

Summary
Ferroptosis is a regulated necrosis process driven by iron-dependent lipid peroxidation. Although ferroptosis and cellular metabolism interplay with each other, whether mitochondria are involved in ferroptosis is under debate. Here we demonstrate that mitochondria play a crucial role in cysteine deprivation-induced ferroptosis but not in that induced by inhibiting glutathione peroxidase-4 (GPX4), the most downstream component of the ferroptosis pathway. Mechanistically, cysteine deprivation leads to mitochondrial membrane potential hyperpolarization and lipid peroxide accumulation. Inhibition of mitochondrial TCA cycle or electron transfer chain (ETC) mitigated mitochondrial membrane potential hyperpolarization, lipid peroxide accumulation, and ferroptosis. Blockage of glutaminolysis had the same inhibitory effect, which was counteracted by supplying downstream TCA cycle intermediates. Importantly, loss of function of fumarate hydratase, a tumor suppressor and TCA cycle component, confers resistance to cysteine deprivation-induced ferroptosis. Collectively, this work demonstrates the crucial role of mitochondria in cysteine deprivation-induced ferroptosis and implicates ferroptosis in tumor suppression.
Graphical Abstract
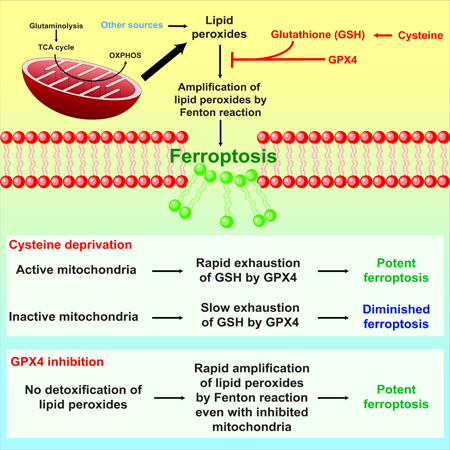
In brief
Gao et al show that mitochondria play a crucial and proactive role in cysteine deprivation-induced ferroptosis but not in GPX4 inhibition-induced ferroptosis. Mechanistically, the mitochondrial TCA cycle and electron transport chain promote cysteine deprivation-induced ferroptosis by serving as the major source for cellular lipid peroxide production. The anaplerotic role of glutaminolysis in replenishing the TCA cycle intermediates explains its involvement in cysteine deprivation-induced ferroptosis. Importantly, mitochondria-mediated ferroptosis might contribute to the antitumor function of fumarate hydratase, a component of the TCA cycle and a tumor suppressor in renal cancer.
Introduction
Ferroptosis, an iron-dependent form of regulated necrosis, has emerged as a new cell death modality highly relevant to disease (Angeli et al., 2017; Gao and Jiang, 2017; Stockwell et al., 2017; Yang and Stockwell, 2016). Ferroptosis results from the accumulation of cellular reactive oxygen species (ROS) that exceed the redox contents maintained by glutathione (GSH) and the phospholipid hydroperoxidases that use GSH as a substrate. The synthetic small molecule compound erastin can trigger ferroptosis by inhibiting the activity of cystine-glutamate antiporter (system Xc-), leading to the depletion of cellular cysteine and GSH, thus the collapse of cellular redox homeostasis (Dixon et al., 2012). Importantly, it is the lipid ROS/peroxides, rather than cytosolic ROS, that unleash ferroptosis. As such, inactivation of glutathione peroxidase 4 (GPX4), an enzyme required for the clearance of lipid ROS, can induce ferroptosis even when cellular cysteine and GSH contents are normal (Friedmann Angeli et al., 2014; Ingold et al., 2018; Yang et al., 2014).
Although the physiological function of ferroptosis is still elusive, its involvement in multiple human diseases has been established. Ferroptosis is a major mechanism for cell death associated with ischemic organ injury, including ischemic heart diseases, brain damage, and kidney failure (Friedmann Angeli et al., 2014; Gao et al., 2015a; Linkermann et al., 2014). A role of ferroptosis in neurodegeneration has also been implicated (Chen et al., 2015; Do Van et al., 2016; Skouta et al., 2014). In cancer, ferroptosis has been shown to contribute to the tumor suppressive function of p53 (Galluzzi et al., 2015; Jennis et al., 2016; Jiang et al., 2015; Wang et al., 2016); and many types of cancer cells that are resistant to chemotherapy and certain targeted therapies appear to be sensitive to ferroptosis induced by GPX4 inhibition (Hangauer et al., 2017; Viswanathan et al., 2017). These findings suggest that modulating ferroptosis might be potential therapeutic approaches in treating cancer or other diseases.
Cellular metabolism is essential for ferroptosis, presumably because lipid ROS is mainly generated from various steps of cellular metabolism. Mounting evidence has demonstrated that diverse cellular metabolic processes, including lipid metabolism and amino acid metabolism (particularly that involves cysteine and glutamine), contribute to ferroptosis (Gao and Jiang, 2017; Stockwell et al., 2017). Interestingly, the stress-responsive catabolic pathway, autophagy, can also promote ferroptosis by degrading iron-storage protein ferritin and thus increase cellular iron concentration (Gao et al., 2016; Hou et al., 2016). Iron, as an essential cofactor of a plethora of metabolic enzymes and as a catalyst of lipid peroxide-generating Fenton reaction, drives oxygen and redox-based metabolism and cellular ROS production.
Despite the central role of mitochondria in oxidative metabolism, it remains unclear whether mitochondria play a central role in ferroptosis. Supporting this possibility, ferroptosis is associated with dramatic morphological changes of mitochondria, including mitochondrial fragmentation and cristae enlargement (Dixon et al., 2012; Doll et al., 2017); and some potent ferroptosis inhibitors appear to be exquisitely targeted to mitochondria (Krainz et al., 2016). Furthermore, glutamine metabolism, known as glutaminolysis, is required for cysteine deprivation-induced ferroptosis (Gao et al., 2015a; Gao et al., 2015b), and one of the major functions of glutaminolysis is to fuel the mitochondrial tricarboxylic acid (TCA) cycle. Intriguingly, mitochondrial glutaminase (GLS), GLS2, instead of cytosolic GLS1, has been shown to be required for ferroptosis, although both enzymes catalyze glutaminolysis (Cassago et al., 2012; Gao et al., 2015a; Jennis et al., 2016). All these observations are consistent with the potential involvement of mitochondria in ferroptosis. However, there is also strong evidence arguing against a major role of mitochondria in ferroptosis. For example, a comparison between a mitochondrial DNA-depleted (ρ0) cancer cell line and its parental line did not show a significant difference in ferroptosis sensitivity (Dixon et al., 2012), and a mechanistic investigation into a cohort of ferrostatin analogs (inhibitors of ferroptosis) failed to establish a correlation between mitochondrial localization of ferrostatins with their anti-death potency (Gaschler et al., 2018). Taken together, the functional relevance of mitochondria in ferroptosis is still highly debatable.
