

Abstract
Neuroendocrine tumors (NETs) originate from the neuroendocrine cell system in the bronchial and gastrointestinal tract and can produce hormones leading to distinct clinical syndromes. Systemic treatment of patients with unresectable NETs aims to control symptoms related to hormonal overproduction and tumor growth. In the last decades prognosis has improved as a result of increased detection of early stage disease and the introduction of somatostatin analogs (SSAs) as well as several new therapeutic options. SSAs are the first-line medical treatment of NETs and can control hormonal production and tumor growth. The development of next-generation multireceptor targeted and radiolabelled somatostatin analogs, as well as target-directed therapies (as second-line treatment options) further improve progression-free survival in NET patients. To date, however, a significant prolongation of overall survival with systemic treatment in NET has not been convincingly demonstrated. Several new medical options and treatment combinations will become available in the upcoming years, and although preliminary results of preclinical and clinical trials are encouraging, large, preferrably randomized clinical studies are required to provide definitive evidence of their effect on survival and symptom control.
Key Points
| Therapeutic options for symptom control in functioning neuroendocrine tumors (NETs) are still limited, especially in progressive disease |
| Novel targeted systemic treatment options have become available for NET patients in the last years, but their effect on overall survival is still controversial. |
| Randomized clinical trials that compare, combine, and sequence current and potential novel therapies are pressingly required. |
Introduction
The endocrine system is composed of neuroendocrine cells dispersed throughout the entire body. These cells can be found in isolation or may form small aggregates [1]. Neuroendocrine tumors (NETs) originate from these cells [1, 2], and this heterogeneous group of neoplasms displays a wide range of biological behavior ranging from benign behavior with a life expectancy of decades to highly malignant clinical behavior with a very limited life expectancy [3]. Tumor heterogeneity is also reflected in the categorization of these tumors, which includes primary tumor localization (e.g., lung, pancreas, stomach, small bowel, colon), tumor grade, functional activity, disease stage, and susceptibility to drug treatment [4, 5]. NETs are histologically graded into well differentiated (grade 1, 2, or 3 NETs) or poorly differentiated (neuroendocrine carcinomas) tumors [5]. Locoregional or oligometastatic well-differentiated NETs are candidates for surgical resection with curative intent. However, a considerable subset of patients presents with irresectable or metastasized disease and consequently requires palliative systemic treatment.
According to the last National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) statistics, the incidence of NETs has increased substantially (1.09/100,000 persons in 1973 to 6.98/100,000 in 2012), especially for lung, small intestinal, and rectum NETs. In this context, the rate of patients with localized disease at the time of diagnosis has increased in the last years, which allowed for improved surgical resection rates and prognosis. However, survival has also improved for patients with advanced stages of gastroenteropancreatic-(GEP-)NETs, which is partially explained by the introduction of somatostatin analogs (SSAs) [6].
SSAs are the cornerstone of systemic treatment of well-differentiated, locally advanced or metastatic NETs [7–9]. SSAs act through binding to G-protein-coupled somatostatin receptors (SSTRs), which are broadly expressed in NETs and can modulate hormone secretion and tumor cell proliferation [10–12]. Although SSAs can induce sustained disease stabilization, resistance to treatment frequently occurs after prolonged use, even when dose-intensification has been pursued. Various alternative approaches can be considered here, e.g., the use of radiolabeled SSAs [13], the application of specific inhibitors of various tumor-cell signal transduction pathways and/or angiogenesis [14, 15], and the use of chemotherapy [16]. Additionally, new therapeutic options for functioning NETs have been developed [17, 18], and several novel treatments are currently under study [19, 20]. Considering the more recent advances in systemic treatment in NETs, an overview of the currently available treatment options for advanced functional as well as non-functional NETs is presented, as well as an overview of novel therapeutic options and clinical trials. We will not discuss systemic chemotherapy or neuroendocrine carcinomas.
Control of Hormonal Overproduction
NETs may cause specific clinical syndromes due to an overproduction of hormones and bioactive peptides. The incidence of functioning NETs ranges from 0.01–8.4 cases per 100,000 per year, depending on the secreted hormone. Carcinoid syndrome is the most frequent hormone-related syndrome within NETs (2–8.4 new cases/100,000/year) and is predominantly encountered in patients with metastasized midgut NETs [21, 22]. SSAs are considered to be the preferred first-line treatment option in functionally active NETs, including those associated with the carcinoid syndrome and functional pancreatic neuroendocrine tumors (PNETs) [11, 23]. The mechanisms of action of current medical options for functioning NETs are depicted in Fig. 1.
Fig. 1.
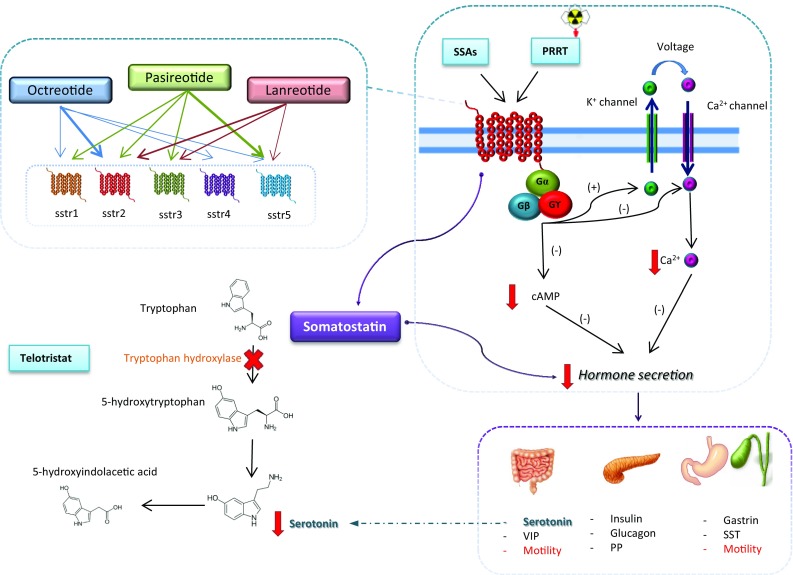
Current medical treatment for symptoms control in neuroendocrine tumors. Short- and long-acting and radiolabeled somatostatin analogs bind to G-protein linked receptors on the cell surface with variable affinity. Decreases in cAMP and intracellular calcium levels inhibit hormone release. Somatostatin influences hormone secretion and motility in the whole gastrointestinal tract. Serotonin production may also be decreased by telotristat, which inhibits the rate-limiting step in the serotonin secretion (the enzyme tryptophan hydroxylase). sstr somatostatin receptor, SSAs somatostatin analog, PRRT peptide receptor radionuclide therapy, cAMP cyclic adenosine monophosphate, VIP vasoactive intestinal peptide, PP pancreatic polypeptide, SST somatostatin
Carcinoid Syndrome (CS)
Carcinoid syndrome (CS) is mediated by several hormones, especially serotonin, and comprises several symptoms including flushing (94%), diarrhea (78%), carcinoid heart disease (CHD) (53%), and abdominal pain (51%) [24]. The role of SSAs on this often highly symptomatic secretory diarrhea and flushes was initially described in 1978 [25–27]. Since then, short-acting octreotide has been considered as a treatment option for carcinoid syndrome. The efficacy of short- and long-acting octreotide is similar once steady-state circulating concentrations of octreotide are achieved [27, 28]. Long-acting preparations of SSAs are widely used, probably due to their efficacy, but also to the comfortable administration every 28 days (in short-acting preparations, drug administration every 8 h may limit treatment compliance). SSAs improve flushes and diarrhea in 53–75% and 45–80% of cases, respectively [29, 30]. Long-acting octreotide and lanreotide similarly reduce urinary 5-hydroxyindoleacetic acid (u5-HIAA) and improve quality of life in NET patients [29]. Both octreotide and lanreotide are generally well tolerated, and side effects include gastrointestinal discomfort, headache, hyperglycemia, and the formation of bile stones [29].
