without disrupting all enhancer activity. (C) This situation is similar to the Z0.3 and Z1.3R enhancers in [20](see Fig. 1). The sequence can be divided into two functional segments as all of the TFBSs necessary for blue expression reside in the left half and all those for yellow expression in the right. It can be argued therefore that this sequences does not represent a single pleiotropic enhancer, but rather two distinct enhancers, a “blue” and a “yellow.” (D) A more complex scenario that involves both pleiotropic TFBSs and TFBSs specific for a single expression pattern. While the left half of the sequence could function as a blue-specific enhancer, the right side by itself is non-functional as it lacks the triangle binding site. A situation similar to this has been observed in the Drosophila stumps locus (ref. [55] and unpublished data).
The founder fallacy has implications that go well beyond the question of enhancer pleiotropy. For example, “shadow enhancer,” a term coined by Levine and colleagues to describe a type of redundant enhancer [22], fell prey at its inception to the founder fallacy by reference to previously-discovered and more proximally-located “primary” enhancers. Although this definition has been walked back to some extent to establish “shadow enhancer” in the lexicon as more-or-less synonymous with “redundant enhancer,” it is almost impossible to separate the connotation of “shadow” from “secondary” and therefore “less important” in cases where one of the pair was discovered earlier than the other. In recognition of this point, Barolo [23] has suggested replacing the term with “distributed enhancer,” but this designation has failed to catch on.
The founder fallacy similarly may have a role in the establishment of “super-enhancers” as a more recently-described new class of regulatory element [24, 25]. Currently, there is much debate over whether or not super-enhancers constitute a bona fide new regulatory type or simply reflect regions of highly clustered, redundant and/or cooperating enhancers [3, 5, 26]. The jury is still out on this question, and while some of the problem stems from the lack of a clear functional or bioinformatic definition [5], some also stems from the founder fallacy: super-enhancers have been defined as large regions by chromatin-based assays and only subsequently broken down functionally into constituent component enhancers. In Drosophila, where extensive enhancer identification has often been performed using reporter gene analysis, the existence of dense clusters of enhancers (with chromatin features similar to super-enhancers, e.g. [13]) has been known for many years without leading to the suggestion that such clustering defines a unique regulatory structure (e.g. [27–30]). In studies where there has been functional testing of the individual component enhancers of super-enhancers, most appear to be typical enhancers that act either additively [31] or redundantly [32] to regulate target gene expression. Had these component enhancers simply been identified first—as was the case in Drosophila—would the larger region still have been proposed after the fact to be a novel regulatory entity?
Validation Creep
A second disturbing trend seen in the enhancer literature can be termed “validation creep.” Validation creep is the tendency to move from considering a set of sequences as “putative enhancers” to accepting them as “enhancers”—sometimes within the context of a single publication—without providing additional evidence of function. Consider, for example, the large-scale cis-regulatory annotation of the mouse genome [33]. The authors conducted RNA-seq and four or more additional ChIP-seq experiments on a set of 13 adult mouse tissues, four embryonic tissues, and two cell lines to predict promoter, enhancer, and insulator sequences across the mouse genome—an experimental and bioinformatics tourde-force that provides an invaluable first-approximation description of the mouse regulatory genome. Using a model incorporating presence of monomethylated histone H3 lysine 4 (H3K4me1) and absence of trimethylated H3K4 (H3K4me3), trained on sites bound by the histone acetyltransferase p300, the authors predicted what they appropriately refer to as 234,764 “potential enhancers” [33]. These were compared to a set of over 700 known, experimentally-identified enhancers, with an 82% validation rate, and luciferase-based reporter gene assays were conducted for a randomly selected eight new predictions, yielding a 75% validation rate. The 75–82% validation range is consistent with earlier studies using similar methodology [34, 35].
By the second figure, however, despite the possible 20–25% false-positive rate, the “potential enhancers” have become the identified set of “enhancers” and remain so for the rest of the paper, where they form the basis for further analysis. This includes development of a new measure for pairing enhancers with their target promoters, motivated by the observation that two existing methods gave poor results. This may well be because those methods, as the authors suggest, are not effective. On the other hand, it is at least possible that the culprit is instead that a high number of non-enhancers in the data set (due to false-positive predictions) negatively affect the results, and the “better” new method is merely overfit to the noisy data. The problem with validation creep is that it eliminates consideration of this possibility, as the predicted enhancer set—false positives and all—has already become the true enhancer set.