In this study, through a series of cellular, molecular, pharmacological, and metabolomic analysis, we demonstrated that the mitochondrion is a crucial player in ferroptosis induced by cysteine deprivation (inhibition of system Xc- by erastin or using culture medium free of cystine). Mechanistically, the canonical metabolic activity of mitochondria, including both the TCA cycle and mitochondrial electron transport chain (ETC) activity, are required for the generation of sufficient lipid ROS to initiate ferroptosis. Furthermore, cancer cells deficient of the mitochondrial tumor suppressor fumarate hydratase (FH) are resistant to cysteine deprivation-induced ferroptosis – this result supports the notion that ferroptosis might be a physiologically relevant tumor suppressive mechanism, and provides insights into potential ferroptosis-inducing cancer therapeutic approaches. Importantly, the role of mitochondria in ferroptosis is context dependent; if the activity of the glutathione-dependent peroxidase, GPX4, is inhibited, cells undergo ferroptosis independent of mitochondrial function.
Results
Mitochondria regulate cysteine deprivation-induced ferroptosis
To determine unambiguously whether mitochondria play a role in ferroptosis, we engineered human fibrosarcoma HT1080 cells depleted of mitochondria. Mitochondrial depletion was achieved by activating mitochondria-targeted autophagy, known as mitophagy (Youle and Narendra, 2011). First, we treated control and Parkin (tagged with mCherry)-overexpressing HT1080 cells with the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazine (CCCP). Under this condition, mitochondria are depolarized by CCCP and consequently subjected to clearance via Parkin-mediated mitophagy (Narendra et al., 2008). As shown in Figures 1A and 1B, upon CCCP treatment for 24 or 48 hours followed by recovery in CCCP-free culture medium for 24 hours, mitochondria were significantly eliminated in Parkin-overexpressed cells (see the disappearance of MitoTracker signal, and mitochondrial protein TOM20 and TIM23). Subsequently, cells were incubated with cystine (CC)-free medium or a pharmacological inducer of ferroptosis, erastin. Compared to control cells, Parkin-overxpressing, mitochondria-depleted cells were less sensitive to ferroptosis triggered by CC starvation or erastin (Figure 1C), confirming that mitochondria promote cysteine deprivation-induced ferroptosis.
Figure 1. Mitochondria regulate cysteine deprivation-induced ferroptosis.

(A-D) Mitochondrial depletion by Parkin-mediated mitophagy inhibits cysteine deprivation-induced ferroptosis. Wild type (Control) or mCherry-Parkin overexpressing (Parkin OE) HT1080 cells were treated with 10 μM CCCP (Carbonyl cyanide m-chlorophenyl hydrazone) for 48 h, unless otherwise indicated (as in panel B, 24 or 48 h), to induce mitochondrial depletion. After recovery from CCCP treatment by incubating cells in CCCP-free normal culture medium for 24 h, the following experiments were performed: the presence of mitochondria was assessed by MitoTracker staining (Scale bar = 10 μM) (A) or by Western blot for endogenous TOM20 and TIM23 (B); Cells were treated with erastin or cystine (CC) starvation for 12 h for cell death measurement (C) or 8 h for lipid ROS measurement (D). Cell death was measured with Sytox Green staining coupled with flow cytometry. The accumulation of lipid ROS was assessed by BODIPY C11 staining coupled with flow cytometry analysis.
All quantitative data in this and other figures are presented as mean ± SD from three independent experiments; and p values were calculated with unpaired t test (same for other figures. *, P<0.05; **, P<0.01; ***, p < 0.001; ****, p<0.0001).
(E) Co-localization of oxidized lipid and mitochondria. MEFs were treated as indicated for 4 h, and then cells were co-stained with BODIPY C11 and MitoTracker. Oxidized BODIPY C11 (Green) indicating lipid ROS and mitochondrial signals (Red) were imaged by fluorescent microscope.
Scale bar = 10 μm.
See also Figure S1.
Lipid ROS accumulation is a hallmark of ferroptosis, but where and how they are generated during ferroptosis has not been defined. Since the mitochondrion is a major organelle for cellular ROS production, does this organelle contribute to lipid ROS generation upon cysteine deprivation? To test this possibility, we monitored lipid ROS accumulation using C11 BODIPY 581/591, a fluorescent probe for membrane-localized ROS. Indeed, upon CC starvation or erastin treatment, mitochondria-depleted cells had a decreased level of lipid ROS accumulation (Figure 1D). Further, when we examined subcellular localization of the lipid ROS probe by confocal imaging (the probe changes fluorescence from red to green upon oxidation), we found in both HT1080 cells and mouse embryonic fibroblasts (MEFs) treated with erastin, the oxidized probe first appeared in a distribution that significantly colocalized with mitochondria, and then colocalized with the plasma membrane at later time points (Figure 1E and Figure S1).
Mitochondrial TCA cycle participates in cysteine deprivation-induced ferroptosis
Glutaminolysis has been demonstrated to be required for cysteine deprivation-induced ferroptosis (Gao et al., 2015a). In the absence of glutamine, neither CC starvation nor pharmacological inhibition of CC transporter subunit SLC7A11 by erastin can induce ferroptosis. Since a major function of glutaminolysis is to maintain the mitochondrial TCA cycle through anaplerosis (Figure 2A), is the TCA cycle required for ferroptosis? To test this possibility, we examined whether TCA cycle metabolites downstream of glutaminolysis can recapitulate the function of glutamine in ferroptosis. Indeed, alpha-Ketoglutaric acid (αKG), which is the TCA cycle metabolite immediately downstream of glutaminolysis, can replace glutamine for cysteine deprivation-induced lipid ROS accumulation and ferroptosis (Figures 2B and 2C). Further, TCA metabolites downstream of αKG, including succinate (Suc), fumarate (Fum) and malate (Mal) can all replace the role of glutamine in lipid ROS accumulation and ferroptosis induced by CC starvation or system Xc- inhibition, in both MEFs and HT1080 cells (Figures 2D-F and Figure S2). To further confirm that glutamine supports ferroptosis via the TCA cycle, we measured TCA metabolites under different conditions by GC/MS metabolomic analysis. The results (Figure 2G) showed that levels of TCA cycle metabolites were not changed by CC starvation alone, within the time range of our analysis. In the absence of glutamine, levels of multiple TCA cycle metabolites, including αKG, fumarate, and malate were significantly decreased. In contrast, TCA cycle metabolites upstream of αKG, such as citrate, were affected more by glucose status than by glutamine status.