Despite the effectiveness of SSAs, loss of response can occur after prolonged use. Tachyphylaxis, downregulation of cell surface SSTRs, and development of antibodies to SSAs have been hypothesized to underlie treatment resistance to SSAs [31–33]. In patients with refractory symptoms, intensified schedules consisting of either increased dose or increased frequency of administration have been found to control symptoms in about 40–81% of patients, without additional adverse events [34, 35]. Addition of short-acting octreotide (ranging from 100 to 500 µg every 8 h) is an alternative approach in case of treatment-refractory disease [36, 37]. In addition, favorable clinical response on carcinoid syndrome-related symptoms has also been reported in patients after treatment with peptide receptor radionuclide therapy (PRRT) [38, 39].
The use of serotonin-3-receptor antagonists (ondansetron) [40] and antidiarrheal drugs (loperamide) in combination with SSAs may improve episodes of diarrhea [41]. Pasireotide, a SSA with affinity to multiple SSTRs, has also been tested in patients with octreotide LAR-resistant tumors. Pasireotide showed efficacy in 33% of patients when administered at 150 µg twice daily, escalated to a maximum dose of 1200 µg per day [42]. However, a randomized phase III study of pasireotide LAR versus high-dose (40 mg) octreotide LAR for symptom control in patients with advanced GEP-NETs, whose disease-related symptoms were uncontrolled by first-generation SSAs at maximum approved doses, failed to show superiority of pasireotide LAR [43]. α-Interferon in combination with octreotide has been suggested as an effective treatment for symptom control, but unfortunately the use of this combination is limited due to the high rate of adverse effects attributed to α-interferon [44].
Recently, telotristate etiprate, a novel inhibitor of tryptophan hydroxylase, the rate-limiting enzyme in the biosynthesis of serotonin, has been developed [45]. This drug is able to suppress serotonin production in patients with NETs, as indicated by a decrease in u5-HIAA, with a concomitant partial improvement of diarrhea [17, 18, 46]. An international, multicenter, randomized, double-blind, placebo-controlled phase III trial (TELESTAR) reported a reduction of approximately 40% of bowel movements per day using telotristat etiprate doses of 750–1500 mg [45]. Furthermore, reductions of ≥ 30% in u5-HIAA levels have been reported with this drug [18, 47]. In this sense, a higher decrease in u5-HIAA has been associated with better improvement in bowel movements [18]. Recently, the phase III companion study TELECAST reported a statistically significant reduction in bowel movements in about 40% of patients, even in patients who were not on SSAs therapy. Unfortunately, the lack of patients in this last category (or that were assigned to the placebo group) limits conclusions in this respect [17]. Importantly, stool form improved in 20% and flushing in 27% of cases [18]. It seems therefore that a subset of patients with midgut NETs treated with SSAs may benefit from telotristat etiprate. Some adverse effects have been reported (including increase in transaminases and gastrointestinal symptoms, especially nausea) not resulting in treatment discontinuation [18, 45]. Some preclinical studies suggest that telotristat etiprate does not cross the blood-brain barrier ([Lexicon, unpublished observations in [18]). The TELESTAR study reported a higher incidence of depression-related events in patients receiving telotristat ethyl 500 mg three times per day, but not in those patients receiving 250 mg three times per day (similar incidence to placebo) [45]. This finding was not observed in the TELECAST study, however [17]. Finally, lowering circulating serotonin levels by telotristat etiprate may also be important to counteract the development of NET-associated cardiac and mesenterial fibrosis, in which serotonin is thought to play a major role [48, 49].
Carcinoid Heart Disease
Tumor-secreted vasoactive hormones may also produce the deposition of plaques on the endocardial surfaces of valve leaflets, the subvalvular area, and cardiac chambers, affecting especially the right side of the heart, and resulting in carcinoid heart disease (CHD) [50]. CHD affects about 50% of patients with CS [51]. SSAs and/or tumor-debulking procedures may improve the hemodynamic impact of tumor vasoactive agents on CHD [28, 52, 53], but there is no convincing evidence that these treatment options can stop the progression of CHD [54].
The initial treatment of decompensated CHD consists of loop diuretics, fluid and salt restriction, and compression stockings. However, in advanced stages these treatments may become ineffective [55]. In those cases, cardiac valve surgery or balloon valvuloplasty is recommended [50]. When surgery is required, continuous octreotide infusion (50–100 μg/h) should be administered 2 h before until 24 h after the surgical procedure in order to prevent carcinoid crisis and its complications [56]. Antihistaminic drugs and corticosteroids may also be used before surgery [55], whereas the use of drugs (opioids, dopamine, adrenaline/epinephrine) that precipitate the release of vasoactive products should be avoided [56, 57]. In addition, the effect of telotristat etiprate in CHD should also be evaluated.
Insulinoma
Most insulinomas are small, benign tumors that can be surgically cured. Unfortunately, the clinical management of patients with metastatic insulinomas may be difficult. Apart from oral or intravenous glucose administration, several medical options are available to treat hypoglycemia in the preoperative setting or in case of metastatic disease. Diazoxide is a well-known drug to treat hypoglycemia in patients with insulinomas. It is a benzothiadiazide derivative that inhibits insulin secretion via ATP-dependent potassium channels in pancreatic β-cells. It also increases hepatic glucose production and inhibits tissue glucose uptake, improving hypoglycemia in 50–60% of cases [58–62]. It should be initiated in low doses (150–200 mg/day) administered two or three times daily until a maximum of 600–800 mg. If after 2–3 weeks of treatment any clinical improvement is observed, diazoxide may be stopped [63, 64]. Adverse effects are observed in about the half of patients and include water retention, hirsutism, weight gain, nausea, emesis, diarrhea, abdominal pain, headache, and rash [58, 59, 65].