A similar example of validation creep can be seen in another landmark genomics study, the “atlas of active enhancers across human cell types and tissues” based on the FANTOM5 cap analysis of gene expression (CAGE) data [36]. CAGE [37] detects capped RNAs and was used to show that bi-directional capped RNAs could serve as a signature for active enhancers. In the paper’s first figure, the enhancers are “candidates” and 123 sequences spanning both strong and weak predictions are tested in reporter gene assays. 67–74% of these had reporter gene activity, whereas a smaller set of potential enhancers predicted based on different genomic criteria only gave a 20–33% validation rate. In subsequent figures, CAGEdefined enhancers are shown to strongly overlap features previously proposed to mark active enhancers, such as H3K4me1 + H3K27ac (71%) and accessible chromatin (87%). However, these chromatin features were poor predictors of CAGE-defined enhancers (11% and 4% respectively). In vivo reporter gene assays in zebrafish successfully validated three out of five (60%) CAGE-predicted enhancers. By all these criteria, the CAGE bidirectional RNAs appear to be strong predictors of enhancer activity—but seemingly not perfect ones. Nevertheless, by the second figure all reference to the enhancers as “candidates” has been dropped.
This study [36] illustrates once again how validation creep allows a predictive method with an experimental validation rate in the 60–80% range to morph into an accepted data set within a single publication. Interestingly, the higher-than-typical amount of experimental validation conducted by the authors suggests that other common methods for determining active enhancers, such as presence of H3K27ac or open-chromatin profiling, may be weak predictors, with validation rates equivalent to the 33% seen in other extensively-validated studies [38]. Nevertheless, validation creep results in enhancer annotations based on these methods being used as “true” data sets, such that there are multiple competing versions of regulatory annotations and continued new analyses based on one or the other of these “defined” sets of enhancers. Abandoning the qualification of “predicted” or “putative” enhancers thus creates tremendous potential for confusion, as well as opens the door to founder fallacy errors as newer predictions based on refined criteria potentially supersede older ones.
A major concern with validation creep is not so much a fear that the broad outlines of the discoveries reported in these papers is incorrect—as stated previously, they represent useful first-approximation descriptions of the regulatory genome—but rather the circularity in enhancer definition that stems from the fact that “known” enhancers frequently serve as the basis for ascribing new enhancer characteristics. Tracing the route to the mouse enhancer predictions reveals how self-reinforcing the definition can be. Earlier work [35] noticed a correlation between enhancers and p300 binding, although it was unknown what percentage of confirmed enhancers bound p300 and what percentage of p300-bound sites are not enhancers. From this it was observed that H3K4me1 was enriched “at nearly all enhancers” (defining enhancers as p300-bound sites) and that enhancers “generally lack” H3K4me3. Shen et al. [33] then used just these three criteria—p300 binding, H3K4me1 enrichment, and H3K4me3 depletion—to build their model for enhancer prediction. While there is no reason to believe that, within their apparent 20–25% false positive rate, they failed to successfully identify a large number of enhancers, the circular approach effectively precludes the discovery of any enhancers that do not meet these criteria. As long as the result set is firmly considered as a set of predictions, this is not a problem. However, the validation creep tends to elide the fact that the results are not a comprehensive catalog of all enhancers, but only those based on a narrow set of characteristics. Thus reduced sensitivity as much as low specificity becomes a serious potential consequence of validation creep. By the same token, Andersson et al. [36], by making their active enhancer definition exclusive to transcribed sequences, propagate a narrow active-enhancer definition that risks leading others to pass over potentially important regulatory sequences.