Figure 2. Mitochondrial TCA cycle promotes cysteine deprivation-induced ferroptosis.

(A) An overview of mammalian Tricarboxylic Acid Cycle.
(B-C) α-ketoglutarate can mimic the death-inducing activity of L-Glutamine (L-Gln). (B) Microscopy showing cell death. MEFs were treated as indicated for 10 h. Upper panel: phase-contrast; lower panel, propidium iodide (PI) staining for dead cells (Scale bar = 100 μM). (C) For cell death measurement, MEFs were subjected to the same treatment as in panel (B) and cell death was determined by PI staining coupled with flow cytometry; for lipid ROS measurement, MEFs were treated as in panel (B) for 8 h and then lipid ROS was determined by BODIPY C11 staining coupled with flow cytometry.
(D-F) TCA metabolites succinate (D), fumarate (E) and Malate (F) can mimic the death inducing activity of L-Gln. (G) Glutamine is required for the maintenance of the normal level of multiple TCA cycle metabolites during ferroptosis. MEFs were treated as indicated for 8 hours, and samples were collected and measured by GC/MS metabolomic analysis as detailed in Methods. For all experiments, 10% (V/V) dialyzed FBS was used in the assay medium. Q: L-glutamine (1 mM); CC: L-cystine (0.2 mM); αKG: Dimethyl-α−2-oxoglutarate (mM); Suc: Dimethyl succinate (mM); Fum: Dimethyl fumarate (μM); Mal: Dimethyl (S)-(−)-malate (mM); Fer-1: ferrostatin-1 (1 μM).
See also Figure S2.
Mitochondrial electron transport chain (ETC) regulates cysteine deprivation-induced ferroptosis
One of the most important functions of the TCA cycle is to support electron transport activity of protein complexes embedded in the inner-membrane of mitochondria (Figure 3A). Since the TCA cycle is required for ferroptosis induced by cellular cysteine deprivation, are components of the electron transport chain (ETC) also required? To address this question, we utilized an array of ETC inhibitors. As shown in Figures 3B-E, inhibitors of mitochondrial complex I (rotenone), complex II (DBM), complex III (antimycin), and complex IV (NaN3) all suppressed lipid ROS accumulation and ferroptosis induced by CC starvation or erastin. Taken together, these results indicate that ETC contributes to ferroptosis induced by cellular cysteine deprivation. Notably, although all tested ETC inhibitors significantly blocked ferroptosis at earlier time points (12 hours), some of them (antimycin and NaN3) maintained their inhibitory effect at later time points but others (inhibitors for complex I and complex II) gradually lost such effect (Figure S3). This might be due to different stability of these compounds; alternatively, it may be that inhibition of complex I or complex II alone is not sufficient to block electron transfer to the downstream complex III.
Figure 3. ETC activity promotes cysteine deprivation-induced ferroptosis.

(A) Scheme shows mitochondrial electron transport chain (ETC) complexes and their inhibitors.
(B) Inhibitors of ETC can inhibit CC starvation-induced ferroptosis. MEFs were treated with CC starvation for 12 h, in the absence or presence of ETC inhibitors as indicated. Cell death was determined by PI staining coupled with flow cytometry. Complex I inhibitor rotenone (Rot), 10 μM; Complex II inhibitor diethyl butylmalonate (DBM), 2 mM; Complex III inhibitor antimycin A (Anti A), 50 μM; Complex IV inhibitor NaN3, 15 mM.
(C) ETC inhibitors can inhibit CC starvation-induced lipid ROS accumulation. MEFs were treated with CC for 6 h, in the absence or presence of ETC inhibitors as indicated. Accumulation of lipid ROS was determined by BODIPY C11 staining coupled with flow cytometry.
(D) ETC inhibitors can inhibit erastin-induced ferroptosis. MEFs were treated with 1 μM erastin for 12 h, in the absence or presence of ETC inhibitors as indicated. Cell death was determined by PI staining coupled with flow cytometry.
(E) ETC inhibitors can inhibit erastin-induced lipid ROS accumulation. MEFs were treated with 1 μM erastin for 6 h, in the absence or presence of ETC inhibitors as indicated. Accumulation of lipid ROS was determined by BODIPY C11 coupled with flow cytometry.
See also Figure S3.
Mitochondrial membrane potential (MMP) hyperpolarization is associated with cysteine deprivation-induced lipid ROS accumulation and ferroptosis.
Mitochondria produce ATP by utilizing the proton electrochemical gradient potential across the mitochondrial membrane, which is the result of the action of the TCA cycle and ETC. Since the TCA cycle and ETC promote cysteine deprivation-induced ferroptosis, is ferroptosis associated with a change of MMP? To test this possibility, we used MitoTracker (tetramethylrhodamine ethyl ester, or TMRE) staining to monitor MMP upon induction of ferroptosis and as a control, of apoptosis. As expected, during mitochondria-mediated apoptosis, a decrease of MMP was observed prior to eventual cell death (Figure S4A and Supplementary Movie S1). This decrease of MMP is indicative of mitochondrial outer membrane permeabilization (MOMP), a crucial characteristic of mitochondria-mediated apoptosis (Green and Reed, 1998; Kroemer and Reed, 2000). In striking contrast, ferroptosis inducers such as CC starvation (Figures 4A and 4B, and Supplementary Movie S2), amino acid-free medium plus full serum (Gao et al., 2015a) (Figure S4B and Supplementary Movie S3), erastin (Figure S4C), or glutamate (Figure S4D), can all induce MMP hyperpolarization.
Figure 4. Mitochondrial membrane potential hyperpolarization is associated with cysteine deprivation-induced lipid ROS accumulation and ferroptosis.

