

Abstract
Metastasis is the leading cause of human cancer deaths. Unfortunately, no approved drugs are available for anti-metastatic treatment. In our study, high-throughput sequencing-based high-throughput screening (HTS2) and a breast cancer lung metastasis (BCLM)-associated gene signature were combined to discover anti-metastatic drugs. After screening of thousands of compounds, we identified Ponatinib as a BCLM inhibitor. Ponatinib significantly inhibited the migration and mammosphere formation of breast cancer cells in vitro and blocked BCLM in multiple mouse models. Mechanistically, Ponatinib represses the expression of BCLM-associated genes mainly through the ERK/c-Jun signaling pathway by inhibiting the transcription of JUN and accelerating the degradation of c-Jun protein. Notably, JUN expression levels were positively correlated with BCLM-associated gene expression and lung metastases in breast cancer patients. Collectively, we established a novel approach for the discovery of anti-metastatic drugs, identified Ponatinib as a new drug to inhibit BCLM and revealed c-Jun as a crucial factor and potential drug target for BCLM. Our study may facilitate the therapeutic treatment of BCLM as well as other metastases.
Electronic supplementary material
The online version of this article (10.1007/s13238-018-0533-8) contains supplementary material, which is available to authorized users.
Keywords: anti-metastatic drug discovery, gene expression signature, high-throughput sequencing-based high-throughput screening, Ponatinib, breast cancer lung metastasis, c-Jun
Introduction
The absence of effective and high-throughput metastatic cell models in vitro has obstructed anti-metastatic drug discovery. Metastasis represents a multistep cascade of events, including local invasion, intravasation, survival in the circulation, extravasation and colonization (Nguyen et al., 2009). Few in vitro cell assays can be used to represent a particular step of metastasis. For example, Transwell migration or wound healing assays can be used to study cell migration or invasion; and soft-agar colony-formation assays are a well-established method for characterizing cell colonization capability. However, these assays cannot completely reflect the cancer metastatic process and are not suitable for anti-metastatic drug discovery.
Breast cancer is the most common invasive cancer in women (Siegel et al., 2017), and the lungs are one of the most common metastatic organs (Lee, 1983; Quiet et al., 1995). Despite enormous efforts in academia and industry to develop specifically anti-metastasis medications for breast cancer, no drugs are available on the market. Therefore, there is an urgent needed for effective medical treatments for breast cancer metastasis.
The gene expression signatures of cells or tissues can be used as “fingerprints” to define cancer subtyping (Perou et al., 2000), predict cancer metastasis (Kang et al., 2003; Minn et al., 2005; Bos et al., 2009) and determine the clinical outcome of patients (van’t Veer et al., 2002). Gene expression profiling of cellular perturbations has been used to effectively predict drug sensitivity (Chambers et al., 1999; Ayers et al., 2004) and compound mechanism-of-action (MoA) (Iorio et al., 2010), as well as for anti-cancer drug discovery (Stegmaier et al., 2004; Lamb et al., 2006; Li et al., 2012b; Lee et al., 2016). Compared with common technologies for detecting gene expression, such as microarray (Lamb et al., 2006) and Luminex beads (Subramanian et al., 2017), high-throughput sequencing-based high-throughput screening (HTS2), which takes advantage of powerful next-generation sequencing technologies and greatly enhances the parallel processing of samples and genes (Li et al., 2012b), has a huge advantage in terms of throughput, required labor and costs. Thus, the combination of metastasis-associated gene signatures and HTS2 might be an appropriate approach for anti-metastatic drug discovery.
c-Jun is a protein encoded by the proto-oncogene JUN, the cellular homolog of the transforming viral oncogene v-JUN in humans. Based on accumulating evidence, c-Jun is involved in a wide variety of cellular processes, including proliferation, differentiation, growth, apoptosis, cell migration and transformation (Lopez-Bergami et al., 2010). c-Jun’s activity is regulated by post-translational modifications that are largely controlled by components of mitogen-activated protein kinase (MAPK) family kinases, including c-Jun N-terminal kinase (JNK), extracellular-signal-regulated kinase (ERK) and p38 kinase. However, few reports have identified a role for c-Jun in breast cancer organ-specific metastasis, especially breast cancer lung metastasis (BCLM).
The aim of the present study was to explore the possibility of using metastasis-associated gene signature-based high-throughput screening to discover anti-BCLM drugs. We used in vitro cell migration and colonization assays and in vivo mouse models to characterize screening hits. Among thousands of compounds, we determined that Ponatinib, a tyrosine-kinase inhibitor, represses BCLM-associated gene expression via the ERK/c-Jun signaling pathway and inhibits BCLM in mouse models. Our study not only provided new insights into anti-metastatic drug discovery but also revealed c-Jun as a crucial factor and potential drug target for BCLM.
Results
Ponatinib is identified by HTS2 screening as a potential BCLM-inhibiting compound
We set up an approach to profile the mRNA levels of 46 genes in the breast cancer MDA-MB-231 cell line using the HTS2 method. Among the 46 genes, 13 genes representing the BCLM gene signature (Minn et al., 2005) were chosen from published literature, and 33 genes stably expressed in breast cancer were selected as an internal control (Casey et al., 2009; Clarke et al., 2013). The MDA-MB-231 cell line has been widely used to study breast cancer metastasis and is therefore an appropriate cell model for this screening (Kang et al., 2003; Minn et al., 2005; Bos et al., 2009). The assay had high sensitivity and excellent reproducibility, which were validated in a pilot screen (Fig. S1A and S1B). MDA-MB-231 cells were treated for 24 h with thousands of compounds, including US Food and Drug Administration (FDA)-approved drugs. The activity of each compound was scored according to its ability to reverse gene expression in the BCLM gene signature (Subramanian et al., 2005) (Fig. 1A and 1B). Among the top 5 hit compounds, 2 compounds targeted the aurora kinase, which induce mammary cell migration and breast cancer metastasis via the cofilin-F-actin pathway (Wang et al., 2010). These results demonstrated that our approach can effectively identify potential anti-metastatic drugs. Ponatinib, an FDA-approved drug for leukemia, has not been previously reported to inhibit breast cancer metastasis (O’Hare et al., 2009) (Fig. 1C and 1D). The genes most affected by Ponatinib were chosen to validate the screening results. By RT-qPCR, we confirmed that Ponatinib indeed dose- and time- dependently affected the expression of 6 BCLM-associated genes, including ANGPTL4, MMP1, PTGS2, TNC, LY6E and RARRES3 (Fig. 1F and 1G). These genes exhibited similar expression in human breast cancer LM2 cells and mouse breast cancer 4T1 cells (Fig. 1E) when treated with Ponatinib. These findings indicated that the effect of Ponatinib on these 6 BCLM-associated genes may be general. Thus, based on HTS2 screening, Ponatinib was selected as an anti-metastatic drug for further study.
Figure 1.
