

Abstract
While stress-induced synthesis of mono ADP-ribose (mADPr) and poly ADP-ribose (pADPr) conjugates by PARP enzymes has been studied extensively, the removal and degradation of pADPr, and the fate of ADPr metabolites has received less attention. The observations that stress-induced pADPr undergoes rapid turnover and deficiencies in ADPr degradation phenocopy loss of pADPr synthesis suggest that ADPr degradation is fundamentally important to the cellular stress response. Recent work has identified several distinct families of pADPr hydrolyases that can degrade pADPr to release pADPr or mADPr into the cytoplasm. Further, many stress response proteins contain ADPr binding domains that can interact with these metabolites. Here, we will discuss how pADPr metabolites generated during pADPr degradation can function as signaling intermediates in processes such as inflammation, apoptosis and DNA damage responses. These studies highlight that the full cycle of ADPr metabolism, including both synthesis and degradation, is necessary for responses to genotoxic stress.
Conjugation of ADP-ribose (ADPr) to proteins plays a critical signaling function in gene transcription, chromatin organization and stress responses. Poly-ADPr polymerase (PARP1) was originally thought responsible for producing all cellular ADPr (1). However, PARP1 is one of 18 human enzymes (also known as diphtheria toxin-type ADP-ribose transferases (ARTDs, See Glossary), reviewed in (2)), which can ADP ribosylate proteins. Further, families of ADPr hydrolases which cleave pADPr chains, protein-ADPr bonds or which remove the terminal mADPr from pADPr chains have been identified. In addition, multiple ADPr binding domains have been described. There are therefore both writers and erasers of ADPr as well as protein modules that can read mADPr and pADPr. Protein-mADPr, pADPr polymers (both protein-linked and soluble) and free mADPr may therefore represent distinct functional signaling effectors in the cell.
THE PARP FAMILY: Generating pADPr.
The 18 PARP proteins share a common catalytic domain and can be classified into three groups based on their enzymatic activity. PARP1, PARP2, PARP5a and PARP5b create branching poly-ADPr (pADPr) chains ranging from 2-200 ADPr units in length (3). Notably, PARP1 produces ~90% of cellular pADPr following genotoxic stress. PARP9 and PARP13 lack catalytic activity, and PARP18 is currently uncharacterized. The remaining 11 PARP enzymes add a single ADP-ribose (mADPr) unit onto their targets (3). An early experiment in rat liver estimated that there are ~1000 times more mADPr amino acids than pADPr amino acids (4), demonstrating the ubiquity of mADPr modification. PARP family members therefore specialize in adding either mADPr or in creating pADPr, often during stress responses.
PARPs and stress responses:
PARP1, PARP2 and PARP3 are DNA-dependent PARPs (5) which can be activated by single and double-strand DNA breaks (SSB and DSB (6, 7)) or by breaks in unstable DNA structures such as G-quadruplexes (8). During DSB repair, rapid (seconds to minutes) formation of pADPr (9) serves to concentrate many chromatin regulatory proteins at the damaged sites. This includes histones macroH2A1.1 and H3.3 (10–12), remodeling complexes (ALC1 and CHD2 (12, 13)) and many chromatin binding proteins (14, 15). The accumulation of proteins promotes PARP-mediated chromatin re-organization at DNA breaks and access to and repair of damaged chromatin (16, 17). Many of these repair proteins contain pADPr-binding modules, which aid in their recruitment and retention. PARP enzymes predominantly modify acidic amino acids, including aspartate and glutamate, although serine may be the major target during DSB repair (18). Further, even though PARP1 generates pADPr and PARP3 generates mADPr, they exhibit significant substrate overlap (19), suggesting that PARP3 may create the initial protein-mADPr while PARP1 extends the pADPr chain. Further, the exact protein target(s) for PARP proteins during repair are unclear – histones, chromatin binding proteins and auto-ADPr of PARP 1 at DNA breaks have all been reported (2). In addition, DNA-dependent PARPs can also add mADPr and pADPr directly to the DNA ends (9, 20–22). While protein-ADPr has been well studied, how cells control the extent of pADPr and remove mADPr and pADPr chains has received much less attention. Here, we will discuss a new class of enzymes which function to degrade pADPr chains and discuss emerging evidence that pADPr and mADPr produced by degradation of protein-pADPr can function as signaling intermediates.
pADPr HYDROLASES: Removing pADPr.
Because ADPr conjugated to proteins can regulate activity, it is important to dePARylate these targets and allow a return to the basal state. pADPr is rapidly turned over during DNA repair (23), suggesting that dynamic turnover is biologically important. Further, deletion of PARP 1 or the dePARylating enzyme PARG both have similar effects on DNA repair (24), suggesting that PARylation and dePARylation are tightly coupled. pADPr degradation is complex because of the difference between the chemical bonds linking ADPr to protein (via e.g. aspartate, glutamate, or serine) and those linking successive ADPr units (Figure 1, Key Figure). Several structurally distinct families of pADPr hydrolases have now been identified (Figure 1, Key Figure), including the macrodomain containing enzymes MacrodDl, MacroD2 and TARG1, which cleave the protein-ADPr bond; ARH1-3 (which contain a Dra-G like fold) and PARG, which cleave the ADPr polymer chain; and the recently identified members of the NUDIX and ENPP families, which can act directly on released mADPr. Metabolism of pADPr chains can therefore have multiple outcomes, including: (i) complete hydrolysis of pADPr chains by e.g. PARG, leading to free, soluble mADPr; (ii) release of mixed populations containing large, soluble nuclear or cytoplasmic pADPr polymers following endo-glycohydrolylase activity; (iii) trimming of pADPr chains to leave mADPr-protein (Figure 1, Key Figure).
Figure 1. Key Figure – DePARylation.
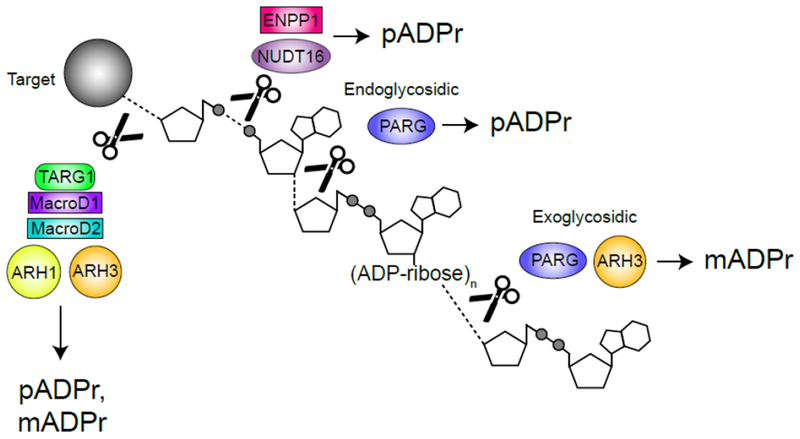
ADP(ribose) (ADPr) hydrolases cleave specific chemical linkages to release poly(ADP)ribose (pADPr) or mono(ADP)ribose (pADPr) from protein and DNA targets. PARG cleaves within the pADPr chain (endoglycosidically) to yield free pADPr and at the end of the chain (exoglycosidically) to produce mADPr (25, 26). ARH3 cleaves exoglycosidically to produce mADPr from the end of pADPr chains and from MARylated serines (28). ENPP1 and NUDT16 cleave phosphodiester bonds to produce protein-conjugated ribose-5’-phosphate and pADPr (37). TARG1, MacroD1 and MacroD2 cleave the terminal ADPr bond to release pADPr or mADPr from glutamate while ARH1 cleaves the terminal bond but only for targets MARylated on arginine (2, 30). ADP-ribosen represents chains of n = 2-100.
pADPr hydrolases.