Is this concern valid or merely hypothetical? I would argue that the problem is real. A growing number of studies have adopted one or the other of these enhancer definitions, and there is increasing evidence that each provides only a partial characterization of the enhancer landscape (Figure 3). While there is general agreement, for instance, that enhancer transcription correlates with enhancer activity, several studies (e.g. [39, 40]) suggest that enhancers can be active without the transcriptional signature defined in [36]. Strikingly, Henriques et al. [13] recently showed that rather than being excluded from enhancers, H3K4me3 is found at enhancers—but only at those with the strongest activity, suggesting that previous enhancer definitions explicitly excluding H3K4me3-marked sequences are leaving out the most active enhancers. Other studies indicate that characterized enhancer regions can contain a wide variety of histone modifications in many different combinations, and without necessarily bearing the “canonical” H3K4me1 or H3K27ac marks (e.g., [41–44]). Moreover, accumulating evidence points to chromatin marks such as H3K4me1 being associated with but not required for enhancer activity [45, 46], while chromatin modifications caused by the Hairy long-range repressor may often be the result of “errant targeting” that have little or no consequence on gene regulation [47]. Taken together, the available data suggest that the chromatin and transcriptional characteristics of enhancers are complex and varied, and potentially differ based on enhancer activity, cell type, role of target gene, or other yet-to-be-determined criteria. Validation creep, by promoting circular definitions and confusing predicted elements with established ones, masks much of this complexity.
Figure 3: Enhancer “definitions” are not consistent.
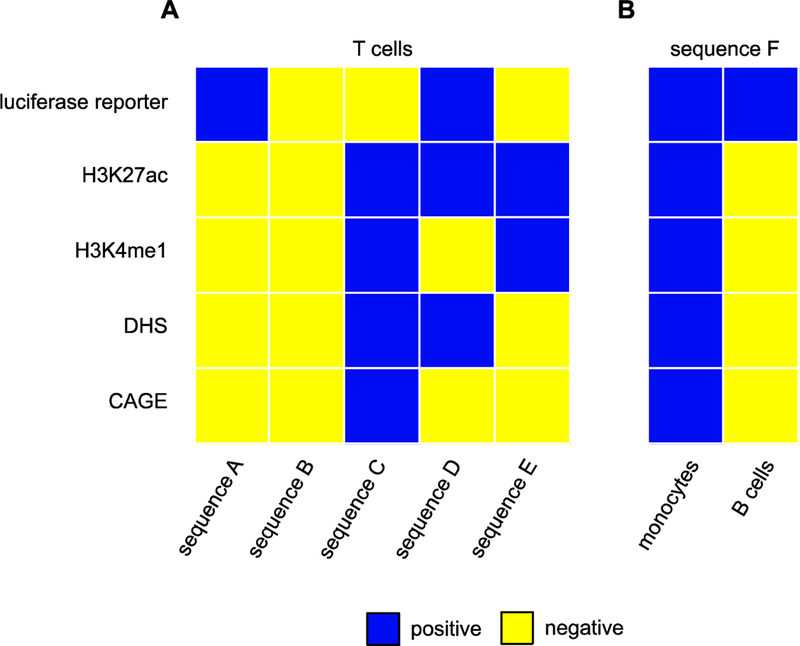
Data from [36] illustrate how defining enhancers based on reporter gene activity, histone modifications, chromatin accessibility, and enhancer transcription provides inconsistent and/or contradictory results. (A) Five different sequences have a different pattern of possible enhancer characteristics when tested in a common cell type (T cells). Blue squares indicate a positive assay result, yellow negative. Each sequence was assessed for: its ability to drive a luciferase reporter gene; H3K27 acteylation; H3K4 monomethylation; chromatin accessibility in the form of DNAseI hypersensitive sites (DHS); and bidirectional transcription (CAGE). No clear trend emerges from the set of assays. (B) The identical sequence can drive reporter gene expression in two different cell lines, but in one cell type can be positive for a set of several possible enhancer characteristics whereas in the other it can be negative for the entire same set. [Data for this figure were adapted from Figure S17 of [36]; sequences A-F correspond to the eighth, ninth, twenty-eighth, first, thirteenth, and eighteenth columns, respectively.]
The large scale/small scale bias
The tendency toward validation creep is understandable and perhaps even inevitable given genome-scale assays, where validation of all results is impossible and data must often be interpreted broadly based on statistical arguments. However, there is still something troubling about taking an experiment in which only 75% of tested sequences are shown to have enhancer function and then accepting all the sequences as enhancers, regardless. Consider the experiment in Figure 4, which depicts a hypothetical old-school “enhancer bashing” assay in which eight sequences were tested for enhancer function by reporter gene assays. The six red fragments had activity, whereas the two black ones did not: a 75% validation rate. It would be unheard of to proceed with an analysis on the basis that all eight sequences are enhancers, and no credible peer reviewer would go on to allow the authors to assert, absent empirical testing, that a similar set of eight sequences from another locus were all enhancers, having just seen a 25% failure-to-validate rate in a preceding figure. Yet, this is exactly the situation we see occurring time and again with respect to genome-scale enhancer studies.