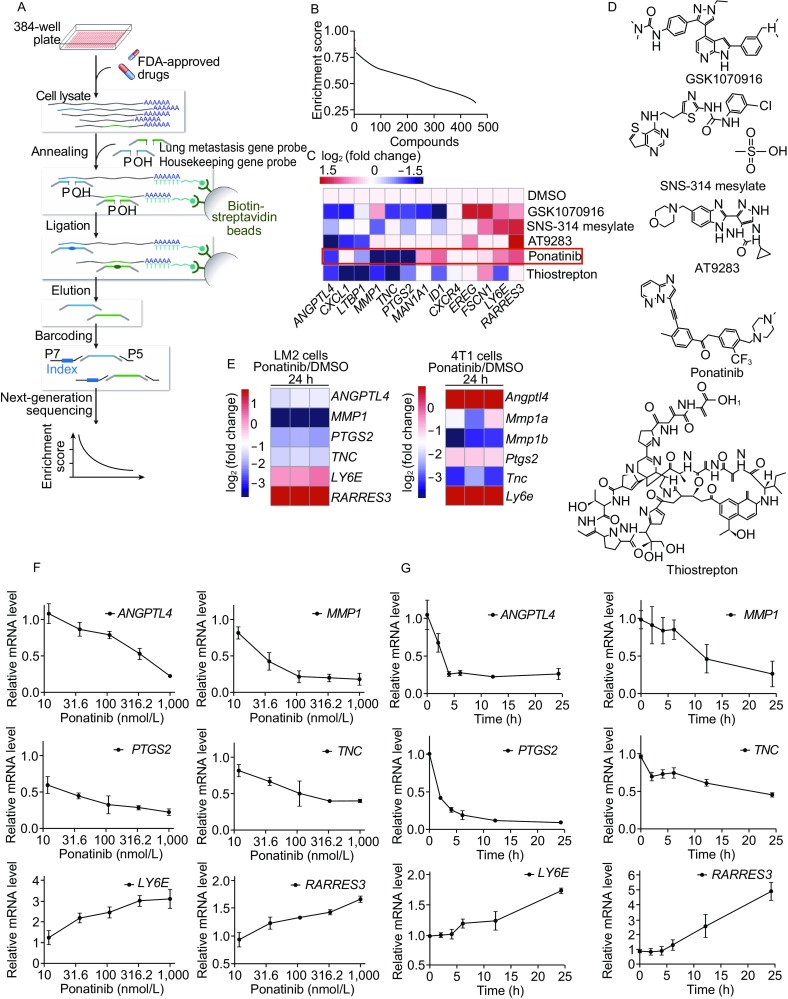
Ponatinib reverses the expression of certain BCLM-associated genes. (A) The scheme of the experimental process. (B) Potential anti-metastatic compounds are sorted by the activity score of reversing the BCLM-associated gene signature. The top 5 hit compounds are shown as red dots. (C) Heat map representing the expression of 13 metastatic-related genes (up-regulated, red; down-regulated, blue) in MDA-MB-231 cells treated with the top 5 hit compounds in the screen data, the log2 (fold change) is used to scale the differences in gene expression. (D) The top 5 chemical structures are shown. (E) LM2 cells or mouse breast cancer 4T1 cells were treated with 1 µmol/L Ponatinib at 24 h, heat map representing the expression of 6 metastatic-related genes in three independent experiments. (F and G) RT-qPCR analysis showed that Ponatinib was capable of reversing the expression of 6 BCLM-associated genes in a dose- and time-dependent manner. Cells were treated with a serial dilution of Ponatinib at 24 h (F) or with 1 µmol/L Ponatinib at various time points (0, 2, 4, 6, 12 and 24 h) (G). Data present mean ± SD of three independent experiments and normalized to DMSO control
Ponatinib inhibits breast cancer cell migration and colonization in vitro
The 4 genes regulated by Ponatinib are involved in cell migration and colonization; thus, we first examined the effect of Ponatinib on cell migration ability. Ponatinib significantly inhibited the migration of MDA-MB-231 cells and LM2 cells (P < 0.001) (Fig. 2A and 2B), which was confirmed by another standard assay for testing cell migration, the wound healing assay (P < 0.001) (Fig. 2C and 2D). Moreover, mammosphere formation in MDA-MB-231 cells and LM2 cells was significantly inhibited by Ponatinib treatment in an ultralow attachment microplate (Fig. 2E). In addition to the cell migration, our result indicated that Ponatinib could also repress the anchorage-independent growth and colonization, which is also one of the hallmarks of cell transformation.
Figure 2.
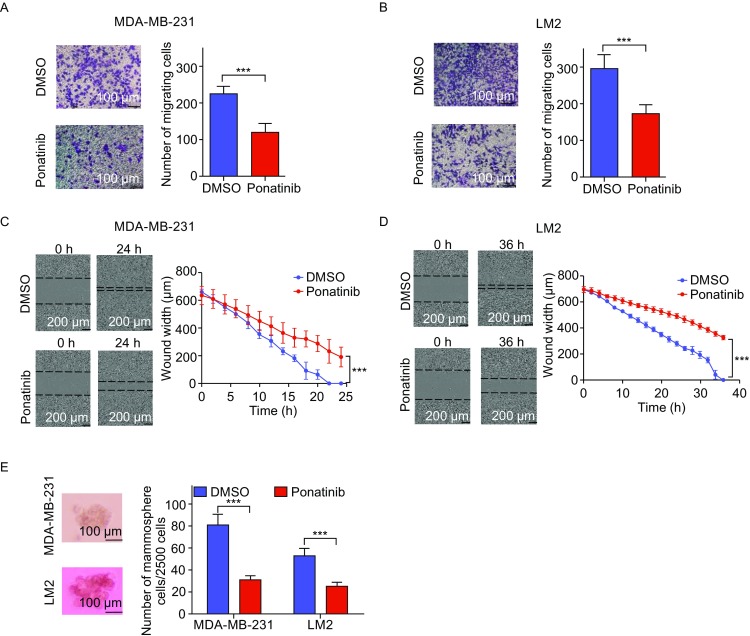
Ponatinib inhibits breast cancer migration in vitro. (A and B) The effect of Ponatinib on the migration of MDA-MB-231 cells (A) and LM2 cells (B) was tested by a Transwell migration assay. Cells were treated with 1 µmol/L Ponatinib for 8 h, the migrating cells were stained with crystal violet (left panel) and quantified (right panel). ***P < 0.001 by two-sided Student’s t-test. Scale bar, 100 µm. (C and D) The effect of Ponatinib on the migration of MDA-MB-231 cells (C) and LM2 cells (D) was tested by a wound healing assay. Cells were treated with 1 µmol/L Ponatinib, the wound width was photographed after scratching (left panel) and quantified (right panel). P value by one-tailed Mann-Whitney U test. Scale bar, 200 µm. (E) The effect of Ponatinib on mammosphere formation of MDA-MB-231 cells and LM2 cells was tested by an ultralow attachment microplate. Cells were treated with 1 µmol/L Ponatinib. ***P < 0.001 by two-sided Student’s t-test. Scale bars, 100 µm. Representative images of one of three independent experiments are shown. Error bars represent mean ± SD of 6 samples in each group (A–E)
Ponatinib inhibits breast cancer metastasis to the lungs in vivo
To examine whether Ponatinib effectively prevents breast cancer lung metastasis in vivo, we pretreated BALB/c-nude mice with Ponatinib or vehicle at a dose of 30 mg/kg/day through oral gavage for 2 days. Then, LM2 cells, which are derived from MDA-MB-231 cells and show high lung metastatic capability (Minn et al., 2005), were injected into the mouse tail vein, and lung metastatic progression was monitored with bioluminescence imaging (BLI) (Fig. 3A). At 4 weeks post-LM2 cell injection, 8 of 9 mice in the vehicle-treated group presented luciferase activity in the lung, while only 1 of 9 mice in the Ponatinib-treated group exhibited luciferase activity in the lung. Compared with vehicle treatment, Ponatinib treatment inhibited > 90% of cancer cell colonization in the lung as demonstrated by bioluminescence signals (mean 0.35 × 106 vs. 5.46 × 106, P = 0.003) (Fig. 3B and 3C) and H&E staining (Fig. 3D).
Figure 3.