PARG is a macrodomain-containing pADPr hydrolase that rapidly degrades pADPr chains following DNA damage. PARG exhibits both endo- (cleavage within chains) and exo- (cleavage from the end) hydrolase activity (25, 26) (Figure 1, Key Figure), giving it the potential to release large branched pADPr molecules and to trim pADPr chains. PARG functions primarily as an exo-glycohydrolase, although endo cleavage can occur when cells contain high levels of pADPr (26), with the released pADPr acting as a signal for apoptosis (discussed further below). ARH3, which is structurally unrelated to PARG, can also degrade pADPr, although it prefers longer pADPr chains and has lower activity (27). However, PARG cannot cleave the protein-ADPr bond. Instead, ARH3 preferentially cleaves ADPr attached to serine (28, 29), TARG1, MacroD1 or MacroD2 remove mADPr from aspartate and glutamate (30) and ARH1 cleaves arginine-ADPr. This requirement for multiple protein-ADPr hydrolases may reflect the distinct cellular locations (e.g. MacroD1 is enriched in the mitochondria (31), Figure 2) and the need to remove ADPr from different amino acids or from DNA (31).
Figure 2 – pADPr signaling beyond the nucleus.
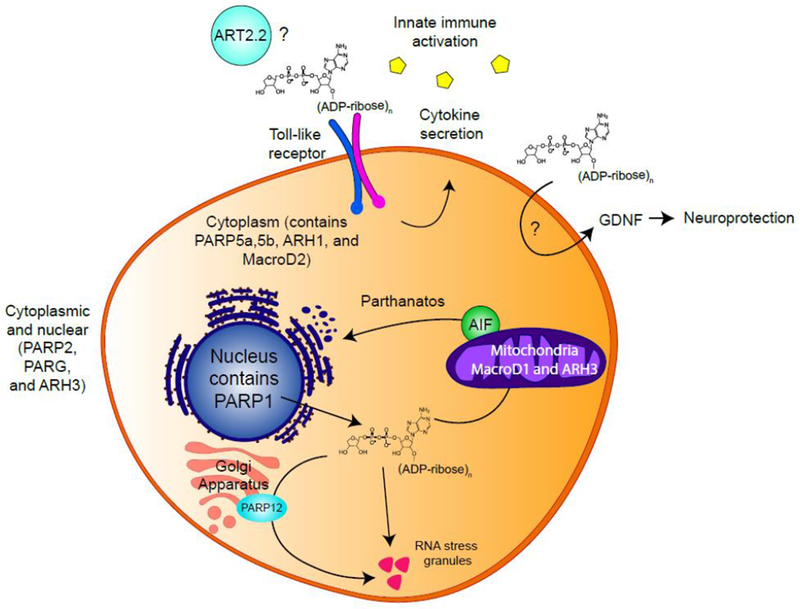
Different subsets of PARPs and pADPr hydrolases are found in different subcellular compartments including the nucleus, cytoplasm and mitochrondria. In the immune system, ART2.2 might catalyze pADPr synthesis extracellularly (43, 44). Extracellular pADPr activates TLR signaling, inducing macrophages to secrete cytokines (40). In the nervous system, extracellular pADPr increase Glial-cell Derived Neurotrophic Factor (GDNF) in astrocytes, playing roles in neuroprotection (41). Free cytoplasmic pADPr binds AIF triggering its release from the mitochondria into the cytoplasm, inducing parthanatos (47). Cytoplasmic pADPr also induces PARP12 to translocate from the Golgi apparatus to RNA stress granules, promoting stress granule function (53).
Complete removal of pADPr from proteins will therefore require the coordinated action of PARG, which hydrolyses ADPr chains, and a specific protein-ADPr hydrolase to remove the protein-ADPr link. Currently, it is unclear if TARG1 (or other protein-ADPr hydrolases) can remove the entire pADPr chain on its own (by cleaving the link to protein), or if it requires initial trimming of pADPr by PARG to allow access to the last ADPr. The most commonly used pADPr antibody preferentially binds long pADPr chains over short ones, and cannot detect mADPr. The rapid appearance and loss of pADPr during processes like DNA repair monitored using immunostaining approaches may therefore fail to detect mADPr proteins or short pADPr chains remaining following PARG processing of pADPr. Given the large number of DNA damage response (DDR) proteins that can associate with ADPr, residual mADPr may recruit or retain macrodomain proteins such as macroH2A1.1 (32, 33) or ALC1 (13) at sites of DNA damage. In fact, deficiency of the serine-ADPr hydrolase TARG1 is associated with increased sensitivity to DNA damage (34) and TARG1 is recruited to DNA breaks. This implies that removal of both pADPr and protein-ADPr are critical for completion of DNA repair, and that this process may be tightly linked to specific ADPr hydrolases. However, a better understanding of the substrate specificity and cellular location of these enzymes is still needed.
Other ADPr hydrolases:
Two recently identified enzymes, NUDT16 (35, 36) and ENPP1 (37) can also cleave protein linked ADPr, but leave a ribose-5-phosphate attached to the protein (Figure 1, Key Figure). These novel pADPr hydrolases have not been characterized nor has the influence of ribose-5-phosphate on protein function been explored.
EXTRACELLULAR mADPr/pADPr and SIGNALING:
ENPP1 is an extracellular protein, suggesting that pADPr signaling can occur in the extracellular space. Serum antibodies against pADPr and/or PARG have been found in patients with Lupus, Ulcerative Colitis and Alzheimer’s disease (38, 39), indicating that extracellular pADPr might play an active role in disease pathology. Extracellular pADPr (but not mADPr) can activate toll-like receptor (TLR) signaling, which induces macrophages to secrete cytokines and promotes innate immune activation (40) (Figure 2). In the nervous system, extracellular pADPr (but not mADPr) increased Glial-cell Derived Neurotrophic Factor (GDNF) in astrocytes (41). Consistent with the neuroprotective functions of GDNF, injection of pADPr into brain striatum mitigated disease phenotypes in a rodent model of Parkinson’s disease. Both studies (40, 41) indicate that uptake of extracellular pADPr can regulate signaling in macrophages and neuronal cells. The origin of extracellular pADPr is unclear as PARP enzymes are largely intracellular (2), suggesting that pADPr synthesized following oxidative stress may be released into the extracellular space. Alternatively, ecto-ADP ribosyl transferases (ecto-ARTs), a poorly characterized family of ribosyl transferases with extracellular catalytic domains (42), may be responsible. ART2.2, an ecto-ART anchored to the surface of T cells and lymphocytes is a potential candidate, although its ability to generate pADPr in vivo requires additional characterization (43, 44). Further work identifying extracellular enzymes that can synthesize and degrade pADPr and determining whether mADPr and pADPr are exported from cells as soluble signaling molecules is needed to address this.
CYTOPLASMIC mADPr/pADPr and SIGNALING:
In the cytoplasm, free pADPr is important for the parthanatos cell death pathway (Figure 2), a PARP1-dependent pathway activated following ischemia-reperfusion injury (45, 46). pADPr binds directly to apoptosis-inducing factor (AIF) (47), releasing AIF from the mitochondria into the cytoplasm (45, 46). AIF then chaperones Macrophage Migratory Inhibitory Factor (MIF), a nuclease, into the nucleus, leading to MIF-dependent fragmentation of the genome and parthanatos (48). Delivering synthetic pADPr to the cytoplasm can activate parthanatos and free pADPr of increasing length and complexity is the most potent trigger (45, 46). Further, ARH3 and PARG function together to degrade free, cytoplasmic pADPr and protect cells from parthanatos (49).