Figure 4: A typical small-scale “enhancer bashing” experiment.
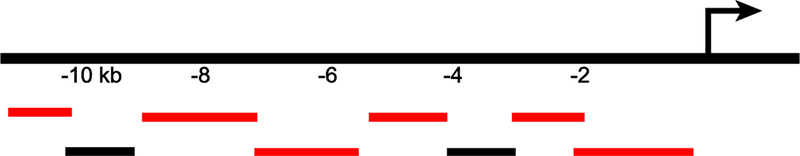
A hypothetical enhancer bashing experiment with tested sequence fragments illustrated below their location in a 10 kb region upstream of a gene. Fragments that showed activity in a reporter assay are shown in red, those that failed to show activity are depicted in black. Six out of eight (75%) of the tested sequences in this experiment had positive regulatory activity.
The empirical result bias
I suspect that validation creep is also abetted by what I term “empirical result bias.” This is the tendency to give greater weight to results obtained through experiment versus, for instance, from computational modeling. In cases where experimental results are dispositive, of course, this makes perfect sense—but this is not the case for many genomics studies, where the experimental results are not individually controlled, and the results are mainly correlative with support from bulk statistical analysis. Enhancer identification is a prime example of this (although hardly exclusive). When enhancers are predicted strictly computationally, whether by analysis of potential binding sites, machine learning methods trained on subsequence (k-mer) composition, or some other means [7, 48], the results are acknowledged as merely predictive and validation creep is minimal. However, when enhancers are predicted by virtue of histone modification, chromatin accessibility, or bidirectional transcription, for instance, validation creep is common, as discussed above— even though these results, too, are merely predictive. The imprimatur of “experiment” seems to lead to a misplaced acceptance of prediction for proof. As a result, validation rates that would be considered low for many computational prediction methods are often accepted when considering experimental methods, and computational methods with higher apparent true-positive rates are given less credibility.
Interestingly, empirical result bias cuts both ways, as it also impacts what is accepted as “validation” for enhancer predictions—a longstanding problem that deserves more attention than it usually receives. The primary accepted validation method for enhancers is the reporter gene assay, often performed in cell culture. Empirical result bias implies that if there is a 75% validation rate of enhancer prediction by reporter assay, then there is a 25% false positive prediction rate. But this is of course not true: like any negative experimental result, failure of validation cannot be treated as a basis to refute a hypothesis. This is particularly true due to the fact that reporter gene assays, despite being the gold standard, have a number of serious flaws. Differences in cell type, in choice of promoter, in genomic context of the reporter gene as compared to the endogenous gene, and myriad other issues can all affect observed enhancer activity, leading to false positive (ectopic enhancer activity) as well as to false negative results [5, 7, 8, 49]. This is true both for traditional enhancer-bashing experiments and for contemporary genomic methods. However, other validation methods also have their shortcomings. For instance, while it is now possible to mutate or silence an endogenous enhancer using CRISPR [50], difficulties in assaying the effect on gene expression due to enhancer redundancy, ambiguities as to the correct target gene, failure to remove a sufficient portion of enhancer sequences, and other experimental considerations can all lead to false negative results. The best way to counter this will be to insist on increased validation experiments using both ectopic (reporter gene) and endogenous (enhancer deletion) experiments [8]—and to remember that just because a result is experimentally derived, it is not necessarily accurate.
Concluding remarks: is it all just a house of cards?
