

Abstract
Important differences in the biology of focal and diffuse traumatic brain injury (TBI) subtypes may result in unique pathophysiological responses to shared molecular mechanisms. Interleukin-1 (IL-1) signaling has been tested as a potential therapeutic target in preclinical models of cerebral contusion and diffuse TBI, and in a phase II clinical trial, but no published studies have examined IL-1 signaling in an impact/acceleration closed head injury (CHI) model. We hypothesized that genetic deletion of IL-1 receptor-1 (IL-1R1 KO) would be beneficial in focal (contusion) and CHI in mice. Wild type and IL-1R1 KO mice were subjected to controlled cortical impact (CCI), or to CHI. CCI produced brain leukocyte infiltration, HMGB1 translocation and release, edema, cell death, and cognitive deficits. CHI induced peak rotational acceleration of 9.7 × 105 ± 8.1 × 104 rad/s2, delayed time to righting reflex, and robust Morris water maze deficits without deficits in tests of anxiety, locomotion, sensorimotor function, or depression. CHI produced no discernable acute plasmalemma damage or cell death, blood-brain barrier permeability to IgG, or brain edema and only a modest increase in brain leukocyte infiltration at 72 h. In both models, mature (17 kDa) interleukin-1 beta (IL-1β) was induced by 24 h in CD31+ endothelial cells isolated from injured brain but was not induced in CD11b+ cells in either model. High mobility group box protein-1 was released from injured brain cells in CCI but not CHI. Surprisingly, cognitive outcome in mice with global deletion of IL-1R1 was improved in CHI, but worse after CCI without affecting lesion size, edema, or infiltration of CD11b+/CD45+ leukocytes in CCI. IL-1R1 may induce unique biological responses, beneficial or detrimental to cognitive outcome, after TBI depending on the pathoanatomical subtype. Brain endothelium is a hitherto unrecognized source of mature IL-1β in both models.
Keywords: controlled cortical impact, interleukin-1, mice, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a significant public health burden, with over 1.3 million emergency department visits, 275,000 hospitalizations, and 50,000 deaths per year and annual costs estimated at over $70 billion.1 To date, clinical trials have not identified therapies that improve functional outcome in TBI patients.
TBI is a heterogeneous disease with different phenotypes.2 Focal closed head injury (CHI) with intraparenchymal bleeding may cause widespread axonal injury, blood–brain barrier (BBB) damage, and acute and chronic neuronal cell death with rapid deterioration in mental status. In many of these patients, progressive brain edema causes life-threatening intracranial hypertension. In contrast, patients with mild diffuse CHI can have prolonged, debilitating neurological dysfunction in the absence of edema or brain lesions on routine imaging studies. A National Institutes of Health (NIH) workshop on TBI classification concluded that different pathoanatomical lesions may have unique mechanisms of injury that require tailored therapy, rather than a “one-size-fits-all” approach in which patients are treated regardless of lesion type.2
One mechanism hypothesized to be a therapeutic target in severe TBI is interleukin-1 beta (IL-1β). IL-1β is a proinflammatory cytokine that is activated as part of the innate immune response to central nervous system trauma, infection, and other types of injury.3 IL-1β exists as a precursor molecule that is cleaved and activated canonically by caspase-1 in a cytosolic multi-protein inflammasome complex.3 Alternatively, IL-1β can be activated by caspase-8 in a ripoptosome, or by neutrophil elastase, independent of an inflammasome.4,5 IL-1β binding to the IL-1 receptor-1 (IL-1R1)/IL-1R1 accessory protein complex (AcP) leads to activation of mitogen-activated protein kinases and nuclear factor kappa B, resulting in proinflammatory gene expression.6 A truncated IL-1R1 splice variant, IL-1R3, binds IL-1β in neurons and signals by the neuron-specific AcPb, which induces protein kinase B (Akt) and mediates neuroprotection.7 IL-1β messenger RNA and protein is induced within hours and may persist for several months after controlled cortical impact (CCI) in rodents, and is increased in cerebrospinal fluid and brain tissue of patients with severe TBI (defined as Glasgow Coma Scale score ≤8).8–11 Whether IL-1β is involved in the pathogenesis of mild TBI is unknown. This is an important knowledge gap because mechanisms driving neurological deficits in patients with concussion syndrome are unknown, and no specific therapy exists to mitigate neurological dysfunction in these individuals.
To test the role of IL-1β in a model of mild TBI, we modified a mouse CHI model with biomechanical features of impact and head acceleration, with a greater weight drop than in our previous studies12 to produce cognitive deficits similar in magnitude to those in our CCI cerebral contusion model.13 We tested the hypothesis that genetic antagonism of IL-1R1 would show beneficial effects on functional outcome in CCI and CHI. Unexpectedly, we found that although processing and maturation of IL-1β occurs in cerebral endothelial cells in both models, IL-1R1 knockout (KO) produces divergent effects on neurological function in CHI versus CCI, underscoring the importance of studying IL-1R1 mechanisms in the context of specific pathoanatomical TBI subtypes.
Methods
Experiments were performed and animals were treated humanely according to ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. All procedures were performed in accord with the NIH Guide for Care and Use of Laboratory Animals and followed protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. Mice had access to food and water ad libitum and were housed on a 12-h day-night cycle in laminar flow racks in a temperature-controlled room (25°C). Mice were randomized to sham and injury groups except for sham-injured CCI mice that were done as groups separate from CCI mice. Investigators blinded to study groups performed brain injury models and all behavioral experiments. Behavior testing was performed during the same time of day (7:00 am to 11:30 am) to ensure consistent results.
Experimental animals
Studies were performed using adult, 3- to 6-month-old male IL-1 receptor 1 knockout (IL-1R1 KO) mice (B6.129S7-Il1r1tm1Imx; The Jackson Laboratory, Bar Harbor, ME) and the appropriate age-matched wild-type (WT) controls (C57/BL6J, stock #000664; The Jackson Laboratory). For biomechanical assessments, C57Bl/6N mice (stock #027; Charles River Laboratories, Wilmington, DE) were used.
Closed head injury
A modified CHI model was used as previously described.12 Mice were anesthetized with 2.5% isoflourane in 70% N2O and 30% O2 for 90 sec. Anesthetized mice were placed on a custom wooden support holder and the head rested on a taught KimWipe napkin. The brass guide tube was 60 inches long and nine-sixteenths of an inch in diameter. A half-inch diameter, 81-g lead cylindrical weight with a flat, unbuffered surface was dropped onto the vertex of the head between the ears. After impact mice were placed supine, and loss of consciousness (LOC) time was recorded as time to righting reflex. Sham-injured mice received anesthesia but no injury.
Controlled cortical impact
A CCI model was used as described previously,13 with velocity 6 m/sec, depth 0.6 mm, and duration 100 ms.