These studies imply that free pADPr is sufficient to activate parthanatos. However, whether the physiological substrate is free, soluble pADPr or a (soluble) protein-pADPr remains an open question. It is also unclear how nuclear pADPr exits the nucleus. Additionally, there are indications that pADPr may have cytoplasmic functions during normal cell metabolism. In mitotic and meiotic cells, cytoplasmic pADPr is important for spindle formation and positioning (50, 51). Five PARP enzymes and PARG are recruited to and support the function of RNA stress granules upon cellular stress (52). pADPr causes PARP12 to translocate from the Golgi apparatus to RNA stress granules to promote stress granule function (53) (Figure 2). Taken together, these data indicate that the accumulation of free pADPr in the cytoplasm can mediate cellular stress responses such as parthanatos and RNA stress granule formation. Tight control of the synthesis and degradation of protein-pADPr chains is therefore important for regulating stress and apoptotic responses in cells.
READERS of mADPr and pADPr BINDING DOMAINS.
At least five different pADPr binding domains have been described (Table 1). Distinct ADPr reader domains can discriminate between pADPr and mADPr and between soluble and protein-linked ADPr. pADPr and mADPr may therefore regulate distinct sets of proteins depending on the specificity of the ADPr domain on the target protein.
Table 1 –
Non-covalent ADPr binding motifs
| Motif | Abbreviation | Description | Moiety Read | Examples |
|---|---|---|---|---|
| Poly(ADP-ribose)-binding motif | PBM | ~20 aa motif (hydrophobic aa spaced by basic aa) | Unknown | AIF (47), WRN (57), histone H2A/H2B/H3/H4 (119), p53 (59) XPA, p21, XRCC1/5/6, DNA-PKcs, TOPI, DNA ligase 3 (54) |
| WWE | ~80–100 aa | iso-ADP-ribose | Iduna/RNF146 (72), HUWE1, ULF, Deltex1, Deltex2, Deltex4, PARP11 (71) | |
| Macro | 130-190 amino acid motif | terminal ADP-ribose | ALC1 (63, 69), MacroH2A (120), PARP9, PARP14, PARP15, PARG, TARG1, MacroD1, MacroD2 (60) | |
| PAR-binding zinc finger | PBZ | ~30 aa Cys2-His2 (C8-C8-H8-H) | Two adjacent ADP-ribose moieties | APLF (121), CHFR (122), CTCF (123) |
| PAR-binding regulatory motif | PbR | Cys2-His2 (C8-C6-H8-H) | Unknown | CHK1 (124) |
PAR binding motif:
The PAR binding motif (PBM) features hydrophobic amino acids interspersed with charged basic residues (54, 55). The net positive charge of PBMs may promote interaction with negatively charged pADPr chains by electrostatic interaction (56). PBMs are found in AIF, which regulates parthanatos (47) and many DNA damage response proteins (Table 1). PBMs often overlap with other functionally important domains (54), indicating that besides recruiting proteins to pADPr chains, they may also play roles in regulating protein function. The PBM of the Werner syndrome protein (WRN) overlaps with its exonuclease and DNA binding domains and its binding of short or long chains of free pADPr interferes with helicase and exonuclease activities during DSB repair (57). Whether WRN interacts with protein-linked or free pADPr remains an open question, since both forms are inhibitory in biochemical assays (57). Similarly, the PBMs of XPA and p53 overlap with their DNA binding domains and interactions with pADPr abrogate their DNA binding activity during nucleotide excision repair and transcriptional activation, respectively (58, 59) (Figure 3).
Figure 3 – pADPr signaling in the nucleus.
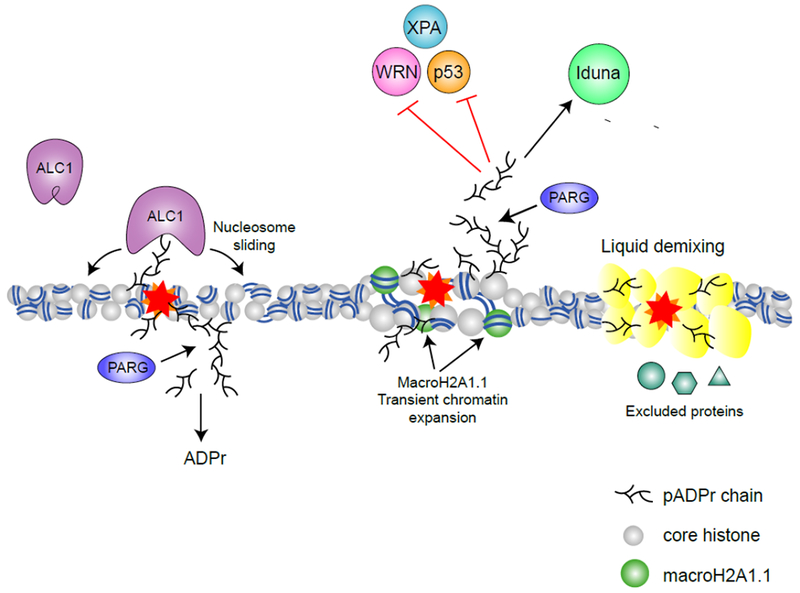
Through binding of pADPr, reader proteins are recruited and functionally regulated during DNA damage response. The chromatin remodeler ALC1 is recruited and activated by pADPr binding, promoting nucleosome sliding (63). MacroH2A1.1 is also recruited to DNA damage sites by pADPr and promotes transient chromatin expansion (32). pADPr binding can regulate protein functions either negatively (e.g. inhibiting nuclease activity of WRN (57), NER activity of XPA (58) and DNA binding activity of p53 (59)) or positively (e.g. activating the ATPase activity of ALC1 (63) and the E3 ligase activity of Iduna (73)). pADPr can also cause liquid demixing, forming a membrane-less organelle that retain some proteins but excludes others (76).
Macrodomains:
Macrodomains are globular pADPr binding modules found in diverse proteins, including macroPARPs (PARP9, PARP14 and PARP15), ADPr hydrolases (PARG, TARG1, MacroD1 and MacroD2) (60), macroH2A1.1 (61, 62) and the chromatin remodeler ALC1 (63). Macrodomains bind mADPr or the terminal ADPr residue in pADPr chains (2). MacroH2A, a histone variant important for maintaining chromatin organization, is encoded by two genes: MacroH2A1 and MacroH2A2. MacroH2A1 has two transcriptional variants: macroH2A1.1 and macroH2A1.2. Although all three proteins contain macrodomains, only macroH2A1.1 binds pADRr and mADPr (33). Upon DNA damage, macroH2A1.1 undergoes rapid, pADPr-dependent recruitment to damaged chromatin (Figure 3) (32, 33) while macroH2A1.2 is transiently excluded (11). At later time points, which coincide with degradation of pADPr to mADPr, macroH2A1.2 re-accumulates to promote homologous recombination and chromatin re-condensation (11). The sequential binding of macroH2A1.1 followed by macroH2A1.2 also coincides with the initial chromatin relaxation and subsequent chromatin condensation after DNA damage (11). This has led to the idea that recruitment of macroH2A1.1 to damaged chromatin initially promotes open chromatin and facilitates repair. In support of this model, in undamaged cells, binding of macroH2A1.1 to pADPr facilitates its localization to acetylated chromatin and even promotes H2B-acetylation, a mark associated with relaxed chromatin (64). Subsequent removal of pADPr and release of macroH2A1.1 following repair then allows for chromatin repacking.