Given the biases in the enhancer literature I have illustrated here, and the circularity in enhancer feature characterization, the reader might wonder if our current understanding of enhancers is just a house of cards, propped up by the thinnest evidence and ready to topple with the next clear experiment. Such a view would be a mistake, and not what I am trying to suggest here. Indeed, as mentioned above, the papers cited here are all important and well-conducted studies, and provide crucial clues to determining how enhancers work and what features they possess. But it is important to realize that much about enhancers still remains a mystery, and there is much we still need to learn (see Outstanding Questions). Among these questions are how enhancers are organized in the genome; what chromatin features and modifications are associated with them, and to what purpose; what the role of transcription at enhancers is; and how to define enhancers and their possible different types and subtypes. The latter may be a particularly important question. While it is not known if there are functional enhancer subtypes, it is possible that different regulatory roles—e.g. RNA polII recruitment, release of paused RNA polII, targeting of genes to “transcription factories,” etc.—are carried out by separate classes of enhancers with distinct properties. Such a scenario could explain some of the discrepancies seen with the presence and absence of various chromatin marks and eRNAs; it also highlights the potential risk of methods that predict enhancers by integrating a wide assortment of chromatin features [e.g. 51, 52], and provides a possible explanation for why such methods have shown low rates of empirical validation [38].
To answer these and other questions effectively, we need to acknowledge the limitations in both traditional and newer methods for investigating enhancers. We need to begin as a field to use a wider and more diverse range of assays to establish enhancer function, and to insisit on higher levels of proof before accepting new defining enhancer characteristics. Importantly, we must be cognizant of the various biases that color our interpretation of experimental and analytical results (Box 2). This will help to avoid being thrown off course by founder fallacies, falling into narrow and circular definitions through validation creep, and over-prioritizing certain sets of results merely because they were obtained through large-scale experimental approaches.
Box 2: Avoiding common traps and biases when studying enhancers.
Competing definitions and disagreements over the most conclusive assays can make deciding which sequences should be considered enhancers a challenge. These guidelines will help in avoiding some common biases and pitfalls:
Remember that statistical arguments don’t apply to individual specific sequences. Various characteristics might suggest that a sequence is more or less likely to be an enhancer, but only a functional assay can demonstrate function.
Beware of circularity when defining enhancer characteristics. Ascribing new characteristics to a set of sequences themselves not known definitively to function as enhancers can lead to narrow and self-reinforcing definitions that omit important features.
Don’t be afraid to use terms such as “putative” and “candidate.” There’s no shame in admitting that we can’t yet validate the function of every sequence in the genome, in every cell type, under every condition.
Think twice before defining new functional classes of enhancers and new terms to describe them. Sometimes an enhancer is just an enhancer.
Use the most up-to-date genomic data and genome annotations available, and update or revise previous analyses as necessary.
Validate, validate, validate! Know the limitations of each assay, and recognize that no single perfect assay yet exists. Use multiple methods, including those that test both the necessity and sufficiency of a sequence to regulate transcription. Weigh each assay according to its strength and reliability, without regard to whether it is genomic or locus-specific in scale, or experimental, computational, or statistical in nature.
Remember that what we don’t know far outweights what we do know. As always, treasure your exceptions!
HIGHLIGHTS.
Historically imprecise along with recently shifting criteria for how we define transcriptional enhancers pose unique challenges for the study of enhancer biology
The recent enhancer literature reveals a number of concerning trends to look out for and guard against
The “founder fallacy” occurs when older enhancer sequences are used for analysis despite newer data refining the functional enhancer boundaries
“Validation creep” is the tendency to designate a set of predicted enhancer sequences as a set of confirmed enhancers without any additional data justifying the switch
“Large scale/small scale bias” and “empirical result bias” reflect a trend to be more accepting of genome-scale experimental data than would be typical for small-scale studies or data derived primarily from computational analysis
OUTSTANDING QUESTIONS.
How should enhancers be defined? Should we rely only on functional criteria or also incorporate chromatin and/or transcriptional features into the definition?
Given the difficulties inherent in accurately defining enhancers, how can we maintain an inclusive enhancer definition without falling back on circular and overly narrow definitions?
Is there a good way to reflect different degrees of confidence in a sequence’s designation as an enhancer in the genome annotation?
What are the best ways to measure enhancer activity and cooperativity?
Is it useful to try to classify enhancers into different types and subtypes? Can we do this effectively without running afoul of the founder fallacy?
ACKNOWLEDGEMENTS
I am grateful to Sat Sinha and David Arnosti for their insightful comments on the manuscript. This work was supported by funding from the National Institutes of Health (R01 GM114067) and the National Science Foundation (DBI 1758252).
GLOSSARY
- CAGE:
Cap Analysis of Gene Expression. CAGE enables both gene expression profiling and determination of the transcription start site (TSS) of each transcript by sequencing fragments (“tags”) derived from trapping the 5’ cap of mRNAs.