SSAs (octreotide LAR and lanreotide autogel) are preferably used in malignant insulinomas, where the activation of SSTR (if expressed) decreases insulin release [66]. Unfortunately if SSTR expression is low or absent, SSAs may paradoxically lower blood glucose levels by suppressing glucagon release [60]. Insulin secretion is also inhibited via the SSTR type 5 (SSTR5) [67]. Since the affinity of pasireotide for SSTR5 is 30- to 40-fold higher when compared with octreotide [68], pasireotide may be an alternative therapeutic option in malignant insulinomas. Some cases reports described an appropriate glucose control with pasireotide in insulinomas resistant to other treatment options including octreotide LAR, everolimus, or chemotherapy [69, 70].
Blood glucose normalization in patients with malignant insulinomas has also been reported after treatment with everolimus when previous treatment options (octreotide LAR, chemotherapy, PRRT, or radiofrequency ablation) failed to achieve appropriate glucose control [65, 71, 72]. Clinical effect may be achieved after 2 weeks of treatment with 10 mg everolimus per day [63]. Everolimus can probably decrease insulin release through the AMP-activated protein kinase (AMPK)/c-Jun N-terminal kinase (JNK)/FoxO pathway, and it may also induce peripheral insulin resistance by downregulation of glucose transporter 1 (GLUT1) via reduced transcription and translation [71, 73–75].
PRRT can also be a successful therapeutic alternative in metastatic insulinomas with SSTR expression [76]. In a small patient series PRRT induced stable disease for 18–50 months and no hypoglycemic episodes occurred during that period [76]. Clinical improvement despite the presence of the tumor suggested a sustained effect of PRRT on insulin secreting machinery [60].
Glucagonoma
This rare tumor originating from α-pancreatic cells may cause the glucagonoma syndrome, which is characterized by the presence of necrolytic migratory erythema (NME), diabetes mellitus, and weight loss; other symptoms include anemia, glossitis, steatorrhea, diarrhea, venous thrombosis, and neuropsychiatric disturbances [77]. If surgery is performed, resection of the tumor load of at least 70–90% is necessary for symptom control, which may be difficult in several cases. Clinical symptoms may improve with SSAs (octreotide and lanreotide) [77]. Specifically, the NME may improve despite the persistence of elevated serum glucagon levels [78]. Pasireotide has also been suggested as an appropriate therapeutic option in octreotide-resistant tumors [77, 79]. In addition, sunitinib and everolimus have been reported as successful treatment options in these functioning tumors and both have been associated with increased progression-free survival (PFS) when compared to placebo [80, 81]. PRRT with 90Yttrium-DOTATOC or 177Lu-DOTATATE may induce disease stabilization or regression with subsequent symptom control [82], but series focusing only on glucagonomas are still lacking.
Gastrinoma
Gastrinomas are sporadic in 75% of cases. In these tumors, surgery is curative only in about 60% [83]. Surgical resection is also controversial in patients with multiple endocrine neoplasia syndrome type I due to a low probability of cure [84]. Consequently, medical treatment is necessary in a considerable number of patients [85].
Gastric hypersecretion and related symptoms should be treated with high-dose H+-K+-ATPase proton-pump inhibitors (PPIs) [83]. Serum levels of vitamin B12 and iron should be monitored at least once a year [86, 87].
Since SSTRs are widely expressed in gastrinomas [88], SSAs (octreotide and lanreotide) effectively suppress gastrin secretion and decrease or normalize gastric acid secretion in over half (50–100%) of gastrinoma patients [86, 89–91]. Their use has been associated with tumor stabilization in 47–75% of patients [91, 92], and SSAs may be combined with chemotherapy in metastasized gastrinomas [93]. In addition, SSAs may prevent the enterochromaffin-like cell hyperplasia or the development of gastric type 2 NETs, which are related to hypergastrinemia [94]. IFN-α (5 × 106 IU/day) improved clinical symptoms caused by hypergastrinemia only in stabilized tumors. Unfortunately, side effects frequently require dose reduction or drug withdrawal [85]. Furthermore, PRRT with radiolabeled beta emitting SSAs can lead to long-lasting (> 1 year) tumor responses in a very high number of patients and also leads to symptomatic improvement [50, 95].
Vasoactive Intestinal Polypeptide (VIP)oma, Somatostatinoma
Functioning NETs that release vasoactive intestinal polypeptide (VIP) have an incidence of 1:10 million persons/year. These tumors produce watery diarrhea, accompanied by severe hypokalemia, and hypo- or achlorhydria. In contrast, somatostatinomas may be asymptomatic or present with diabetes mellitus, cholelithiasis, weight loss, steatorrhea, and diarrhea; their annual incidence is 1 in 40 million [96–98]. Initial treatment in VIPomas and symptomatic somatostatinomas is based on fluid loss replacement and electrolyte correction. Octreotide and lanreotide may control symptoms in the majority of patients [96, 99, 100], but in refractory cases glucocorticoids may be used as adjuvant therapy [96]. Equivalent to other pancreatic NETs, molecular targeted therapy and PRRT may be useful in metastasized cases, but due to their low incidence, subgroup analyses on the efficacy of these novel therapeutic alternatives in these NETs are not available [13, 80–82, 101, 102].
Ectopic Hormone Production Syndromes
Ectopic hormone production is rare in NETs. Among them, adrenocorticotropic hormone (ACTH)-producing tumors are most commonly observed. Ectopic release of other peptides includes corticotropin-releasing hormone (CRH), growth hormone-releasing hormone (GHRH), antidiuretic hormone, parathyroid hormone-related peptide (PTHrP), and gonadotropins [103–105]. The aim of treatment of these patients includes symptomatic long-term control, tumor stabilization or reduction, and prolongation of (progression-free) survival [104].
Adrenocorticotropic Hormone (ACTH)
The ectopic ACTH syndrome (EAS) causes approximately 10% of all cases of Cushing syndrome [106, 107]; clinical evolution is usually faster and characterized by mineralocorticoid effects (hypertension, hypokalemia, and edema), thromboembolic disease, and opportunistic infections [108]. Curative surgery is the primary treatment option but is often not possible [106, 109]. EAS can result in a critical condition for which aggressive medical therapy or life-saving bilateral adrenalectomy is necessary [108]. Medical treatment options for EAS include: (1) Tumor-directed drugs including somatostatin analogs (octreotide, pasireotide) and dopamine agonists that decrease tumoral ACTH secretion [108, 110–113]. The identification of SSTR expression in the tumor using radiolabeled somatostatin analogs may also help to identify suitable patients who might benefit from PRRT [95, 110, 114]. In addition, the tyrosine kinase inhibitors vandetanib and sorafenib may have antisecretory effects in selected cases with EAS [115, 116]. (2) Steroid synthesis inhibitors, which directly suppress adrenal cortisol production. In this sense, a combination of ketoconazole, metyrapone, and mitotane was shown to be effective in critically ill patients with EAS [117]. Additionally, the anesthetic drug etomidate can also rapidly suppress cortisol levels in an ICU setting [118]. (3) Glucocorticoid receptor antagonists. Mifepristone has a short onset of action and was shown to reverse morbidity of EAS in several cases [119].
