

SUMMARY
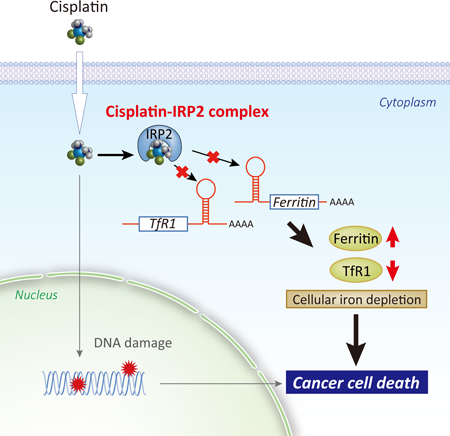
Cisplatin is classically known to exhibit anticancer activity through DNA damage in the nucleus. Here we found a mechanism by which cisplatin affects iron metabolism, leading to toxicity and cell death. Cisplatin causes intracellular iron deficiency through direct inhibition of the master regulator of iron metabolism, iron regulatory protein 2 (IRP2) with marginal effects on IRP1. Cisplatin, but not carboplatin or transplatin, binds human IRP2 at Cys512 and Cys516 and impairs IRP2 binding to iron responsive elements (IREs) of ferritin and transferrin receptor-1 (TfR1) mRNAs. IRP2 inhibition by cisplatin caused ferritin upregulation and TfR1 downregulation leading to sustained intracellular iron deficiency. Cys512/516Ala mutant IRP2 made cells more resistant to cisplatin. Furthermore, combination of cisplatin and the iron chelator desferrioxamine enhanced cytotoxicity through augmented iron depletion in culture and xenograft mouse model. Collectively, cisplatin is an inhibitor of IRP2 that induces intracellular iron deficiency.
Keywords: Cisplatin, Carboplatin, Ferritin, Iron, IRP2, Transferrin Receptor
eTOC paragraph
Short summary
Miyazawa et al. found that the platinum-based anticancer compound cisplatin disrupts the function of iron regulatory protein 2 (IRP2) by adducting to Cys512 and Cys516. Cisplatin-mediated IRP2 inactivation leads to dysregulated iron metabolism and sustained cellular iron deficiency, resulting in cancer cell death in vitro and in mouse tumor models.
Graphical Abstract
INTRODUCTION
Iron is essential for various key enzymes involved in energy metabolism, DNA synthesis, and cell division. Iron deficiency or overload is therefore detrimental to cells and tissues. Iron deficiency impairs iron-dependent enzymes and iron-sulfur clusters- and heme-containing proteins, while iron excess increases a risk of production of reactive oxygen species (ROS) through Fenton reaction (Dixon and Stockwell, 2014). Therefore, cellular iron homeostasis has to be tightly regulated by coordinated expression of genes involved in iron transport and storage, such as transferrin receptor-1 (TfR1) and ferritin (Hentze et al., 2010; MacKenzie et al., 2008). These genes are primarily regulated by iron at the post-transcriptional level through interaction between iron regulatory proteins 1, 2 (IRP1, IRP2) and iron-responsive element (IRE) located in the 3’-untranslated region (UTR) of TfR1 mRNA and 5’-UTR of ferritin mRNA (Anderson et al., 2012; Kuhn, 2015). The binding of IRPs to the IREs is inversely correlated with intracellular iron levels: iron overload disrupts and iron deficiency promotes the binding of IRPs to the IREs (Anderson et al., 2012; Kuhn, 2015). In iron deficient conditions, the binding of IRPs to 3’-TfR1 IRE increases the stability of TfR1 mRNA, resulting in increased iron transport via TfR1 (Mullner et al., 1989). Concomitantly, the binding of IRPs to the 5’-ferritin IRE results in ferritin translational block, resulting in decreased iron storage into ferritin (Goossen et al., 1990; Muckenthaler et al., 1998). Through this coordinated reciprocal regulation of iron transport and storage by the IRP-IRE regulatory system, cells can also adapt to iron overload conditions that induce dissociation of IRPs from IREs, resulting in decreased TfR1 mRNA stability and increased ferritin translation (Bogdan et al., 2016; Wang and Pantopoulos, 2011).
Iron is intimately linked with carcinogenesis and tumor progression (Thompson et al., 1991; Toyokuni, 2014). Tumor cells generally require more iron for keeping the active status of proliferation and DNA synthesis (Torti and Torti, 2013). In addition, high iron may cause increased production of ROS that can stimulate growth factor signaling pathways (Ray et al., 2012) along with DNA oxidation and mutations associated with tumor development (Toyokuni, 2014). Indeed, iron overload has been characterized as a risk factor of human carcinogenesis (Selby and Friedman, 1988; Stevens et al., 1988; Toyokuni, 2014). These results suggest the important roles of IRPs (IRP1 and IRP2) in determining cellular iron availability and proliferation capability. Of note, the majority of IRP1 contains stable 4Fe-4S clusters that do not allow IRP1 to bind IREs, instead serves as a cytosolic aconitase in physiologic conditions (Meyron-Holtz et al., 2004). Unlike IRP1, IRP2 has no iron-sulfur cluster and was reported to be the dominant IRE-binding protein (Meyron-Holtz et al., 2004). However, it should be noted that IRP1 plays important roles in systemic iron homeostasis by regulating the expression of hypoxia inducible factor 2α (HIF2α) (Wilkinson and Pantopoulos, 2013),intestinal iron metabolism (Galy et al., 2008), and mouse embryonic development evidenced by the early lethality of IRP1−/− IRP2 −/− embryos (Smith et al., 2006). IRP2 binding to IRE in physiologic condition is correlated with IRP2 expression levels, in which IRP2 protein is subject to degradation by iron-induced accumulation of the E3 ubiquitin ligase FBXL5 (Salahudeen et al., 2009; Vashisht et al., 2009). Consistently, IRP2, but not IRP1, plays a growth-promoting role in breast cancer cells by elevating intracellular labile iron pool (LIP) (Wang et al., 2014). To deplete iron in cancer cells, evaluation of clinically approved iron chelators, such as desferrioxamine (DFO), a siderophore produced by the Streptomyces pilosus (Wilson et al., 2016) as well as newer chelator compounds such as 3-AP have been underway for potential application of human cancer chemotherapy (Lui et al., 2015; Torti and Torti, 2013).
Platinum-based drugs, such as cisplatin and carboplatin, have been widely used for treatment of solid tumors such as breast, ovarian, testicular, head and neck, and bladder cancers (Dasari and Tchounwou, 2014; Kelland, 2007). General understanding of the anticancer mechanism of these platinum compounds is DNA cross-linking coupled with inhibition of DNA replication and apoptotic cell death after they pass the nuclear membrane (Dasari and Tchounwou, 2014; Kelland, 2007). Additionally, some studies demonstrated the direct interaction between cisplatin and proteins (Karasawa et al., 2013; Will et al., 2008); however, it remains largely unknown whether cisplatin-protein interactions play any important biological roles. In this study, we found that cisplatin, but not carboplatin, transplatin, or platinum chloride, binds to human IRP2 at Cys512 and Cys516 in the IRE-binding cleft and impairs IRP2 binding to IREs. Inhibition of IRP2 by cisplatin caused sustained upregulation of ferritin and downregulation of TfR1, resulting in cellular iron deficiency leading to growth inhibition and cell death. Importantly the cisplatin toxicity was ameliorated when iron was supplied or the cisplatin binding sites of IRP2 were mutated. The notable difference between iron chelators and cisplatin, although both cause cellular iron deficiency, is that iron deficiency induced by iron chelation allows cells to stimulate feedback activation of IRP2 and the IRP-IRE system to recover iron levels, whereas cisplatin does not allow cells to do so due to the direct inhibition of IRP2. Furthermore, combination of cisplatin and the iron chelator DFO enhanced anticancer activity in a mouse xenograft model through augmented iron depletion. These insights into the inhibition mechanism of IRP2 by cisplatin and its impact on cell growth should shed light on the IRE-IRP system, particularly IRP2, as a molecular target of xenobiotic toxicity and anticancer approach as well.
RESULTS
Cisplatin inactivates the IRP/IRE system.
During the course of experiments testing whether some chemotherapeutic agents affect expression of iron metabolism genes, we found that cisplatin is a potent inducer of ferritin heavy chain (ferritin H) in SW480 human colon adenocarcinoma cells (Fig. 1A) and several other human cell types (Supplemental Fig. 1). This effect was seen in cisplatin treatment but not in carboplatin, transplatin (Fig. 1A), or platinum chloride (PtCl2) treatment (Supplement Fig. 1A). Ferritin H and NQO1 are transcriptionally regulated via the Nrf2-ARE system (Iwasaki et al., 2006); however, we observed no induction of NQO1 protein (Fig. 1A) and no increase in ferritin H mRNA levels under cisplatin treatment (Fig. 1B), ruling out the involvement of the Nrf2-ARE system. Concomitantly, we observed downregulation of transferrin receptor-1 (TfR1) protein (Fig. 1A) and mRNA (Fig. 1B) only by cisplatin treatment. Expression of one isoform of divalent metal transporter 1 (DMT1) mRNA containing a 3’-IRE loop was also suppressed by cisplatin treatment in SW480 cells (Supplemental Fig. 1B).
Figure 1. Cisplatin inactivates the IRP/IRE system.