Biomechanics of closed head injury
Biomechanical measurements of CHI were made in 3-month-old male mice (n = 7). Immediately preceding CHI, an 0.8-g triaxial accelerometer (3133A1; Dytran Instruments, Inc., Chatsworth, CA) with sensitivity of 10 mV/g was taped to the ventral jaw of the anesthetized mouse. The lead of the accelerometer was taped along the mouse's body to prevent tugging on the lead at the time of impact. Data were acquired at a rate of 20 kHz (USB-4432; National Instruments, Woburn, MA) during CHI. Linear acceleration was quantified as the magnitude of the three-dimensional acceleration vector. The corresponding rotational acceleration was obtained by dividing linear acceleration by the radius of rotation—measured for each mouse as the distance between the center of the skull and the point located midway between the cervical axis and the scapula. Impact time was defined as the duration between the time at which acceleration increased above 10g (ignoring <10g noise) to the first local minimum below 200g after the peak acceleration. To compare to published thresholds for injury in humans with concussion syndrome, we assumed thathuman and mouse brains have equal stiffness, and that equal strains are required in both brains to achieve the same injury. A characteristic mass scaling ratio, 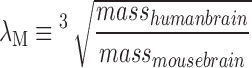 , was used to convert measured values to equivalent scaled human values,14,15 assuming an average human adult male brain mass of 1336 g.
, was used to convert measured values to equivalent scaled human values,14,15 assuming an average human adult male brain mass of 1336 g.
Genotyping of mutant mice
Mice were genotyped according to protocols published by The Jackson Laboratory. Primers used for IL-1R1 mice are as follows – Mutant Forward: CTCGTGCTTTACGGTATCGC, Wild Type Forward: GGTGCAACTTCATAGAGAGATGA, Common: TTCTGTGCATGCTGGAAAAC.
Preparation of brain tissue for TUNEL, Fluoro Jade B, and immunoglobulin G histochemistry
Mice were deeply anesthetized with isoflurane and decapitated. Brains were removed and frozen in liquid nitrogen before making coronal sections (14 μm) on a poly-ι-lysine-coated slide (Thermo Fisher Scientific, Waltham, MA) using cryostat. Brains were cut at 0.5-mm intervals from the anterior to the posterior of the brain. For analyses using paraformaldehyde (PFA)-fixed tissue, mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 4% PFA and brains were post-fixed overnight in 4% PFA, cryoprotected in 30% sucrose overnight, frozen at −80°C, and cut on a cryostat as above. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Roche Diagnostics, Indianapolis, IN) was performed on frozen sections and Fluoro Jade B (FJB; EMD Millipore, Burlington, MA) on fixed brain sections according to the manufacturer's instructions. BBB damage was assessed by immunoglobulin G (IgG) immunohistochemistry (rabbit antimouse IgG-Alexa Fluor 488, 1:200; Jackson ImmunoResearch, West Grove, PA). IgG+ brain regions were assessed qualitatively by comparing to corresponding brain regions of sham-injured mice.
Quantitative assessment of cell counts
TUNEL+, FJB+, propidium iodide (PI)+, CD45+, and degenerating (hematoxylin and eosin; H&E) cell counts were performed in fields randomly chosen from cortex, hippocampus, or striatum in five brain sections spanning anterior to posterior brain. A total of six fields per section were counted for cortex, two to four fields per section for hippocampus, and two to four fields per section for striatum in each hemisphere.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was performed using the Mouse IL-1β Quantikine Kit (R&D Systems, Minneapolis, MN), according to manufacturer's protocol.
Western blot
Brain tissues were prepared for western blot as previously described.16 After blocking in 5% bovine serum albumin for 1 h, membranes were incubated at 4°C overnight in primary antibodies (rabbit antimouse Il-1β, 1:1000 [Abcam, Cambridge, MA] and rabbit antimouse HMGB1 [high mobility group box 1; 1:1000; Abcam]). Horseradish peroxidase–conjugated secondary antibody (1:2000; Cell Signaling Technology, Danvers, MA) was used for enhanced chemiluminescence detection. Results were normalized to β-actin (1:5000; Cell Signaling Technology). Densitometry was performed using image analysis (ImageJ; NIH, Bethesda, MD).
Assessment of brain edema
Brain edema was assessed by the wet-dry/wet weight method. Bisected brains from CCI mice were weighed on a pre-weighed weigh boat and then dried at 80°C overnight. Dried brains were weighed and wet-dry/wet percentage was calculated. For CHI brains, CHI or sham-injured brains were weighed wet, dried, and weighed as for CCI, but were not bisected into separate hemispheres.
Assessment of brain tissue loss after controlled cortical impact
Morphometric image analysis (ImageJ; NIH) was used to determine volume of brain tissue loss as previously described.13
Foot fault test
Mice were placed on an elevated grid (30 × 20 cm). Openings in the grid were 1 cm2. Mice were recorded from below, and front paw steps were counted while the mouse was moving around on the grid. A blinded observer analyzed the video for time to 100 steps and foot faults per side.
Wire grip test
Vestibular-motor function was assessed between 1 and 28 days post-injury using a wire-grip test as previously described.13
Morris water maze
Cognitive function was assessed using the Morris water maze (MWM) as previously described.17 One hidden platform trial was performed on day 1 of MWM, and two hidden platform trials were performed on following days up to seven hidden platform trials. Seven hidden platform trials were performed rather than five because we found that seven trials resulted in more spatial search strategies in probe trials. Probe trials and visible platform trials were performed 24 h after the last hidden trial. MWM was performed on days 3–6 post-injury for CHI mice and days 10–14 for CCI mice. Mice were given 30–60 minutes of rest in between the trials. Reversal MWM was performed at 3 weeks after initial the MWM using the same parameters, but the hidden platform was relocated to a different quadrant.
Elevated plus maze
Anxiety-related behavior was assessed using elevated plus maze. Mice were placed in the center and allowed to walk freely in each arm (30 × 5 cm). The closed arms had walls of 15 cm high. The plus maze was elevated 40 cm from the floor. Times spent in open arms, closed arms, and the center were analyzed using Any-maze (Stoelting Co., Wood Dale, IL).
Y-maze
A Y-maze consisting of three arms (35 × 15 × 6 cm) with an angle of 120 degrees between each of the two arms was used with visual cues (black symbols) on each end of the arms. The Y-maze test consisted of one 5-min trial per mouse. Mice were placed in the center of the Y-maze and had free access to all three arms. Trials were recorded using a ceiling-mounted camera, and the number of entries into each separate arm was analyzed. Alternation was assessed by the sequential pattern of arm entry. Continuous alternation was scored as the number (proportion) of arm choices that differed from the previous two choices. Data are expressed as percentage of alternating choices compared to total choices during the 5 min.