ALC1/CHD1L is an ATP-dependent chromatin remodeler that functions during DNA repair (Figure 3) (65). ALC1 contains a macrodomain that two distinct functions: recruitment of ALC1 to damage sites and activation of ALC1’s ATP-dependent nucleosome sliding activity (13, 66). In the basal (“off’) state, ALC1’s macrodomain interacts with the ATPase domain to auto-inhibit its activity. Binding of pADPr relieves inhibition of the ATPase domain and activates ALC1’s chromatin remodeling activity (63, 67). ALC1 binds pADPr chains, rather than the mADPr recognized by canonical macrodomains (68). This difference may reside in a stretch of basic amino acids adjacent to the ADPr binding pocket (69), suggesting that electrostatic interactions, such as those seen in PAR motifs, may contribute to ALC1-pADPr interaction. Further, nucleosome sliding by ALC1 involves significant motion relative to the underlying DNA. If ALC1 is tethered to protein-pADPr on the nucleosome, this may restrict its motion, and therefore the extent of nucleosome sliding relative to the site of damage. Alternatively, binding of free pADPr to ALC1’s macrodomain would allow ALC1 to move freely along the chromatin. Switching between these two modes of operation may allow cells to control the extent of chromatin remodeling during DNA repair.
WWE and PBZ domains:
The E3 Ligase Iduna/RNF146 binds pADPr through its WWE domain (Figure 3). WWE domains recognize the base-ribose link between two ADPr units, termed iso-ADPr (70, 71), so that WWE domains require pADPr for binding. Iso-ADPr binding induces a conformational change in Iduna which converts it to an active E3 ligase. Iduna then ubiquitinates DNA repair factors, including PARP1, XRCC1, KU70, and LIG3 (72, 73). Because several E3 ligases contain WWE domains, this may represent a general mechanism of activation for these enzymes (71). The PAR-binding zinc finger (PBZ) domain is ~30 amino acids long with a Cys2-His2 (C2H2) zinc finger motif. PBZ domains were first described in the NHEJ factor APLF (74) in which tandem PBZ domains are required for phosphorylation by ATM (75), indicating that pADPr signaling can drive crosstalk with other post-translational modifications.
Liquid demixing.
Local increases in pADPr during stress responses or DNA repair can trigger liquid demixing. In liquid demixing, pADPr (either protein-pADPr or soluble pADPr chains released by PARG) initiates the formation of liquid droplets by seeding aggregation of intrinsically disordered proteins, such as FUS and EWS (76). These aggregates of pADPr are localized regions with a high density of negative charge, and form a membrane-less organelle that retains some proteins but excludes others (76). The ability of pADPr-dependent liquid droplets to preferentially solubilize certain proteins (e.g. MDC1) may explain the striking number of proteins (>100) that accumulate on damaged chromatin in a PARP-dependent manner (77). Soluble pADPr can nucleate droplet formation in vitro (76), raising the question of whether free or protein-bound pADPr (or both) can promote liquid demixing in vivo. Liquid demixing by pADPr may compartmentalize damaged chromatin, maintaining damaged DNA ends in close proximity and promoting repair (Figure 3). Once repair is complete, degradation of pADPr dissolves these transient membrane-less organelles, to relieve the temporal exclusion. Liquid demixing has also been invoked to describe pADPr aggregation at spindles (50) and in RNA stress granules (52). Moreover, the recent findings that heterochromatin protein 1 alpha (HP1α) undergoes liquid demixing to drive heterochromatin formation (78, 79) are intriguing because HP1α is also found to be PARylated (80) and recruited to chromatin in a PAR-dependent manner (81).
The Fate of mADPr.
TRPM2 and cell death:
pADPr degradation by PARG following DNA damage creates a large pool of free mADPr (Figure 4) which can function as a signaling molecule. Transient receptor potential melastatin 2 (TRPM2), a Ca2+-ion channel, binds mADPr via its cytoplasmic NUDT9H domain (82–84) (Figure 4). Ca2+ flux through the TRPM2 channel depends on the relative balance of PARP1 and PARG, with mADPr arising from pADPr degradation during oxidative stress promoting Ca2+ influx (85). This allows TRPM2 to function as a sensor of oxidative stress. However, high levels of mADPr can fully activate TRPM2 channels, leading to toxic intracellular Ca2+ levels and cell death (86). Catabolism of mADPr is therefore critical to turn off mADPr signaling via TRPM2 and other proteins.
Figure 4 – The fate of free mADPr.
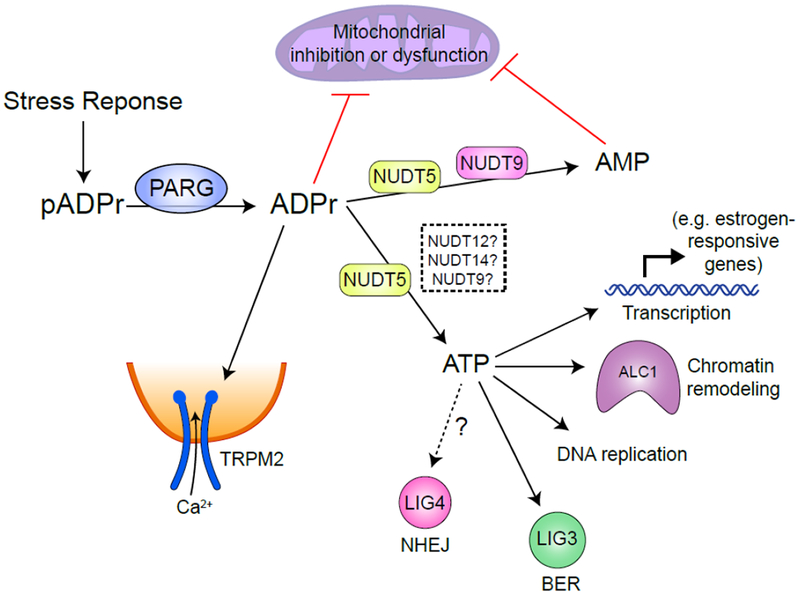
Following a stress response, a large pool of free mADPr is generated by PARP and PARG catalyzed PAR polymerization and degradation in the nucleus. Free mADPr activates TRPM2 cation channel, inducing Ca2+ influx into cells (84). NUDT9 catalyzes mADPr to AMP (91), while NUDT5 possess two catalytical activities to convert mADPr to either AMP or ATP (94). Over-production of mADPr or AMP can lead to mitochondrial dysfunction (92). ATP generated by NUDT5 (whether other Nudix family proteins also catalyze this process is undetermined) may be used locally for transcription, chromatin remodeling during DNA replication or repair.
NUDIX family and mADPr degradation:
The rapid synthesis and degradation of pADPr can deplete ATP and NAD+ in cells (87). pADPr can also bind to and inhibit hexokinase, which catalyzes the initial step of glycolysis (88, 89). Two members of the NUDIX (nucleoside diphosphates linked to other moiety, X) hydrolase family, NUDT5 (90) and NUDT9 (91) can further hydrolyze mADPr to adenosine monophosphate (AMP) and D-ribose 5-phosphate. Elevated AMP can lead to mitochondrial dysfunction (92). Thus sustained activation of PARP during stress responses and degradation of pADPr can lead to bioenergetic collapse through ATP and NAD+ depletion and cell death. Interestingly, NUDT5 (Figure 4) also possess mADPr pyrophosphorylase activity, which can convert mADPr into ATP (93). This provides a potential mechanism for regenerating or maintaining local ATP levels and limiting the rapid depletion of ATP which is a hallmark of sustained PARP activation.