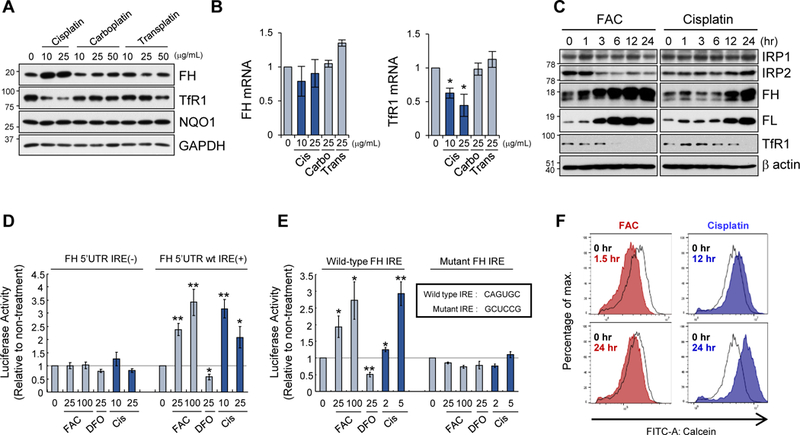
A, B) Expression of ferritin heavy chain (FH) and transferrin receptor 1 (TfR1) proteins (A) and mRNAs (B) was measured by Western blotting and qPCR (normalized with GAPDH mRNA), respectively, in SW480 cells treated with cisplatin, carboplatin, or transplatin for 24 h. NADPH quinone oxidoreductase 1 (NQO1), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) proteins were also measured in A. GAPDH as a protein loading control. Means ± SD are shown (n = 3) in B. * P < 0.001 vs. untreated cells. C) Expression of IRP1, IRP2, ferritin H (FH), ferritin L (FL), TfR1, and β-actin (protein loading control) was measured by Western blotting in SW480 cells treated with 100 µM ferric ammonium citrate (FAC) or 10 µg/mL cisplatin for 0–24 h. D) Luciferase reporter containing 5’-human ferritin H untranslated region (UTR) with or without IRE was transfected into SW480 cells. One day after transfection, cells were treated with 0, 25, 100 µM FAC, 25 µM DFO, or 10, 25 µg/mL cisplatin for 12–24 h, and harvested for luciferase assays. Means ± SD are shown (n = 5–6). * P < 0.05, P < 0.01 vs. untreated cells. E) The same luciferase assays as performed in D to compare FH wild-type IRE to mutant IRE. Means ± SD are shown (n = 5–6). * P < 0.05, ** P < 0.01 vs. untreated cells. F) Calcein-AM staining was performed in SW480 cells treated with 250 µM FAC for 1.5 h and 24 h (left), 10 µg/mL cisplatin for 12 h and 24 h (right). Representative histograms are shown (n = 4). 1.5 h cisplatin treatment showed no shift of the calcein fluorescent peak (not shown).
As both ferritin and TfR1 genes are posttranscriptionally regulated by iron through the IRP-IRE system (Anderson et al., 2012; Kuhn, 2015), we next compared cisplatin with iron during 24 h treatment in the expression of ferritin, TfR1, and IRP1 and IRP2. Ferric ammonium citrate (FAC, 100 µM) induced both ferritin H and L proteins and diminished TfR1 protein by 6 h along with concomitant decrease in IRP2 protein as previously characterized (Salahudeen et al., 2009; Vashisht et al., 2009) (Fig. 1C). Cisplatin also induced ferritin H and L by 12 h, and diminished TfR1 by 24 h; however IRP2 protein levels were slightly increased (Fig. 1C). The upregulation of ferritin H and downregulation of TfR1 by cisplatin without IRP2 protein degradation was also observed in human HeLa, MCF7, and K562 cells (Supplemental Fig. 1C). Ferroportin, a 5’-IRE loop-containing iron efflux gene primarily expressed in macrophages, hepatocytes, and enterocytes (Drakesmith et al., 2015), demonstrated negligible basal expression that was unaffected by iron- or cisplatin-treatment in all tested cell lines (Supplemental Fig 1D); therefore, we consider it unlikely that ferroportin contributes to the observed effects. Furthermore, expression of the 5’-ferritin H IRE-luciferase was induced by cisplatin treatment in SW480 cells (Fig. 1D) and HepG2 (Supplemental Fig. 2) and the loss of function by IRE mutation abolished the effect by FAC and cisplatin (Fig. 1E), suggesting that cisplatin, like FAC, activates ferritin translation via the 5’-UTR IRE. As IRPs are translational repressors of ferritin 5’-UTR-IRE, increased luciferase expression suggests that IRPs and/or the IRP-IRE system was inhibited by cisplatin.
Since cisplatin mimicked most of the iron effects except for downregulation of IRP2 (Fig. 1C), we tested whether cisplatin caused iron overload in the cells. We measured intracellular labile iron pool (LIP) using calcein-AM, a widely adopted fluorescent probe for monitoring iron levels (Ma et al., 2015). The non-fluorescent calcein-AM is converted to a green-fluorescent calcein in cells and its fluorescence is quenched upon binding to intracellular iron. As expected, 100 µM FAC diminished calcein florescence in 1.5 h but returned to untreated levels by 24 h, indicating that LIP was transiently increased (Fig. 1F). Unexpectedly, the calcein fluorescence was significantly enhanced by cisplatin treatment for 12 and further for 24 h (no change at 1.5 h, not shown), indicating that cisplatin causes sustained LIP depletion rather than iron overload (Fig. 1F).
Binding of IRP2 to IRE is inhibited in cisplatin-treated cells.
Given that the expression of the ferritin 5’-IRE-driven luciferase was increased following cisplatin treatment (Fig. 1D, 1E), we tested the possibility of inhibition of the IRP/IRE system as the mechanism of the LIP decrease by cisplatin. First, we tested whether cisplatin inhibits binding of IRPs to the IRE by an RNA-protein pull-down assay. Cell lysates from SW480 treated with cisplatin at 5, 10, and 25 µg/ml for 24 h were incubated with biotinylated ferritin H IRE RNA, followed by precipitation of the IRE binding complex with streptavidin-agarose and Western blotting with anti-IRP1 and anti-IRP2 antibodies. Cells treated with FAC and the iron chelator DFO were included as controls of decreased and increased IRP-IRE interactions, respectively. Indeed, cisplatin treatment caused decreased IRP2 binding to the IRE in a dose-dependent manner, whereas IRP1 binding to the IRE was not inhibited, but rather slightly increased (Fig. 2A). To verify this observation in live cells treated with cisplatin, we performed an RNA immunoprecipitation (RIP) assay. Due to poor immunoprecipitation of endogenous IRP1- and IRP2-mRNA complexes with our IRP1 and IRP2 antibodies, we needed transfection of HA-tagged IRP1 or IRP2 into SW480, followed by treatment with cisplatin at 25 µg/ml, immunoprecipitation of HA-IRP/mRNA complex with anti-HA antibody, and RT-PCR to measure co-precipitated ferritin H mRNA. The anti-HA antibody precipitated ferritin H mRNA 10-times more efficiently in cells expressing HA-IRP2 than HA-IRP1 (Fig. 2B, (−) cisplatin), suggesting that IRP2 is the major ferritin H IRE binding protein. Consistent with the pull-down results in Fig. 2A, cisplatin treatment slightly increased IRP1 binding but significantly decreased IRP2 binding to the ferritin H mRNA (Fig. 2B). In addition, we coincidentally observed that expression of IRP2 in HEK293 cells was undetectable with our anti-IRP2 antibody, in which ferritin expression was unchanged by cisplatin treatment (Supplemental Fig. 3). These results suggest that the inhibition of IRP2 binding to the IRE by cisplatin caused ferritin upregulation and TfR1 downregulation observed in Fig. 1. Indeed, endogenous IRP1 knockdown had a much weaker effect on endogenous ferritin H, ferritin L, and TfR1 expression compared to IRP2 knockdown showing significant increase in ferritin H and L along with pronounced decrease in TfR1 expression (Fig. 2C). IRP1 has been reported to have important roles in systemic iron homeostasis (Galy et al., 2008; Smith et al., 2006; Wilkinson and Pantopoulos, 2013); however, our results suggest that IRP2 is the major regulator of cellular iron homeostasis in our system, consistent with previous studies (Meyron-Holtz et al., 2004; Schalinske et al., 1997). Collectively, these results suggest that cisplatin inhibits IRP2 binding to IRE and the IRP-IRE system.
Figure 2. IRP2 binding to IRE is inhibited by cisplatin.