- CRM:
cis-regulatory module, a generic term covering enhancers as well as other similar types of transcriptional regulatory sequences.
- Enhancer:
a cis-regulatory sequence that binds transcription factors and acts in conjunction with a gene’s promoter to positively activate gene expression. By formal definition, enhancers act independently of position, orientation, and distance relative to their target gene, although these characteristics are rarely tested in a rigorous fashion.
- Pleiotropy:
pleiotropy refers to when a gene affects multiple traits. With respect to regulatory sequences, enhancer pleiotropy refers to an enhancer that regulates more than one expression pattern. Site pleiotropy refers to a transcription factor binding site within an enhancer that regulates a pleiotropic enhancer in more than one regulatory context.
- Reporter gene assay:
A traditional way of testing DNA sequences for regulatory activity. The sequence to be tested is cloned upstream of a minimally-active promoter driving the “reporter gene,” a gene whose activity is easy to monitor, e.g., the bacterial lacZ gene encoding β-galactosidase or a fluorescent protein.
- Shadow enhancer:
one of a pair (or small group) of enhancers driving similar patterns of expression of the same gene.
- Super-enhancer:
a regulatory region consisting of a cluster of enhancers and highly enriched for binding of Mediator and activating chromatin marks. This terms is used differently by different authors and remains controversial as to whether or not super-enhancers constitute a true separate class of regulatory feature.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Banerji J et al. (1981) Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27 (2 Pt 1), 299–308. [DOI] [PubMed] [Google Scholar]
- 2.Smith E and Shilatifard A (2014) Enhancer biology and enhanceropathies. Nat Struct MolBiol 21 (3), 210–9. [DOI] [PubMed] [Google Scholar]
- 3.Buffry AD et al. (2016) The Functionality and Evolution of Eukaryotic Transcriptional Enhancers. Adv Genet 96, 143–206. [DOI] [PubMed] [Google Scholar]
- 4.Murakawa Y et al. (2016) Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends Genet 32 (2), 76–88. [DOI] [PubMed] [Google Scholar]
- 5.Rickels R and Shilatifard A (2018) Enhancer Logic and Mechanics in Development and Disease. Trends Cell Biol [DOI] [PubMed]
- 6.Shlyueva D et al. (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15 (4), 272–86. [DOI] [PubMed] [Google Scholar]
- 7.Suryamohan K and Halfon MS (2015) Identifying transcriptional cis-regulatory modulesin animal genomes. Wiley Interdisciplinary Reviews: Developmental Biology 4 (2), 59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catarino RR and Stark A (2018) Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev 32 (3–4), 202–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long HK et al. (2016) Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167 (5), 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerstein MB et al. (2007) What is a gene, post-ENCODE? History and updated definition. Genome Res 17 (6), 669–81. [DOI] [PubMed] [Google Scholar]
- 11.Pesole G (2008) What is a gene? An updated operational definition. Gene 417 (1–2), 1–4. [DOI] [PubMed] [Google Scholar]
- 12.Portin P and Wilkins A (2017) The Evolving Definition of the Term “Gene”. Genetics 205 (4), 1353–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henriques T et al. (2018) Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32 (1), 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klingler M et al. (1996) Disperse versus Compact Elements for the Regulation ofruntStripes inDrosophila. Developmental Biology 177 (1), 73–84. [DOI] [PubMed] [Google Scholar]
- 15.Ludwig MZ et al. (2005) Functional evolution of a cis-regulatory module. PLoS Biol 3 (4), e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuh CH et al. (1998) Genomic cis-regulatory logic: experimental and computation alanalysis of a sea urchin gene. Science 279 (5358), 1896–902. [DOI] [PubMed] [Google Scholar]
- 17.Janssens H et al. (2006) Quantitative and predictive model of transcriptional control ofthe Drosophila melanogaster even skipped gene. Nat Genet 38, 1159–1165. [DOI] [PubMed] [Google Scholar]
- 18.Halfon MS (2006) (Re)modeling the transcriptional enhancer. Nat Genet 38 (10), 11021103. [DOI] [PubMed] [Google Scholar]
- 19.Li L et al. (2007) Large-scale analysis of transcriptional cis-regulatory modules reveals both common features and distinct subclasses. Genome Biol 8 (6), R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Preger-Ben Noon E et al. (2018) Comprehensive Analysis of a cis-Regulatory Region Reveals Pleiotropy in Enhancer Function. Cell Rep 22 (11), 3021–3031. [DOI] [PubMed] [Google Scholar]