Porsolt forced swim test
The Porsolt forced swim test was performed as previously described,18 with minor modifications. Mice were placed in a cylindrical transparent glass tank of 30.5 × 20.3 cm (height × diameter) that was filled with water up to a height of 20 cm. A white styrofoam box provided visual shielding on three sides. Mice were videotaped for 6 min and put back into their cages after the trial. Videos were analyzed for escape-related mobility behavior that was quantified by a blinded observer by comparing the time the mouse spent struggling versus the time spent floating during the last 4 min of the video.
Open-field test
Mice were individually placed in housing cages with clean bedding covered by a thin wire grid. Movements were tracked and recorded by ceiling-mounted cameras and Any-maze (Stoelting Co.). Recording started between 7:00 am and 8:00 pm, 3 h after mice had been placed in their cages, and continued for 8 h. Mice were tested at 6 weeks post-injury.
Isolation of brain leukocytes by fluorescence activated cell sorting
Microglia and brain leukocytes were isolated by MACS dissociation and fluorescence activated cell sorting (FACS) as previously described.19,20 Mice were transcardially perfused with PBS, and brains were processed using gentleMACS Dissociator (Miltenyi Biotech, Bergisch Gladbach, Germany) in an RPMI solution containing 0.2% Collagenase Type 3 (Worthington Biochemical Corporation, Lakewood, NJ) and Dispase (2 U/mL; Worthington Biochemical Corporation). DNase I (40 U/mL) was added after dissociation and incubated at 37°C for 10 min. Five percent fetal bovine serum in PBS/ethylenediaminetetraacetic acid solution was added to inactivate the digestion enzymes and passed through a 100-μm filter and centrifuged. Cell pellets were resuspended and mixed gently with physiological Percoll and centrifuged. Red blood cells (RBCs) were lysed with RBC lysis buffer. Cells were filtered through a 40-μm filter and stained with Alexa 488–labeled CD45 (BioLegend, San Diego, CA) and Alexa 647–labeled CD11b (BioLegend) antibodies. Cells were sorted based on CD11b/CD45 expression using FACS ARIA (BD, Franklin Lakes, NJ).
Isolation of brain CD31+ endothelial cells and CD11b+ cells
Adult mouse brain endothelial cells (ECs) were isolated as previously described using enzymatic dissociation and anti-CD31–conjugated or anti-CD11b–conjugated magnetic beads.21 Mice were transcardially perfused with PBS, and brains were homogenized in a 1-mg/mL collagenase A (Roche) solution and incubated at 37°C for 30 min. Samples were triturated with a 14-gauge metal cannula and passed through a 40-μm filter and centrifuged at 400g for 8 min. Cell pellets were resuspended in PBS and incubated with either anti-CD31–conjugated (BD) or anti-CD11b–conjugated (BioLegend) Dynabeads (Thermo Fisher Scientific). Samples were incubated at 4°C for 30 min and isolated using Dynamag (Thermo Fisher Scientific). Cells were frozen at −80°C until processing for western blot.
Quantification and statistical analysis
Data are represented as mean ± standard error of the mean (SEM). All data with n = 5 or more per group passed normality tests (Anderson-Darling test and others). Data with n = 4/group were densitometry or cell counts (continuous) and expected to be normally distributed. Data were analyzed using GraphPad PRISM software (version V; GraphPad Software Inc., La Jolla, CA). Wire-grip, overnight open field, and MWM were analyzed using two-factor repeated measures of analysis of variance (ANOVA; group × time). Foot fault, brain edema, lesion volume, probe trial data, elevated plus maze, forced swim test, Y-maze, western blot densitometry data, and percentage of leukocytes were analyzed by t-test. For all comparisons, p < 0.05 was considered significant.
Results
Mortality
A total of 335 mice were used in the studies. All CCI mice survived the post-injury period. For the CHI model, 1 of 12 WT mice in the model development group was euthanized for persistent rolling behavior within a short time post-injury. No mice died in the post-injury period in the groups used for gene knockout studies.
Characterization of a mouse closed head injury model
Compared to sham injury, CHI induced significant LOC (Fig. 1A; p < 0.001 vs. sham) without skull fractures or gross brain injury. Mean velocity change was 4.8 ± 0.3 m/s. Peak linear acceleration was 990 ± 82g across an average impact time of 1.2 ± 0.2 ms, which corresponded to a peak rotational acceleration 9.7 × 105 ± 8.1 × 104 rad/s2. There was no evidence for acute cell membrane damage (PI labeling), or cell death by TUNEL, FJB staining, or degenerating cells on H&E staining at 24 or 48 h (0 ± 0 PI+, TUNEL+, FJB+, IgG+, or degenerating cells/ × 200 field; p = not significant [ns] vs. sham; Fig. 1B). CHI did not induce brain edema at 24 h (sham, 78.1 ± 0.06%, CHI 77.9 ± 0.11% brain water content; p = 0.2; n = 6/group) or BBB damage by IgG immunohistochemistry (Fig. 1B), confirming a histologically mild CHI model. Flow cytometric analyses showed no infiltration of CD11b+/CD45+ leukocytes at 24 h, but a modest increase (twice sham levels) at 72 h post-CHI (Fig. 1C,D).
FIG. 1.

Histopathology of closed head injury (CHI). (A) Time to righting reflex (*p < 0.001; n = 10–12/group). (B) Representative histopathology of CHI at 48 h post-injury (n = 4–6/group, all photomicrographs were taken from fields injured parietal cortex except for H&E stain sections, which were taken from dentate gyrus); controlled cortical impact (CCI) tissue shows positive staining for all markers assessed. H&E stain shows argyrophilic cells in dentate gyrus of hippocampus in CCI, but not in CHI. All other analyses showed negative staining in CHI. Scale bars, 25 μm. (C) Representative images from fluorescence activated cell sorting analyses of Lymphocyte (L) and Macrophage + Neutrophil (M+N) populations after CHI. (D) No quantitative change in percentages of (L) and (M+N) populations at 24 h after CHI, but slight increase at 72 h after CHI (*p < 0.05 vs. sham; n = 3–4/group). Data are mean ± SEM. FJB, Fluoro Jade B; H&E, hematoxylin and eosin; IgG, immunoglobulin G; PI, propidium iodide; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Supplementary Figure 1 (see online supplementary material at http://www.liebertpub.com) shows results of behavioral analyses post-CHI. No difference in wire grip performance was noted between sham and CHI mice (Supplementary Fig. 1A,B) (see online supplementary material at http://www.liebertpub.com). Similarly, no differences in foot faults were noted between groups on day 1 and week 2 between sham and CHI groups (Supplementary Fig. 1C) (see online supplementary material at http://www.liebertpub.com) further suggesting that the CHI model does not produce significant acute sensorimotor deficits that might confound cognitive function testing. In the MWM (days 3–6), compared to sham-injured mice, CHI mice performed significantly worse on tests of hidden platform (p < 0.001 for group), visible platform (p < 0.001 for group), and probe trials (p < 0.001; Supplementary Fig. 1D,E) (see online supplementary material at http://www.liebertpub.com). In a reverse MWM performed 3 weeks post-injury, in which spatial learning is again tested with the goal platform moved to a new quadrant, CHI mice still performed worse than sham in hidden platform (p < 0.05 for group) and in probe trials (p < 0.01; Supplementary Fig. 1F,G) (see online supplementary material at http://www.liebertpub.com), despite no differences in locomotor activity (6 weeks; Supplementary Fig. 1H) (see online supplementary material at http://www.liebertpub.com), and probe trial swim patterns appeared less spatial for CHI mice (Supplementary Fig. 1I) (see online supplementary material at http://www.liebertpub.com) with no differences between groups in swim speed (p = ns for group; not shown). No differences were observed between sham and CHI in elevated plus maze (a test of anxiety/impulsivity; 2 weeks; p = 0.5; not shown) or performance in a forced swim test that assesses a depression phenotype (7 weeks; p = 0.6; not shown). Thus, CHI in mice produced cognitive deficits in MWM testing associated with hippocampal function that were not attributable to sensorimotor deficits or influenced by anxiety, impulsivity, hypolocomotion, or depression phenotypes.