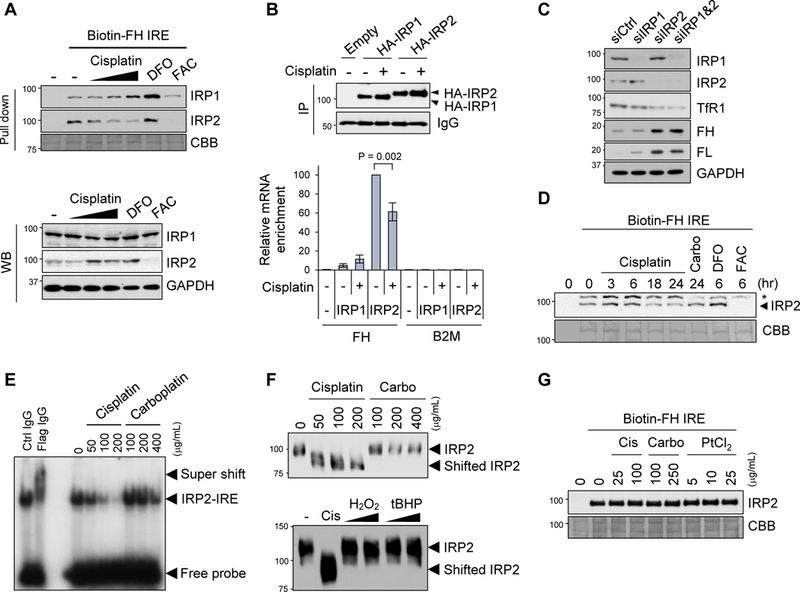
A) Binding of IRPs to IRE was assessed by pull-down assays using whole cell lysates from SW480 cells incubated with cisplatin (0, 5, 10, 25 µg/mL) for 24 h, DFO (100 µM) or FAC (250 µM) for 10 h. Whole cell lysates were incubated with biotinylated ferritin H IRE probe (Biotin-FH IRE) and precipitated with streptavidin beads, followed by Western blotting with IRP1 and IRP2 specific antibodies. Coomassie Brilliant Blue (CBB) staining for verification of equal protein loading. IRP1, IRP2, and GAPDH protein levels in the whole cell lysates were measured by Western blotting (WB). B) Binding of IRPs to ferritin IRE was assessed by RIP assays. SW480 cells stably transfected with pcDNA3.1/C-HA (Empty), HA-IRP1, or HA-IRP2 plasmid were treated with 25 µg/mL cisplatin for 22 h. Whole cell lysates were immunoprecipitated with anti-HA antibody (IP) and 10% of IP samples were subjected to Western blot with anti-HA antibody to check IP efficiency (top Western blots). IgG signal as a loading control. The same IP samples were used for RIP assays for detection of ferritin H (FH) or β2-microglobulin (B2M, negative control) mRNA enrichment by qPCR (bottom graph). mRNA enrichment was normalized by input total RNA, and the results were shown as relative mRNA enrichment by IRP2 (- cisplatin, 100%). Mean ± SD (n = 3). C) siRNA for control (siCtrl), IRP1, or/and IRP2 was transfected into SW480 cells and whole cell lysates were used for Western blots with IRP1, IRP2, TfR1, ferritin H (FH), ferritin L (FL) or GAPDH antibodies. D) Whole cell lysates were incubated with 10 µg/mL cisplatin for 0, 3, 6, 18, 24 h, 25 µg/mL carboplatin for 24 h, 100 µg/mL DFO for 6 h, or 250 µg/mL FAC for 6 h in test tubes, and pull-down assay was performed using a biotin-FH IRE probe for detection of IRE-bound IRP2 by Western blotting. * indicates a non-specific band. CBB staining is shown for verification of equal loading. E) Human Myc-Flag-tagged IRP2 protein was incubated with cisplatin or carboplatin in the presence of 32P-labeled ferritin H IRE probe. IRP2-IRE complex was separated from the free IRE probe by electrophoresis and visualized by autoradiography. Control IgG or anti-Flag antibody was incubated with samples of Myc-Flag-IRP2 mixed with 32P-IRE probe for verification of the IRP2-IRE interaction as a super shift (left two lanes). F) Human Myc-Flag-tagged IRP2 protein was incubated with indicated concentrations of cisplain, carboplatin (top), or cisplatin (200 µg/mL), H2O2 (0.1 mM, 1 mM) or tert-butylhydroperoxide (tBHP, 0.1 mM, 1 mM) (bottom) overnight at room temperature. Samples were loaded on native polyacrylamide gel electrophoresis and subjected to Western Blotting with anti-IRP2 antibody. G) A biotin-FH IRE probe mixed with streptavidin beads was pre-incubated with cisplatin, carboplatin or PtCl2 overnight. Then, the complex was washed to remove unbound platinum compounds and re-incubated with SW480 whole cell lysates, and pull down assay was performed for detection of IRP2-IRE interaction by Western blotting. CBB staining for verification of equal protein loading.
Cisplatin directly inhibits IRP2 for interaction with IRE.
If cisplatin directly inhibits the binding of IRP2 to IRE, there are two targets; IRP2 and the IRE stem-loop RNA. First, to test the possibility of direct interaction between cisplatin and IRP2, whole cell lysates were pre-incubated with cisplatin for 3–24 h or carboplatin for 24 h in test tubes prior to incubation with a biotinylated ferritin IRE RNA probe and pull-down with streptavidin-agarose beads. We observed that the amount of IRP2 precipitated with the IRE probe was decreased at 18 and 24 h (Fig. 2D). To confirm this result, we performed electrophoresis mobility shift assay (EMSA) by pre-incubation of purified Myc-Flag-IRP2 protein with either cisplatin or carboplatin in test tubes, followed by incubation with radiolabeled ferritin H IRE RNA probe and acrylamide gel electrophoresis. We observed that cisplatin but not carboplatin significantly inhibited IRP2 binding to the IRE probe in a dose-dependent manner (Fig. 2E). In addition, we analyzed the EMSA samples of Myc-Flag-IRP2 incubated with cisplatin or carboplatin on native polyacrylamide gel electrophoresis (without 2-mercaptoethanol) and Western blotting with anti-Flag antibody. We observed that the IRP2 protein incubated with cisplatin exhibited migration shift (faster migration) than untreated or carboplatin-treated samples (Fig. 2F). Furthermore, this migration shift was not likely due to oxidation of IRP2 by cisplatin because incubation of the purified IRP2 with various oxidizing agents such as 0.1 and 1 mM hydrogen peroxide (H2O2) and tert-butyl hydroperoxide (tBHP) did not induce the faster migration (Fig. 2F).
Given the propensity of cisplatin to covalently bind to nucleic acid, we also assessed the possibility of the inhibition of the IRP-IRE system through direct binding of cisplatin to the IRE RNA. To this end, we pre-incubated the ferritin H IRE RNA probe with cisplatin, carboplatin, or PtCl2 for 14 h, and precipitated the IRE probe with streptavidin agarose to remove unbound platinum compounds. Then, we incubated the pre-incubated ferritin H IRE RNA probe with whole cell lysates and pulled down the IRE-protein binding complexes with streptavidin agarose and Western blots with anti-IRP2 antibody. As shown in Fig. 2G, pre-incubation of cisplatin with the IRE RNA probe did not affect the IRP2 binding, suggesting that direct interaction of cisplatin with the IRE RNA stem loop is not the mechanism of the inhibition of IRP2-IRE interaction. Collectively, these results suggest that cisplatin directly inhibits IRP2 for the binding to the IRE.
Cisplatin binding at Cys512 and Cys516 of human IRP2
To identify cisplatin binding sites on IRP2, purified human IRP2 protein incubated with cisplatin was analyzed using the liquid chromatography-mass spectrometry LC-MS/MS. The sequence coverage of the entire IRP2 protein was 98% (Supplemental Fig. 4A). Cisplatin modification sites in the IRP2 protein were searched in peptide fragments Mass spectrum, in which the peptide L499~P518 (LSHGSVVIAAVISCTNNCNP) was found as a platinated fragment (Supplemental Fig. 4B). Further analyses with Proteome Discoverer 1.4 software (Thermo Scientific) identified the major cisplatin modification site at Cys512 indicated by a mass increase of cisplatin 246.03 [Pt(NH3)2(H2O)] on b14-H2O2+ ion (Fig. 3A, Supplemental Fig. 4C).
Figure 3. Identification of cisplatin-IRP2 adduct formation by LC/MS/MS.
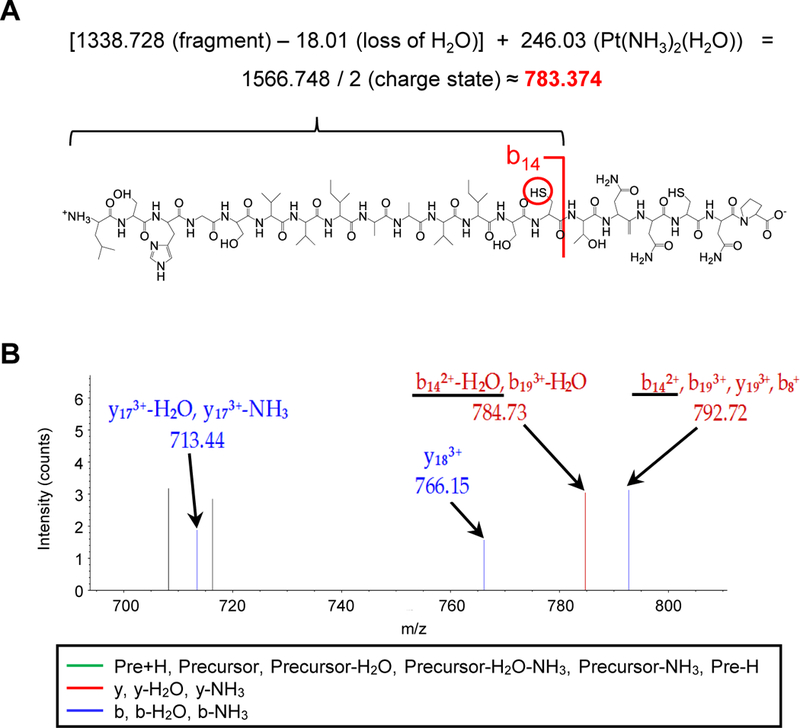
A) Human Myc-Flag-tagged IRP2 protein was incubated with 500 µg/mL cisplatin for 24 h at 4 °C and LC-MS/MS was performed. Platination of Cys512 was manually confirmed by the calculation based on b14-H2O2+ ion of the peptide L499-P518 (LSHGSVVIAAVISCTNNCNP). B) Zoomed spectrum to Cys512 (b142+-H2O and b142+ underlined) is shown.
We were also able to manually inspect the spectra and confirmed the platination at Cys512 (Fig. 3B). In addition to Cys512, we detected the second platination site at Cys516 based on the mass data with a loss of water or ammonia of the b and y ions from the L499~P518 peptide (Supplemental Fig. 4D, 4E). Collectively, we concluded that cisplatin directly interacts with the human IRP2 primarily at Cys512 and additionally at Cys516.