NUDT5 forms phosphorylated dimers which predominantly convert mADPr to AMP and pyrophosphate. However, dephosphorylation of NUDT5 destabilizes the dimer and favors synthesis of ATP, suggesting that NUDT5 metabolism of mADPr to AMP or ATP is dictated by phosphorylation (94). However, the NUDT5 kinase or phosphatase responsible for this functional switch is not known. PARylation is also important for regulating chromatin structure and gene transcription. In breast cancer cells, NUDT5 is frequently over-expressed and promotes estrogen-dependent chromatin remodeling and transcription by increasing local ATP levels (94). NUDT5 associates with PARP1 and PARG and coupled synthesis and catabolism of pADPr by PARP1/PARG/NUDT5 complexes can generate a local source of ATP for hormone-induced chromatin remodeling. The ATP generated by NUDT5 plays a critical role in maintaining displacement of histones at estrogen-responsive genes and thereby supporting transcription and cell growth. NUDT5 can therefore provide ATP to support chromatin reorganization by remodeling ATPases which promote transcription. Many aspects of the DDR also require energy. For example, ATP-dependent chromatin remodelers such as ALC1 or p400 (13, 16, 17) reorganize chromatin during DNA repair. ATP from pADPr has also been reported to fuel DNA replication (95), the ligation step of base excision repair (93) and repair by non-homologous end joining (5). In addition, cells lacking NUDT5 show increased sensitivity to DNA damage (94). The ability of NUDT5 to convert mADPr to ATP during DNA repair may maintain local ATP concentrations for kinase or ligase activity and ATP-dependent chromatin remodeling. However, the recent discovery that pADPr can be directly coupled to DNA (9) indicates that there are significant complexities to mADPr metabolism during DNA repair which require further experimental investigation. Locally generated ATP may also be funneled to the NAD+ salvage pathway during PARP1 hyper-activation to replenish pools of NAD+ (96). Further, ATP has hydrotropic properties (97) which may help redissolve hydrophobic proteins that had undergone PAR-dependent liquid demixing (76). NUDT5, and NUDT12 and NUDT14, which may also use ADPr as a substrate (98), may remove mADPr to terminate signaling, and recycle a fraction of mADPr to increase local ATP levels which were depleted by the initial pADPr generation (Figure 4). Understanding how NUDT5 and other proteins participate in pADPr metabolism during both transcriptional activation and DNA repair is an exciting area for further study.
Targeting pADPr metabolism for therapy:
The key role of PARP enzymes in DNA repair motivated the development of PARP inhibitors, which produce synthetic lethality in tumors with deficiency in homologous recombination (HR). Inhibition of PARP enzymes shows promise as a monotherapy in DNA repair-deficient (BRCA1/BRCA2) tumors and can sensitize cells to ionizing radiation (IR) (99). The past five years have seen the first FDA approvals of the PARP inhibitor Olaparib for patients with germline BRCA1/BRCA2 mutations. However, one of the central challenges of PARP inhibitors is therapeutic resistance that develops either as a result of re-activation of the HR pathway or unknown mechanisms. A genetic screen identified loss of PARG as a mechanism to confer PARP inhibitor resistance in BRCA2−/− cells and demonstrated mosaic loss of PARG in human breast and ovarian cancers, suggesting that PARG−/− clones might be selected by treatment with PARP inhibitors. Despite conferring PARP inhibitor resistance, loss of PARG led to new vulnerabilities, including enhanced sensitivity to IR and the alkylating chemotherapeutic agent temozolomide (100). Further study is needed to determine whether PARG suppression is a physiological mechanism of PARP inhibitor resistance in human tumors.
Both synthesis and degradation of pADPr are important for functional DNA repair processes (101, 102). Therefore, targeting PARG or other enzymes involved in pADPr degradation may also have clinical applications. PARG inhibitors also sensitize cells to IR, suggesting an additional strategy to disrupt pADPr homeostasis (103, 104). The PARG dependency of HR-deficient tumors is less straightforward. Recent reports examining genetic susceptibilities between PARG and BRCA1/2 have shown contradictory results, suggesting that the broader cellular context or PARG expression level may be important. (105–107). Further, PARG inhibitor sensitivity was independent of BRCA1/BRCA2 status, suggesting that PARG inhibitors could have different clinical applications from PARP inhibitors (108). For example, PARG inhibitors synergize with Ibrutinib, a clinically useful kinase inhibitor, suggesting the possibility of new vulnerabilities (108). Other possible PARG inhibitor synergies await discovery. Other therapeutic strategies to disrupt pADPr homeostasis are in earlier stages of development. A novel screening platform identified inhibitors of MacroD1 (109), which may be effective in treating lung, breast and pancreatic cancers which overexpress MacroD1 (110). A NUDT5 inhibitor which inhibits formation of ATP from mADPr blocks the proliferation of breast cancer cells (111), providing a third potential approach for interfering with pADPr metabolism.
Finally, small molecules that disrupt pADPr metabolism may have applications beyond oncology (reviewed (112)). For example, PARP inhibitors are being examined for prevention of reperfusion injury following heart attack and pulmonary hypertension (113, 114) and may have applications in stroke and neurodegeneration (112). Recent studies have identified familial mutations in TARG and ARH3 that lead to accumulation of pADPr and cause rare neurodegenerative diseases (34, 115, 116). Several more common neurodegenerative diseases including Alzheimer’s and Parkinson’s (AD and PD) are characterized by hyper-activation of PARP1 leading to pADPr accumulation in neurons. Interestingly, neuronal cells are particularly vulnerable to mitochondrial toxicity associated with PARP1 hyperactivation and pADPr accumulation. Recent data from C. elegcms and mice shows that inhibiting pADPr accumulation, either by inhibiting PARP enzymes or expressing PARG, improves neuronal regenerative potential (117). Further, in mouse models of AD and PD, PARP1-deficiency mitigates the disease phenotype (118). Taken together, these data suggest that disrupting pADPr metabolism is neurodegenerative diseases may be a promising strategy.
CONCLUDING REMARKS
Since the discovery of ADPr addition to proteins, there have been significant advances in our understanding of the role of this modification in cell function and cell stress. In addition to a family of PARPs that can write this modification, a second, diverse group of erasers has been identified. The coupled action of these readers and writers leads to both the rapid accumulation of complex pADPr chains on proteins, as well as the degradation of these chains into both mADPr and large, soluble pADPr complexes. Much work remains to be done to determine how modification of different amino-acid residues through mADPr alters target protein function (See Outstanding Questions). In particular, further studies are needed to reveal the roles and regulation of different mADPr hydrolases which remove ADPr from specific residues in response to stress. In addition to writers and erasers of ADPr, there are several families of ADPr readers which can bind to mADPr or pADPr, providing the potential for regulation of protein targets, either by direct protein tethering to soluble mADPr/pADPr, or by interaction with ADPr bound to proteins. Finally, turnover of free mADPr in the cell has received the least attention, but removal of mADPr is critical for limiting apoptotic events and negative impacts on ATP production in the cell. Further, the potential that mADPr can be recycled back to ATP provides a novel method for maintaining local ATP levels during energy intensive pADPr signaling and stress. ADPr provides a rich signaling mechanism for controlling stress responses, from directly modifying protein function, to promoting protein interaction and the potential for pADPr degradation to create a new signaling molecule, mADPr. pADPr and its metabolites therefore represent both a flexible post-translational modification and a new signaling molecule which provides rapid and dynamic regulation of cell function during cellular stress and related events. Unraveling the specificity of this signaling pathway and the function of its diverse writers, readers and erasers during stress is needed to fully appreciate the importance of this signaling pathway.