(A) Ferroptosis is accompanied by mitochondrial membrane potential (MMP) hyperpolarization. Representative still images from time-lapse imaging of mitochondrial membrane potential in MEFs undergoing CC starvation-induced ferroptosis. Cells were incubated with TMRE (200 nM), and subjected to CC starvation for indicated time (Scale bar = 5 μM).
(B) Quantification of MMP during CC starvation-induced ferroptosis. MEFs were treated with CC starvation, and 500 nM TMRE was added 30 min before each indicated time point. MMP was measured by flow cytometry and mean fluorescence was calculated.
(C) CCCP can inhibit ferroptosis-associated MMP hyperpolarization. MEFs were treated with CC starvation plus or minus 10 μM CCCP for indicated time, and 500 nM TMRE was added 30 min before each indicated time point. MMP was measured by flow cytometry and mean fluorescence was calculated.
(D) CCCP can block ferroptosis induced by CC starvation or erastin. MEFs were treated as indicated for 12 h and cell death was determined by PI staining coupled with flow cytometry.
(E) CCCP can block lipid ROS accumulation induced by CC starvation or erastin. MEFs were treated as indicated for 8 h and lipid ROS was measured by BODIPY C11 staining coupled with flow cytometry.
(F) Cells protected by CCCP from erastin-induced ferroptosis maintain viability. MEFs and HT1080 cells were seeded at low density in 12-well dishes and treated with erastin for 16 h, in the absence or presence of CCCP as indicated. Cells were then allowed to recover in drug-free DMEM medium for 3 days and subsequently fixed and stained with crystal violet.
(G-H) Glutaminolysis and TCA cycle drive ferroptosis-associated MMP hyperpolarization in MEFs.(G) Glutamine is required for CC starvation-induced MMP hyperpolarization.(H) Transaminase inhibitor AOA (Aminooxyacetic acid, 0.5 mM) can block CC starvation-induced MMP hyperpolarization.
(I-L) TCA intermediates α-ketoglutarate (I) succinate (J), fumarate (K) and Malate (L) can replace the requirement of glutamine for CC starvation-induced MMP hyperpolarization. αKG: Dimethyl-α−2-oxoglutarate (8 mM); Suc: Dimethyl succinate (8 mM); Fum: Dimethyl fumarate (5 μM); Mal: Dimethyl (S)-(−)-malate (32 mM).
To further investigate the role of MMP in cysteine deprivation-induced ferroptosis, we examined the effect of the mitochondrial uncoupler CCCP on ferroptosis. As shown in Figures 4C-4D and S4E-S4F, CCCP disrupted MMP, and completely blocked cysteine deprivation-induced lipid ROS accumulation and ferroptosis. Moreover, proliferation can be recovered after removing both erastin and CCCP, as assessed by cell colony formation assay (Figure 4F), indicating that temporarily disrupting MMP by CCCP can prevent ferroptotic cell death without compromising long-term cell viability. This conclusion is consistent with the effect of pharmacological inhibition of ETC on ferroptosis (Figure 3 and Figures S4 G-J).
Glutaminolysis and TCA cycle drive ferroptosis-associated MMP hyperpolarization
We subsequently determined the role of glutaminolysis and the TCA cycle in ferroptosis-associated MMP hyperpolarization. In the absence of glutamine, CC starvation failed to induce MMP hyperpolarization (Figure 4G). Similarly, inhibition of glutaminolysis by aminooxyacetic acid (AOA) blocked CC starvation-induced MMP hyperpolarization (Figure 4H). Therefore, glutamine metabolism is essential for CC starvation-induced MMP hyperpolarization. Furthermore, TCA cycle intermediates downstream of glutaminolysis, including αKG, succinate, fumarate, and malate were all able to substitute glutamine for cystine starvation-induced MMP hyperpolarization (Figures 4I-4L). Therefore, glutaminolysis and TCA cycle are essential for ferroptosis-associated MMP hyperpolarization. Presumably, MMP hyperpolarization reflects an increase of mitochondrial ETC activity and subsequent lipid ROS generation, which explains its role in ferroptosis. However, the mechanisms by which cysteine deprivation triggers MMP hyperpolarization are not clear and warrant further investigation.
Mitochondrial function is dispensable for GPX4 inhibition-induced ferroptosis
Importantly, although glutaminolysis and mitochondrial TCA cycle/ETC activity contribute to cysteine deprivation-induced ferroptosis, ferroptosis can still be triggered by pharmacological inhibition or genetic elimination of GPX4 when these mitochondrial activities are disrupted. Mitochondria-depleted HT1080 cells did not show significant resistance when ferroptosis was triggered by the GPX4 inhibitor, RSL3 (Figures 5A and5B). ETC inhibitors also failed to impinge on lipid ROS accumulation and ferroptosis upon RSL3 treatment (Figures 5C and 5D and Figure S5A). Further, we generated GPX4-knockout (KO) HT1080 cells by CRIPSR/Cas9 technology and found that ETC inhibitors could not block ferroptosis induced by GPX4-KO (Figures 5E-5G). Consistently, although RSL3 treatment can induce MMP hyperpolarization, the mitochondrial uncoupler CCCP can completely ablate RSL3-induced MMP hyperpolarization (Figure S5B) without inhibiting RSL3-induced lipid ROS accumulation or ferroptosis in both MEFs and HT1080 cells (Figures 5C-5D and S5A-S5B). Moreover, we found GPX4 inhibition-induced ferroptosis can occur even in the absence of glutamine (Figure 5G). Therefore, glutamine and mitochondria are dispensable for GPX4 inhibition-induced ferroptosis.
Figure 5. Mitochondria are dispensable for GPX4 inhibition-induced ferroptosis.

(A-B) Mitochondrial depletion by Parkin-mediated mitophagy cannot inhibit RSL3-induced ferroptosis. Mitochondria-depleted HT1080 cells were created as described in Figure 1. Cells were treated with RSL3 for 6 h for cell death measurement (A) or 4 h for lipid ROS measurement (B). Cell death was measured with DAPI staining coupled with flow cytometry. The accumulation of lipid ROS was assessed by BODIPY C11 staining coupled with flow cytometry analysis.
(C-D) Mitochondrial ETC inhibitors cannot inhibit RSL3-induced ferroptosis and lipid ROS accumulation.
(E) Western blotting confirmed CRISPR-Cas9 mediated knockout of GPX4 in HT1080 cells (see Methods for detail).