Cys512 and Cys516 mutations restore IRP2 function under cisplatin treatment
To determine whether cisplatin needs both Cys512 and Cys516 for inhibition of IRP2, we isolated SW480 cells stably transfected with HA-tagged human wild-type IRP2 and Cys to Ala mutant IRP2 (C512A, C516A, and C512A/C516A) plasmids and tested whether Cys512 and/or Cys516 mutations affects their bindings to IRE and ferritin expression under cisplatin treatment. We also isolated cells stably expressing C578A/C581A IRP2, which is Cys to Ala mutated in the CXXC sequence we found in the human IRP2. This is to test another potential cisplatin adduct site according to the previous report that cisplatin adducts cysteines in the CXXC sequence of the copper chaperon Atox1 protein (Calandrini et al., 2014). They expressed wild-type and mutant IRP2 proteins equally (Fig. 4A, top). Whole cell lysates were incubated with cisplatin and analyzed for their interactions with the biotin-labeled ferritin IRE probe by pull-down with streptavidin-agarose and Western with anti-HA antibody. Without incubation with cisplatin, all IRP2 mutants exhibited equivalent binding to the IRE probe compared with wild-type IRP2; however, binding of wild-type IRP2 and C578A/C581A mutant to the IRE probe was significantly diminished when incubated with cisplatin (Fig. 4A, bottom). In contrast, C512A, C516A, and C512A/C516A IRP2 were resistant to cisplatin and able to fully bind to the IRE probe (Fig. 4A, bottom). These results suggest that the interaction of cisplatin at both Cys512 and Cys516 of IRP2 is necessary to impair the binding of IRP2 to IRE. Similar results were obtained in cell lysates of K562 cells transiently expressing non-tagged IRP2 (Supplemental Fig. 5A).
Figure 4. Cys512 and Cys516 mutations restore IRP2 function under cisplatin treatment.
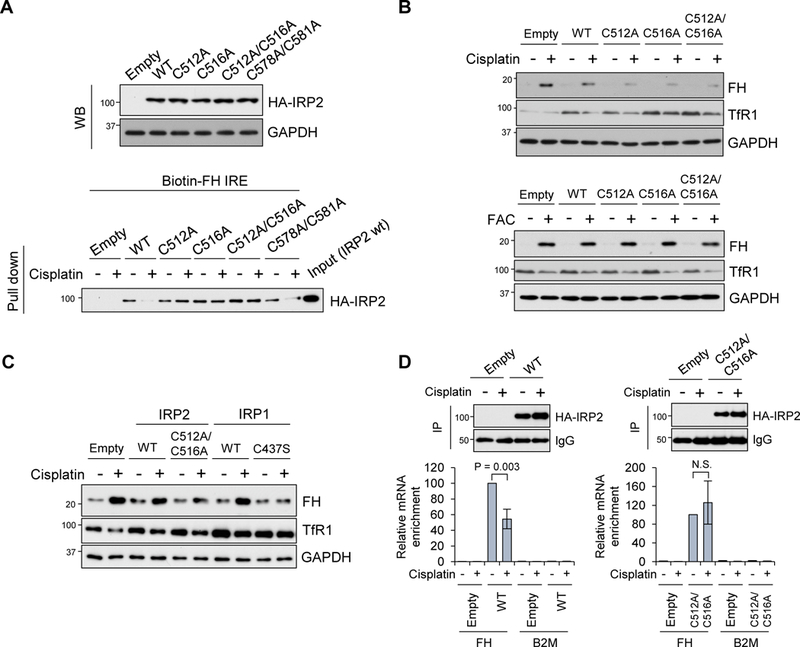
A) HA-tagged full length human IRP2 wild-type and Cys to Ala mutants (C512A, C516A, C512A/C516A, C578A/C581A) in pcDNA3.1/C-HA plasmids were stably transfected into SW480 cells. Expression of transfected HA-IRP2 was detected by Western blot with anti-HA antibody, and GAPDH as a loading control (WB, top). These cell lysates were also incubated with 20 µg/mL cisplatin and subjected to pull down assay using a biotin-FH IRE probe. IRP2 binding to IRE was detected by Western blot with anti-HA antibody (Pull down, bottom). B) SW480 cells stably transfected with IRP2 wt and mutants were treated with 10 µg/mL cisplatin for 36 h (top) or 100 µg/mL FAC for 16 h (bottom), and Western blots were performed with ferritin H (FH), TfR1 and GAPDH antibodies. C) SW48 cells stably transfected with empty vector, IRP2 (WT, C512A/C516A) and IRP1 (WT, C437S) were treated with 10 µg/ml cisplatin for 20 hr and Western blots were performed with ferritin H (FH), TfR1 and GAPDH antibodies. D) pcDNA3 (Empty), IRP2WT, or C512A/C516A expressing SW480 cell were treated with 25 µg/mL cisplatin for 22 h, subjected to RIP assay using anti-HA antibody, followed by qPCR using ferritin H (FH) or B2M primers. 10% of IP samples were subjected to Western blot with anti-HA antibody to check IP efficiency (top Western blots), and IgG signal as a loading control. RNA enrichment was normalized by input total RNA and results were shown as relative RNA enrichment by WT or C512A/C516A (- cisplatin, 100%). Mean ± SD (n = 3) (bottom graphs).
Instead of incubation of cisplatin and cell lysates in the test tube, we treated these stable wild-type and mutant IRP2 expressing cells with cisplatin and measured expression of endogenous ferritin H and TfR1 proteins by Western blotting. Consistently, cisplatin treatment highly induced ferritin H expression in pcDNA3 empty vector or wild-type IRP2 expressing cells, while cells expressing C512A, C516A, and C512A/C516A IRP2 proteins significantly lost the induction of ferritin H synthesis in response to cisplatin treatment (Fig. 4B, top). Downregulation of TfR1 by cisplatin was partially rescued in cells expressing these IRP2 mutants (Fig. 4B).
These results suggest that protection of either Cys512 or Cys516 by Ala substitution makes the IRP2 mutants resistant to inactivation by cisplatin. In other words, inhibition of IRP2 by cisplatin needs both Cys512 and Cys516. Notably, all these wild-type and mutant IRP2 proteins showed normal response to iron treatment for ferritin upregulation and TfR1 downregulation (Fig. 4B, bottom) and normal IRP2 protein stabilization in iron (FAC) or iron chelator (DFO) treatment (Supplemental Fig. 5B). If the inactivation of IRP2 by cisplatin is responsible for ferritin upregulation and TfR1 downregulation, forced expression of an IRP1 mutant that constitutively binds to IREs (C437S) (DeRusso et al., 1995; Wang and Pantopoulos, 2002) should rescue the effects of cisplatin-mediated IRP2 inactivation. To address this question, we treated stably transfected SW480 (empty, wtIRP2, C512A/C516A-IRP2, wtIRP1, and C437S-IRP1) with cisplatin followed by ferritin H and TfR1 Western blotting. We found that the IRP1 mutant C437S rescued cisplatin-induced ferritin upregulation and TfR1 downregulation (Fig 4C), suggesting that the IRE-binding form of IRP1 can compensate for cisplatin-induced inactivation of IRP2.
To assess the inhibitory effect of cisplatin on IRP2 through attacking Cys512 and Cys516 in the cells, we performed RIP assays to measure the ferritin IRE and IRP2 interaction in stable wild-type or C512A/C516A IRP2 expressing cells after treatment with cisplatin. As expected, interaction between the IRE-containing ferritin mRNA and wild-type IRP2 was significantly diminished by cisplatin treatment, whereas the interaction with C512A/C516A mutant IRP2 was unchanged after cisplatin treatment (Fig. 4D). Taken all together, we concluded that cisplatin directly binds to human IRP2 both at Cys512 and Cys516 and inhibits binding of IRP2 to the IREs, resulting in the release of ferritin translation suppression and simultaneous destabilization of TfR1 mRNA as observed in Fig. 1A and 1C. The increased ferritin and decreased TfR1 by inhibition of IRP2 can drive more iron storage into ferritin and less iron transport through TfR1 leading to iron deficiency as observed in Fig. 1F. This is consistent with the results of IRP2 knockout cells and mouse models (Jeong et al., 2011; LaVaute et al., 2001; Meyron-Holtz et al., 2004; Zumbrennen-Bullough et al., 2014).
Protection of IRP2 Cys512 and Cys516 from the cisplatin attack makes cells resistant to cisplatin cytotoxicity
Given that IRP2 has growth-promoting and oncogenic functions (Maffettone et al., 2010; Wang et al., 2014), we next tested whether expression of the cisplatin-resistant IRP2 is sufficient for making cells resistant to cisplatin toxicity. To this end, SW480 cells stably expressing HA-tagged IRP2 wild-type or C512A/C516A equally (Fig. 5A) were treated with 20 µg/ml cisplatin for 24 h and cultured for additional 48 h in the normal growth media, and viable cells were counted. Overexpression of wild-type IRP2 made cells slightly resistant to cisplatin toxicity (Fig. 5B), and C512A/C516A mutant IRP2 made more resistant to cisplatin toxicity, 1.6 folds higher than the pcDNA3 empty vector transfectants (Fig. 5B). This was correlated with the weakest activation of caspase 3 by cisplatin treatment in C512A/C516A mutant IRP2 expressing cells (Fig. 5C). Cisplatin treatment depleted intracellular iron levels in all three cell types, in which expression of C512A/C516A mutant IRP2 alleviated the iron deficiency more effectively than wild-type IRP2 (Fig. 5D). To further verify the involvement of iron deficiency in cisplatin toxicity, cell viability was measured in cisplatin-treated cells for 1–3 days with or without additional iron in the media. This assay showed that cisplatin cytotoxicity was alleviated by iron supply (FAC) (Fig. 5E). In fact, iron chelator inducible and pro-apoptotic genes such as GADD45β, p21 and DUSP1 (Saletta et al., 2011; Yu and Richardson, 2011) were remarkably enhanced by cisplatin treatment (Fig. 5F). Taken together, iron deficiency induced by cisplatin is involved in its anti-proliferative and apoptotic toxicity mechanisms.
Figure 5. Protection of IRP2 Cys512 and Cys516 from the cisplatin attack makes cells resistant to cisplatin cytotoxicity.