HIGHLIGHTS.
DNA damage promotes rapid production of poly(ADP-ribose) (pADPr) chains on damaged chromatin and is critical for initiating recruitment of DNA repair proteins.
pADPr then undergoes rapid (seconds-minutes) degradation by PARG and related enzymes, releasing monoADP-ribose (mADPr) and soluble pADPr molecules into the nucleoplasm and cytoplasm.
Many stress response proteins contain ADP-ribose binding modules with specificity for mADPr or pADPr, so that binding of specific pADPr metabolites can regulate DNA repair proteins and apoptosis during stress.
A family of hydrolases, including NUDT5, which degrade mADPr, can remove mADPr and terminate stress responses while generating useful metabolites.
pADPr and its metabolites may function as novel stress induced signaling molecules which regulate the function of stress and DNA repair proteins.
Outstanding Questions.
NUDT5 is not the only NUDIX enzyme that could convert ADPr into AMP or ATP. What are the cellular roles of NUDT9, 12, and 14 and could they also participate in stress response?
Even though they have opposing activities in the cell, deletion of PARP1 or PARG results in similar phenotypes of sensitivity to genotoxic stress. It remains to be explored whether PARP1 and PARG act in concert in some way that contributes to the similarity of the phenotype.
Are there other ATP-dependent chromatin remodelers that derive ATP from similar recycling mechanisms? For example, FACT and ISWI have genetic interactions with PARP1 – could they also use ATP that comes from recycled pADPr?
Are enzymes that require mobility on DNA restricted by being tethered to pADPr chains or does cleavage of the pADPr chain grant them mobility whilst maintaining the activating interaction?
Does the NUTD9H domain in the TRPM2 Ca2+ channel define another ADPr-binding domain that might be found in other proteins as well?
Acknowledgements:
Supported by NIH grants CA177884 and CA64585 (BDP) and K01AG056554 (TAD).
Glossary
- DNA Damage Response (DDR)
A complicated network of pathways that mediate the detection, signaling and repair of DNA damage
- ARTD
ADP-ribose transferases with diphtheria toxin homology that transfer one or more ADP-ribose groups from NAD+ to target proteins
- G-quadruplexes
Nucleic acid secondary structures in which two or more G-tetrads (four G residues linked by the sugar–phosphate backbone and connected through Hoogsteen-type hydrogen bonds) stack on top of each other
- PARylation
Poly(ADP-ribose)ylation, a post-translational modification with linear or branched polymeric chains of ADPr subunits
- DePARylation
Removal of poly(ADP-ribose) chains from target proteins or DNA catalyzed by poly(ADP-ribose) hydrolases
- Ecto-ART/ARTC
ADP ribosyl transferases located on cell surface or extracellular compartment that transfer one ADPr group from extracellular NAD+ to target proteins
- Parthanatos
A programmed cell death process which is mediated by pADPr
- Stress Granule
Dense aggregations of proteins and RNAs formed in the cytosol under stress
- Homologous Recombination (HR)
The error-free DNA repair pathway that occurs through homologous strand exchange and takes place in the late S-G2 phases of the cell cycle
- Chromatin Remodeling
The dynamic rearrangement of chromatin architecture to allow access of condensed genomic DNA, including histone variant incorporation, histone depletion and/or histone sliding
- Liquid Demixing
An aggregation of intrinsically disordered proteins by phase separation from the soluble intracellular space
- Intrinsically Disordered Protein (IDP)
Dynamically disordered proteins that feature biased amino acid composition and low sequence complexity
- Mitochondrial Dysfunction
Loss of function in mitochondria caused by the reduction of oxidative phosphorylation efficiency and ATP production
- Base Excision Repair (BER)
The predominant DNA damage repair pathway for the processing of small base lesions, derived from oxidation, deamination, and alkylation damage. It is initiated by a DNA glycosylase that recognizes and removes the damaged base and completed by short- or long-patch repair
- Non-Homologous End Joining (NHEJ)
The predominant pathway to repair DNA double stand breaks by religation of DNA ends with limited processing
- NAD+ salvage pathway
Production of NAD+ from nicotinamide, an end product of NAD+ consumption, catalyzed by nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide mononucleotide adenylyltransferase (NMNAT)
- Monotherapy
The treatment of disease or disorder with use of only one type of treatment such as radiation therapy. In drug therapy it refers to treatment with a single drug molecule
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chambon P, Weill JD, & Mandel P (1963) Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochemical and biophysical research communications 11:39–43. [DOI] [PubMed] [Google Scholar]
- 2.Hottiger MO (2015) Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annual review of biochemistry 84:227–263. [DOI] [PubMed] [Google Scholar]
- 3.Vyas S, et al. (2014) Family-wide analysis of poly(ADP-ribose) polymerase activity. Nature communications 5:4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bredehorst R, Wielckens K, Adamietz P, Steinhagen-Thiessen E, & Hilz H (1981) Mono(ADP-ribosyl)ation and poly(ADP-ribosyl)ation of proteins in developing liver and in hepatomas: relation of conjugate subfractions to metabolic competence and proliferation rates. European journal of biochemistry 120(2):267–274. [DOI] [PubMed] [Google Scholar]
- 5.Langelier MF, Riccio AA, & Pascal JM (2014) PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic acids research 42(12):7762–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eustermann S, et al. (2015) Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Molecular cell 60(5):742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck C, Robert I, Reina-San-Martin B, Schreiber V, & Dantzer F (2014) Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Experimental cell research 329(1):18–25. [DOI] [PubMed] [Google Scholar]
- 8.Day TA, et al. (2017) PARP3 is a promoter of chromosomal rearrangements and limits G4 DNA. Nature communications 8:15110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munnur D & Ahel I (2017) Reversible mono-ADP-ribosylation of DNA breaks. The FEBS journal 284(23):4002–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu C, Xu Y, Gursoy-Yuzugullu O, & Price BD (2012) The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. Febs Letters 586(21):3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khurana S, et al. (2014) A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell reports 8(4):1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luijsterburg MS, et al. (2016) PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol Cell 61(4):547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahel D, et al. (2009) Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325(5945):1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou DM, et al. (2010) A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 107(43):18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gursoy-Yuzugullu O, Carman C, & Price BD (2017) Spatially restricted loading of BRD2 at DNA double-strand breaks protects H4 acetylation domains and promotes DNA repair. Scientific reports 7(1):12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhar S, Gursoy-Yuzugullu O, Parasuram R, & Price BD (2017) The tale of a tail: histone H4 acetylation and the repair of DNA breaks. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 372(1731). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price BD & D’Andrea AD (2013) Chromatin remodeling at DNA double-strand breaks. Cell 152(6):1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palazzo L, et al. (2018) Serine is the major residue for ADP-ribosylation upon DNA damage. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson BA, et al. (2016) Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353(6294):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belousova EA, Ishchenko capital A C, & Lavrik OI (2018) Dna is a New Target of Parp3. Scientific reports 8(1):4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarkovic G, et al. (2018) Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic acids research 46(5):2417–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talhaoui I, et al. (2016) Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic acids research 44(19):9279–9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortusewicz O, Fouquerel E, Ame JC, Leonhardt H, & Schreiber V (2011) PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic acids research 39(12):5045–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirai H, et al. (2013) PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell death & disease 4:e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slade D, et al. (2011) The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477(7366):616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barkauskaite E, et al. (2013) Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nature communications 4:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oka S, Kato J, & Moss J (2006) Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. The Journal of biological chemistry 281(2):705–713. [DOI] [PubMed] [Google Scholar]