Closed cortical impact and closed head injury induce interleukin-1 beta expression/processing and high mobility group box 1 translocation
Post-CCI, expression of IL-1β (ELISA) increased in brain tissue homogenates versus sham-injured mice (p < 0.0001; Fig. 2A). Post-CHI, IL-1β was not detected in homogenates of cortex, hippocampus, or striatum at 6 or 24 h post-injury, either by ELISA (6 h data, not shown) or western blot (not shown). However, in CD31+ endothelial cells isolated from mouse brain, IL-1β (pro-form, 31 kDa) was decreased and mature (17 kDa) IL-1β was increased at 24 h post-CHI (p < 0.01 vs. sham; Fig. 2B,C). At 24 h post-CCI, mature IL-1β was detected in isolated brain endothelial cells, and pro-IL-1β was also markedly increased compared to sham injury (Fig. 2D,E). In CD11b+ cells isolated by immunopanning (macrophages, neutrophils, and microglia), pro-IL-1β was increased in CHI at 24 h post-injury, but no change was detected for mature-IL-1β (Fig. 2F,G). Similarly, in CCI, immunopanned CD11b+ cells had an increase of pro-IL-1β at 24 h post-injury and no change of mature-IL-1β (Fig. 2H,I). Neither the pro- nor mature forms of IL-1β were detected by western blot when antibody was pre-incubated with recombinant mouse IL-1β, confirming specificity of the reagents used to detect IL-1β (not shown).
FIG. 2.

Detection of interleukin-1 beta (Il-1β) protein in brain after controlled cortical impact (CCI) and closed head injury (CHI). (A) ELISA showing total Il-1β changes at 24 h after CCI (*p < 0.0001 vs. WT sham; n = 6/group). IL-1β did not change in brain homogenates at 24 h after CHI versus sham or in sham-injured mice across all groups. (B and C) Decreased pro-Il-1β and increased mature Il-1β in isolated brain CD31+ endothelial cells at 24 h after CHI (*p < 0.01 vs. sham; n = 4/group). (D and E) Both pro-Il-1β and mature Il-1β were increased in isolated brain CD31+ endothelial cells at 24 h after CCI (*p < 0.001 vs. sham; n = 4/group). (F and G) Increased pro-Il-1β, but no change in mature Il-1β CD11b+ cells isolated by immunopanning at 24 h after CHI (*p < 0.05 vs. sham; n = 3/group). (H and I) Increased pro-Il-1β, but no changes in mature Il-1β in CD11b+ cells isolated by immunopanning at 24 h after CCI (*p < 0.05 vs. sham; n = 3/group). Nonspecific western blot bands were cropped for clarity in panels (B), (D), (F), and (H). Data are mean ± SEM. ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean; WT, wild type.
HMGB1 release from contused brain tissue was apparent at 24 h post-CCI (Supplementary Fig. 2A,B) (see online supplementary material at http://www.liebertpub.com), consistent with cytokine signaling in cells with plasmalemma damage, whereas HMGB1 levels remained similar in cortex, hippocampus, and striatum in sham and CHI mice at 24 and 48 h (Supplementary Fig. 2C,D) (see online supplementary material at http://www.liebertpub.com). However, post-CHI, nuclear to cytosol translocation, but not release, of HMGB1 was apparent in cortical and hippocampal brain regions in all CHI animals studied compared to sham (Supplementary Fig. 2E) (see online supplementary material at http://www.liebertpub.com). Taken together, the data suggest a possible role for brain endothelium in IL-1β signaling post-CHI despite overall less brain inflammation compared to CCI.
Detrimental role for interleukin-1 receptor 1 in closed head injury
We next interrogated a possible functional role for IL-1R1 signaling in CHI using the MWM. Sham-injured mice deficient in IL-1R1 (IL-1R1 KO) had similar performance as WT in hidden and visible platform trials of the MWM, but injured IL-1R1 KO mice had improved performance in hidden (p < 0.01 for group) and visible platform trials (p < 0.05 for group; Fig. 3A) versus injured WT mice. Sham and CHI IL-1R1 KO mice performed similarly in probe trials whereas WT CHI mice performed worse than WT shams (p < 0.01; Fig. 3B), suggesting preserved spatial memory in CHI IL-1R1 KO mice. At 3 months post-injury, IL-1R1 KO mice continued to perform better in a reverse MWM paradigm (p < 0.05 for group; Fig. 3C), but swim patterns (Fig. 3D), probe trial latencies, swim speed, and elevated plus maze performance (data not shown) did not differ between sham and injured WT and IL-1R1 KO mice. In the Y-maze, injured WT mice performed worse than shams (p < 0.001) whereas injured IL-1R1 KO performed similarly to their respective shams, and performance was significantly better in injured IL-1R1 KO versus CHI WT mice (8 weeks; p < 0.0001; Fig. 3E), showing evidence of improved working memory in injured IL-1R1 KO mice. Locomotor data at 4 months showed no differences among sham and CHI WT and IL-1R1 KO mice (Fig. 3F). Taken together, the data suggest that hippocampal learning and memory dysfunction post-CHI is decreased in IL-1R1 KO mice.
FIG. 3.