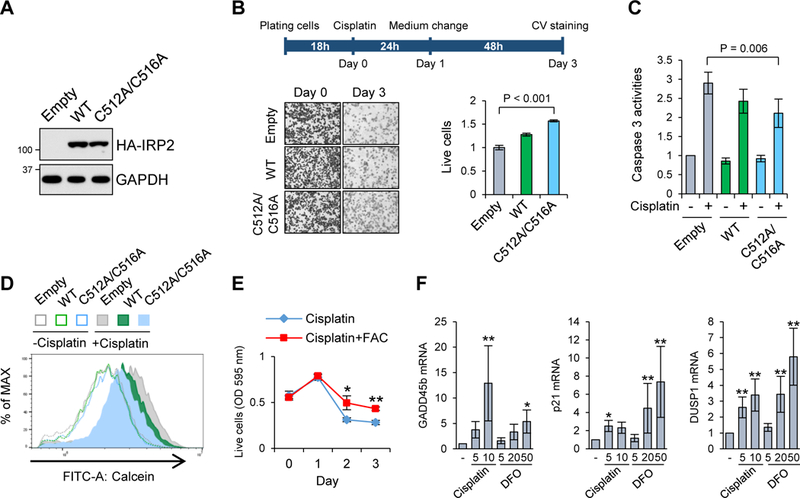
A) Expression of stably transfected IRP2WT and C512A/C516A IRP2 mutant in SW480 cells were assessed by Western blot with anti-HA antibody. B) They were treated with 20 µg/mL cisplatin for 24 h, and cultured for additional 48 h in the normal growth media. Left panels represent microscopic photographs of cells stained with crystal violet (CV) at day 0 and day 3. CV staining at day 3 was quantitated by normalization with day 0, and the ratio in pcDNA3 transfected cells (Empty) was defined as 1. Means ± SD are shown (n = 4). C, D) They were treated with 10 µg/mL cisplatin for 16–18 h and the results of caspase 3 assay (in C, means ± SD, n = 6) or calcein-AM staining (in D, a representative histogram from four independent experiments) are shown. E) Cell viability was measured by CV staining in SW480 cells treated with 10 µg/mL cisplatin with or without 100 µM FAC for 1–3 days. Means ± SD are shown (n = 3). *P = 0.016, **P < 0.001. F) mRNA expression of GADD45β,p21 and DUSP1 was measured by qPCR (normalized by GAPDH or B2M mRNA) in SW480 cells treated with 5 and 10 µg/mL cisplatin, 5, 20 and 50 µM DFO for 20–24 h,shown as means ± SD (n = 8–16). *P < 0.05, **P < 0.001 vs. untreated cells.
IRP2 dominates cancer cell survival and proliferation
Various tumors demand more iron to maintain their rapid cell proliferation and DNA synthesis (Torti and Torti, 2013). TfR1 is overexpressed in many types of cancers (Chan et al., 2014; Habashy et al., 2010; Magro et al., 2011; Prutki et al., 2006). In addition to the transcriptional activation of the TfR1 gene in cancer cells (O’Donnell et al., 2006), TfR1 mRNA can be stabilized by the IRP-IRE system (Miyazawa et al., 2018; Mullner et al., 1989). To validate this possibility in human cancer samples, we searched the relationship between IRPs and TfR1 mRNA expression using RNA-Seq V2 data set of the breast invasive carcinoma patients (817 samples, (Ciriello et al., 2015)) in The Cancer Genome Atlas (TCGA) database. In contrast to poor correlation between IRP1 and TfR1 mRNA levels (Pearson’s chi-squared test: 0.048, Spearman’s rank correlation: 0.102), IRP2 expression was well correlated with TfR1 mRNA levels (Pearson’s chi-squared test: 0.217, Spearman’s rank correlation: 0.339) (Fig. 6A, 6B). The correlation in the expression levels of TfR1 mRNA and IRP2 better than IRP1 seems to be consistent with the fact that the majority of IRP1 forms a stable iron-sulfur cluster serving as a cytoplasmic aconitase therefore does not participate in binding to IREs in animal tissues (Meyron-Holtz et al., 2004). We also analyzed the relationship between overall survival rates and IRPs mRNA expression in the same breast invasive carcinoma patient data. We found that IRP1 expression levels had marginal correlation with patient’s survival rates (Fig. 6C), contrasting that high IRP2 expression group had significantly shorter survival rates than lower IRP2 expression group (Fig. 6D). We also observed that IRP2 contributed to cell survival and proliferation in culture cells. IRP1 knockdown or overexpression in SW480 cells showed no effect on TfR1 protein expression and cell viability (Fig. 6E). By contrast, IRP2 knockdown significantly decreased cell viability versus control (Fig. 6F), which was associated with caspase-3 activation compared with siControl (Supplemental Fig. 6). Conversely, IRP2 overexpression increased TfR1 protein and viable cell numbers as well (Fig. 6F). These results support the previous reports (Maffettone et al., 2010; Wang et al., 2014) and suggest that IRP2 dominates survival and proliferation of some human cancer cells.
Figure 6. IRP2 dominates cancer cell survival and proliferation.
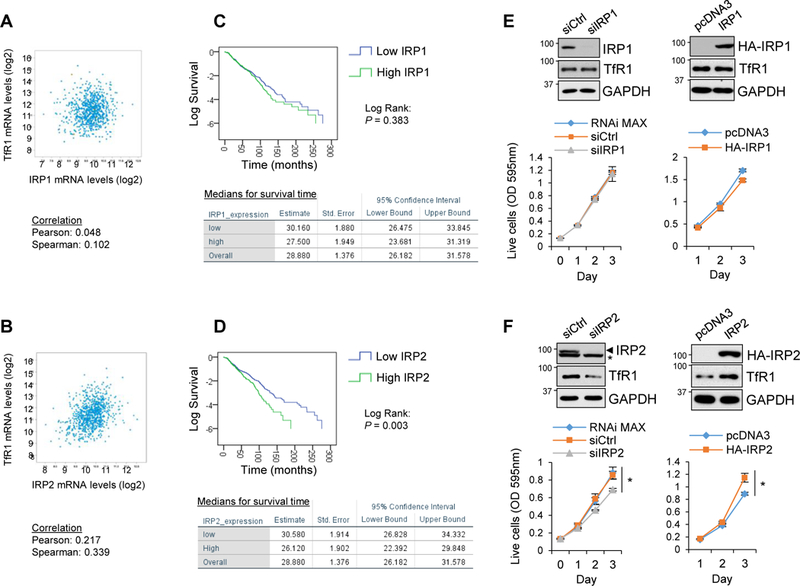
A, B) Scatter plots of expression levels of IRP1, IRP2, and TfR1 mRNAs are shown in breast invasive carcinoma patients [817 samples in The Cancer Genome Atlas (TCGA) database in cBioPortal (Ciriello et al., 2015). C, D) Relationship between IRP1, IRP2 mRNA levels and survival rates of the same breast invasive carcinoma patients was calculated by IBM SPSS 24 statistic software. Data from all 817 samples were classified (top 50%: high expression, bottom 50%: low expression group), and Kaplan-Meier estimation and Log Rank (Mantel-Cox) test were performed between high and low groups (IRP1: P = 0.383, IRP2: P = 0.003). E, F) IRP1 or IRP2 siRNA or HA-tagged wild-type IRP1 or IRP2 expression plasmid was transfected into SW480 cells and protein expression levels of IRPs, TfR1 and GAPDH were measured by Western blotting (top panels). Cell viability/growth was also measured by CV staining from day 0 to day 3 after transfection and quantitated (bottom graphs). Means ± SD are shown (n = 4–6). *P < 0.001.
Sustained iron depletion and enhanced cytotoxicity by combination of cisplatin and iron chelator DFO in vitro and in vivo
Chemotherapeutic strategies using iron chelators for depletion of iron in cancer cells and microenvironment have been elucidated for antineoplastic potential in various human cancers (Lui et al., 2015; Torti and Torti, 2013). Besides the development of several new iron chelators, there were mixed outcomes in regard to their anticancer activities in clinical trials (Lui et al., 2015). The major drawback of iron chelators is that iron deficiency induced by iron chelation is transient by allowing cells to stimulate feedback activation of IRP2 and the IRP-IRE system, resulting in more iron transport and less iron storage to recover from iron deficiency. Given that cisplatin depletes LIP by inactivation of IRP2, we hypothesized that the combination treatment of cisplatin and an iron chelator may decrease LIP more efficiently because the inactivation of IPR2 by cisplatin does not allow cells to reboot the IRP-IRE system. To test this possibility first in cell culture, we measured live cell numbers after treatment with cisplatin, DFO, or cisplatin plus DFO for 5 days. Indeed, cisplatin plus DFO enhanced the cytotoxicity compared to cisplatin or DFO alone, which was ameliorated partially but significantly by adding iron (Fig. 7A). Cisplatin and DFO co-treatment further reduced the LIP level compared with cisplatin or DFO alone, which was reverted partially by supplying iron with FAC treatment (Fig. 7B).
Figure 7. Sustained and enhanced iron deficiency and toxicity by combination of cisplatin and iron chelator DFO in vitro and in vivo.
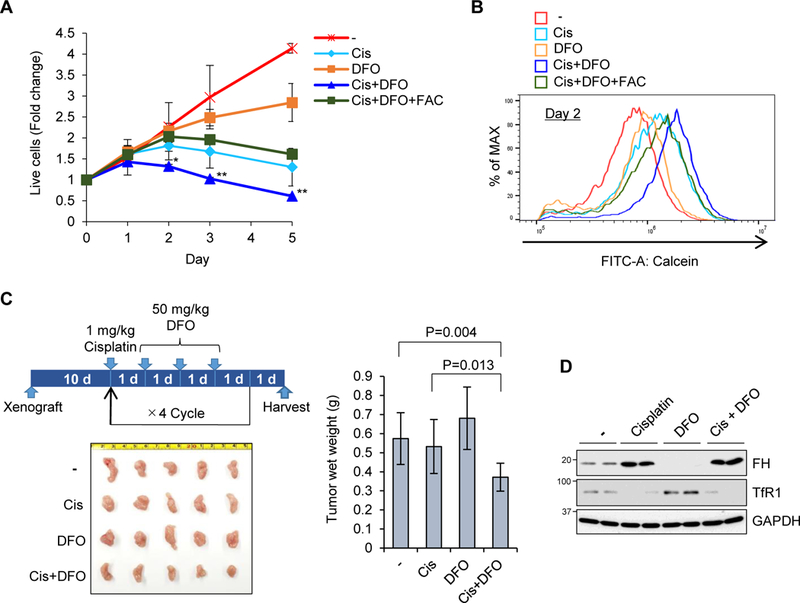
A) SW480 cells were treated with 5 µg/mL cisplatin (Cis), 5 µM DFO, 10 µM FAC, or their combinations for 5 days, and stained with crystal violet. OD 595 was measured and normalized with day 0 as 1 in each condition. Means ± SD are shown (n = 3–6). *P < 0.01, **P < 0.001. B) The same treatment as A) for 2 days and calcein-AM staining for LIP were shown (n =3–6). C) SW480 cells were grafted into NSG mice subcutaneously, and 1 mg/kg/day cisplatin and 50 mg/kg/day DFO for 3 days were injected into abdominal cavity. After four cycles of the treatment, these tumors were isolated and wet weight was measured. Means ± SD are shown (n = 6–8). D) One day after injection, these tumors were isolated and used for Western blot with ferritin H (FH), TfR1 and GAPDH antibodies.