Growth Hormone-Releasing Hormone (GHRH)
Ectopic GHRH production is predominantly encountered in patients with lung carcinoids [120]. Although some patients have evident clinical features of increased GH production, GHRH-staining may be positive in asymptomatic NET patients [121]. SSAs are widely used for the treatment of acromegaly due to GH-secreting pituitary adenomas, but medical treatment of acromegaly due to ectopic GHRH secretion is less well explored [122]. SSAs (octreotide and lanreotide) lower ectopic tumoral production of GHRH with a subsequent decrease in circulating GH and insulin-like growth factor-1 (IGF-1) levels [123, 124]. In some cases, tumor control and clinical improvement may be achieved, but circulating GHRH may continue to be elevated [125].
Parathyroid Hormone-Related Protein (PTHrp)
PTHrp production is predominantly associated with PNETs; overall survival in patients with PTHrp production is significantly shorter compared to pNET patients with normocalcemia [104, 126]. In PTHrp-producing NETs, hypercalcemia needs to be controlled. Treatment options for hypercalcemia include intravenous isotonic saline (corrects volume depletion), bisphosphonates (interfere with the osteoclast-mediated bone resorption), and denosumab (reduces the formation, function, and survival of osteoclasts via the receptor activator of nuclear factor κB (RANK) pathway) [104, 127, 128]. SSAs may help to improve symptom control but might be insufficient in patients with tumor progression [88, 104]. Tumor stabilization with parallel calcium control has been described in three of five patients after receiving PRRT with 177Lu-DOTATATE [104].
Control of Tumor Growth
According to the latest analysis of the SEER database, 27.4% of NETs have distant metastases at diagnosis and 20% have regional infiltration [6]. Survival in NETs is related to tumor localization, tumor load, and grading, and these factors should be considered when selecting the appropriate medical treatment. Current and promising novel medical options for tumor growth control in NETs are presented in Fig. 2. The landmark clinical trials proving efficacy of the current medical options for tumor growth control in NETs are depicted in Table 1. Additionally, some current clinical trials for tumor growth control in NETs are depicted in Table 2.
Fig. 2.
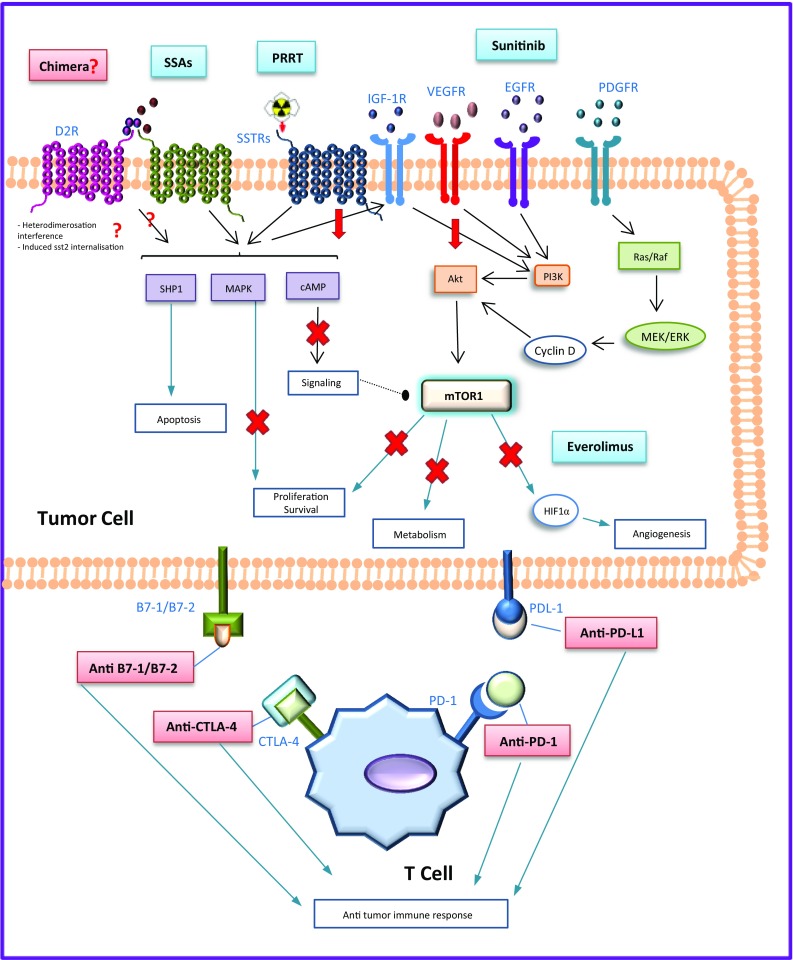
Current and future medical options for tumor control in neuroendocrine tumors. Current therapeutic options are presented in blue, possible novel therapeutic options are presented in red. SSAs and PRRT: increase apoptosis by activating the protein tyrosine phosphatase SHP1; decrease cell proliferation and survival through the mitogen-activated protein kinase (MAPK) and cyclic adenosine monophosphate (cAMP); and inhibit the signaling of the insulin-like growth factor receptor type 1 (IGFR-1); additionally, PRRT produces DNA double strand breaks induced by β-irradiation, consequently leading to apoptosis. Sunitinib is a multikinase inhibitor that modulates the phosphoinositate-3-kinase/Akt pathway (it blocks the vascular endothelial growth factor receptors (VEGFR) 1-3, the platelet-derived growth factor receptors (PDGFR) α and β, and the epidermal growth factor receptor (EGFR)). Everolimus decreases tumor cell proliferation, metabolism, survival, and angiogenesis through the mammalian target of rapamycin complex-1. The indirect inhibition of mTOR through the phosphoinositate-3-kinase/Akt produced by the SSAs seems to increase sensitivity to mTOR inhibition. Multi-receptor chimeras may bind SSTR and D2R, and may enhance the signaling of the cAMP and JNK pathways; induced SST2R internalization and SST2R/D2R heterodimerisation interference have also been hypothesized. The interaction between some receptors expressed on the surface of cytotoxic T-cells (PD-1, CTLA-4) with ligands expressed on the tumor cells (PDL-1, B7-1/B7-2) downregulates the immune response to tumor cells; novel drugs that target these specific immune checkpoints inhibit this interaction allowing the immune system to maximize an efficient antitumor response. SSAs somatostatin analogs, PRRT peptide receptor radionuclide therapy, IGF-1R insulin-growth factor receptor type 1, VEGFR vascular endothelial growth factor, EGFR epidermal growth factor receptor, PDGFR platelet-derived growth factor receptors, mTOR mammalian Target of Rapamycin, CTL4 cytotoxic T-lymphocyte antigen-4, PDL-1 Programmed death-ligand 1
Table 1.