(F) Mitochondrial ETC inhibitors cannot inhibit GPX4 Knockout-induced ferroptosis. GPX4 KO HT1080 cells were seeded and subsequently grew in DMEM medium containing 0.2 mM Trolox. Ferroptosis induction was initiated by switching cells to Trolox-free medium, in the presence or absence of indicated ETC complex inhibitors for 8 h. Complex I inhibitor rotenone (Rot), 10 μM; Complex II inhibitor Diethyl butylmalonate (DBM), 2 mM; Complex III inhibitor Myxothiazol (Myxo), 15 μM; Complex IV inhibitor NaN3, 15 mM.
(G) Glutamine is dispensable for GPX4 Knockout-induced ferroptosis. GPX4 KO HT1080 cells were seeded and subsequently grew in DMEM medium containing 0.2 mM Trolox. Ferroptosis induction was initiated by switching cells to Trolox-free medium, in the presence or absence of 1 mM glutamine for 8 h.
Scale bar = 100 μM. See also Figure S5.
Loss of function of mitochondrial tumor suppressor fumarate hydratase confers resistance to ferroptosis in renal cancer cells
Genetic mutation of the gene encoding for the TCA cycle enzyme fumarate hydratase (FH, also known as fumarase) has been identified in benign and malignant renal cancer lesions, and has been demonstrated to be a bona fide tumor suppressor in renal cancer (Alam et al., 2005; Chuang et al., 2005; Gottlieb and Tomlinson, 2005; King et al., 2006; Tomlinson et al., 2002).
In light of our finding that mitochondrial TCA cycle and respiration drive cysteine deprivation-induced ferroptosis, we examined whether loss of FH function renders cells more resistant to ferroptosis – thus contributing to its tumor suppressive function. We tested this possibility in two pairs of patient-derived isogenic renal cancer cell lines, FH-mutant UOK262 and UOK262 reconstituted with wild-type (WT) FH, and FH-mutant UOK268 and UOK268 reconstituted with WT FH (Tong et al., 2011). Compared to cells expressing WT FH, FH-mutant renal cancer cells were less sensitive to CC starvation-induced ferroptosis (Figures 6A-6C), and cell death in these mutant cells could be restored by supplying exogenous malate, which is the metabolite downstream of FH in the TCA cycle (Figures 6A and 6B). Importantly, in a cell colony formation experiment, FH-mutant renal cancer cells remained viable and proliferative upon CC starvation, whereas the isogenic cells expressing wild-type FH failed to proliferate under this condition (Figure 6D). These results suggest that under oxidative stress (common for growth environment of solid tumors), loss of FH function can make cancer cells more resistant to ferroptotic death, thus conferring tumorigenic advantage.
Figure 6. Loss of function of mitochondrial tumor suppressor fumarate hydratase confers resistance to ferroptosis.

(A-C) Fumarase (FH)-mutant cancer cells are more resistant to CC starvation-induced ferroptosis than isogenic cells expressing WT FH. (A) Microscopy showing cell death. Two pairs of FH-mutant cancer cells and their isogenic cells expressing WT FH were treated as indicated for 36 h (UOK262 and UOK262+WT FH) or 60 h (UOK268 and UOK268+WT FH). Upper panel: phase-contrast; lower panel, PI staining for dead cells (Scale bar = 100 μM). Western blot shows the expression level of FH in these cell lines. (B) Cell death was determined by PI staining coupled with flow cytometry. (C) lipid ROS was determined by BODIPY C11 staining coupled with flow cytometry
(D) FH-mutant cancer cells recovered and proliferated after CC starvation. Cells were treated with CC starvation for 24 h (UOK262 and UOK262+WT FH) or 60 h (UOK268 and UOK268+WT FH), and then cells were incubated in full DMEM medium for 3 days. Colony formation was measured by crystal violet staining.
See also Figure S6.
Discussion
Cellular metabolism and ferroptosis closely interact with each other; and lipid ROS, mainly a product of oxygen/iron-driven metabolism, is essential for the execution of ferroptosis. For these reasons, it appears logical that mitochondria should play a central role in ferroptosis. Nonetheless, whether the mitochondria are an important component in ferroptosis remains highly controversial. In this study, we present evidence to demonstrate that the mitochondrion is indeed a crucial player in ferroptotic cell death induced by cysteine deprivation. When mitochondria are depleted via Parkin-dependent mitophagy, cells become more resistant to ferroptosis upon CC starvation or pharmacological inhibition of CC import (Figure 1). Mechanistically, we showed that the role of the mitochondrion in ferroptosis is due to its metabolic function, and both mitochondrial TCA cycle and the action of ETC are required for a potent ferroptosis (Figures 2, 3, S2, and S3). Ferroptotic function of TCA cycle is the reason why glutaminolysis, a major source of anaplerosis, is required for ferroptosis (Figures 2 and S2).
Why, then, is the role of mitochondria in ferroptosis so controversial? The following three reasons can reconcile this discrepancy. First, our study indicates that the role of mitochondria in ferroptosis is context-dependent: blockage of mitochondrial function potently inhibits cysteine deprivation-induced ferroptosis; however, upon elimination or pharmacological inhibition of GPX4, the very downstream component of the ferroptosis pathway responsible for lipid ROS clearance, cells can commit ferroptosis independently of mitochondria (Figures 5 and S5). When cysteine is scarce, mitochondrial metabolism contributes significantly to rapid glutathione depletion, and subsequent lipid ROS generation and ferroptosis. On the other hand, upon GPX4 inhibition, although one would predict that inhibition of mitochondria should also attenuate lipid ROS generation and thus may at least delay ferroptosis, we did not observe a measurable effect of mitochondrial ETC inhibitors on slowing down ferroptosis in GPX4-KO HT1080 cells (Figure S5D). It is likely that once GPX4 is eliminated, the low amount of lipid ROS produced by other mechanisms will be rapidly amplified through the Fenton chain reaction, leading to full-blown ferroptotic cell death even when mitochondrial activity is diminished. It is also possible that GPX4 inactivation may transduce a signal to, and thus hyperactivate, certain enzymes responsible for lipid ROS generation. Future investigation is needed to clarify this issue. Second, since ferroptosis is dictated by cellular metabolism, long-term alteration of cellular metabolism may rewire the molecular processes underlying ferroptosis. For example, in a type of experimental model cells known as ρ0 cells, mitochondrial DNA is depleted permanently (King and Attardi, 1989, 1996). To compensate the long-term lack of mitochondria, these cells need to be cultured with nutrients different from mitochondria-containing cells (King and Attardi, 1989, 1996). Therefore, metabolism in ρ0 cells, including lipid ROS generation (mainly a consequence of metabolism), is fundamentally rewired. For this reason, the mechanism governing the mitochondria-independent ferroptosis in ρ0 cells may differ significantly from that in normal, mitochondria-containing cells. Thirdly, the methods for cell death measurement need to be carefully considered. For example, although cell viability assays are often used to infer cell death, they are not suitable for the study of the role of mitochondria in ferroptosis. This is because that manipulation of mitochondrial function may impact the outcome of these assays (many of them are ATP content-based or metabolic activity-based), regardless of cell death status. Assays directly measuring cell death are preferable for this purpose.