To evaluate the efficacy of cisplatin and DFO combination in tumor growth in vivo, we used a mouse xenograft model by subcutaneous injection of SW480 cells in NOD scid gamma (NSG) immunodeficient mice. After tumor implantation for 10 days, intraperitoneal injection of cisplatin (1 mg/kg) was followed by daily injection of DFO (50 mg/kg) consecutively for 3 days because of short plasma half-life of DFO (Lui et al., 2015). The same administration protocol was repeated 4 times before harvesting solid tumors (Fig. 7C). In this chemotherapy protocol, we observed that cisplatin or DFO alone failed but combination of cisplatin and DFO inhibited the solid tumor growth (Fig. 7C). In solid tumors harvested from each treatment group, we found significant ferritin induction and TfR1 downregulation in cisplatin and cisplatin plus DFO treated tumors (Fig. 7D). Collectively, cisplatin is an inhibitor of IRP2 causing iron depletion and cytotoxicity in a different mechanism from iron chelators and their combination results in an enhanced cytotoxic response due to disruptions in iron metabolism.
DISCUSSION
Cisplatin binds proteins covalently (Karasawa et al., 2013; Will et al., 2008) but little was known about their roles in cisplatin cytotoxicity. In this study we found that cisplatin is an inhibitor of IRP2 through a site-specific interaction that impairs the key regulatory mechanism of cellular iron homeostasis, the IRP-IRE system. Cisplatin causes cytotoxicity through iron deprivation via the IRP2 inhibition, supported by the results that iron supply (Fig. 5E, 7A) or expression of cisplatin-resistant mutant IRP2 (Fig. 5A-5D) ameliorated the cisplatin cytotoxicity. According to the previous report of cisplatin binding to a Cys-X-X-Cys sequence in the copper chaperone Atox protein (Calandrini et al., 2014), we also tested this sequence as potential cisplatin binding sites in the human IRP2 (Cys578 and Cys581) (Fig. 4). These cysteines are conserved in IRP1 at Cys503 and Cys506 located in the region required for IRP1 iron-sulfur cluster formation (Zumbrennen et al., 2009). However, IRP1 was not inactivated by cisplatin (Fig. 2A). Furthermore, the C578A/C581A mutant IRP2 behaved similarly to wild-type IRP2 and lost IRE binding ability in response to cisplatin treatment (Fig. 4A), indicating that these Cys residues are not the cisplatin interaction sites. Instead, our mass spectrometry analyses identified the human IRP2 Cys512 and Cys516 as cisplatin interaction sites (Fig. 3 and Supplemental Fig. 4). These sites are located in the RNA binding cleft of IRP2 (Zumbrennen et al., 2009). It should be noted that these cysteines are able to form a disulfide bond under oxidative stress conditions, which decreased binding of IRP2 to IRE leading to destabilization of TfR1 mRNA (Zumbrennen et al., 2009). In addition, a recent report suggested that Cys512 and Cys516 are potential sites for succination (Kerins et al., 2017). The human IRP2 Cys512 is conserved in IRP1 at Cys437 but IRP2 Cys516 is not conserved, replaced with Ser441 in the human IRP1. Our IRP2-IRE binding assays demonstrated that cisplatin needed both Cys512 and Cys516 for the inactivation of IRP2 (Fig. 4B). Although IRP1 has a minor role in the regulation of the IRP-IRE system in our system (Fig. 2C), we anticipate that IRP1 may be resistant to cisplatin for binding to IRE even if Cys437 is modified with cisplatin because of the lack of the second cysteine in IRP1. Notably, mutating IRP1 Cys437 to serine (C437S) creates a form of IRP1 that constitutively binds to IREs and can compensate for IRP2 inactivation by cisplatin (Fig 4C).
No inactivation of IRP2 by carboplatin, transplatin, or platinum chloride (Fig. 1A, Supplemental Fig. 1A) may be consistent with the propensity that DNA adduct formation by carboplatin is weaker and slower than cisplatin (Kelland, 2007). Other second-generation platinum compounds such as Nedaplatin, Oxaliplatin, Lobaplatin, and Heptaplatin are structural derivatives of carboplatin having less reactivity and toxicity (Dasari and Tchounwou, 2014; Kelland, 2007). Therefore, like carboplatin, we postulate that these platinum compounds may not cause iron deprivation via the inhibition of IRP2, although it would be necessary to test their effects on IRP2. The lack of IRP2 inhibition may be a part of reasons for generally less cytotoxicity and less side effects by these second-generation platinum compounds.
Inhibition of IRP2 by cisplatin caused downregulation of TfR1 and upregulation of ferritin, resulting in significant decrease in LIP (Fig. 1F, 7B). This is consistent with the results in IRP2 knockout mice displaying dysregulation of iron metabolism with iron deficiency due to overexpression of ferritin and downregulation of TfR1 in various tissues including brain (LaVaute et al., 2001), which contributed to the late-onset behavioral impairments or neurodegeneration (Jeong et al., 2011; LaVaute et al., 2001; Zumbrennen-Bullough et al., 2014). Thus, iron-deficiency induced by knocking out the IRP2 gene or IRP2 inhibition by cisplatin can cause cytotoxicity. However, the possibility of brain neuronal damage by cisplatin is less likely in vivo because cisplatin (and other platinum compounds) poorly crosses the blood brain barrier (Gregg et al., 1992).
Cancer cells generally require more iron for their aggressive proliferation (Torti and Torti, 2013). This is consistent with the report that increased IRP2 but not IRP1 is responsible for upregulation of TfR1 and downregulation of ferritin in human breast cancer cells, in which IRP2 knockdown decreased LIP and inhibited tumor cell growth in a xenograft model (Wang et al., 2014). Thus, higher expression of IRP2 increases LIP and promotes cell proliferation, and vice versa. Furthermore, increased expression of IRP2 was correlated with high-grade human breast cancer (Fig. 6D) (Wang et al., 2014). Conversely, another study showed that forced expression of IRP2 promoted growth of lung cancer cells in a xenograft model (Maffettone et al., 2010). These results support the idea that IRP2 is a promising anticancer target. Limiting iron availability using iron chelators is another reasonable antineoplastic approach, in particular using those clinically approved for iron overload diseases (Lui et al., 2015; Torti and Torti, 2013). However, thus far, no iron chelator has been approved for tumor chemotherapy because of inconclusive outcomes of their effects in clinical trials. The major drawback of iron chelators is that iron deficiency induced by iron chelation allows feedback activation of the IRP-IRE system to transport more iron and adjust the imbalance of iron homeostasis accordingly. By contrast, cisplatin inhibits IRP2 and the IRP-IRE system but does not allow cells to recover from the iron deficiency (Fig. 1F). As shown in Fig. 7, combination of lower doses of cisplatin and DFO depletes more intracellular iron than each agent alone. This can be explained by the mechanism that cisplatin inhibits the IRP-IRE system and blocks cellular adjustment of the iron deficiency caused by DFO, which can ultimately enhance the anti-cancer efficacy of each agent. This study sheds light on the IRP-IRE system, particularly IRP2, as a cellular target for cisplatin and probably some other endobiotics, xenobiotics, and their metabolites.
Significance
Traditionally, cisplatin anticancer activity was understood to be a function of cisplatin-DNA adduct formation in the nucleus. In addition to this, many proteins bind to cisplatin at specific amino acid sites, but the biological significance of these cisplatin-protein adducts remains poorly defined. Here, we have characterized the effect of a cisplatin-protein adduct associated with cellular iron homeostasis using an in vitro culture system as well as mouse xenograft models. We demonstrate that cisplatin directly inhibits the master regulator of iron metabolism, the cytoplasmic protein iron regulatory protein 2 (IRP2), through covalent binding on IRP2 Cys512 and Cys516, which induces intracellular iron deficiency. The cisplatin-induced, IRP2-inactivation-mediated intracellular iron deficiency emerged as a major mechanism of cytotoxicity after cisplatin treatment. These findings help expand our understanding of cisplatin anticancer effects due to protein adduct formation and iron metabolism dysregulation in the cytoplasm.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yoshiaki Tsuji (ytsuji@ncsu.edu).
Experimental Model and Subject Details
Animal Studies
The protocol of the xenograft study was approved by North Carolina State University Institutional Animal Care and Use Committee (IACUC) (Protocol # 16–089). For this experiment, 3–6 months male NSG mice were used.