Clinical trials supporting the current systemic strategies for tumor growth control in NETs
| Drug/study | Study characteristics | Outcome results/safety |
|---|---|---|
| Somatostatin analogs | ||
| CLARINET [134] | Randomized, double-blind, placebo-controlled, multicenter study. Advanced, well-/moderately differentiated, non-functioning, SSTR positive NETs (pancreas, midgut, hindgut, or of unknown origin). Placebo (n = 103) or 120 mg of lanreotide autogel once every 28 days (n = 101) for 96 weeks | PFS was significantly increased at 24 months in the lanreotide group 65.1% (95% CI 54.0–74.1) compared to 33.0% (95% CI 23.0–43.3) in the placebo group. Differences in quality of life were not statistically different between the two groups. The most common treatment-related adverse event was diarrhea (26% of the lanreotide group vs. 9% in the placebo group); 8 serious adverse events were considered to be related to the study drug (7 for lanreotide and 1 for placebo) |
| PROMID [132, 133] | Randomized, double-blind, placebo-controlled, multicenter study. Treatment-naive patients (well-differentiated midgut NET or unknown origin believed to be of midgut origin). Placebo (n = 43) or octreotide-LAR 30 mg intramuscularly monthly (n = 42) until tumor progression or death | PFS was increased in the octreotide group (14.3 months; 95% CI 11.0–28.8 months) compared to 6.0 months (95% CI 3.7–9.4 months) in the placebo group. Both groups had comparable levels of quality of life. The long-term tumor-related overall survival was similar in both groups. The most common treatment-related adverse events were gastrointestinal (6 patients in the octreotide-LAR group; 5 patients receiving placebo). Five patients treated with octreotide-LAR discontinued the treatment |
| Radionuclide therapy | ||
| NETTER-1[166] | Open-label, randomized, controlled, multicenter study. Well-differentiated, metastatic midgut NETs progressive on octreotide LAR 30 mg every 4 weeks received 7.4 GBq of 177 Lu-Dotatate every 8 weeks [(four intravenous infusions, plus best supportive care including octreotide LAR) (n = 116)] or octreotide LAR 60 mg every 4 weeks (n = 113) | PFS at month 20 was 65.2% (95% CI 50.0–76.8) in the 177Lu-Dotatate group vs. 10.8% (95% CI 3.5–23.0) in the control group. HR for death with 177 Lu-Dotatate group vs. control, 0.40; p = 0.004. OS data is pending. Seven patients receiving 177Lu-Dotatate and 10 in the control group stopped their participation in the trial due to adverse events. Nausea and vomiting were the most frequent adverse events in the 177 Lu-Dotatate group (59% and 47%, respectively). Grade 3–4 adverse events were similar in the two groups, but grade 3 or 4 neutropenia, thrombocytopenia, and lymphopenia were reported in 1%, 2%, and 9% in the 177 Lu-Dotatate group, respectively. No renal toxicity was reported |
| mTOR inhibitors | ||
| RADIANT-3 [81] | Open-label, randomized, placebo-controlled study. Advanced, progressive, low- or intermediate-grade PNETs. Everolimus 10 mg/day (n = 207) or placebo (n = 203) | PFS was 11.0 months in the everolimus group compared to 4.6 months in the placebo group (HR 0.35; 95% CI 0.27–0.45; p < 0.001). A non-significant OS benefit of 6.3 months was observed. Stomatitis was the most common adverse event (64% in the everolimus group vs. 17% in the placebo group). Infections and pneumonitis occurred in 12% of the patients in the everolimus group; grade 3 or 4 drug-related adverse events included anemia, hyperglycemia, stomatitis, thrombocytopenia, diarrhea, hypophosphatemia, and neutropenia. One death was related to the use of everolimus |
| RADIANT-4 [187] | Open-label, randomized, placebo-controlled study. Advanced, progressive, well-differentiated, non-functional lung, gastrointestinal NETs. Everolimus 10 mg/day (n = 205) or placebo (n = 97) | Everolimus was associated with a 52% reduction of the risk of progression or tumor-related death (HR 0.48; 95% CI 0.35–0.67; p < 0.00001). OS did not significantly improve in the everolimus group. Stomatitis was the most common adverse event (63% in the everolimus group vs. 19% in the placebo group). Grade 3 or 4 drug-related adverse events included anemia, stomatitis, diarrhea, fatigue, and infections. Non-infectious pneumonitis occurred in 16% of the everolimus group |
| Tyrosine kinase inhibitors | ||
| SUN 1111 [80, 195] | Multinational, randomized, double-blind, placebo-controlled study. Advanced, well-differentiated PNETs. 37.5 mg sunitinib (n = 86) or placebo (n = 85) | PFS was 11.4 months in the sunitinib group compared to 5.5 months in the placebo group (HR 0.42; 95% CI 0.26–0.66; p < 0.001). The objective response rate was higher in the sunitinib group (9.3% vs. 0%). OS non-significantly improved by 10 months. Dose-limiting toxicities included grade 3 fatigue, grade 3 hypertension, and grade 2 bullous skin toxicity, which were reversible |
SSTR somatostatin receptor, NETs neuroendocrine tumors, PFS progression-free survival, CI confidence interval, HR hazard ratio, PRRT peptide-receptor radionuclide therapy, OS overall survival, PNETs pancreatic neuroendocrine tumors
Table 2.