Mitochondria are also involved in other modalities of programmed cell death, particularly apoptosis. Intriguingly, the roles of mitochondria in apoptosis and ferroptosis are fundamentally different. In mitochondria-mediated apoptosis, the mitochondrion serves as a reservoir, storing apoptosis-regulatory proteins such as cytochrome c and Smac, which upon release to the cytoplasm, promote caspase activation (Jiang and Wang, 2004; Wang, 2001). Notably, the cytosolic apoptotic activities of these mitochondrial proteins are independent of, and can be molecularly dissected from, their mitochondrial functions. As such, a compromise of mitochondrial integrity and loss of mitochondrial membrane potential, known as mitochondrial outer membrane permeability (MOMP), precedes the release of these proteins and subsequent caspase-9 activation. Interestingly, in cysteine deprivation-induced ferroptosis, it is the canonical metabolic function of mitochondria that actively contributes to lipid ROS generation, a prerequisite of ferroptosis; and inhibition of TCA cycle or ETC function, or disruption of MMP, can all abrogate ferroptosis under this condition. Strikingly, a transient hyperpolarization of MMP can be observed during cysteine deprivation-induced ferroptosis, followed by the eventual collapse of MMP (Figures 4 and S4, and Supplementary videos). This unique feature is an indicative for the crucial role of mitochondrial metabolic activity in cysteine deprivation-induced ferroptosis. Taken together, while mitochondria play a passive role in apoptosis by storing various pro-apoptotic proteins, they play a crucial and proactive role in cysteine deprivation-induced ferroptosis by fueling metabolism and lipid ROS production.
Further, the findings presented in this work provide novel mechanistic insights into some enigmatic observations in cancer biology. Fumarate hydratase (FH), a metabolic enzyme in TCA cycle, has been demonstrated to be a tumor suppressor. This is counterintuitive, because one would assume that defects in the TCA cycle or ETC would be disadvantageous for cancer cell growth. Indeed, under normal culture condition, reconstitution of wild-type FH into UOK262/268 cancer cells rendered them proliferating better (Figure S6). So what is the molecular basis underlying the tumor suppressive activity of FH? A pseudo-hypoxic pathway was proposed to be the link between FH mutation and tumorigenesis. FH defect leads to aberrant accumulation of fumarate, which in turn inactivates prolyl hydroxylases and thus stabilizes hypoxia-inducible factor (HIF)-1α (Isaacs et al., 2005; Selak et al., 2005). However, both HIF-1α and HIF-2α are dispensable for the formation of hyperplastic renal cysts in mice lacking expression of FH in the kidney (Adam et al., 2011), suggesting additional tumorigenic consequences caused by FH mutation. Our finding that loss of FH function renders cells more resistant to cysteine deprivation-induced ferroptosis (Figure 6) provides a novel mechanistic explanation of how dysfunction of FH contributes to malignancy independent of hypoxic signaling – we hypothesize that stronger resistance to ferroptosis caused by FH dysfunction contributes to tumorigenesis, although this is most likely not sufficient on its own. It remains to be determined why FH mutation preferentially drives tumorigenesis only in certain specific tissue types.
Conceptually, our finding that ferroptosis contributes to the tumor suppressive function of FH is important. It has been reported recently that p53 can suppress cancer development via its ferroptosis-promoting activity even when it loses its function to promote apoptosis, senescence, and growth arrest (Jiang et al., 2015; Wang et al., 2016). Based on these novel findings about FH and p53, we hypothesize that ferroptosis is a natural tumor suppressive mechanism under diverse biological conditions. The significance of this hypothesis in cancer biology is obvious. As for the biology of ferroptosis, tumor suppression might represent an authentic physiological role of this unique modality of cell death, which, until now, remains to be defined.
STAR * Methods
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| γ-tubulin | Sigma | Cat# T6557 |
| Tom20 | Santa Cruz | Cat#SC11415 |
| Tim23 | Cell Signaling | Cat# 3130S |
| Fumarase | Cell signaling | Cat#4567S |
| Vinculin | Cell signaling | Cat#13901S |
|
Chemicals and Reagents | ||
| erastin | Sigma | Cat#E7781 |
| RSL3 | Selleckchem | Cat# S8155 |
| Dimethyl 2-oxoglutarete | Sigma | Cat#349631 |
| Dimethyl succinate | Sigma | Cat#W239607 |
| Dimethyl fumarate | Sigma | Cat#242926 |
| Dimethyl (S)-(-)-malate | Sigma | Cat#374318 |
| Carbonyl cyanide m-chlorophenyl hydrazone | Sigma | Cat#C2759 |
| Rotenone | Sigma | Cat#R8875 |
| Diethyl butylmalonate | Sigma | Cat#112038 |
| Antimycin A | Sigma | Cat#A8674 |
| Sodium azide | Sigma | Cat# S2002 |
| Oligomycin A | Sigma | Cat#75351 |
| Myxothiazol | Sigma | Cat# T5580 |
| Ferrostatin-1 | Sigma | Cat# M60042 |
| TMRE | Enzo Life Sciences | Cat#ENZ-52309 |
| BODIPY C11 | Thermo Fisher | Cat# D3861 |
| Lipofectamine 2000 | Life Technologies | Cat# 11668027 |
| Propidium Iodide Staining Solution | BD Pharmingen™ | Cat# 556463 |
| SYTOX™ Green Nucleic Acid Stain | Thermo Fisher | S7020 |
|
Deposited Data | ||
| Raw Data | Mendeley | DOI: 10.17632/95zbjbj95x.1 |
|
Experimental Models: Cell Lines | ||
| HT1080 | ATCC | |
| HT1080 mCherry Parkin | Dr. Xuejun Jiang Lab | |
| MEFs | ATCC | |
| UOK262 | Dr. W. Marston Linehan Lab | |
| UOK262 WT | Dr. W. Marston Linehan Lab | |
| UOK268 | Dr. W. Marston Linehan Lab | |
| UOK268 WT | Dr. W. Marston Linehan Lab | |
|
Plasmid and sgRNA sequence | ||
| pLx-gRNA | Addgene | #50662 |
| GPX4-CACGCCCGATACGCTGAGTG | NA | NA |
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources, reagents and further information should be directed to, and will be fulfilled by Xuejun Jiang (jiangx@mskcc.org) and Minghui Gao (gaominghui@hit.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Unless specified otherwise, all mammalian cells are maintained in DMEM with high glucose, sodium pyruvate (1 mM), glutamine (2 mM), penicillin (100 U/ml), streptomycin (0.1 mg/ml) and 10% (v/v) FBS at 37 °C and 5 % CO2. Cell death was analyzed by PI (100 ng/ml), Sytox (5 nM) or DAPI (1 μg/ml) staining coupled with microscopy or flow cytometry.