Method Details
Cell Culture and Chemicals
SW480 human colon adenocarcinoma (established from 50 years old male), MCF7 human breast adenocarcinoma (from 69 years old female), HeLa human cervix adenocarcinoma (from 31 years old female), HepG2 human hepatocellular carcinoma (from 15 years old male), K562 human erythroleukemia (from 53 years old female), HEK293 immortalized human embryonic kidney cells (from fetus female) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). SW480 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (50–003-PC; Corning, Manassas, VA) containing 10% fetal bovine serum (FBS) (35–010-CV; Mediatech, Manassas, VA). MCF7 cells were cultured in Minimum Essential Medium (MEM) (50–011-PC; Corning) containing 10% FBS with 10 µg/mL bovine insulin, and 1 mM sodium pyruvate. HeLa, HepG2, and HEK293 cells were cultured in MEM containing 10% FBS. K562 cells were cultured in RPMI1640 (50–020-PC; Corning) containing 10% FBS. They were cultured in a humidified 95% incubator (37°C, 5% CO2). Cisplatin (three different cisplatin suppliers, Calbiochem, La Jolla, CA, TSZChem, Framingham, MA, and Alexis Biochemicals, San Diego, CA, all of which showed the same effects), carboplatin (Alexis Biochemicals), PtCl2 (Acros Organics, Geel-Belgium, NJ), ferric ammonium citrate (FAC, Sigma-Aldrich, St. Louis, MO), desferrioxamine (DFO, Sigma, St. Louis, MO), Hydrogen peroxide (H2O2) (Calbiochem), and t-butyl hydroperoxide (tBHP) (Sigma-Aldrich) were dissolved in distilled ultrapure water. Transplatin (Aldrich, St. Louis, MO) was dissolved in dimethyl sulfoxide at 10 mg/mL for stock solution.
Western blot and Antibodies
Western blot analysis was performed as previously described (Miyazawa and Tsuji, 2014) except for no boiling of samples for detection of ferroportin. Antibodies used in this work were anti-ferritin H (sc-25617), anti-NQO1 (sc-32793), anti-IRP1 (sc-14216), anti-IRP2 (sc-33682) from Santa Cruz Biotechnology (Santa Cruz, CA), anti-TfR1 (ab84036) from Abcam (Cambridge, MA); anti-ferroportin 1 (NBP1–21502) from Novus (Littleton, CO); anti-β-actin (A2066) and anti-ferritin L (F5012) from Sigma (St. Louis, MO); anti-GAPDH (MAB374) from Chemicon (Temecula, CA); Anti-HA Tag (HA.11, 901514) from BioLegend (San Diego, CA).
Real time PCR
Total RNA was isolated with TRI Reagent RT (Molecular Research Center, Cincinnati, OH) and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) with 500 ng of total RNA as a template. Equal amounts of cDNA were amplified by 30–45 cycles of denaturing for 10 sec at 95°C and annealing and extension for 45 sec at 60°C in a CFX96 Real-Time PCR System with iTaq Universal SYBR Green Supermix (Bio-Rad). Sequences of specific human primer pairs are shown in supplemental table 1.
Luciferase assay
Human ferritin H 5’-UTR with IRE (0.35kb) luciferase plasmid was constructed by promoter luciferase without IRE (0.15kb (Tsuji, 2005)) in the pBluescriptSK(−) plasmid. The luciferase reporter plasmid containing mutant ferritin H IRE (Henderson et al., 1996) was constructed by QuikChange II Site-Directed Mutagenesis (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s protocol. SW480 or HepG2 cells were transfected with these reporter plasmids with Polyethylenimine (linear, ~25kDa, Polysciences, Inc) and treated with cisplatin, transplatin, carboplatin, PtCl2, FAC, or DFO for 18–24 h. Cell lysates in luciferase cell culture lysis reagent (Promega, Madison, WI) were incubated with the luciferase assay substrate (E1501; Promega). Luciferase activates were measured by GloMax 20/20 (Promega).
Pull-down mRNA-protein binding assay
200 µg whole-cell lysates were incubated with 10 µg biotin-labeled ferritin H IRE probes (Biotin-5’-GGUUUCCUGCUUCAACAGUGCUUGGACGGAAC-3’) and streptavidin-agarose beads (Invitrogen, Carlsbad, CA) in PBS- containing 0.5% NP40 and inhibitors of protease and RNase (buffer A) at 4°C. Precipitates with beads were washed with buffer A, and subjected to Western blot with anti-HA, anti-IRP1 or anti-IRP2 antibody.
Electrophoresis Mobility Shift Assay (EMSA)
A human ferritin H IRE probe was end-labeled with [γ−32P] ATP and T4 polynucleotide kinase at 37°C for 10 min. Human Myc-Flag-tagged IRP2 protein (50 ng/µL, Origene, Rockville, MD) pre-incubated with cisplatin or carboplatin at room temperature for 16 h were incubated with 32P-labeled ferritin H IRE probe at room temperature for 1 h. Protein-RNA complexes were separated on 4% polyacrylamide-0.5% TBE gel electrophoresis and subjected to autoradiography. Mouse normal IgG (sc-2025; Santa Cruz) and anti-Flag antibody (F3165; Sigma) were used to verify the IRP2-IRE binding complex.
Expression plasmids of IRP1 and IRP2 mutants
pCMV-SPORT6 IRP1 (human) and pCR4-TOPO-IRP2 (human) were purchased from Open Biosystems. The IRP1 and IRP2 cDNAs were cloned into pcDNA3 or pcDNA3-cHA vector. IRP2 C512A and C516A mutations were introduced by QuikChange II Site-Directed Mutagenesis with a specific mutation primer set (Supplemental Table I). IRP2 C512A/516A double mutations were created by using the IRP2 C512A mutant as a template with the IRP2 C516A primer set. IRP1 C437S mutation was introduced according to the primers listed in (Zumbrennen et al., 2009).
Crystal violet staining
Cells treated with cisplatin or DFO in 12-well plates were fixed with 4% formaldehyde-PBS for 10 min, stained with 0.1% crystal violet (EM Science) solution for 15 min, and washed with tap water five times. The dye was eluted with 50% ethanol, 0.1% sodium citrate (pH4.0) solution and measured at 595 nm using NonoDrop2000 (Thermo Scientific).
siRNA transfection
Cells were grown in 6-well plate (6×105 cells/well) for 18h, and transfected with 20 pmol of siRNA with Lipofectamin RNAiMAX (Invitrogen) according to the manufacture’s instruction. siRNA sequences are shown in supplemental table I.
Intracellular free iron measurement
Cells treated with cisplatin, DFO, FAC or their combinations were incubated with 100 nM Calcein-AM (BioLegend) for 20 min at 37°C and trypsinized. Fluorescence intensity was measured by BD Accuri C6 Plus flow cytometer (BD Biosciences, San Jose, CA).
Caspase 3 activity assay
Caspase 3 assay was performed using Caspase 3/7 Glo assay kit (Promega). 2 µg of whole cell lysates were incubated with 20 ul of Caspase Glo reagent for 30 min at room temperature. The luminescence was measured by GLOMAX 20/20 Luminometer with preset program of “Promega CASPASE” protocol.
RNA immunoprecipitation (RIP) assay
Cells expressing HA-tagged IRPs were lysed in PBS containing 1% NP40, 0.5% sodium deoxycholate lysis buffer with inhibitors of proteases and RNase. Whole cell lysates were incubated with anti-HA antibody and protein G magnetic beads (Bio-Rad) for 4 h at 4 °C. The immunoprecipitates were washed with PBS containing 0.5% NP40 and total RNAs were isolated using RNAqueous®-Micro Total RNA Isolation Kit (Ambion). cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad) and equal amounts of cDNA were amplified by qPCR as described above using human ferritin H and B2M primer sets (supplemental table I).
Identification of cisplatin-IRP2 adduct sites
10 µg of human IRP2 recombinant protein (Origene) was incubated with 500 µg/mL isplatin for 24 h at 4 °C, and LC-MS/MS analysis was performed at ITSI Biosciences (Johnstown, PA). Briefly, IRP2 and cisplatin mixture was reduced/alkylated, suspended in 100 mM triethyl ammonium bicarbonate buffer and digested overnight with trypsin. The digest was dried down and resuspended in 0.1% formic acid. Peptides were desalted using zip tip (EMD Millipore, Billerica, MA), dried and reconstituted in a solution containing 2% acetonitrile and 0.1% formic acid. The peptide samples were loaded onto a PicoFrit C18 nanospray column using a Thermo Scientific Surveyor Autosampler and eluted from the column using a linear 2–30% acetonitrile gradient over 90 minutes for an LTQ XL mass spectrometer analysis (Thermo Scientific, Rockford, IL). Raw data files were searched against the most recent Uniprot database for Human IREB2 (Accession #A0A0A6YY96).
Mice Xenograft
NOD scid gamma (NSG) mice were obtained from Jackson Laboratory (Bar Harbor,ME). SW480 cells were injected under the skin of NSG mice (5×106 cells per site, two sites per mouse, single flank injection in 50% matrigel/PBS, 100µL injection by 21G needle). Tumor growth was detectable within 7 days post implantation. After that, cisplatin and iron chelator treatment were started by intraperitoneal injection.
Quantification and Statistical Analysis
Data are presented as means ± SD. All experimental replicates are biological replicates. Results were analyzed by one-way analysis of variance (ANOVA), and P values were assigned by using the Fisher’s Least Significant Difference (LSD) test. Differences among means were considered statistically significant when the P value is less than 0.05. SPSS Statistics 24 software (IBM, Armonk, NY) was used for all statistics analysis.
Supplementary Material
Highlights.