- 28.Abplanalp J, et al. (2017) Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nature communications 8(1):2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fontana P, et al. (2017) Serine ADP-ribosylation reversal by the hydrolase ARH3. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenthal F, et al. (2013) Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nature structural & molecular biology 20(4):502–507. [DOI] [PubMed] [Google Scholar]
- 31.Agnew T, et al. (2018) MacroD1 Is a Promiscuous ADP-Ribosyl Hydrolase Localized to Mitochondria. Frontiers in microbiology 9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timinszky G, et al. (2009) A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nature structural & molecular biology 16(9):923–929. [DOI] [PubMed] [Google Scholar]
- 33.Xu C, Xu Y, Gursoy-Yuzugullu O, & Price BD (2012) The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS letters 586(21):3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharifi R, et al. (2013) Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. The EMBO journal 32(9):1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palazzo L, et al. (2015) Processing of protein ADP-ribosylation by Nudix hydrolases. Biochem J 468(2):293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniels CM, Thirawatananond P, Ong SE, Gabelli SB, & Leung AK (2015) Nudix hydrolases degrade protein-conjugated ADP-ribose. Scientific reports 5:18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palazzo L, et al. (2016) ENPP1 processes protein ADP-ribosylation in vitro. The FEBS journal 283(18):3371–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamanaka S, et al. (2018) Investigation of novel biomarkers for predicting the clinical course in patients with ulcerative colitis. Journal of gastroenterology and hepatology. [DOI] [PubMed] [Google Scholar]
- 39.Kanai Y, Akatsu H, Iizuka H, & Morimoto C (2007) Could serum antibody to poly(ADP-ribose) and/or histone H1 be marker for senile dementia of Alzheimer type? Annals of the New York Academy of Sciences 1109:338–344. [DOI] [PubMed] [Google Scholar]
- 40.Krukenberg KA, Kim S, Tan ES, Maliga Z, & Mitchison TJ (2015) Extracellular poly(ADP-ribose) is a pro-inflammatory signal for macrophages. Chemistry & biology 22(4):446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakajima H, et al. (2017) Extracellular poly(ADP-ribose) is a neurotrophic signal that upregulates glial cell line-derived neurotrophic factor (GDNF) levels in vitro and in vivo. Biochemical and biophysical research communications 484(2):385–389. [DOI] [PubMed] [Google Scholar]
- 42.Cohen MS & Chang P (2018) Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nature chemical biology 14(3):236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrison AR, et al. (2006) ART2, a T cell surface mono-ADP-ribosyltransferase, generates extracellular poly(ADP-ribose). The Journal of biological chemistry 281(44):33363–33372. [DOI] [PubMed] [Google Scholar]
- 44.Rissiek B, et al. (2017) Ecto-ADP-ribosyltransferase ARTC2.1 functionally modulates FcgammaR1 and FcgammaR2B on murine microglia. Scientific reports 7(1):16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu SW, et al. (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proceedings of the National Academy of Sciences of the United States of America 103(48):18314–18319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrabi SA, et al. (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proceedings of the National Academy of Sciences of the United States of America 103(48):18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. (2011) Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Science signaling 4(167):ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, et al. (2016) A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354(6308). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mashimo M, Kato J, & Moss J (2013) ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proceedings of the National Academy of Sciences of the United States of America 110(47):18964–18969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie B, et al. (2018) Poly(ADP-ribose) mediates asymmetric division of mouse oocyte. Cell research 28(4):462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang P, Jacobson MK, & Mitchison TJ (2004) Poly(ADP-ribose) is required for spindle assembly and structure. Nature 432(7017):645–649. [DOI] [PubMed] [Google Scholar]
- 52.Leung AK, et al. (2011) Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular cell 42(4):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Catara G, et al. (2017) PARP1-produced poly-ADP-ribose causes the PARP12 translocation to stress granules and impairment of Golgi complex functions. Scientific reports 7(1):14035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pleschke JM, Kleczkowska HE, Strohm M, & Althaus FR (2000) Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. The Journal of biological chemistry 275(52):40974–40980. [DOI] [PubMed] [Google Scholar]
- 55.Gagne JP, et al. (2008) Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic acids research 36(22):6959–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teloni F & Altmeyer M (2016) Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic acids research 44(3):993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Popp O, et al. (2013) Site-specific noncovalent interaction of the biopolymer poly(ADP-ribose) with the Werner syndrome protein regulates protein functions. ACS chemical biology 8(1):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fischer JM, et al. (2014) Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. The FEBS journal 281(16):3625–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischbach A, et al. (2018) The C-terminal domain of p53 orchestrates the interplay between non-covalent and covalent poly(ADP-ribosyl)ation of p53 by PARP1. Nucleic acids research 46(2):804–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rack JG, Perina D, & Ahel I (2016) Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annual review of biochemistry 85:431–454. [DOI] [PubMed] [Google Scholar]
- 61.Karras GI, et al. (2005) The macro domain is an ADP-ribose binding module. The EMBO journal 24(11):1911–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Douet J, et al. (2017) MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. Journal of cell science 130(9):1570–1582. [DOI] [PubMed] [Google Scholar]
- 63.Singh HR, et al. (2017) A Poly-ADP-Ribose Trigger Releases the Auto-Inhibition of a Chromatin Remodeling Oncogene. Molecular cell 68(5):860–871 e867. [DOI] [PubMed] [Google Scholar]
- 64.Chen H, et al. (2014) MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nature structural & molecular biology 21(11):981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sellou H, et al. (2016) The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Molecular biology of the cell 27(24):3791–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottschalk AJ, et al. (2009) Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proceedings of the National Academy of Sciences of the United States of America 106(33):13770–13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vilchez Larrea SC, et al. (2013) Host cell poly(ADP-ribose) glycohydrolase is crucial for Trypanosoma cruzi infection cycle. PloS one 8(6):e67356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gottschalk AJ, Trivedi RD, Conaway JW, & Conaway RC (2012) Activation of the SNF2 family ATPase ALC1 by poly(ADP-ribose) in a stable ALC1.PARP1.nucleosome intermediate. The Journal of biological chemistry 287(52):43527–43532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lehmann LC, et al. (2017) Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1. Molecular cell 68(5):847–859 e847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andrabi SA, et al. (2011) Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nature medicine 17(6):692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Z, et al. (2012) Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes & development 26(3):235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang HC, et al. (2011) Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proceedings of the National Academy of Sciences of the United States of America 108(34):14103–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DaRosa PA, et al. (2015) Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 517(7533):223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ahel I, et al. (2008) Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451(7174):81–85. [DOI] [PubMed] [Google Scholar]