Measurement of mitochondrial membrane potential
Time-lapse imaging: cells were pre-loaded with 200 nm TMRE (Enzo Life Sciences, Cat#ENZ-52309) for 1 h. Subsequently, cells were washed with PBS twice to remove traces of TMRE from the media and treated as indicated. Fluorescence and differential interference contrast (DIC) images were acquired every 5–7 min, and images were analyzed using NIS elements software (Nikon) and ImageJ software (NIH).
Flow cytometry: Cells were treated as indicated, and then 0.5 μM TMRE was added and incubated for 30 min. Excess TMRE was removed by washing the cells with PBS. Labeled cells were trypsinized and resuspended in PBS plus 2% FBS. Fluorescence at Ex/Em = 549/575 nm was analyzed using a flow cytometer.
Measurement of lipid ROS
Lipid ROS was analyzed by flow cytometry: Cells were seeded at a density of 2.5×105 per well in a 6-well dish and grown overnight in DMEM. 5 μM BODIPY C11 (Thermo Fisher, Cat# D3861) was added into cell culture medium and incubated for 30 min after indicated treatment. Excess BODIPY C11 was then removed by washing the cells with PBS twice. Labeled cells were trypsinized and resuspended in PBS plus 2% FBS. Oxidation of BODIPY C11 resulted in a shift of the fluorescence emission peak from 590 nm to 510 nm proportional to lipid ROS generation and was analyzed using a flow cytometer.
Lipid ROS imaging: Cells were seeded at a density of 2.5×105 per well on coverslips placed in a 6-well dish and grown overnight in DMEM. Cells were washed twice in HBSS and treated as indicated. Coverslips were washed in HBSS and incubated in HBSS containing 2 μM BODIPY 581/591 C11 (Invitrogen) and 200 nM MitoTracker Deep Red FM (Invitrogen) for 20 minutes. Coverslips were then inverted onto microscope slides. Slides were imaged using a Nikon Eclipse Ti-U inverted microscope and images were processed in Photoshop (Adobe).
CRISPR-mediated GPX4 deletion.
Guide RNA sequence CACGCCCGATACGCTGAGTG targeting human GPX4 was used. pLx-gRNA (Addgene#50662) expression plasmid encoding guide RNA was generated and co-transfected with Streptococcus pyogenes Cas9 expression plasmid and transfected into HT1080 cells in the presence of 0.2 mM Trolox using Lipofectamine 2000 (Life Technologies). 24 h after the transfection, the cells were trypsinized and about 200 cells were seeded into a 10-cm plate and cultured in the presence of 0.2 mM Trolox. After cell clones were formed and expanded, western Blot was performed to screening for GPX4 knockout clones.
Fluorescence microscopy
MEFs stably expressing SU9-GFP or HT1080 cells stably expressing mCherry-Parkin were grown on glass cover slips in a 6-well plate. 24 h later, cells were treated as indicated. Cover slips were then fixed with 3.7% (vol/vol) paraformaldehyde (PFA) in 20 mM HEPES pH 7.5 for 30 min at room temperature. Cover slips were then mounted on microscope slides for visualization using Nikon Eclipse Ti-U Microscope with a 60 × magnification objective.
Metabolite analysis by gas chromatography-mass spectrometry (GC-MS)
Metabolite analysis by GC-MS was performed as previously described (Carey et al., 2015). In brief, cells were seeded in standard culture medium in 6-well plates and the next day were changed into experimental medium. Metabolites were extracted with ice-cold 80% methanol supplemented with 20 μM deuterated 2-hydroxyglutarate (D-2-hydroxyglutaric2,3,3,4,4-d5 acid (d5–2HG)) as an internal standard. After overnight incubation at −80°C, lysates were harvested and centrifuged at 21,000 g for 20 min to remove protein. Extracts were dried in an evaporator (Genevac EZ-2 Elite) and resuspended by incubation at 30°C for 90 min in 50 μl of 40 mg/ml methoxyamine hydrochloride in pyridine. Metabolites were further derivatized by addition of 80 μl of MSTFA plus 1% TMCS (Thermo Scientific) and 70 μl ethyl acetate (Sigma) and incubated at 37°C for 30 min. Samples were analyzed using an Agilent 7890A GC coupled to Agilent 5975C mass selective detector. The GC was operated in splitless mode with constant helium gas flow at 1 ml/min. One microliter of derivatized metabolites was injected onto an HP-5MS column and the GC oven temperature ramped from 60°C to 290°C in 25 min. Peaks representing compounds of interest were extracted and integrated using MassHunter software (Agilent Technologies) and then normalized to both the internal standard (d5–2HG) peak area and the relative total biomass as determined by total cell number × mean cell volume. Ions used for quantification of metabolite levels are as follows: d5–2HG m/z 354; Citrate, m/z 465; αKG, m/z 304; fumarate, m/z 245 and malate, m/z 335. All peaks were manually inspected and verified relative to known spectra for each metabolite.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using Prism 7.0c GraphPad Software. Data are presented as mean ± SD from 3 independent experiments. p-values were calculated with Student’s unpaired t test, except for the measurement of mitochondrial membrane potential for which a two-way ANOVA test was used (*P<0.05, **P<0.01, ***p < 0.001, ****p<0.0001, see figure legend for further detail).