Cognitive outcome in interleukin-1 receptor knockout (IL-1R1 KO) mice after closed head injury (CHI). (A) Results of Morris water maze (MWM) testing in sham (n = 10/group) and injured (n = 20/group) wild-type (WT) and IL-1R1 KO mice. Sham IL-1R1 KO and sham WT and injured IL-1R1 KO mice performed similarly, whereas injured WT performed significantly worse than injured IL-1R1 KO in hidden (p < 0.01 for group) and visible (p < 0.05 for group × time interaction) platform trials. (B) Injured IL-1R1 KO mice performed similarly to sham IL-1R1 KO and sham WT mice in probe trials (*p < 0.01 between WT sham and WT CHI). (C) IL-1R1 KO mice had significantly better performance in hidden platform trials of a reverse MWM (p < 0.05 for group vs. WT CHI). (D) Swim patterns of WT CHI and IL-1R1 KO CHI mice do not suggest an obvious difference between groups (n = 14/group). (E) Y-maze performance was worse in WT CHI versus sham (*p < 0.0001) and WT CHI versus IL-1R1 KO CHI (#p < 0.0001). (F) No differences were observed in open-field tests among sham and injured WT and IL-1R1 KO mice assessed at 6 weeks post-injury. Data are mean ± SEM. SEM, standard error of the mean.
Beneficial role for interleukin-1 receptor 1 in controlled cortical impact
We next assessed a role for IL-1R1 in the pathogenesis of CCI. Post-CCI, brain edema (24 h) and lesion volume (5 months) did not differ between IL-1R1 KO and WT mice (Fig. 4A,B). However, IL-1R1 KO mice had improved wire-grip performance post-CCI compared to WT mice (p < 0.05 for group for score and p < 0.0001 for group for latency; Fig. 4C,D). However, injured IL-1R1 KO mice still performed worse in hidden (p < 0.05 for group) and visible (p < 0.05 for group) platform trials of the MWM versus injured WT mice (Fig. 4E), but no differences were observed between groups in probe trials (not shown) or Y-maze performance (p = 0.8; Fig. 4F). Both WT and IL-1R1 KO mice performed significantly worse post-CCI compared to their respective sham-injured mice (p < 0.001 for group, hidden; p < 0.05 for group, visible; Fig. 4E). Differences in MWM performance were not attributable to swim speeds, which did not differ between groups (not shown). In reverse MWM, IL-1R1 KO mice again had worse hidden platform performance versus WT mice (p < 0.05 for group; Fig. 4G), but swim patterns in probe trials appeared similar to those of WT mice (Fig. 4H), arguing against differences in hippocampal-dependent spatial memory. Taken together, the data suggest that nonspatial aspects of MWM learning (such as striatal-dependent procedural learning) are exacerbated post-CCI by genetic deletion of IL-1R1.
FIG. 4.

Histological and behavioral outcome of wild-type (WT) and interleukin-1 receptor knockout (IL-1R1 KO) mice after controlled cortical impact (CCI). (A) No difference in brain water content between WT versus IL-1R1 KO in uninjured (Contra) and injured (Ipsi) hemispheres after CCI (n = 6–8/group). (B) No differences between groups were observed for lesion volume. Representative image of lesion from a WT mouse is shown. (C) Wire grip score (p < 0.05 for group) and (D) wire grip latency (p < 0.0001 for group) differed between WT and IL-1R1 KO after CCI. (E) Performance in hidden and visible platform trials was significantly better in WT versus IL-1R1 KO mice after CCI (p < 0.05 for group for both comparisons). WT CCI and IL-1R1 KO CCI mice performed significantly worse in the hidden platform trials (p < 0.001 for group) and in the visible platform trials (p < 0.05 for group) compared to their respective sham-injured mice. (F) Percent alternating decisions in the Y-maze did not differ between WT and IL-1R1 KO mice post-CCI. (G) IL-1R1 KO mice also performed worse in reverse water maze trials (p < 0.05 for group vs. WT). (H) Swim patterns of WT and IL-1R1 KO mice suggest no obvious differences in spatial learning in IL-1R1 KO mice subjected to CCI. n = 12 WT CCI mice and n = 15 IL-1R1 KO CCI mice for panels (B)–(F) and n = 10–13 for panels (G) and (H). n = 12 WT sham CCI mice and n = 10 IL-1R1 KO sham CCI mice for panel (E). Data are mean ± SEM. SEM, standard error of the mean.
One possible mechanism for the difference in outcome between IL-1R1 KO and WT mice in CCI could be differences in brain leukocyte accumulation. Figure 5 shows FACS data for CCI mice. Compared to sham, WT and IL-1R1 KO CCI mice had similar robust (>10-fold) increases in brain CD11b+/CD45+ leukocyte accumulation at 48 h (p < 0.01 vs. respective sham groups). However, IL-1R1 KO, but not WT, mice had a modest increase in brain lymphocytes versus their respective shams, but injured IL-1R1 KO and WT mice did not differ with respect to brain lymphocytes.
FIG. 5.

Brain leukocyte infiltration observed by fluorescence activate cell sorting (FACS) at 48 h after controlled cortical impact (CCI) in IL-1R1 KO and WT mice. (A) Representative images of Lymphocyte (L) and Macrophage + Neutrophil (M+N) populations from FACS of contused mouse brain (n = 4 per group). (B) A significant increase of M+N population was observed after CCI in WT and IL-1R1 KO versus WT sham and IL-1R1 KO sham, respectively, but no differences were observed between injured IL-1R1 KO and injured WT (*p < 0.01 vs. sham). Compared to their respective sham groups, lymphocytes were increased in injured IL-1R1 KO mice, but not WT mice, but no differences in brain lymphocyte counts were observed between injured IL-1R1 KO and WT mice. Data are mean ± SEM. SEM, standard error of the mean.
Discussion
To our knowledge, this is the first study of IL-1R1 deletion in a clinically relevant model of cerebral contusion or mild TBI. Although CHI and CCI were calibrated to induce cognitive deficits of similar magnitude, as expected the two models produced markedly different gross structural, cellular, and biochemical responses of distinct pathoanatomical TBI phenotypes.2 Nonetheless, post-injury mature IL-1β was induced in brain endothelial cells in both models; however, genetic deficiency of IL-1R1 had opposite effects on neurological outcome, detrimental in CCI but beneficial in CHI. The data provide evidence for the significance of important distinctions between injury types that contribute to pathobiology and functional outcome in TBI models that could impact treatment trials in discrete subgroups of TBI patients.22
To address the ethical limitations of studying responses to mild TBI in human brains, we used a mouse CHI model that recapitulates impact and head acceleration mechanisms of humans with concussion syndrome.12 The scaled rotational acceleration generated by CHI (3800 ± 320 rad/s2) is just below published thresholds observed in human concussion (4600–8200 rad/s2).14 CHI produced LOC, but did not result in skull fractures or significant mortality (1 mouse), overt brain damage, edema, BBB permeability to IgG, or acute cell death, and produced only a modest, delayed leukocyte infiltration at 3 days. In contrast, CCI induced brain edema, BBB damage, acute cell death, HMGB1 release, and early and robust brain leukocyte infiltration. HMGB1 promotes brain inflammation after cerebral contusion,23 but a role for HMGB1 has not been addressed in a (nonfocal) CHI model; thus, it was unknown whether HMGB1 release from brain cells contributes to the inflammatory response post-CHI. This is an important question because HMGB1 has been hypothesized to be a therapeutic target to improve outcome after focal brain injury.23 The finding that HMGB1 translocated from the nucleus to cytoplasm, but was not released from brain cells post-CHI, may, in part, explain the relative lack of leukocytosis at 24 h post-CHI. Taken together, the model differences could imply that targeting inflammation might have more impact in CCI, yet IL-1R1 KO produced significant neurological phenotypes in both models.