Cisplatin covalently binds to human IRP2 at Cys512 and Cys516
Cisplatin represses IRP2 binding on ferritin and TfR1 mRNAs (iron metabolism genes)
IRP2 dysregulation by cisplatin promotes intracellular iron deficiency
Cisplatin/iron chelator co-treatment potentiates iron deficiency and cancer cell death
Acknowledgments
We are grateful to Dr. Michael Bereman for their initial Mass Spectrometry analyses and helpful discussion to characterize cisplatin and IRP2 interaction. We thank Drs. Susan Sumner and James Harrington at RTI International for ICP-MS analyses to measure platinum for preliminary characterization of direct interaction between cisplatin and IRP2. We also thank Dr. Jun Ninomiya-Tsuji for helping our xenograft mouse experiments. This work was supported by the NIH grants R01GM088392 and R01GM095550 from the National Institute of General Medical Sciences to Y. Tsuji and also in part by P30ES025128 from the National Institute of Environmental Health Sciences to Center for Human Health and the Environment (CHHE). Alexander R. Bogdan was supported by NIH Training Grant T32ES007046 from the National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Anderson CP, Shen M, Eisenstein RS, and Leibold EA (2012). Mammalian iron metabolism and its control by iron regulatory proteins. Biochimica et biophysica acta 1823, 1468–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan AR, Miyazawa M, Hashimoto K, and Tsuji Y (2016). Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends in biochemical sciences 41, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandrini V, Nguyen TH, Arnesano F, Galliani A, Ippoliti E, Carloni P, and Natile G (2014). Structural biology of cisplatin complexes with cellular targets: the adduct with human copper chaperone atox1 in aqueous solution. Chemistry 20, 11719–11725. [DOI] [PubMed] [Google Scholar]
- Chan KT, Choi MY, Lai KK, Tan W, Tung LN, Lam HY, Tong DK, Lee NP, and Law S (2014). Overexpression of transferrin receptor CD71 and its tumorigenic properties in esophageal squamous cell carcinoma. Oncology reports 31, 1296–1304. [DOI] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. (2015). Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari S, and Tchounwou PB (2014). Cisplatin in cancer therapy: molecular mechanisms of action. European journal of pharmacology 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRusso PA, Philpott CC, Iwai K, Mostowski HS, Klausner RD, and Rouault TA (1995). Expression of a constitutive mutant of iron regulatory protein 1 abolishes iron homeostasis in mammalian cells. The Journal of biological chemistry 270, 15451–15454. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2014). The role of iron and reactive oxygen species in cell death. Nature chemical biology 10, 9–17. [DOI] [PubMed] [Google Scholar]
- Drakesmith H, Nemeth E, and Ganz T (2015). Ironing out Ferroportin. Cell metabolism 22, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy B, Ferring-Appel D, Kaden S, Grone HJ, and Hentze MW (2008). Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell metabolism 7, 79–85. [DOI] [PubMed] [Google Scholar]
- Goossen B, Caughman SW, Harford JB, Klausner RD, and Hentze MW (1990). Translational repression by a complex between the iron-responsive element of ferritin mRNA and its specific cytoplasmic binding protein is position-dependent in vivo. The EMBO journal 9, 4127–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg RW, Molepo JM, Monpetit VJ, Mikael NZ, Redmond D, Gadia M, and Stewart DJ (1992). Cisplatin neurotoxicity: the relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 10, 795–803. [DOI] [PubMed] [Google Scholar]
- Habashy HO, Powe DG, Staka CM, Rakha EA, Ball G, Green AR, Aleskandarany M, Paish EC, Douglas Macmillan R., Nicholson RI, et al. (2010). Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast cancer research and treatment 119, 283–293. [DOI] [PubMed] [Google Scholar]
- Henderson BR, Menotti E, and Kuhn LC (1996). Iron regulatory proteins 1 and 2 bind distinct sets of RNA target sequences. The Journal of biological chemistry 271, 4900–4908. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Galy B, and Camaschella C (2010). Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38. [DOI] [PubMed] [Google Scholar]
- Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, and Tsuji Y (2006). Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol Cell Biol 26, 2845–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, Crooks DR, Wilson-Ollivierre H, Ghosh MC, Sougrat R, Lee J, Cooperman S, Mitchell JB, Beaumont C, and Rouault TA (2011). Iron insufficiency compromises motor neurons and their mitochondrial function in Irp2-null mice. PloS one 6, e25404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa T, Sibrian-Vazquez M, Strongin RM, and Steyger PS (2013). Identification of cisplatin-binding proteins using agarose conjugates of platinum compounds. PloS one 8, e66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland L (2007). The resurgence of platinum-based cancer chemotherapy. Nature reviews Cancer 7, 573–584. [DOI] [PubMed] [Google Scholar]
- Kerins MJ, Vashisht AA, Liang BX, Duckworth SJ, Praslicka BJ, Wohlschlegel JA, and Ooi A (2017). Fumarate Mediates a Chronic Proliferative Signal in Fumarate Hydratase-Inactivated Cancer Cells by Increasing Transcription and Translation of Ferritin Genes. Molecular and cellular biology 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn LC (2015). Iron regulatory proteins and their role in controlling iron metabolism. Metallomics : integrated biometal science 7, 232–243. [DOI] [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, et al. (2001). Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nature genetics 27, 209–214. [DOI] [PubMed] [Google Scholar]
- Lui GY, Kovacevic Z, Richardson V, Merlot AM, Kalinowski DS, and Richardson DR (2015). Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 6, 18748–18779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Abbate V, and Hider RC (2015). Iron-sensitive fluorescent probes: monitoring intracellular iron pools. Metallomics : integrated biometal science 7, 212–222. [DOI] [PubMed] [Google Scholar]
- MacKenzie EL, Iwasaki K, and Tsuji Y (2008). Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxidants & redox signaling 10, 997–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffettone C, Chen G, Drozdov I, Ouzounis C, and Pantopoulos K (2010). Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PloS one 5, e10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magro G, Cataldo I, Amico P, Torrisi A, Vecchio GM, Parenti R, Asioli S, Recupero D, D’Agata V, Mucignat MT, et al. (2011). Aberrant expression of TfR1/CD71 in thyroid carcinomas identifies a novel potential diagnostic marker and therapeutic target. Thyroid : official journal of the American Thyroid Association 21, 267–277. [DOI] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, et al. (2004). Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. The EMBO journal 23, 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa M, Bogdan AR, Hashimoto K, and Tsuji Y (2018). Regulation of transferrin receptor-1 mRNA by the interplay between IRE-binding proteins and miR-7/miR-141 in the 3’-IRE stem-loops. Rna 24, 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa M, and Tsuji Y (2014). Evidence for a novel antioxidant function and isoform-specific regulation of the human p66Shc gene. Molecular biology of the cell 25, 2116–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckenthaler M, Gray NK, and Hentze MW (1998). IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Molecular cell 2, 383–388. [DOI] [PubMed] [Google Scholar]
- Mullner EW, Neupert B, and Kuhn LC (1989). A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell 58, 373–382. [DOI] [PubMed] [Google Scholar]
- O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, and Dang CV (2006). Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Molecular and cellular biology 26, 2373–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prutki M, Poljak-Blazi M, Jakopovic M, Tomas D, Stipancic I, and Zarkovic N (2006). Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer letters 238, 188–196. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, and Tsuji Y (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, and Bruick RK (2009). An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletta F, Suryo Rahmanto Y., Siafakas AR, and Richardson DR (2011). Cellular iron depletion and the mechanisms involved in the iron-dependent regulation of the growth arrest and DNA damage family of genes. The Journal of biological chemistry 286, 35396–35406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalinske KL, Blemings KP, Steffen DW, Chen OS, and Eisenstein RS (1997). Iron regulatory protein 1 is not required for the modulation of ferritin and transferrin receptor expression by iron in a murine pro-B lymphocyte cell line. Proceedings of the National Academy of Sciences of the United States of America 94, 10681–10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby JV, and Friedman GD (1988). Epidemiologic evidence of an association between body iron stores and risk of cancer. Int J Cancer 41, 677–682. [DOI] [PubMed] [Google Scholar]
- Smith SR, Ghosh MC, Ollivierre-Wilson H, Hang Tong W., and Rouault TA (2006). Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol Dis 36, 283–287. [DOI] [PubMed] [Google Scholar]
- Stevens RG, Jones DY, Micozzi MS, and Taylor PR (1988). Body iron stores and the risk of cancer. N Engl J Med 319, 1047–1052. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Kennedy K, Witt M, and Juzefyk J (1991). Effect of dietary iron deficiency or excess on the induction of mammary carcinogenesis by 1-methyl-1-nitrosourea. Carcinogenesis 12, 111–114. [DOI] [PubMed] [Google Scholar]
- Torti SV, and Torti FM (2013). Iron and cancer: more ore to be mined. Nature reviews Cancer 13, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S (2014). Iron and thiols as two major players in carcinogenesis: friends or foes? Frontiers in pharmacology 5, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y (2005). JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 24, 7567–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, et al. (2009). Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Pantopoulos K (2002). Conditional derepression of ferritin synthesis in cells expressing a constitutive IRP1 mutant. Molecular and cellular biology 22, 4638–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Pantopoulos K (2011). Regulation of cellular iron metabolism. The Biochemical journal 434, 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Deng Z, Hatcher H, Miller LD, Di X, Tesfay L, Sui G, D’Agostino RB Jr., Torti FM, and Torti SV (2014). IRP2 regulates breast tumor growth. Cancer research 74, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson N, and Pantopoulos K (2013). IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood 122, 1658–1668. [DOI] [PubMed] [Google Scholar]
- Will J, Wolters DA, and Sheldrick WS (2008). Characterisation of cisplatin binding sites in human serum proteins using hyphenated multidimensional liquid chromatography and ESI tandem mass spectrometry. ChemMedChem 3, 1696–1707. [DOI] [PubMed] [Google Scholar]
- Wilson BR, Bogdan AR, Miyazawa M, Hashimoto K, and Tsuji Y (2016). Siderophores in Iron Metabolism: From Mechanism to Therapy Potential. Trends in molecular medicine 22, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, and Richardson DR (2011). Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. The Journal of biological chemistry 286, 15413–15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrennen KB, Wallander ML, Romney SJ, and Leibold EA (2009). Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Molecular and cellular biology 29, 2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrennen-Bullough KB, Becker L, Garrett L, Holter SM, Calzada-Wack J, Mossbrugger I, Quintanilla-Fend L, Racz I, Rathkolb B, Klopstock T, et al. (2014). Abnormal brain iron metabolism in Irp2 deficient mice is associated with mild neurological and behavioral impairments. PloS one 9, e98072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.