Supplementary Material
Time-lapse video of mitochondrial membrane potential (MMP) in MEFs undergoing apoptosis induced by amino acid/serum-double starvation, related to Figure 4.
Time-lapse video of MMP in MEFs undergoing cystine starvation-induced ferroptosis, related to Figure 4.
Time-lapse video of MMP in MEFs undergoing ferroptosis induced by amino acid starvation in the presence of full serum, related to Figure 4.
Highlights.
Mitochondria play a pivotal role in cysteine deprivation-induced (CDI) ferroptosis
Mitochondrial membrane potential hyperpolarization is associated with CDI ferroptosis
TCA cycle, electron transport chain, and glutaminolysis function in CDI ferroptosis
Mutation of tumor suppressor fumarate hydratase confers resistance to CDI ferroptosis
Acknowledgements
The authors thank Dr. W. Marston Linehan for kindly providing the FH-mutant UOK262 and UOK268 cell lines, as well as their wild-type FH reconstituted counterparts. The authors thank members of the Jiang lab and the Thompson lab for critical reading and suggestions. This work is supported by NIH R01CA204232 (to XJ), NIH R01CA201318 (to CBT), a Geoffrey Beene Cancer Research fund (to XJ), National Natural Science Foundation of China 31871388 (to MG), a Leukemia and Lymphoma Society Fellowship (to JZ), and NIH T32 fellowship 5T32GM008539–23 (to AM). This work is also supported by NCI cancer center core grant P30CA008748 to Memorial Sloan Kettering Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests (CBT is a founder of Agio Pharmaceuticals and a member of its scientific advisory board. Agio does not hold financial interests in the work reported in this paper).
References
- Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, et al. (2011). Renal Cyst Formation in Fh1-Deficient Mice Is Independent of the Hif/Phd Pathway: Roles for Fumarate in KEAP1 Succination and Nrf2 Signaling. Cancer cell 20, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam NA, Olpin S, Rowan A, Kelsell D, Leigh IM, Tomlinson IP, and Weaver T (2005). Missense mutations in fumarate hydratase in multiple cutaneous and uterine leiomyomatosis and renal cell cancer. The Journal of molecular diagnostics : JMD 7, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JPF, Shah R, Pratt DA, and Conrad M (2017). Ferroptosis Inhibition: Mechanisms and Opportunities. Trends in pharmacological sciences 38, 489–498. [DOI] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, Pereira HM, Garratt RC, Dias SM, and Ambrosio AL (2012). Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proceedings of the National Academy of Sciences of the United States of America 109, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LJ, Hambright WS, Na R, and Ran QT (2015). Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. Journal of Biological Chemistry 290, 28097–28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang GS, Martinez-Mir A, Geyer A, Engler DE, Glaser B, Cserhalmi-Friedman PB, Gordon D, Horev L, Lukash B, Herman E, et al. (2005). Germline fumarate hydratase mutations and evidence for a founder mutation underlying multiple cutaneous and uterine leiomyomata. Journal of the American Academy of Dermatology 52, 410–416. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, et al. (2016). Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiology of disease 94, 169–178. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature chemical biology 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, and Kroemer G (2015). Ferroptosis in p53-dependent oncosuppression and organismal homeostasis. Cell Death Differ 22, 1237–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, and Jiang X (2017). To eat or not to eat-the metabolic flavor of ferroptosis. Current opinion in cell biology 51, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015a). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MH, Monian P, and Jiang XJ (2015b). Metabolism and iron signaling in ferroptotic cell death. Oncotarget 6, 35145–35146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MH, Monian P, Pan QH, Zhang W, Xiang J, and Jiang XJ (2016). Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018). Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. Acs Chem Biol 13, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, and Tomlinson IP (2005). Mitochondrial tumour suppressors: a genetic and biochemical update. Nature reviews. Cancer 5, 857–866. [DOI] [PubMed] [Google Scholar]
- Green DR, and Reed JC (1998). Mitochondria and apoptosis. Science 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, and Tang D (2016). Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng XX, Freitas FP, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409-+. [DOI] [PubMed] [Google Scholar]
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, et al. (2005). HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer cell 8, 143–153. [DOI] [PubMed] [Google Scholar]
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. (2016). An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30, 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li TY, Wang SJ, Su T, Hibshoosh H, Baer R, and Gu W (2015). erroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XJ, and Wang XD (2004). Cytochrome C-mediated apoptosis. Annual Review of Biochemistry 73, 87–106. [DOI] [PubMed] [Google Scholar]
- King A, Selak MA, and Gottlieb E (2006). Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682. [DOI] [PubMed] [Google Scholar]
- King MP, and Attardi G (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. [DOI] [PubMed] [Google Scholar]
- King MP, and Attardi G (1996). Isolation of human cell lines lacking mitochondrial DNA. Methods in enzymology 264, 304–313. [DOI] [PubMed] [Google Scholar]
- Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, and Wipf P (2016). A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS central science 2, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, and Reed JC (2000). Mitochondrial control of cell death. Nature medicine 6, 513–519. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, et al. (2014). Synchronized renal tubular cell death involves ferroptosis. Proceedings of the National Academy of Sciences of the United States of America 111, 16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, and Youle RJ (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell 7, 77–85. [DOI] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang JL, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, et al. (2014). Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J Am Chem Soc 136, 4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30, 406–410. [DOI] [PubMed] [Google Scholar]
- Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, Romero VV, Sougrat R, Vaulont S, Viollet B, et al. (2011). The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer cell 20, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SJ, Li DW, Ou Y, Jiang L, Chen Y, Zhao YM, and Gu W (2016). Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell reports 17, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD (2001). The expanding role of mitochondria in apoptosis. Genes & development 15, 2922–2933. [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2016). Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 26, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, and Narendra DP (2011). Mechanisms of mitophagy. Nature reviews. Molecular cell biology 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse video of mitochondrial membrane potential (MMP) in MEFs undergoing apoptosis induced by amino acid/serum-double starvation, related to Figure 4.
Time-lapse video of MMP in MEFs undergoing cystine starvation-induced ferroptosis, related to Figure 4.
Time-lapse video of MMP in MEFs undergoing ferroptosis induced by amino acid starvation in the presence of full serum, related to Figure 4.