To date, most pre-clinical TBI studies of anti-IL-1β antibodies or IL-1R1 antagonist protein in rodents have been done in cerebral contusion models, with only one study examining IL-1 blocking antibodies in a midline fluid percussion model of diffuse TBI.24 These studies have shown beneficial effects on histopathological and functional outcome measures, as well as seizure development,25–27 leading to a phase II clinical trial of Anakinra, an IL-1 receptor antagonist protein, in TBI patients.28 The relatively modest benefit of IL-1 antibodies in midline fluid percussion injury24 and lack of studies in a CHI model with impact and head rotational acceleration leave open the question as to whether IL-1R1-targeted therapies might be effective in patients with concussion syndrome—a much larger patient population than that modeled by CCI.
Our results are in contrast to those showing histological and behavioral protection with pharmacological inhibition of IL-1β in CCI models.25–27 However, it is not surprising that in CCI, IL-1R1 KO mice did not recapitulate findings using IL-1β antagonists. Unlike pharmacological studies, brain development in the absence of IL-1R1 might induce compensatory mechanisms that affect outcome post-CCI (and CHI). IL-1R1 may also exert pleiotropic effects in TBI,29 with harmful effects in the acute phase, such as propagation of inflammation and cell death, but beneficial effects on repair and recovery in later stages, such as upregulation of growth factors,30,31 astrocyte activation,32 and promotion of angiogenesis.33 Time-dependent dual effects of tumor necrosis factor alpha (TNF-α) KO have also been reported in experimental TBI.34 Thus, pharmacological inhibition of IL-1R1 can be targeted to early detrimental functions whereas later reparative functions could still occur after treatment. We used a genetic strategy to inhibit IL-1R1 uniformly in CHI and CCI models because it would be challenging to achieve equal drug concentrations in brain regions affected by CCI and CHI, because of differences in BBB damage between the two models, making it impossible to attribute differences in drug effects solely to pharmacodynamics. Temporally restricted, cell-specific IL-1R1 knockout/knock-in strategies are needed to better establish differential effects of IL-1R1 in CHI and CCI.
Previous studies from our laboratory support the idea that specific inflammatory mechanisms may exert opposite effects in CHI versus CCI. Mice deficient in TNF/Fas receptor had improved cognitive outcome post-CCI, but worse outcome in a CHI model.12,13 Similarly, pharmacological antagonism of Akt/mammalian target of rapamycin improved post-injury cognitive outcome in CCI, but worsened it in a CHI model.16,35 Results from the current and aforementioned studies offer direct support for the NIH guidelines that pre-clinical studies of injury mechanisms and putative therapies should incorporate multiple TBI models to understand whether targeting a given molecular mechanism has broadly beneficial effects, so that robust therapeutic targets may be chosen for clinical trials.2 Such strategies have been successfully applied to pharmacological agents in contusion and penetrating TBI models,36 but contusion and diffuse TBI models have not been examined together to nearly the same extent.16,35
Why might IL-1R1 KO behave differently in CCI versus CHI? One possibility is that IL-1R1 KO may affect injury and repair mechanisms unique to each model. CCI elicits early infiltration of C-C chemokine receptor type 2–positive macrophages that promote brain tissue damage and neurological dysfunction,37 whereas brain macrophage infiltration is modest and more delayed in CHI. Post-CCI, IL-1R1KO mice had similar influx of macrophages + neutrophils as WT mice, but IL-1R KO might also influence macrophage polarization, and hence function, differently in CCI versus CHI.38 IL-1β contributes to resolution of brain edema and BBB damage in experimental stroke,39 but IL-1R1 KO mice had similar brain edema as WT post-CCI.
Different outcomes in the two models could additionally depend on the relative importance of IL-1β and IL-1α in each. In vitro, IL1R1 can induce angiogenesis33 and neurogenesis through IL-1α, whereas IL-1β inhibits neurogenesis in vivo.40 Moreover, the precursor form of IL-1α (but not IL-1β) is biologically active and can act independently of IL-1R1 by localizing to the nucleus and inducing transcription.41,42 In addition, neurons from WT and IL-1R1 KO mice used herein43 express IL-1R37 that may account for previously unexplained IL-1R1-independent, paradoxical activity of IL-1β in cultured neurons and in the brain.44 It is possible that in the absence of IL-1R1, IL-1R3 signaling exerts a beneficial effect on neurological function in CHI, but not CCI.
A major finding of the current study was robust induction of mature IL-1β in CD31+ endothelial cells, but not CD11b+ cells (microglia and infiltrating macrophages/neutrophils) isolated from brain after CCI and CHI, suggesting a vascular source of IL-1β in both models. In vitro, cultured brain endothelial cells respond to percussive trauma by secreting IL-1β45; however, no previous in vivo studies have demonstrated IL-1β signaling in brain endothelium in a TBI model. Our data do not rule out other sources of IL-1β, but raise the possibility that the brain endothelium might be an important source of IL-1β signaling in CCI and CHI. Interestingly, CHI did not produce BBB permeability to endogenous IgG, given that IL-1 is known to mediate vascular permeability.46 However, it is also not clear from our experiments whether brain endothelium produces sufficient mature IL-1β to activate IL-1R1 in endothelium or other cell types—experiments examining IL-1R1 activation in both models are ongoing in our lab, but are challenging because of the often-transient nature of IL-1β signaling.29 In brain, Il-1R1 expression is maximal in endothelial cells,47 thus autocrine (or paracrine) IL-1/IL-1R1 signaling might be a mechanism of cognitive and cerebrovascular48 deficits common to multiple pathoanatomic subtypes of TBI. IL-1β is expressed in the cerebral endothelium and is associated with cognitive impairment in Alzheimer's disease models,49 and several recent studies have highlighted the potential importance of the cerebrovasculature in the pathogenesis of cognitive deficits in CCI models.50 Additional studies are needed to test whether IL-1β/IL-1R1 signaling is a possible link between TBI and development of vascular dementia.