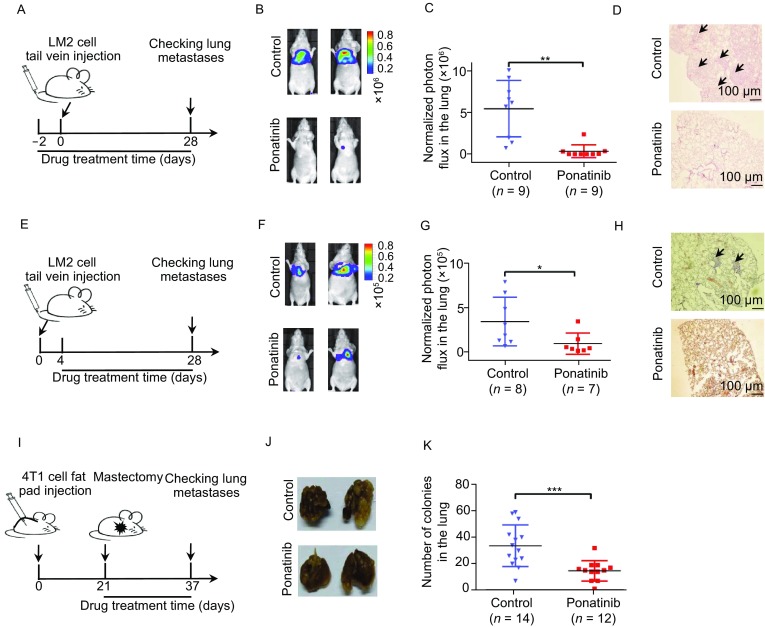
Ponatinib inhibits breast cancer metastasis to the lung in mouse models. (A) Schematic representation of mouse experimental design. At day 0, LM2 cells (2 × 105 cells) were injected into the tail veins of mice, and lung colonization was assayed by bioluminescence imaging (BLI). Mice were treated with vehicle or Ponatinib (30 mg/kg/day) for up to 30 consecutive days. (B) Representative bioluminescence images of lung colonization in the mice. (C) Plots show normalized photon flux in the lungs of each group of mice at day 28. n = 9 mice per group. **P < 0.01 by two-sided Student’s t-test. Error bars, SEM. (D) Representative H&E staining of lung sections are shown. Arrow reflects the metastatic foci detected in lung section. (E) LM2 cells (2 × 105 cells) were injected into the tail veins of mice. After 4 days, the mice received vehicle or Ponatinib (30 mg/kg/day) via oral gavage each day, and lung colonization was quantified. (F) Representative bioluminescence images of lung colonization in the mice. (G) Plots show normalized photon flux in the lungs of each group of mice at day 28. n = 8 mice for the control group and n = 7 for the Ponatinib treatment group. *P < 0.05 by two-sided Student’s t-test. Error bars, SEM. (H) Representative immunohistochemistry images of metastasis cells in mouse lung section. (I) Schematic representation of mouse experimental design. At day 0, 4T1 cells (1 × 106 cells) were injected into the fat pads of mice. When the tumor volume reached 200 mm3, the tumors were resected, and the mice were treated with vehicle or Ponatinib (30 mg/kg/day) for up to 16 consecutive days. At day 37, the mice were sacrificed, and metastatic colonies were counted. (J) Representative images of metastases in the lung; arrows indicate metastatic foci. (K) Plots show the metastatic colonies in the lungs of each group of mice. n = 14 mice for the control group and n = 12 for the drug treatment group. ***P < 0.001 by two-sided Student’s t-test. Error bars, SEM
To further examine whether Ponatinib could inhibit the growth of micro-metastases of breast cancer in mouse lungs, we injected LM2 cells into the tail veins of nude mice and waited for 4 days to let the form of micro-metastases in the lungs (Padua et al., 2008). Then, these mice were treated with Ponatinib or vehicle daily through oral gavage. Lung colonization was examined by lung bioluminescence signals 24 days later (Fig. 3E). Compared with controls, Ponatinib decreased breast cancer cell growth in the lung by as much as 80% according to BLI (mean photon flux of 0.89 × 105 vs. 3.40 × 105, P < 0.05) (Fig. 3F and 3G) and H&E staining (Fig. 3H). Hence, Ponatinib treatment significantly inhibited the growth of micro-metastases in the lungs. To evaluate the general effect of Ponatinib on cancer metastasis, we used a spontaneous metastasis mouse model, which is the gold standard for mimicking the full metastatic process in mice. Mouse breast cancer 4T1 cells were injected into the mammary fat pads of mice to induce primary breast tumor growth. When the volume of primary tumors reached 200 mm3, the primary tumors were surgically resected. These mice were randomly grouped and treated with vehicle or Ponatinib by oral gavage. After 16 days of drug treatment, breast cancer metastases were assayed by BLI (Fig. 3I). Quantification of metastatic colonies in the lungs of each mouse established the significant inhibitory effect of Ponatinib on BCLM (mean 16 vs. 33, P = 0.0003) (Fig. 3J and 3K).
To investigate whether the inhibition of Ponatinib on breast cancer metastasis was caused by the decreased growth of primary tumor, we orthotopically injected LM2 cells into the mammary fat pads of BALB/c-nude mice, and tumor volume was measured during the three weeks course of drug treatment. The tumor volume at the primary site was not significantly different between mice treated with vehicle control and Ponatinib (P = 0.730) (Fig. S2B). Meanwhile, the proliferation of MDA-MB-231 cells and LM2 cells was not sensitive to Ponatinib treatment, the IC50 of Ponatinib with these two cells was as high as 1 µmol/L (Fig. S2A). All these data suggested that the anti-metastatic effect of Ponatinib is not due to the growth inhibition of primary tumors in vivo and in vitro.
Collectively, supported by two human xenograft models and a spontaneous mouse model, our work clearly indicated that Ponatinib significantly prevents and inhibits breast cancer metastasis to the lungs in mouse models.
Ponatinib inhibits the expression of BCLM-associated genes only partially via its reported target ABL
To investigate how Ponatinib affected the expression of BCLM-associated genes, we first examined whether Ponatinib reversed the expression of these 6 genes through ABL, a well-known target of Ponatinib (O’Hare et al., 2009). The ABL gene family has 2 members, ABL1 and ABL2. We knocked down the expression of ABL1 and/or ABL2 by shRNAs in LM2 cells and measured the expression changes in these BCLM-associated genes by RT-qPCR. The expression of only half of these 6 genes was significantly reversed (Figs. 4A, 4B, S3A and S3B). This result was consistent with a previous report in which the expression of ABL1 and/or ABL2 was knocked down in 1833 cells that were also derived from MDA-MB-231 cells; the expression of only 1 of these 6 BCLM-associated genes was reversed (Wang et al., 2016) (Fig. S3C).
Figure 4.
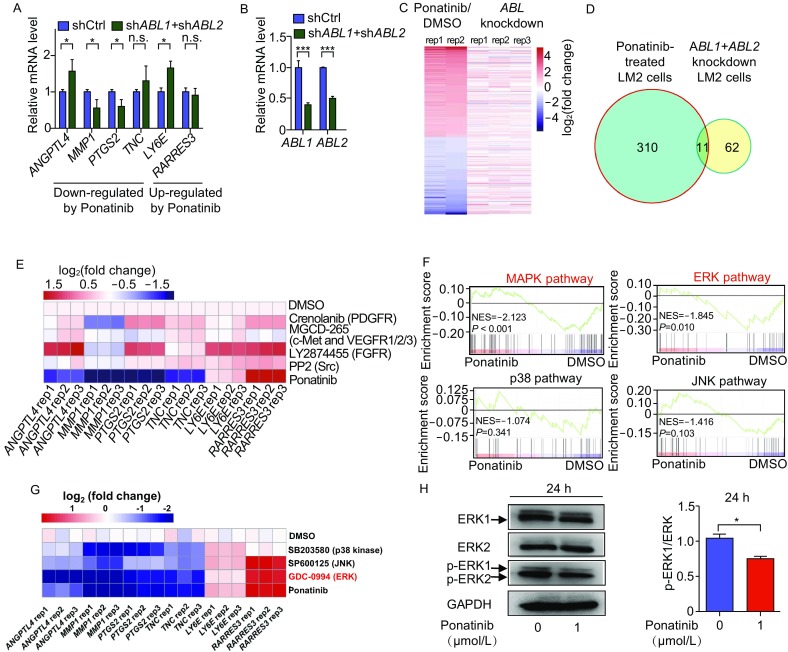
Ponatinib inhibits the expression of 4 BCLM related genes mainly through the ERK pathway. (A and B) The 6 BCLM-associated gene expression in control and ABL1/ABL2 double-knockdown LM2 cells is shown in (A), and the knock down efficiency of ABL1/ABL2 is shown in (B). Data are mean ± SD of three independent experiments. *P < 0.05, ***P < 0.001 by two-sided Student’s t-test. (C) Heat map of genes differently expressed in Ponatinib-treated LM2 cells, the related genes expression in ABL1/ABL2 or double-knockdown in LM2 cells were also shown. Blue and red colors represent down- and up-regulated genes, respectively. (D) Venn diagrams of overlapped genes with Ponatinib treatment and ABL1/ABL2 double-knockdown in LM2 cells. The differential expressed genes were defined by fold-change > 2 and P < 0.05. (E) The common targets of Ponatinib were inhibited with specific inhibitors. LM2 cells were treated with 1 µmol/L Crenolanib (PDGFR inhibitor), 1 µmol/L MGCD-265 (c-Met and VEGFR1/2/3 inhibitor), 1 µmol/L LY2874455 (FGFR inhibitor), 1 µmol/L PP2 (Src inhibitor) individually for 24 h, 6 BCLM-associated gene expression is detected by RT-qPCR. Three independent experiments were performed. (F) GSEA analysis of the indicated gene signatures in DMSO- or Ponatinib-treated cells. NES, normalized enrichment score. P value represents false discovery rate adjusted P-value. (G) LM2 cells were treated with 20 µmol/L SP600125 (JNK inhibitor), 20 µmol/L SB203580 (p38 MAPK inhibitor), 1 µmol/L GDC-0994 (ERK inhibitor) individually. Gene expression was detected by RT-qPCR. Three independent experiments were performed. (H) LM2 cells were treated with DMSO or 1 µmol/L Ponatinib for 24 h, Western blot analysis of the indicated protein level and phosphorylation level (left panel), related p-ERK level were quantified (right panel). Data represent mean ± SD of three independent experiments. Two-tailed Student’s t-test were performed, *P < 0.05
To further determine the role of ABL in Ponatinib-mediated inhibition of BCLM, we used RNA sequencing (RNA-seq) to obtain a genome-wide profile of differentially expressed genes (DEGs) in LM2 cells, which were treated with Ponatinib as well as ABL1 and/or ABL2 shRNAs (Fig. 4C). As expected, the expression changes in these 6 BCLM-associated genes as well as ABL1 and ABL2 genes after Ponatinib treatment or ABL1 and ABL2 knockdown were consistent with our RT-qPCR results (Fig. S4A and S4B). RNA-seq also revealed 321 and 73 DEGs (fold change >2, P < 0.05) after Ponatinib treatment and ABL1/ABL2 double knockdown, respectively. However, only 15% of ABL1/ABL2 double-knockdown-affected genes were also affected by Ponatinib treatment (11/73 genes) (Fig. 4D). This genome-wide analysis again suggested that ABL might not be the major mediator of Ponatinib’s inhibitory effect on the expression of BCLM-associated genes. Notably, a recent report also mentioned that loss of function of ABL1 and/or ABL2 does not inhibit breast cancer metastasis to the lung (Wang et al., 2016), which further demonstrated that ABL does not play crucial roles in the anti-metastatic function of Ponatinib. Ponatinib is a multitarget inhibitor of ABL, PDGFRα, VEGFR2, FGFR1 and Src, with a low IC50 (O’Hare et al., 2009). Unfortunately, when inhibiting these targets of Ponatinib, we did not identify a target similar to Ponatinib that regulates the expression of these genes (Fig. 4E).