- 21.Monteiro A and Podlaha O (2009) Wings, horns, and butterfly eyespots: how do complex traits evolve? PLoS Biol 7 (2), e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong JW et al. (2008) Shadow enhancers as a source of evolutionary novelty. Science 321 (5894), 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barolo S (2012) Shadow enhancers: frequently asked questions about distributed cis-regulatory information and enhancer redundancy. Bioessays 34 (2), 135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hnisz D et al. (2013) Super-enhancers in the control of cell identity and disease. Cell 155 (4), 934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whyte WA et al. (2013) Master transcription factors and mediator establish super enhancers at key cell identity genes. Cell 153 (2), 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pott S and Lieb JD (2015) What are super-enhancers? Nat Genet 47 (1), 8–12. [DOI] [PubMed] [Google Scholar]
- 27.Barrio R et al. (1999) Identification of regulatory regions driving the expression of the Drosophila spalt complex at different developmental stages. Dev Biol 215 (1), 33–47. [DOI] [PubMed] [Google Scholar]
- 28.Fujioka M et al. (1999) Analysis of an even-skipped rescue transgene reveals both composite and discrete neuronal and early blastoderm enhancers, and multi-stripe positioning by gap gene repressor gradients. Development 126 (11), 2527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujioka M and Jaynes JB (2012) Regulation of a duplicated locus: Drosophila sloppy paired is replete with functionally overlapping enhancers. Dev Biol 362 (2), 309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorberbaum DS et al. (2016) An ancient yet flexible cis-regulatory architecture allows localized Hedgehog tuning by patched/Ptch1. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hay D et al. (2016) Genetic dissection of the alpha-globin super-enhancer in vivo. Nat Genet 48 (8), 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moorthy SD et al. (2017) Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res 27 (2), 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Y et al. (2012) A map of the cis-regulatory sequences in the mouse genome. Nature 488 (7409), 116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heintzman ND et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459 (7243), 108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heintzman ND et al. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics 39 (3), 311–318. [DOI] [PubMed] [Google Scholar]
- 36.Andersson R et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature 507 (7493), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiraki T et al. (2003) Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci U S A 100 (26), 15776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwasnieski JC et al. (2014) High-throughput functional testing of ENCODE segmentation predictions. Genome Res [DOI] [PMC free article] [PubMed]
- 39.Cheng JH et al. (2015) Genome-wide analysis of enhancer RNA in gene regulation across12 mouse tissues. Sci Rep 5, 12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young RS et al. (2017) Bidirectional transcription initiation marks accessible chromatin and is not specific to enhancers. Genome Biol 18 (1), 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonn S et al. (2012) Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 44 (2), 14856. [DOI] [PubMed] [Google Scholar]
- 42.Pradeepa MM et al. (2016) Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet 48 (6), 681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor GC et al. (2013) H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res 23 (12), 2053–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z et al. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40 (7), 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dorighi KM et al. (2017) Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol Cell 66 (4), 568–576 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rickels R et al. (2017) Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat Genet 49 (11), 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kok K et al. (2015) Genome-wide errant targeting by Hairy. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleftogiannis D et al. (2016) Progress and challenges in bioinformatics approaches for enhancer identification. Brief Bioinform 17 (6), 967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atkinson TJ and Halfon MS (2014) Regulation of Gene Expression in the Genomic Context. Comput Struct Biotechnol J 9, e201401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shukla A and Huangfu D (2018) Decoding the noncoding genome via large-scaleCRISPR screens. Curr Opin Genet Dev 52, 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ernst J and Kellis M (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9 (3), 215–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffman MM et al. (2012) Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 9 (5), 473–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gramates LS et al. (2017) FlyBase at 25: looking to the future. Nucleic Acids Res 45 (D1),D663–D671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallo SM et al. (2011) REDfly v3.0: toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res 39 (Database issue), D118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halfon MS et al. (2002) Computation-based discovery of related transcriptional regulatory modules and motifs using an experimentally validated combinatorial model. Genome Res 12 (7), 1019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]