Our study has several limitations that must be considered. IL-1R1 KO mice have a number of phenotypes that could impact outcome in either CCI or CHI, including altered innate immunity and inflammatory responses, wound healing, glucose homeostasis, and insulin sensitivity.51 In addition, global tissue knockout of IL-1R1 may induce compensatory mechanisms during development that can have indirect effects on outcome post-TBI. A better tool for examining IL-1R1 function in CCI and CHI would be a cell-specific, inducible IL-1R1 KO that could be used to examine the effect of IL-1R1 deletion without developmental effects. Third, we did not characterize axonal injury in the CHI model because our focus was on vascular mechanisms, but axonal injury is an important correlate of mild TBI and further studies are needed to characterize axonal injury in the CHI model used herein. These studies will be important inasmuch as axonal injury in the vision circuitry and elsewhere might explain the differences in the initial trials of the MWM in CHI mice, as well as their worse visible platform times. However, these findings can also be readily explained by striatal injury, given that lesioning studies demonstrate the involvement of the dorsolateral striatum in acquisition of the procedural aspects of hidden and visible platform tasks.52
In conclusion, CCI and CHI produce long-lasting cognitive deficits with significantly different histopathological lesions. Although the pathogenesis of each TBI model involves endothelial IL-1β processing, IL-1R1 KO mice have divergent outcomes in CHI and CCI. The exact clinical relevance of these findings must await further studies using more-precise genetic tools. However, the data suggest that clinical trials examining IL-1R1 antagonists could yield variable effects on neurological outcome in patients with distinct pathoanatomical TBI subtypes. Future studies are needed to test this possibility using pharmacological IL-1R1 antagonists in multiple pre-clinical TBI models.
Supplementary Material
Acknowledgments
The work was funded by NIH/NINDS 1RO1NS092847-01 (to D.K.), 5RO1NSO91573 (to J.L.), R21HD086385 (to M.J.W.), and NIA RF1AG051506 (to J.E.K.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1. Faul M., Xu L., Wald M., and Coronado V.G. (2010). Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths, 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta, GA [Google Scholar]
- 2. Saatman K.E., Duhaime A.C., Bullock R., Maas A.I., Valadka A., and Manley G.T. (2008). Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 25, 719–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walsh J.G., Muruve D.A., and Power C. (2014). Inflammasomes in the CNS. Nat. Rev. Neurosci. 15, 84–97 [DOI] [PubMed] [Google Scholar]
- 4. Moriwaki K., and Chan F.K. (2016). Necroptosis-independent signaling by the RIP kinases in inflammation. Cell. Mol. Life Sci. 73, 2325–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer-Hoffert U. (2009). Neutrophil-derived serine proteases modulate innate immune responses. Front. Biosci. 14, 3409–3418 [DOI] [PubMed] [Google Scholar]
- 6. Parker L.C., Luheshi G.N., Rothwell N.J., and Pinteaux E. (2002). IL-1 beta signalling in glial cells in wildtype and IL-1RI deficient mice. Br. J. Pharmac. 136, 312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith D.E., Lipsky B.P., Russell C., Ketchem R.R., Kirchner J., Hensley K., Huang Y., Friedman W.J., Boissonneault V., Plante M.M., Rivest S., and Sims J.E. (2009). A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 30, 817–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Helmy A., Carpenter K.L., Menon D.K., Pickard J.D., and Hutchinson P.J. (2011). The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab. 31, 658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hutchinson P.J., O'Connell M.T., Rothwell N.J., Hopkins S.J., Nortje J., Carpenter K.L., Timofeev I., Al-Rawi P.G., Menon D.K., and Pickard J.D. (2007). Inflammation in human brain injury: intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. J. Neurotrauma 24, 1545–1557 [DOI] [PubMed] [Google Scholar]
- 10. Holmin S., and Hojeberg B. (2004). In situ detection of intracerebral cytokine expression after human brain contusion. Neurosci. Lett. 369, 108–14 [DOI] [PubMed] [Google Scholar]
- 11. Clark R.S., Kochanek P.M., Chen M., Watkins S.C., Marion D.W., Chen J., Hamilton R.L., Loeffert J.E., and Graham S.H. (1999). Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 13, 813–821 [DOI] [PubMed] [Google Scholar]
- 12. Khuman J., Meehan W.P. Zhu X., III, Qiu J., Hoffmann U., Zhang J., Giovannone E., Lo E.H., and Whalen M.J. (2011). Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 31, 778–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bermpohl D., You Z., Lo E.H., Kim H.H., and Whalen M.J. (2007). TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 27, 1806–1818 [DOI] [PubMed] [Google Scholar]
- 14. Ommaya A.K., Yarnell P., Hirsch A.E., and Harris E.H. (1967). Scaling of experimental data on cerebral concussion in sub-human primates to concussion threshold for man. Stapp Car Crash J. 11, 73–80 [Google Scholar]
- 15. Viano D.C., Hamberger A., Bolouri H., and Saljo A. (2009). Concussion in professional football: animal model of brain injury—part 15. Neurosurgery 64, 1162–1173; discussion, 73 [DOI] [PubMed] [Google Scholar]
- 16. Park J., Zhang J., Qiu J., Zhu X., Degterev A., Lo E.H., and Whalen M.J. (2012). Combination therapy targeting Akt and mammalian target of rapamycin improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 32, 330–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mannix R., Meehan W.P., Mandeville J., Grant P.E., Gray T., Berglass J., Zhang J., Bryant J., Rezaie S., Chung J.Y., Peters N.V., Lee C., Tien L.W., Kaplan D.L., Feany M., and Whalen M. (2013). Clinical correlates in an experimental model of repetitive mild brain injury. Ann. Neurol. 74, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng J.S., Craft R., Yu GQ., Ho K., Wang X., Mohan G., Mangnitsky S., Ponnusamy R., and Mucke L. (2014). Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One 9, e115765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hickman S.E., Kingery N.D., Ohsumi T.K., Borowsky M.L., Wang L.C., Means T.K., and El, Khoury J. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. El Khoury J., Toft M., Hickman S.E., Means T.K., Terada K., Geula C., and Luster A.D. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438 [DOI] [PubMed] [Google Scholar]
- 21. Lim Y.C., Garcia-Cardena G., Allport J.R., Zervoglos M., Connolly A.J., Gimbrone M.A, Jr., and Luscinskas F.W. (2003). Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am. J. Pathol. 162, 1591–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giza C.C., Prins M.L., and Hovda D.A. (2017). It's not all fun and games: sports, concussions, and neuroscience. Neuron 94, 1051–1055 [DOI] [PubMed] [Google Scholar]
- 23. Parker T.M., Nguyen A.H., Rabang J.R., Patil A.A., and Agrawal D.K. (2017). The danger zone: Systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj. 31, 2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ekmark-Lewen S., Flygt J., Fridgeirsdottir G.A., Kiwanuka O., Hanell A., Meyerson B.J., Mir A.K., Gram H., Lewen A., Clausen F., Hillered L., and Marklund N. (2016). Diffuse traumatic axonal injury in mice induces complex behavioural alterations that are normalized by neutralization of interleukin-1beta. Eur. J. Neurosci. 43, 1016–1033 [DOI] [PubMed] [Google Scholar]