Together, our data clearly showed that ABL plays only a minor role in the regulation of BCLM-associated gene expression by Ponatinib. This result suggested that Ponatinib may regulate BCLM-associated gene expression via unknown or multiple targets.
Ponatinib inhibits the expression of BCLM-associated genes mainly through the extracellular signal-regulated kinase (ERK) pathway
Because ABL played only a minor role in the inhibitory effect of Ponatinib on metastasis-associated gene expression, we investigated relevant pathways in drug-treated LM2 cells. We first carried out gene ontology (GO) analysis using the Kyoto encyclopedia of genes and genomes (KEGG) database (Subramanian et al., 2005) with RNA-seq data from Ponatinib-treated LM2 cells. Tumor necrosis factor (TNF) and MAPK signaling pathways were mostly enriched (Fig. S5). As a critical cytokine, TNF can induce a wide range of intracellular signaling pathways, and a substantial part of its function is primarily mediated by the MAPK cascade. Therefore, we focused on the MAPK pathway, which was significantly inhibited by Ponatinib treatment in gene-set enrichment analysis (GSEA) (Fig. 4F). ERK, p38 and JNK are the three main molecules downstream of the MAPK pathway (Morrison, 2012). After further GSEA analysis, only the ERK pathway was significantly inhibited by Ponatinib treatment (P = 0.01) (Fig. 4F). To further validate this result, we treated LM2 cells with specific inhibitors for the ERK, p38 and JNK pathways and tested the expression of BCLM-associated genes. In RT-qPCR results, the pattern of DEG expression initiated by ERK pathway inhibitor GDC-0994 was indeed more similar to that initiated by Ponatinib treatment but not p38 inhibitor SB203580 or JNK inhibitor SP600125 treatment (Fig. 4G). The phosphorylation of ERK 1/2 was decreased in LM2 cells treated with Ponatinib, consistent with our GSEA analysis (Fig. 4H). Thus, the regulatory effect of Ponatinib on BCLM-associated genes might be mainly carried out via the ERK pathway.
Transcription factor c-Jun, which directly binds to and activates BCLM-associated genes, is inhibited by Ponatinib
To understand how these BCLM-associated genes are regulated by Ponatinib treatment, we examined the expression of these 6 BCLM-associated genes in detail. Four genes (ANGPTL4, MMP1, PTGS2 and TNC) and 2 genes (LY6E and RARRES3) from the BCLM gene signature were inhibited and activated by Ponatinib treatment, respectively. This observation suggested that at least two different regulatory mechanisms are involved in the reversal of BCLM-associated gene expression by Ponatinib.
To illustrate how these 4 genes are down-regulated by Ponatinib treatment, we examined which transcription factors (TFs) bind the promoters of these 4 genes. After checking ENCODE ChIP-seq data (Raney et al., 2011), we identified 7 TFs with known putative binding sites for all 4 promoters (Fig. 5A). Analysis of RNA-seq data from Ponatinib-treated LM2 cells revealed that among these 7 TFs, only JUN expression was significantly down-regulated (Fig. 5B), which indicated that c-Jun might contribute to the regulation of these 4 genes by Ponatinib. Next, we knocked down JUN mRNA by 2 shRNAs with different sequences in LM2 cells and found that the expression of 3 genes was indeed down-regulated (Figs. 5C and S6A). Moreover, overexpression of JUN led to significant activation of the expression of these 4 genes (Figs. 5D and S6B). In addition, the expression of JUN and these 4 BCLM-associated genes was higher in lung metastatic LM2 cells than in parental MDA-MB-231 cells (Lu et al., 2010), which indicates that c-Jun may play a role in the process of BCLM. Collectively, our results suggested that c-Jun activates the expression of these 4 BCLM-associated genes.
Figure 5.
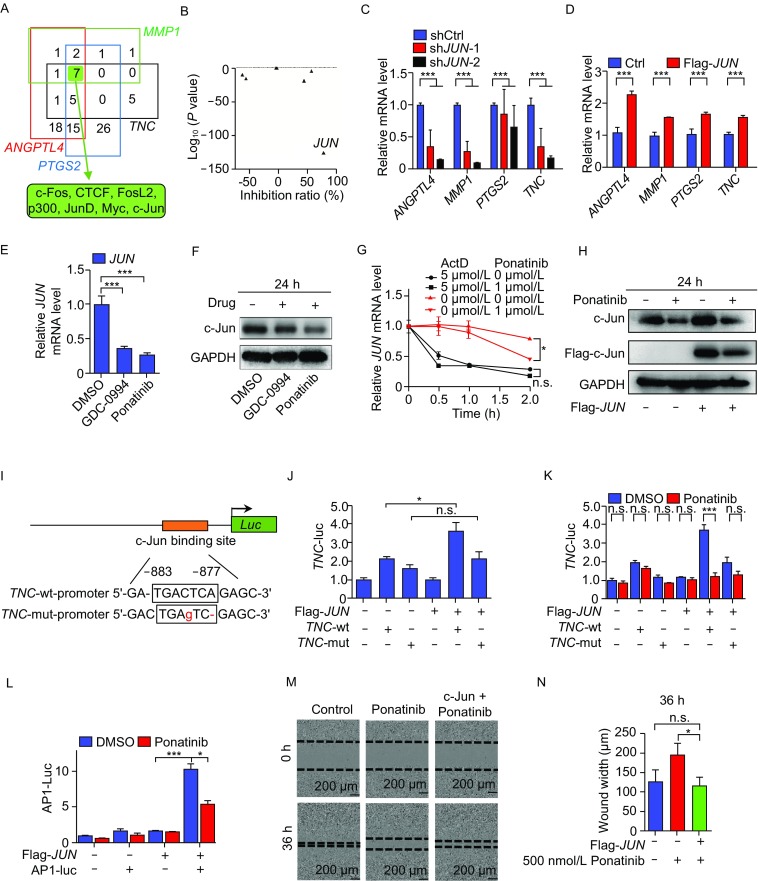
Ponatinib inhibits the expression of certain BCLM-associated genes and cell migration via the transcription factor c-Jun. (A) Transcription factors that bind to the promoters of 4 BCLM-associated genes down-regulated by Ponatinib in ENCODE ChIP-seq data. The numbers in each color box represent the number of transcription factors bound to the promoter region of each BCLM related genes (group), the shared 7 transcription factors in four groups were marked with a green solid box. (B) Gene expression analysis of these 7 transcription factors in (A) when LM2 cells were treated with Ponatinib. (C and D) RT-qPCR analysis of the expression of 4 BCLM-associated genes in LM2 cells with knockdown (C) or overexpression (D) of JUN. (E) RT-qPCR analysis of JUN mRNA expression in LM2 cells treated with 1 µmol/L Ponatinib or 10 µmol/L GDC-0994 for 24 h. (F) Western blot analysis of the protein level of endogenous c-Jun in LM2 cells treated with 1 µmol/L Ponatinib or 10 µmol/L GDC-0994 for 24 h. (G) LM2 cells were treated with 1 µmol/L Ponatinib and 5 µmol/L actD for the indicated time, RT-qPCR was used to analyze the JUN mRNA expression. (H) LM2 cell were transfected with c-Jun or empty vector for 48 h, then treated with DMSO or 1 µmol/L Ponatinib for 24 h, Western blot analysis of the protein level of endogenous or transfected c-Jun. (I) Schematic representation of the location of the c-Jun DNA binding site (AP1) in the TNC promoter. (J and K) LM2 cells were transfected by luciferase reporter vectors driven by TNC wild-type or mutant promoters and JUN overexpression or control vectors. Next, luciferase activity was determined (J); or transfected cells were treated with DMSO or 1 µmol/L Ponatinib for 24 h, then luciferase activity was examined (K). (L) Suppression of AP1 site-Luc activity by Ponatinib in c-Jun-overexpressing LM2 cells. (M and N) MDA-MB-231 cells were transfected with c-Jun or empty vector for 48 h, and treated with DMSO or 500 nmol/L Ponatinib. Cells were scratched and the wound width was photographed. Representative images of one of three independent experiments are shown (M) and quantified (N) among the three groups at 36 h after Ponatinib treatment. Data represent mean ± SD of three independent experiments in (C–H, J–L and N). Two-tailed Student’s t-test were performed in (C–E, J–L and N), ***P < 0.001, **P < 0.01, *P < 0.05
The repression of Ponatinib treatment on the expression of JUN was confirmed by RT-qPCR analysis on mRNA level and Western blot analysis on protein level. As expected, the inhibition of ERK by compound GDC-0994 on the expression of JUN shows the similar trend (Fig. 5E and 5F). To determine how Ponatinib regulates the expression of JUN, we treated LM2 cells with actinomycin D (actD) as well as Ponatinib. ActD is an antibiotic that inhibits DNA-primed RNA polymerase and is widely used as a transcription inhibitor. Our data indicated that Ponatinib significantly inhibited the transcription of JUN (P < 0.05) but had no effect on the stability of JUN mRNA (Fig. 5G). Western blot results showed that Ponatinib treatment accelerated the degradation of c-Jun protein, regardless of whether it was translated from the endogenous or transfected JUN gene (Fig. 5H). These results suggested that Ponatinib inhibits the expression of JUN in mRNA and protein levels.