Registered clinical trials for tumor growth control medical therapies in NETs
| Drug | Study characteristics | Primary outcome/ClinicalTrials.gov Identifier |
|---|---|---|
| Somatostatin analogs | ||
| Lanreotide (CLARINET FORTE) | Open-label single-group clinical trial for evaluating the efficacy and safety of lanreotide 120 mg every 14 days in well-differentiated, metastatic or locally advanced, unresectable pancreatic or midgut NETs with radiological progression with lanreotide 120 mg every 28 days | PFS (102 weeks)/NCT02651987 |
| 177Lu-DOTA0-Tyr3-Octreotate (NETTER-1) | Multi-center, randomized, phase III study comparing 177Lu-DOTA0-Tyr3-Octreotate to Octreotide LAR in patients with inoperable, progressive, somatostatin receptor positive midgut carcinoid tumors | PFS, OS data is pending/NCT02651987 |
| Kinase inhibitors | ||
| Sulfatinib | Randomized, multicenter phase III study to evaluate the efficacy and safety of sulfatinib (angio-immunokinase inhibitor targeting VEGFR, FGFR1, and CSF-1R kinases) vs. placebo in advanced PNETs | PFS (7 months after the last patient enrolled)/NCT02589821 |
| Sulfatinib | Randomized, double-blind, multicenter phase III study to evaluate the efficacy and safety of sulfatinib vs. placebo in advanced PNETs | PFS (9 months after the last patient enrolled)/NCT02588170 |
| Radionuclide therapy | ||
| 177Lu-PRRT vs. 177Lu-PRRT plus capecitabine | Open-label phase II study to compare the efficacy of 177Lu-PRRT vs. 177Lu-PRRT plus capecitabine in SSTR and 18-FDG PET/CT positive, G1-G2-G3 GEP-NETs | PFS (72 months)/NCT02736448 |
| 177Lu-Octreotate -CAPTEM vs. (i) CAPTEM and (ii) 177Lu-Octreotate | Two parallel phase II randomized open-label trials of PRRT with 177Lu-Octreotate and CAPTEM (i) vs. CAPTEM alone in the treatment of low to intermediate grade PNETs (ii) vs. 177Lu-Octreotate alone in the treatment of low to intermediate grade midgut NETs | PFS (12 months in PNETs and 24 months in midgut NETs)/NCT02358356 |
| 177Lu-edotreotide vs. Everolimus | Prospective, randomized, controlled, open-label, multicenter phase III study to evaluate the efficacy and safety of PRRT with 177Lu-edotreotide compared to everolimus in GEP-NETs | PFS assessed up to 24 months/NCT03049189 |
| 177Lu-DOTA0-Tyr3-Octreotate vs. sunitinib | Open-label randomized phase II antitumor efficacy of PRRT with 177Lu-DOTA0-Tyr3-Octreotate vs. sunitinib in unresectable progressive well-differentiated PNETs | PFS (12 months)/NCT02230176 |
| mTOR inhibitors | ||
| Everolimus and LEE011 (Ribociclib) | Open-label study to evaluate the efficacy and safety of the combination LEE011 (inhibitor of cyclin D1/CDK4 and CDK6 pathway) 300 mg once daily for 3 weeks (4th week off) and everolimus 2.5 mg daily in foregut WDNETs | PFS (2 years)/NCT03070301 |
| Everolimus and TMZ | Open-label study to evaluate everolimus and temozolomide as first-line treatment in advanced NEC with a Ki67 of 20–55% | Disease control rate/NCT02248012 |
| Everolimus and bevacizumab | Randomized phase II study of everolimus alone vs. combined with bevacizumab in patients with PNETs (currently active, not recruiting) | PFS (up to 3 years)/NCT01229943 |
| Everolimus and cisplatinum | Open-label phase II study of cisplatinum and everolimus in metastatic or unresectable NEC of extrapulmonary origin | Disease control rate/NCT02695459 |
| Immunotherapy | ||
| Pembrolizumab | Open-label phase II study of monotherapy with pembrolizumab (humanized anti-PD-1 monoclonal antibody) in patients with metastatic high-grade NETs who have failed platinum-based chemotherapy | Objective response rate/NCT02939651 |
| PDR001 | Open-label phase II study to evaluate PDR001 (high-affinity, ligand-blocking, humanized IgG4 antibody directed against PD-1) in advanced or metastatic, well-differentiated, non-functional, thoracic and GEP-NETs or GEP-NECs | Overall response rate/NCT02955069 |
| Durvalumab and tremelimumab | Multicenter open-label phase II study to evaluate the combination therapy between durvalumab (MEDI4736; humanized antibody against PD-1) and tremelimumab (CTLA-4 inhibitor) in advanced/metastatic, grade 1/2 (G1/G2) lung and GEP-NETs, and grade 3 (G3) GEP- tumors or of unknown primary site after progression to previous therapies | Clinical benefit rate/NCT03095274 |
NETs neuroendocrine tumors, PRRT peptide receptor radionuclide therapy, CAPTEM Capecitabine/temozolomide, PFS progression-free survival, OS overall survival, VEGFR vascular endothelial growth factor, FGFR fibroblast growth factor receptor, CSF-1R colony-stimulating factor 1 receptor, PNETs pancreatic neuroendocrine tumors, PRRT peptide receptor radionuclide therapy, 18-FDG PET/CT18-fluorodeoxiglucose positron emission tomography–computed tomography, GEP-NET gastroenteropancreatic neuroendocrine tumors, CDK cyclin-dependent kinases, WDNETs well-differentiated neuroendocrine tumors, TMZ temozolomide, NEC neuroendocrine carcinoma, PDGFR platelet-derived growth factor receptor, PD1 programmed death-1, CTLA-4 cytotoxic T-lymphocyte-associated protein 4
Somatostatin Analogs
The antiproliferative effect of SSAs mostly depends on SSTR tumor expression, although indirect antitumor effects have been described as well [129]. SSAs may inhibit the cell cycle and increase apoptosis, and indirect effects may include immuno-modulation, antiangiogenic effects, and growth factor inhibition [9, 12, 100]. Octreotide and lanreotide bind preferably to SSTR2, and pasireotide has high binding affinity to multiple SSTR, particularly SSTR5 (Fig. 1) [130]. Long-acting preparations of octreotide and lanreotide monthly are usually used for disease stabilization in NETs [9, 131]. The antiproliferative effect of SSAs in NETs was initially evaluated in the PROMID study [132]. In this phase IIIB study, 85 well-differentiated metastatic midgut NETs were included. Patients were randomized to receive placebo or octreotide-LAR 30 mg every 4 weeks. A difference of 8.3 months in tumor progression was observed after comparing the octreotide and the placebo groups. Stable disease after 6 months was observed in 66.7% of patients treated with octreotide-LAR compared to 37.2% in the placebo group [132]. Despite the initial good response to octreotide LAR, the results from the long-term survival analysis revealed that overall survival (OS) was not significantly different in the placebo and the octreotide groups [133]. Interestingly, patients with resected primary tumor and/or lower liver tumor load benefitted more from the initial administration of octreotide [132, 133].