- 25. Clausen F., Hanell A., Bjork M., Hillered L., Mir A.K., Gram H., and Marklund N. (2009). Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 30, 385–396 [DOI] [PubMed] [Google Scholar]
- 26. Clausen F., Hanell A., Israelsson C., Hedin J., Ebendal T., Mir A.K., Gram H., and Marklund N. (2011). Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 34, 110–123 [DOI] [PubMed] [Google Scholar]
- 27. Semple B.D., O'Brien T.J., Gimlin K., Wright D.K., Kim S.E., Casillas-Espinosa P.M., Webster K.M., Petrou S., and Noble-Haeusslein L.J. (2017). Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J. Neurosci. 37, 7864–7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helmy A., Guilfoyle M.R., Carpenter K.L., Pickard J.D., Menon D.K., and Hutchinson P.J. (2014). Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J. Cereb. Blood Flow Metab. 34, 845–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luheshi N.M., Rothwell N.J., and Brough D. (2009). Dual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapy. Br. J. Pharmacol. 157, 1318–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Araujo D.M., and Cotman C.W. (1992). Basic FGF in astroglial, microglial, and neuronal cultures: characterization of binding sites and modulation of release by lymphokines and trophic factors. J. Neurosci. 12, 1668–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basu A., Krady J.K., O'Malley M., Styren S.D., DeKosky S.T., and Levison S.W. (2002). The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J. Neurosci. 22, 6071–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin H.W., Basu A., Druckman C., Cicchese M., Krady J.K., and Levison S.W. (2006). Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J. Neuroinflammation 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salmeron K., Aihara T., Redondo-Castro E., Pinteaux E., and Bix G. (2016). IL-1alpha induces angiogenesis in brain endothelial cells in vitro: implications for brain angiogenesis after acute injury. J. Neurochem. 136, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scherbel U., Raghupathi R., Nakamura M., Saatman K.E., Trojanowski J.Q., Neugebauer E., Marino M.W., and McIntosh T.K. (1999). Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl. Acad. Sci. U. S. A. 96, 8721–8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu X., Park J., Golinski J., Qiu J., Khuman J., Lee C.C., Lo E.H., Degterev A., and Whalen M.J. (2014). Role of Akt and mammalian target of rapamycin in functional outcome after concussive brain injury in mice. J. Cereb. Blood Flow Metab. 34, 1531–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kochanek P.M., Bramlett H.M., Shear D.A., Dixon C.E., Mondello S., Dietrich W.D., Hayes R.L., Wang K.K., Poloyac S.M., Empey P.E., Povlishock J.T., Mountney A., Browning M., Deng-Bryant Y., Yan H.Q., Jackson T.C., Catania M., Glushakova O., Richieri S.P., and Tortella F.C. (2016). Synthesis of findings, current investigations, and future directions: operation brain trauma therapy. J. Neurotrauma 33, 606–614 [DOI] [PubMed] [Google Scholar]
- 37. Morganti J.M., Jopson T.D., Liu S., Riparip L.K., Guandique C.K., Gupta N., Ferguson A.R., and Rosi S. (2015). CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 35, 748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jassam Y.N., Izzy S., Whalen M., McGavern D.B., and El Khoury J. (2017). Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron 95, 1246–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodriguez-Grande B., Swana M., Nguyen L., Englezou P., Maysami S., Allan S.M., Rothwell N.J., Garlanda C., Denes A., and Pinteaux E. (2014). The acute-phase protein PTX3 is an essential mediator of glial scar formation and resolution of brain edema after ischemic injury. J. Cereb. Blood Flow Metab. 34, 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gonzalez-Perez O., Gutierrez-Fernandez F., Lopez-Virgen V., Collas-Aguilar J., Quinones-Hinojosa A., and Garcia-Verdugo J.M. (2012). Immunological regulation of neurogenic niches in the adult brain. Neuroscience 226, 270–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng W., Shivshankar P., Zhong Y., Chen D., Li Z., and Zhong G. (2008). Intracellular interleukin-1alpha mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect. Immun. 76, 942–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Werman A., Werman-Venkert R., White R., Lee J.K., Werman B., Krelin Y., Voronov E., Dinarello C.A., and Apte R.N. (2004). The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. U. S. A. 101, 2434–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Labow M., Shuster D., Zetterstrom M., Nunes P., Terry R., Cullinan E.B., Bartfai T., Solorzano C., Moldawer L.L., Chizzonite R., and McIntyre K.W. (1997). Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J. Immunol. 159, 2452–2461 [PubMed] [Google Scholar]
- 44. Qian J., Zhu L., Li Q., Belevych N., Chen Q., Zhao F., Herness S., and Quan N. (2012). Interleukin-1R3 mediates interleukin-1-induced potassium current increase through fast activation of Akt kinase. Proc. Natl. Acad. Sci. U. S. A. 109, 12189–12194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gourin C.G., and Shackford S.R. (1997). Production of tumor necrosis factor-alpha and interleukin-1beta by human cerebral microvascular endothelium after percussive trauma. J. Trauma 42, 1101–1107 [DOI] [PubMed] [Google Scholar]
- 46. Konsman J.P., Drukarch B., and Van Dam A.M. (2007). (Peri)vascular production and action of pro-inflammatory cytokines in brain pathology. Clin. Sci. 112, 1–25 [DOI] [PubMed] [Google Scholar]
- 47. Liu X., Yamashita T., Chen Q., Belevych N., McKim D.B., Tarr A.J., Coppola V., Nath N., Nemeth D.P., Syed Z.W., Sheridan J.F., Godbout J.P., Zuo J., and Quan N. (2015). Interleukin 1 type 1 receptor restore: a genetic mouse model for studying interleukin 1 receptor-mediated effects in specific cell types. J. Neurosci. 35, 2860–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Churchill N., Hutchison M., Richards D., Leung G., Graham S., and Schweizer T.A. (2017). Brain structure and function associated with a history of sport concussion: a multi-modal magnetic resonance imaging study. J. Neurotrauma 34, 765–771 [DOI] [PubMed] [Google Scholar]
- 49. Grammas P., Martinez J., Sanchez A., Yin X., Riley J., Gay D., Desobry K., Tripathy D., Luo J., Evola M., and Young A. (2014). A new paradigm for the treatment of Alzheimer's disease: targeting vascular activation. J. Alzheimers Dis. 40, 619–630 [DOI] [PubMed] [Google Scholar]
- 50. Badaut J., and Bix G.J. (2014). Vascular neural network phenotypic transformation after traumatic injury: potential role in long-term sequelae. Transl. Stroke Res. 5, 394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Glaccum M.B., Stocking K.L., Charrier K., Smith J.L., Willis C.R., Maliszewski C., Livingston D.J. Peschon J.J., and Morrissey P.J. (1997). Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 159, 3364–3371 [PubMed] [Google Scholar]
- 52. Devan B.D., Goad B., and Petri H.L. (1996). Dissociation of hippocampal and striatal contributions to spatial navigation in the water maze. Neurobiol. Learn. Mem. 66, 305–323 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.