To assess whether c-Jun directly binds to and regulates the expression of these 4 genes, we chose the TNC promoter as an example for a reporter assay. c-Jun regulates the responsive promoter via binding to its canonical TGAG/CTCA motif (AP1) (Angel et al., 1987). We identified one potential c-Jun binding site at −883 to −877 bp upstream of the TNC transcription start site (TSS) (Fig. 5I). To determine whether the putative c-Jun binding site is involved in TNC transcription, we constructed reporter vectors with the DNA sequence of the TNC promoter containing a wild-type (TNC-wt) or mutated (TNC-mut) putative c-Jun binding site (Fig. 5I). These constructs were co-transfected into LM2 cells with or without a JUN expression plasmid. JUN overexpression activated luciferase gene expression, which was driven by the TNC-wt promoter but not the TNC-mut promoter (P < 0.05 and P > 0.05, respectively) (Fig. 5J). Notably, the luciferase gene expression drove by TNC-wt promoter and c-Jun overexpression was diminished significantly by Ponatinib treatment (P < 0.001) (Fig. 5K).
To determine whether Ponatinib treatment inhibits the expression of TNC only or c-Jun target genes in general, we transfected LM2 cells with a reporter vector containing 3 × AP1 sites combined with the luciferase gene with or without a JUN expression plasmid. The overexpression of JUN significantly activated luciferase gene expression (P < 0.001), and this activation was significantly inhibited by Ponatinib treatment (P = 0.011) (Fig. 5L). These results clearly indicated that Ponatinib treatment can effectively inhibit the expression of c-Jun target genes in general.
Together, our findings suggested that Ponatinib treatment represses the expression of c-Jun through inhibiting its transcription as well as accelerating c-Jun protein degradation, which leads to less binding of c-Jun to the promoters of BCLM-associated genes and consequently inhibits the transcription of these genes.
c-Jun rescues the migration of breast cancer cells treated with Ponatinib
To examine whether c-Jun is functionally involved in the migration of breast cancer cells, a wound healing assay was performed. The migration of LM2 cells was significantly inhibited by knocking down JUN expression (Fig. S7A and S7B). This result was consistent with a previous report that c-Jun is associated with cell movement in other breast cancer cell lines (Chen et al., 2009). In addition, overexpressed JUN partially rescued the migration of breast cancer cells treated with Ponatinib (P < 0.001), which indicated that c-Jun might be a major mediator of Ponatinib’s inhibitory effect on cancer cell migration (Figs. 5M, 5N, S6C and S8A). This result was also confirmed by an alternative method Transwell assay, which is also popular for testing cell migration (P < 0.01) (Fig. S8B and S8C)
The expression of JUN is positively correlated with the expression of four BCLM-associated genes in breast cancer tumors
To evaluate whether the regulation of c-Jun on these 4 BCLM-associated genes also occurs in breast cancer patients, we analyzed the transcriptome of 279 breast tumors from a previous publication (GSE41998) (Horak et al., 2013) and found that the expression of JUN was significantly and positively correlated with the expression of the 4 genes (Fig. 6A, upper panel). Notably, the expression of these 4 genes are also positively correlated with the expression of JUN in another dataset, and the correlation from 3 of these 4 genes is significant (P < 0.05), except MMP1 (P < 0.300) (GSE2603) (Minn et al., 2005) (Fig. 6A, lower panel).
Figure 6.
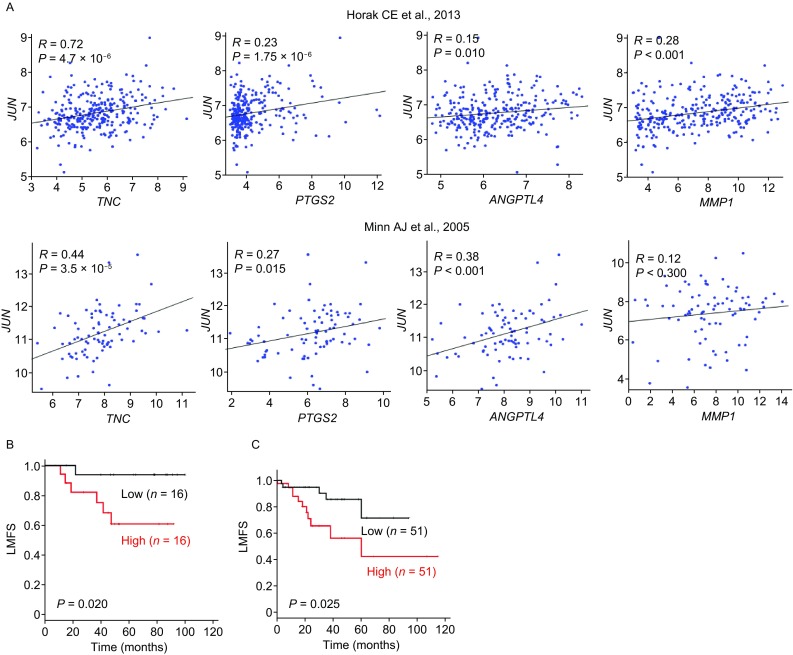
Increased expression of JUN is correlated with BCLM-associated gene expression and BCLM. (A) Positive correlation between the expression of c-Jun and 4 BCLM-associated genes from 2 independently published datasets GSE41998 (upper panel) and GSE2603 (lower panel). Each spot represents a tumor sample. (B and C) Kaplan-Meier analysis of the probability of cumulative lung metastasis-free survival (LMFS) in 82 and 204 breast cancer cases GSE2603 (B) and GSE12276 (C) by low and high JUN expression. P values were calculated by log-rank test
Increased expression of JUN is associated with the incidence of BCLM
To determine whether JUN expression is associated with BCLM we analyzed the correlation of JUN expression in primary breast tumors with patient prognosis. We examined the association between JUN mRNA levels and organ-specific metastasis within 82 tumor samples based on a published dataset (GSE2603) (Minn et al., 2005). Compared with patients with low expression of JUN, patients with high expression of JUN exhibited significantly high chance of lung metastasis (P = 0.02) (Fig. 6B). Furthermore, this result could also be confirmed in other 204 tumor samples from another published dataset (GSE12276) (Bos et al., 2009) (P = 0.025) (Fig. 6C). Together, our findings from clinical samples demonstrated a positive association between the expression of JUN and lung metastasis in patients with breast cancer, and suggested that c-Jun play an important role in BCLM.