Similar to the PROMID study, the CLARINET study revealed that lanreotide (120 mg every 28 days) increased PFS of patients with metastatic well- and moderate-differentiated GEP NETs when compared to placebo (PFS rate of 65.1% in the lanreotide group and 33% in the placebo group) [134]. Usually SSAs induce tumor stabilization, but in selected cases SSAs can cause tumor shrinkage, possibly due to their effects on the perfusion of liver metastases [135]. Representative images of tumor shrinkage in response to lanreotide are depicted in Fig. 3a. According to the current clinical guidelines, SSAs are indicated in grade 1 (G1) non-functional NETs after progression without previous treatment (watch and wait strategy), in G1 NETs with high tumor load, or in non-functional grade 2 (G2) NETs with SSTR expression [23]. Because G1 and G2 NETs can initially show a stable disease course without treatment, markers are needed that can identify those patients in whom early treatment should be evaluated versus those patients in whom a wait-and-see strategy should be considered initially.
Fig. 3.

Peptide receptor radionuclide therapy in neuroendocrine tumors (NETs). a CT imaging of a pancreas neuroendocrine tumor grade 2 with lymphatic and liver metastasis (segment 6); in this case, four cycles of peptide receptor radionuclide therapy (cumulative dose 30 Gbq) was administered resulting in decreased size of the primary tumor (b). After 6 years of partial response and stable disease, the primary tumor increased in size accompanied by new liver and mesenteric metastasis (c). Because of an initial good treatment response, two cycles of PRRT (14.9 GBq) were administered, and a decreased size of primary tumor and liver metastasis were observed (d). Images are of an NET patient evaluated in the ENETS Center of Excellence Erasmus MC, Rotterdam. Informed consent was provided
In addition, increased doses of SSAs seem to exert an apoptotic effect, which is not achieved with the standard doses [136]. These effects have been described in patients showing disease progression under regularly dosed lanreotide and octreotide [35, 137]. Currently, the CLARINET FORTE study is evaluating the safety and antitumor effect of lanreotide 120 mg given every 14 days in patients with pancreatic or midgut NETs with progressive disease under the same dose used every 4 weeks for at least 6 months (NCT02651987).
Pasireotide has also been studied in NETs, in which pasireotide concentrations correlated with tumor shrinkage in a non-significant manner [138]. Other studies have reported predominantly disease stabilization (60%) in treatment-naïve patients with grade 1–2 NETs, but also partial response (4%) and disease progression (36%) have been described [139]. Additionally, pasireotide-LAR has been compared to octreotide-LAR in patients with metastatic NETs and carcinoid symptoms. In these patients, pasireotide tended to increase the tumor control rate after 6 months and was associated with a longer PFS [43]. In the prospective phase II LUNA study in advanced (unresectable or metastatic), progressive, well-differentiated carcinoid tumors of the lung or thymus, pasireotide LAR treatment resulted in an objective tumor response in 39% of patients [140]. In the randomized, open-label, phase II COOPERATE-2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-differentiated, progressive pancreatic neuroendocrine tumors, the addition of pasireotide to everolimus was not associated with improvement in PFS compared with everolimus alone [46]. Further investigation to evaluate the applicability of pasireotide alone or in combination with other therapies is required.
SSAs are generally well tolerated; common adverse events include nausea, abdominal pain, headache, dizziness, fatigue, and back pain; hematopoietic complications and bile stones have been also described [141]. These events may begin shortly after the first administration of the drug and may decrease progressively over the subsequent weeks as treatment continues [88]. In the PROMID study five patients stopped treatment due to an adverse event. In the CLARINET study, 25 of 101 patients had serious adverse events but only three were related to the study treatment [134].
Interferon-Alpha
Interferon-alpha has antiproliferative, pro-apoptotic, cytotoxic/cytostatic, and immunomodulatory effects in NETs [142, 143]. It has been considered as a second-line therapeutic option in progressive NETs under SSAs [23, 144]. Several studies have failed to show a significant additional effect of interferon-alpha on top of that of SSAs [32, 44, 145]. Tumor response rates of about 10% have been reported [146] and its efficacy is similar to other agents including bevacizumab when combined with SSAs [147]. Unfortunately, several adverse effects have been described; a pegylated formulation seems to be associated with decreased side effects, and its combination with octreotide seems to be better tolerated [146, 148]. Despite this, the availability of novel therapeutic options with higher efficacy and lower side effects limit the applicability of this drug for tumor control [149].
Peptide Receptor Radionuclide Therapy (PRRT)
PRRT with a somatostatin analog allows targeted delivery of radionuclides to tumor cells expressing high levels of SSTR. Radiolabeled SSAs consist of a radionuclide isotope (90Y or 177Lu), a carrier molecule (generally octreotide or octreotate), and a chelator (usually DOTA: tetra-azacyclododecane-tetra-acetic acid or DTPA: diethylenetriamine penta-acetic acid) [150]. 177Lu-DOTA-octreotate or 177Lu-Dotatate is the most studied molecule. The 177Lu radionuclide is characterized by the emission of β-rays, which have an intermediate tissue penetration, and γ-rays, which are used for monitoring and dosimetry by post-therapy scintigraphy [151].
Treatment response is directly related to the expression of SSTR in the tumor, making it a predictive marker of response [152], although tissue SSTR immunohistochemistry has no additional value to SSTR scintigraphy in predicting tumor response to PRRT [153]. For PRRT, patients should have a positive NET histology and a positive SSTR scintigraphy with 111In-DTPA-octreotide (OctreoScan; lesion uptake equal or greater than the liver uptake) or a positive 68Ga-DOTA-SSA PET-CT [154]. In 443 bronchial and GEP NET patients PRRT induced an objective tumor response (complete or partial) in 39% and stable disease in 43% of patients. In this cohort PFS was 29 months (range 26–33) and OS 63 months (range 55–72) [155]. Tumor response may differ according to the primary tumor localization and tumor load [156]. OS is also different in NETs of different localizations (pancreas 71 months (95% CI 56–86), midgut 60 months (95% CI 52–68))[155]. In contrast, response rates are lower in patients with larger tumor load and higher liver infiltration [157]. A representative example of tumor response to PRRT is depicted in Fig. 3b.
An OS of 27–95 months and a PFS of 16–29 months have been reported in NET patients receiving treatment with 90Y-DOTATOC [158–160], but regimens employing 90Y have been associated with an increased incidence of renal toxicity. Available studies are difficult to compare since the inclusion criteria and basal characteristics of the included patients differ considerably [150]. Single-center studies evaluating 177Lu-Dotatate reported a median OS of 46 months, median time to progression of 36–40 months, and PFS of 33–36 months [82, 161–163]. Imaging response rates of 18–44% (by RECIST criteria) and improvement in quality of life have also been reported [164, 165]. The pivotal phase III NETTER-1 trial for the first time evaluated the efficacy of PRRT with 177Lu-DOTATATE in a multicenter, randomized clinical trial. This study included 229 patients with well-differentiated, metastatic midgut NETs that were progressive on a standard dose of long-acting SSA. Patients were randomized to receive four cycles of PRRT with 177Lu-Dotatate or a double dose (60 mg/28 days) of octreotide LAR. The primary outcome was an increase in PFS (median not reached vs. 8.4 months) in favor of patients treated with PRRT. This study also reported a 79% reduction in the risk of progression or death compared to octreotide and increased overall response rate (ORR) in the PRRT group (18%) compared to 3% in the control group [166]. Despite PRRT being a promising treatment in GEP-NETs, the application in lung NETs may be limited due to a low expression of SSTR in some cases, especially in some atypical lung carcinoids; notwithstanding, ORR of 15%, disease stabilization of 47%, and OS of 52 months (95% CI 49–55) have been reported [155, 167].