- 75.Fenton AL, Shirodkar P, Macrae CJ, Meng L, & Koch CA (2013) The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic acids research 41(7):4080–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Altmeyer M, et al. (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nature communications 6:8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Izhar L, et al. (2015) A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell reports 11(9):1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Strom AR, et al. (2017) Phase separation drives heterochromatin domain formation. Nature 547(7662):241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Larson AG, et al. (2017) Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547(7662):236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quenet D, et al. (2008) The histone subcode: poly(ADP-ribose) polymerase-1 (Parp-1) and Parp-2 control cell differentiation by regulating the transcriptional intermediary factor TIF1beta and the heterochromatin protein HP1alpha. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 22(11):3853–3865. [DOI] [PubMed] [Google Scholar]
- 81.Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, & Price BD (2014) DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proceedings of the National Academy of Sciences of the United States of America 111(25):9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perraud AL, et al. (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411(6837):595–599. [DOI] [PubMed] [Google Scholar]
- 83.Sano Y, et al. (2001) Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293(5533):1327–1330. [DOI] [PubMed] [Google Scholar]
- 84.Yu P, et al. (2017) Identification of the ADPR binding pocket in the NUDT9 homology domain of TRPM2. The Journal of general physiology 149(2):219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blenn C, Wyrsch P, Bader J, Bollhalder M, & Althaus FR (2011) Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cellular and molecular life sciences : CMLS 68(8):1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hara Y, et al. (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Molecular cell 9(1):163–173. [DOI] [PubMed] [Google Scholar]
- 87.Alano CC, et al. (2010) NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. The Journal of neuroscience : the official journal of the Society for Neuroscience 30(8):2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andrabi SA, et al. (2014) Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proceedings of the National Academy of Sciences of the United States of America 111(28):10209–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fouquerel E, et al. (2014) ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell reports 8(6):1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gasmi L, Cartwright JL, & McLennan AG (1999) Cloning, expression and characterization of YSA1H, a human adenosine 5’-diphosphosugar pyrophosphatase possessing a MutT motif. The Biochemical journal 344 Pt 2:331–337. [PMC free article] [PubMed] [Google Scholar]
- 91.Perraud AL, et al. (2003) NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. The Journal of biological chemistry 278(3):1794–1801. [DOI] [PubMed] [Google Scholar]
- 92.Formentini L, et al. (2009) Poly(ADP-ribose) catabolism triggers AMP-dependent mitochondrial energy failure. The Journal of biological chemistry 284(26):17668–17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oei SL & Ziegler M (2000) ATP for the DNA ligation step in base excision repair is generated from poly(ADP-ribose). The Journal of biological chemistry 275(30):23234–23239. [DOI] [PubMed] [Google Scholar]
- 94.Wright RH, et al. (2016) ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 352(6290):1221–1225. [DOI] [PubMed] [Google Scholar]
- 95.Maruta H, Matsumura N, & Tanuma S (1997) Role of (ADP-ribose)n catabolism in DNA repair. Biochemical and biophysical research communications 236(2):265–269. [DOI] [PubMed] [Google Scholar]
- 96.Kennedy BE, et al. (2016) NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacological research 114:274–283. [DOI] [PubMed] [Google Scholar]
- 97.Patel A, et al. (2017) ATP as a biological hydrotrope. Science 356(6339):753–756. [DOI] [PubMed] [Google Scholar]
- 98.Carreras-Puigvert J, et al. (2017) A comprehensive structural, biochemical and biological profiling of the human NUDIX hydrolase family. Nature communications 8(1):1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robson M, et al. (2017) Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. The New England journal of medicine 377(6):523–533. [DOI] [PubMed] [Google Scholar]
- 100.Gogola E, et al. (2018) Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer cell 33(6):1078–1093 e1012. [DOI] [PubMed] [Google Scholar]
- 101.Ame JC, et al. (2009) Radiation-induced mitotic catastrophe in PARG-deficient cells. Journal of cell science 122(Pt 12):1990–2002. [DOI] [PubMed] [Google Scholar]
- 102.Illuzzi G, et al. (2014) PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic acids research 42(12):7776–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gravells P, et al. (2018) Radiosensitization with an inhibitor of poly(ADP-ribose) glycohydrolase: A comparison with the PARP1/2/3 inhibitor olaparib. DNA repair 61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.James DI, et al. (2016) First-in-Class Chemical Probes against Poly(ADP-ribose) Glycohydrolase (PARG) Inhibit DNA Repair with Differential Pharmacology to Olaparib. ACS chemical biology 11(11):3179–3190. [DOI] [PubMed] [Google Scholar]
- 105.Fathers C, Drayton RM, Solovieva S, & Bryant HE (2012) Inhibition of poly(ADP-ribose) glycohydrolase (PARG) specifically kills BRCA2-deficient tumor cells. Cell cycle 11(5):990–997. [DOI] [PubMed] [Google Scholar]
- 106.Noll A, Illuzzi G, Ame JC, Dantzer F, & Schreiber V (2016) PARG deficiency is neither synthetic lethal with BRCA1 nor PTEN deficiency. Cancer cell international 16:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gravells P, Grant E, Smith KM, James DI, & Bryant HE (2017) Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA repair 52:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rotin LE, et al. (2016) Ibrutinib synergizes with poly(ADP-ribose) glycohydrolase inhibitors to induce cell death in AML cells via a BTK-independent mechanism. Oncotarget 7(3):2765–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haikarainen T, Maksimainen MM, Obaji E, & Lehtio L (2018) Development of an Inhibitor Screening Assay for Mono-ADP-Ribosyl Hydrolyzing Macrodomains Using AlphaScreen Technology. SLAS discovery : advancing life sciences R & D 23(3):255–263. [DOI] [PubMed] [Google Scholar]
- 110.Shao Y, Li X, Lu Y, Liu L, & Zhao P (2015) Aberrant LRP16 protein expression in primary neuroendocrine lung tumors. International journal of clinical and experimental pathology 8(6):6560–6565. [PMC free article] [PubMed] [Google Scholar]
- 111.Page BDG, et al. (2018) Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells. Nature communications 9(1):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Berger NA, et al. (2018) Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. British journal of pharmacology 175(2):192–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Morrow DA, et al. (2009) A randomized, placebo-controlled trial to evaluate the tolerability, safety, pharmacokinetics, and pharmacodynamics of a potent inhibitor of poly(ADP-ribose) polymerase (INO-1001) in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of the TIMI 37 trial. Journal of thrombosis and thrombolysis 27(4):359–364. [DOI] [PubMed] [Google Scholar]
- 114.Anonymous (2017) PARP-1 Inhibition in Pulmonary Arterial Hypertension. [Google Scholar]
- 115.Ghosh SG, et al. (2018) Biallelic Mutations in ADPRHL2, Encoding ADP Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome. American journal of human genetics 103(3):431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Danhauser K, et al. (2018) Bi-allelic ADPRHL2 Mutations Cause Neurodegeneration with Developmental Delay, Ataxia, and Axonal Neuropathy. American journal of human genetics 103(5):817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Byrne AB, et al. (2016) Inhibiting poly(ADP-ribosylation) improves axon regeneration. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martire S, Mosca L, & d’Erme M (2015) PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mechanisms of ageing and development 146-148:53–64. [DOI] [PubMed] [Google Scholar]
- 119.Strickfaden H, et al. (2016) Poly(ADP-ribosyl)ation-dependent Transient Chromatin Decondensation and Histone Displacement following Laser Microirradiation. The Journal of biological chemistry 291(4):1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Posavec Marjanovic M, et al. (2017) MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD(+) consumption. Nature structural & molecular biology 24(11):902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li GY, et al. (2010) Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proceedings of the National Academy of Sciences of the United States of America 107(20):9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oberoi J, et al. (2010) Structural basis of poly(ADP-ribose) recognition by the multizinc binding domain of checkpoint with forkhead-associated and RING Domains (CHFR). The Journal of biological chemistry 285(50):39348–39358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zampieri M, et al. (2012) ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. The Biochemical journal 441(2):645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Min W, et al. (2013) Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nature communications 4:2993. [DOI] [PubMed] [Google Scholar]