Discussion
Approximately 90% of cancer patients die from metastasis (Weigelt et al., 2005), the discovery of anti-metastatic drugs is extremely crucial for the therapeutic treatment of patients with late stage cancers. In this study, to address this critical problem, we used a novel approach by combining HTS2 and metastasis-associated genes for high-throughput screening of anti-metastatic drugs. Notably, the expression of metastasis-associated genes was utilized as a readout of drug screening by HTS2. Using this new approach, we determined that Ponatinib, which can reverse the expression of 6 BCLM-associated genes, inhibits the lung metastasis of breast cancer. Mechanistically, this anti-metastatic function of Ponatinib is mainly acted through inhibition of the expression of transcription factor c-Jun but not ABL (Fig. 7).
Figure 7.
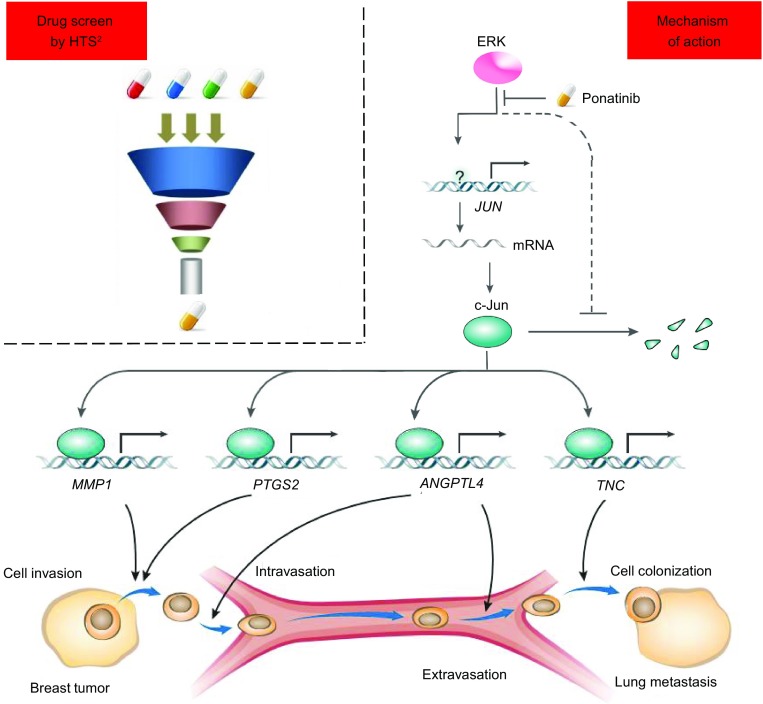
Proposed model for the role of inhibition of BCLM by Ponatinib. By using gene signature based drug screen strategy, a series of potential anti-breast cancer lung metastasis (BCLM) drugs were found. Among these drugs, Ponatinib inhibits c-Jun function by repressing mRNA transcription and accelerating degradation of c-Jun protein via ERK/c-Jun signal pathway. Through c-Jun, Ponatinib down regulates 4 BCLM related gene expression, finally inhibits BCLM in mouse models
To date, the cell phenotype-based screening is a popular approach for drug discovery and contributes significantly for the development of FDA-approved drugs. However, cell phenotype-based screening has drawbacks, include: 1) the requirement of engineering a separate reporter cell line, 2) most screens depend on a single or a limited number of surrogate readouts, 3) toxic components are hard to filter out, 4) many diseases lack good “drug” targets for developing an effective screening strategy. Gene signature based screen could overcome these drawbacks effectively. 1) Because detection of gene expression, engineering cell model is unnecessary. 2) Through analyzing the housekeeping and cytotoxicity-related gene perturbations by drugs, the toxic compounds could be filtered at the early stages of screening. 3) In addition, known drug targets are not needed for drug development. 4) Importantly, HTS2 takes advantage of powerful next-generation sequencing technologies and greatly enhances the parallel processing of samples and genes. These advantages help gene signature based approach to be complementary to cell phenotype-oriented screening in drug discovery.
There have been a number of applications of this approach with a particular focus on drug discovery, such as identifying new therapeutics (Lee et al., 2016), revealing mode of actions for existing chemicals (Saito et al., 2009). For anti-metastatic drug discovery, more works are needed, such as setting up unbiased gene expression signature from publications by other lab (Landemaine et al., 2008; Harrell et al., 2012), or using this approach to other types of organ specific cancer metastasis.
Ponatinib is developed to treat chronic myeloid leukemia (CML) and Philadelphia- chromosome positive (Ph+) acute lymphoblastic leukemia (ALL) with Bcr-Abl T315I mutation. The present findings proved that Ponatinib could prevent and inhibit breast cancer lung metastasis in mouse models, may provide a possibility of new perspectives for this drug. More work is needed to identify the direct target(s) of Ponatinib, which could regulate the expression of c-Jun in breast cancer cells.
In the present study, we demonstrated that c-Jun directly binds to and activates at least 4 BCLM-associated genes, including ANGPTL4, MMP1, PTGS2 and TNC. These 4 genes contribute to multiple steps of the metastatic process, such as endothelial tight junction (Le Jan et al., 2003), cell migration and invasion (Birkedal-Hansen et al., 1993; Howe et al., 2005), cell differentiation and colonization (Oskarsson et al., 2011). Loss of the function of any of these 4 genes could significantly impair breast cancer metastasis in vitro and in vivo (Gupta et al., 2007). Since c-Jun can activate the expression of all of these 4 genes, which strongly suggests that c-Jun might be a key factor in the process of breast cancer cells metastasizing to the lung and a promising drug target for anti-metastasis.
In summary, in this study, we established a novel approach for the discovery of anti-metastasis drugs, identified Ponatinib as a new and effective drug to inhibit breast cancer lung metastasis in mouse models and revealed c-Jun as a crucial factor and potential drug target for BCLM. Our current studies may pave the way to drug discovery and therapeutic treatment for metastasis, the leading cause of cancer patient mortality.
Materials and Methods
Chemicals
The screening library (1,154 FDA approved drugs), GDC-0994 (S7554), Crenolanib (CP-868596), MGCD-265 (S1361), LY2874455 (S7057), PP2 (S7008), GDC-0994 (S7554) and Ponatinib (S1490) were purchased from Selleck. SP600125 (HY-12041), SB203580 (HY-10256) were purchased from MedChem Express. Actinomycin D (A4262), B27 (17504044) and EGF (100-47) were purchased separately from Sigma, Invitrogen and Peprotech. All chemicals were dissolved in dimethyl sulfoxide (DMSO) for in vitro studies.
Antibodies and plasmids
c-Jun antibody (#9165) and GAPDH antibody (#2118) were purchased from Cell Signaling, anti-Flag antibody (F1804) was purchased from Sigma-Aldrich. For gene knockdown, the shRNA sequence for scrambled shRNA is GGTGTATGGGCTACTATAGAA, others plasmids were purchased from sigma including ABL1 (TRCN0000039898), ABL2 (TRCN0000218815) and JUN (TRCN0000039590 and TRCN0000039591). To construct the human JUN ectopic expression plasmid, pcDNA3.1-JUN, a full-length human JUN cDNA was cloned from LM2 cDNA. pGL3-AP1-Luc containing 3× c-Jun binding motif (AP1) was purchased from addgene. The TNC promoter sequence (−1054 to +246 bp related to TSS) was cloned from DNA of LM2 cells, and inserted into the pGL3-vector. To generate the TNC-mutation promoter, multiple site-directed mutagenesis was used to introduce mutations into wild-type TNC promoter.
Cell culture
MDA-MB-231 and 293T cells were obtained from China Infrastructure of Cell Line Resources (Beijing, China), LM2 (Luc+) cells and 4T1 (Luc+) cells were from Dr. Joan Massague (Memorial Sloan Kettering Institute, NY). MDA-MB-231 and 4T1 (Luc+) cells were cultured in RPMI1640 medium (Gibco) containing 10% fetal bovine serum (Gemini) and 100 units/mL streptomycin and penicillin (Gibco), LM2 and 293T cells were maintained in DMEM medium (Gibco) supplemented with 10% FBS and 100 units/mL PS. All the mentioned cell lines were incubated in a 5% CO2 atmosphere at 37 °C. All cell lines were authenticated by using PCR for short tandem repeats and verified to be free of Mycoplasma.
Drug screening
About 3,000 MDA-MB-231 cells were seeded per well in 384-well plates. At 24 h, cells were treated with the small molecules from screening library at 1 µmol/L drugs for 24 h. HTS2 assay was performed to quantify the mRNA level of the target gene as described (Li et al., 2012a). The cells were lysed in GentLys buffer (Nanopure, China).
Screening data processing
Reads were mapped to the probe sequences allowed 3 mismatches, and were normalized by using the expression of 33 stable genes in breast cancer (GSE42568, GSE10797). For each compound treatment, fold-change of gene expression was computed with the normalized signal of drug treatment and averaged normalized signal of DMSO treatment in the same 384-well plate. Genes with fold change >2 and P < 0.05 were considered differently expressed genes (DEGs). The method of calculating the anti-metastatic activity score of each compound was similar with described previously (Subramanian et al., 2005).
Real-time quantitative PCR
Total RNA was extracted by using TRIzol reagent (Invitrogen). Full-length cDNA was synthesized by using the RevertAid first strand cDNA synthesis kit (Thermo). Real-time quantitative PCR were performed by using the KAPA SYBR FAST qPCR Kit (Kapa Biosystems). The gene specific primers are in Table S1.
Transwell migration assay
The assay was based on the published protocol (Li et al., 2016). Briefly, MDA-MB-231 cells or LM2 cells were seeded in the upper chamber of the Transwell insert (Corning) with serum-free medium. 1 µmol/L Ponatinib were added into the chamber. After treatment for 8 h, migrated cells on the lower membrane were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet and counted by using Image J. For rescue experiment, LM2 cells were transfected with c-Jun or empty vector for 48 h, and treated with DMSO or 500 nmol/L Ponatinib, and then the transwell migration assay was performed.
Wound healing assay
The assay was based on the published protocol (Thakur et al., 2015). Briefly, MDA-MB-231 cells or LM2 cells at 80%–90% confluence were seeded in a 96-well plate. The wound was made through the cell layer by using a pin tool. After washing with phosphate buffered saline (PBS), complete media was added with or without 1 µmol/L Ponatinib. The wound width was photographed under a light microscope. The kinetic curve of wound width was used to indicate the cell motility potential. For rescue experiment, MDA-MB-231 cells were transfected with c-Jun or empty vector for 48 h, and treated with DMSO or 500 nmol/L Ponatinib, and then the wound healing assay was performed.
Mammosphere formation assay
The assay was based on the published protocol (Thakur et al., 2015). 1,000 cells (MDA-MB-231, LM2) were seeded in ultra-low attachment microplates (Corning). Cells were grown in a serum-free medium supplemented with 2% B27 (Invitrogen), 20 ng/mL EGF (Peprotech), 1% PS and 1 µmol/L Ponatinib. Media was changed every 3 day for 2 weeks and mammospheres were counted.
Animal studies
BALB/c-nude female mice 5–6 weeks old were used for xenograft experiments. Experimental metastasis assay was performed as described previously (Minn et al., 2005; Padua et al., 2008). Briefly, 2 × 105 LM2 cells were washed and harvested in PBS and then injected into the lateral tail vein of mouse in a volume of 0.1 mL (day 0). Lung metastasis was detected by bioluminescence imaging (BLI) with an IVIS Lumina II instrument (PerkinElmer). Mice underwent oral gavage with Ponatinib (30 mg/kg/day) or vehicle (25 mmol/L citrate buffer, pH 2.75) (O’Hare et al., 2009). For orthotopic metastasis assay, 1 × 106 4T1 cells were injected in 50 µL Matrigel (Corning) and diluted with PBS (1:1), then the cells were injected into the mammary fat pad of BALB/c female mice. The mammary tumor growth was regular measurement using a digital caliper. When the tumor volume reached 200 mm3, tumors were resected and mice were treated once daily by oral gavage with vehicle (25 mmol/L citrate buffer, pH 2.75), or Ponatinib (30 mg/kg/day) for up to 16 consecutive days. At 37 days, mice were sacrificed. The lungs were fixed in 4% paraformaldehyde and statistics for the metastatic colony. For the primary tumor growth model, BALB/c-nude female mice were orthotopically injected with 1 × 106 LM2 cells into the mammary fat pad. Tumors formed by LM2 cells were measured with digital calipers and tumor volume was calculated with the formula: volume (mm3) = [width2 (mm2) × length (mm)]/2.
RNA-sequencing (RNA-seq) analysis
LM2 cells at 3 × 106 were plated in a 10-cm petri dish for 24 h then treated with Ponatinib at 1 µmol/L for 6 h. Cells were harvested and RNA was isolated by using TRIzon (Invitrogen). The libraries were constructed by using the NEBNext Ultra™ RNA Library Prep Kit for Illumina (NEB) and sequenced on HiSeq 2500 sequencing system (Illumina). RNA-seq data were mapped to the reference genome (hg38) by using Bowtie/TopHat. The reads were counted, and the differential expression between experimental groups was quantified by using edgeR (Fold change > 2 and P value < 0.05).
Gene ontology (GO) and gene-set enrichment analysis (GSEA) analysis
GO analysis was performed in DAVID (https://david.ncifcrf.gov/) with 321 DEGs (fold change > 27 and P value < 0.05) by using the KEGG database. For GSEA, we used a ranked gene list by decreasing fold-change expression of genes. The gene signatures of the MAPK, ERK, p38 and JNK pathway were from the BioCarta pathway and canonical pathway database. Genes were aligned to the ranked list and the running sum was calculated and lead to an enrichment score. The P values were calculated on the basis of random permutations.
Viral transduction
For gene knockdown, a lentiviral vector containing shRNA together with psPAX2 packaging and the pMD2.G envelope plasmid were co-transfected into 293T cells at the ratio of 4:3:1 (PLKO.1:psPAX2:pMD2.G) by using DNAFect Transfection Reagent (CW Biotech, Beijing, China). At 48 h, culture media were harvested, filtered and stored at −80 °C. Viral particles were incubated with cells for 48 h, and surviving cells were harvested after selection with puromycin.
Transient transfection
For gene-overexpression experiments, 2 × 105 cells were transfected with pcDNA3.1 (vector) or pcDNA3.1-JUN by using DNAFect Transfection Reagent. At 24 h after transfection, cells underwent RT-qPCR and Western blot to verify transfection efficiency.
Western blot
Cells were harvested by RIPA lysis buffer containing 1 mmol/L PMSF and 1× cocktail (Biotool, Beijing). By BCA protein quantification (Pierce), protein samples were separated by SDA-PAGE gel and transferred to nitrocellulose membranes (Amersham Pharmacia), and probed with the indicated antibodies. The bands were visualized by using Pierce ECL Western blot substrate (Amersham). GAPDH was an internal control.
Actinomycin D assay
Actinomycin D (actD) assay was performed as described previously (Xiu et al., 2017). Briefly, cells were treated with 1 µmol/L Ponatinib and actD at 5 µmol/L for 0, 0.5, 1 or 2 h prior to RNA extraction with TRIzol reagent. Subsequently, RT-qPCR was used to analyze changes in RNA levels.
Luciferase reporter assay
Luciferase assay was performed as described previously (Wu et al., 2010). Briefly, LM2 cells were seeded in 24-well plates in triplicate 1 day before transfection. An amount of 750 ng reporter vector of the pGL3-wt/mut-TNC promoter or empty vector, 150 ng of the internal control plasmid pRL-TK and 750 ng plasmid for the expression of pcDNA3.1-JUN or empty vector was co-transfected as indicated per well. At 24 h after transfection, reporter activity of samples was assayed by using the Dual Luciferase Assay System (Promega). To assess whether the Ponatinib worked through c-Jun regulated expression of TNC, transfected samples were treated with DMSO or Ponatinib for 24 h, then reporter activity was detected by the same method. The pGL3-AP1-Luc reporter assay was performed similar with TNC reporter assay, LM2 cells were co-transfected with pGL3-AP1-Luc or empty vector, pRL-TK and pcDNA3.1-JUN or empty vector for 48 h, and treated with DMSO or Ponatinib for 24 h, and then dual luciferase assay was performed.
Correlation analysis of JUN with four BCLM related genes
For the correlation analysis of JUN with BCLM-related genes including ANGPTL4, MMP1, PTGS2 and TNC in breast cancer samples, datasets GSE41998 (n = 279) and GSE2603 (n = 82) were used. Pearson correlation coefficient was used for correlation analysis.
Clinical data analysis
For survival analysis of the relationship of JUN expression with the breast cancer metastasis organ, the levels of JUN and clinical metastasis data were acquired from the datasets GSE2603 (n = 82) and GSE12276 (n = 204) and the original reports. The ranked expression levels were classified into the top 20/25%, bottom 20/25% and middle 60/50%, respectively, then Kaplan-Meier survival curves were drawn and log-rank test P values were calculated by using the R package “survival”.
Statistical analysis
Comparisons of two groups involved two-tailed unpaired Student’s t-test, log-rank test was used for Kaplan-Meier survival analysis, Pearson’s product-moment correlation test was used for gene correlation analysis, and one-tailed Mann-Whitney test was used for wound healing test. The level of significance was set at *P < 0.05, **P < 0.01, ***P < 0.001.
Data availability
RNA-seq data have been deposited in the gene expression omnibus (GEO) database under access number GSE95079. All relevant data are available from the authors on request.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (PPTX 1,860 kb)
Acknowledgements
We thank Dr. J. Massague (Memorial Sloan Kettering Institute, NY) for providing LM2 cells; Dr. Xuerui Yang and Yusheng Wu (Tsinghua University, China) for help with wound healing assay; Qian Gao for help with HiSeq 2500 operation; Libing Mu for drawing the schematic diagram; Ting Yao (Institute of Biophysics, Chinese Academy of Sciences, Beijing) and Ming Yang for careful reading and editing of the manuscript. This work was supported by a grant from the National Natural Science Foundation of China (Grant No. 81673460) and funding from Tsinghua-Peking Joint Center for Life Sciences and Beijing Municipal Science & Technology Commission.
Abbreviations
ANGPTL4, angiopoietin-like 4; BCLM, breast cancer lung metastasis; ERK, extracellular signal regulated kinase; GO, gene ontology; GSEA, gene set enrichment analysis; HTS2, high-throughput sequencing-based high-throughput screening; JUN, Jun proto-oncogene; JNK, c-Jun N-terminal kinase; KEGG, kyoto encyclopedia of genes and genomes; LMSF, lung metastasis-free survival; LY6E, lymphocyte antigen 6e; MMP1, matrix metalloproteinase-1; PTGS2, prostaglandin-endoperoxide synthase 2; RARRES3, retinoic acid receptor responder protein 3; TNBC, triple negative breast cancer; TNC, tenascin C
Compliance with Ethics Guidelines
Wei Shao, Shasha Li, Lu Li, Kequan Lin, Xinhong Liu, Haiyan Wang, Huili Wang and Dong Wang declare that they have no conflict of interest. All animal studies were approved by the Medical Experimental Animal Care Commission of Tsinghua University. National guidelines for the care and use of laboratory animals were followed.
References
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, Hess K, Lecocke M, Metivier J, Booser D, Ibrahim N, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–2293. doi: 10.1200/JCO.2004.05.166. [DOI] [PubMed] [Google Scholar]
- Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, Eneman J, Crocker A, White J, Tessitore J, et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat. 2009;114:47–62. doi: 10.1007/s10549-008-9982-8. [DOI] [PubMed] [Google Scholar]
- Chambers J, Angulo A, Amaratunga D, Guo H, Jiang Y, Wan JS, Bittner A, Frueh K, Jackson MR, Peterson PA, et al. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J Virol. 1999;73:5757–5766. doi: 10.1128/jvi.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK, Liu K, Huang S. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69:9228–9235. doi: 10.1158/0008-5472.CAN-09-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke C, Madden SF, Doolan P, Aherne ST, Joyce H, O’Driscoll L, Gallagher WM, Hennessy BT, Moriarty M, Crown J, et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013;34:2300–2308. doi: 10.1093/carcin/bgt208. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massague J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, Anders C, Ewend M, Perou CM. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132:523–535. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak CE, Pusztai L, Xing G, Trifan OC, Saura C, Tseng LM, Chan S, Welcher R, Liu D. Biomarker analysis of neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or Paclitaxel in early-stage breast cancer. Clin Cancer Res. 2013;19:1587–1595. doi: 10.1158/1078-0432.CCR-12-1359. [DOI] [PubMed] [Google Scholar]
- Howe LR, Chang SH, Tolle KC, Dillon R, Young LJ, Cardiff RD, Newman RA, Yang P, Thaler HT, Muller WJ, et al. HER2/neu-induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase-2 knockout mice. Cancer Res. 2005;65:10113–10119. doi: 10.1158/0008-5472.CAN-05-1524. [DOI] [PubMed] [Google Scholar]
- Iorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, Ferriero R, Murino L, Tagliaferri R, Brunetti-Pierri N, Isacchi A, et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci USA. 2010;107:14621–14626. doi: 10.1073/pnas.1000138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/S1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Landemaine T, Jackson A, Bellahcene A, Rucci N, Sin S, Abad BM, Sierra A, Boudinet A, Guinebretiere JM, Ricevuto E, et al. A six-gene signature predicting breast cancer lung metastasis. Cancer Res. 2008;68:6092–6099. doi: 10.1158/0008-5472.CAN-08-0436. [DOI] [PubMed] [Google Scholar]
- Le Jan S, Amy C, Cazes A, Monnot C, Lamande N, Favier J, Philippe J, Sibony M, Gasc JM, Corvol P, et al. Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol. 2003;162:1521–1528. doi: 10.1016/S0002-9440(10)64285-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YT. Breast carcinoma: pattern of metastasis at autopsy. J Surg Oncol. 1983;23:175–180. doi: 10.1002/jso.2930230311. [DOI] [PubMed] [Google Scholar]
- Lee J, Liu J, Feng X, Salazar Hernandez MA, Mucka P, Ibi D, Choi JW, Ozcan U. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat Med. 2016;22:1023–1032. doi: 10.1038/nm.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Qiu J, Fu XD. RASL-seq for massively parallel and quantitative analysis of gene expression. Curr Protoc Mol Biol. 2012 doi: 10.1002/0471142727.mb0413s98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhou H, Wang D, Qiu J, Zhou Y, Li X, Rosenfeld MG, Ding S, Fu XD. Versatile pathway-centric approach based on high-throughput sequencing to anticancer drug discovery. Proc Natl Acad Sci USA. 2012;109:4609–4614. doi: 10.1073/pnas.1200305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wang Y, Xiao H, Li Y, Kan X, Wang X, Zhang G, Wang Z, Yang Q, Chen X, et al. Chamaejasmenin B, a novel candidate, inhibits breast tumor metastasis by rebalancing TGF-beta paradox. Oncotarget. 2016;7:48180–48192. doi: 10.18632/oncotarget.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bergami P, Lau E, Ronai Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer. 2010;10:65–76. doi: 10.1038/nrc2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Lu X, Kang Y. Organ-specific enhancement of metastasis by spontaneous ploidy duplication and cell size enlargement. Cell Res. 2010;20:1012–1022. doi: 10.1038/cr.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4:a011254. doi: 10.1101/cshperspect.a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massague J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Quiet CA, Ferguson DJ, Weichselbaum RR, Hellman S. Natural history of node-negative breast cancer: a study of 826 patients with long-term follow-up. J Clin Oncol. 1995;13:1144–1151. doi: 10.1200/JCO.1995.13.5.1144. [DOI] [PubMed] [Google Scholar]
- Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM, et al. ENCODE whole-genome data in the UCSC genome browser (2011 update) Nucleic Acids Res. 2011;39:D871–875. doi: 10.1093/nar/gkq1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-Ya K, Tsuruo T, Tomida A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009;69:4225–4234. doi: 10.1158/0008-5472.CAN-08-2689. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Stegmaier K, Ross KN, Colavito SA, O’Malley S, Stockwell BR, Golub TR. Gene expression-based high-throughput screening (GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36:257–263. doi: 10.1038/ng1305. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur R, Trivedi R, Rastogi N, Singh M, Mishra DP. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci Rep. 2015;5:10194. doi: 10.1038/srep10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang LH, Xiang J, Yan M, Zhang Y, Zhao Y, Yue CF, Xu J, Zheng FM, Chen JN, Kang Z, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 2010;70:9118–9128. doi: 10.1158/0008-5472.CAN-10-1246. [DOI] [PubMed] [Google Scholar]
- Wang J, Rouse C, Jasper JS, Pendergast AM. ABL kinases promote breast cancer osteolytic metastasis by modulating tumor-bone interactions through TAZ and STAT5 signaling. Sci Signal. 2016;9:ra12. doi: 10.1126/scisignal.aad3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Peterse JL, van’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhang Y, Zhang H, Yang X, Wang Y, Ren F, Liu H, Zhai Y, Jia B, Yu J, et al. p15RS attenuates Wnt/β-catenin signaling by disrupting β-catenin-TCF4 interaction. J Biol Chem. 2010;285:34621–34631. doi: 10.1074/jbc.M110.148791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu YL, Sun KX, Chen X, Chen S, Zhao Y, Guo QG, Zong ZH. Upregulation of the lncRNA Meg3 induces autophagy to inhibit tumorigenesis and progression of epithelial ovarian carcinoma by regulating activity of ATG3. Oncotarget. 2017;8:31714–31725. doi: 10.18632/oncotarget.15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1 (PPTX 1,860 kb)
Data Availability Statement
RNA-seq data have been deposited in the gene expression omnibus (GEO) database under access number GSE95079. All relevant data are available from the authors on request.