In the case of tumor progression after an initial good response, retreatment with PRRT represents an alternative. In this sense, disease control rates of 70–85% have been reported, but tumor response is limited [168, 169]. 177Lu-DOTATATE has also been evaluated with radiosensitizing agents; its use in combination with 5-fluorouracil, capecitabine, or temozolomide may increase the response rate (ORR 24–38%), but toxicity should still be evaluated [170–172]. Similar ORR has been reported when combined with everolimus [173]. Some case reports and series have suggested the use of pre-operative PRRT for downstaging NETs [174–176], but further investigation is still required on the efficacy of neoadjuvant PRRT in patients with initially unresectable NETs.
PRRT is generally well tolerated. Nausea, fatigue, or abdominal pain may occur; they have been related to the amino acid infusion given for kidney protection and are self-limiting within 24 h [150]. Other adverse effects include neutropenia (1%), thrombocytopenia (2%), and lymphopenia (9%) [166]. Increased serum levels of liver enzymes and chromogranin A during or directly after therapy may be related to radiation-induced inflammation or progressive disease, and follow-up and repeated measurements are required [177]. Long-term side effects of PRRT may include renal failure and acute leukemia or myelodysplastic syndrome in 1–2% of cases [150, 155, 156]. Some clinical conditions increase PRRT toxicity (hypertension, diabetes, and renal or bone marrow impairment).
Currently several clinical trials aim to further optimize PRRT by evaluating the efficacy of PRRT alone or in combination with chemotherapeutic agents (capecitabine or capecitabine/temozolomide) in patients with undifferentiated NETs (NCT02358356). Novel radioligands include SSTR antagonists, the combination of PRRT with immunotherapy, and the use of α emitters [150]. SSTR antagonists may have higher tumor uptake, longer retention, and decreased radioactivity in healthy organs compared to 177Lu-Dotatate [150, 178]. Alpha emitters, such as 213Bi, can induce more DNA damage; however, toxicity is a problem for alpha-emitting radionuclides, which limits their applicability in the clinical practice. Additionally, other pharmaceutical agents (olaparib) could sensitize NET cells to PRRT according to in vitro models [179]. Predictive response models are also under study. For instance, NET blood gene expression assays have been evaluated in recent years [180]; these results, in combination with tumor grade, may help to predict the response to PRRT [181]. Recently a binary predictive quotient was described based on the use of NET blood gene transcripts combined with Ki67, and this quotient predicted 100% of responders and 84% of non-responders with an accuracy of 95% [182]. These data should, however, be validated in independent cohorts. Soon the results of the COMPETE study will be available. The aim of this multicenter phase III study is to evaluate the efficacy and safety of PRRT (177Lu-Edotreotide), compared to everolimus, in progressive GEP-NETs with positive expression of SSTR (NCT03049189). This comparison aims to provide information on the treatment sequence that should be followed in progressive NETs under SSAs. Finally, the development of new radiopeptides targeting other receptors (e.g., gastrin-releasing peptide) may also represent a therapeutic option in the future [183].
Everolimus
The mTOR pathway plays an important role in the regulation of cell proliferation in NETs [15]. The efficacy of the PI3 K/AKT/mTOR inhibitor everolimus in well-differentiated NETs has been shown in several clinical trials [23]. In the phase III RADIANT 3 trial, everolimus was compared to placebo in patients with low or intermediate-grade PNETs with radiologic progression within the previous year. PFS was longer in the everolimus group. Additionally, only a non-significant 6.3 months OS benefit was observed, and this last result may be related to the switch of patients from the placebo group (85% experienced progressive disease) to the open-label everolimus group [184]. Despite this, the effect of everolimus on PFS and OS was independent of the prior use of chemotherapy or SSAs [184–186]. A similar study was conducted in gastrointestinal and lung NETs (RADIANT 4), where increased PFS (11 vs. 3.9 months) and higher disease control rate (81% vs. 64%) in favor of the everolimus-treated group were reported [187]. However, although everolimus is considered a safe drug, treatment can be accompanied by grade 3 and 4 drug-related adverse events (diarrhea, infections, anemia, fatigue, hyperglycemia) [187], which may limit treatment tolerance and consequently patient adherence. Importantly, the RADIANT 4 study, as the previous ones, failed to demonstrate statistically significant improvements in OS [187], which should be taken into account especially in those patients with poor treatment tolerance.
The combination of everolimus and octreotide LAR has also been compared to octreotide LAR alone in functioning GEP- and lung NETs (carcinoid syndrome; phase III RADIANT 2). PFS was 5.1 months longer in the combination group but the hazard ratio (HR) was not statistically significant between the two groups [188]. In lung NETs, 2.4-fold longer PFS, 28% reduction in the risk of disease progression, and a twofold increase in the proportion of patients with tumor shrinkage were reported in the combination arm [189]. In colorectal NETs similar results have been described, including a fourfold prolonged PFS and increased frequency of tumor shrinkage (67% vs. 37%) [190]. As opposed to the positive results of combination therapy with everolimus and octreotide LAR, the combination of everolimus with pasireotide did not improve PFS or OS when compared to everolimus alone in progressive PNETs [46]. In contrast, in lung and thymic NETs the combination of pasireotide (60 mg every 28 days) with everolimus increased the progression-free rate (58.5%) when compared to pasireotide (39%) or everolimus (33.3%) at 9 months (phase II LUNA trial). Importantly, 27% of patients discontinued the treatment due to progressive disease or adverse events [191]. A synergistic effect of epidermal growth factor receptor (EGFR) and mTOR pathways inhibitors has been suggested in bronchial carcinoids [192]. For this reason the combination with sunitinib/erlonitib is currently under study.
Everolimus is considered as first-line therapy in progressive atypical lung carcinoids, SSTR-negative lung NETs, and in well-differentiated midgut SSTR-negative NETs [23]. Currently several studies are evaluating the combination of everolimus with other therapies including chemotherapeutic agents, SSAs, molecular targeted therapies, radiotherapy, and PRRT.
Sunitinib
Because NETs are generally hypervascularized tumors, treatment with antiangiogenic drugs seems a rational approach. Sunitinib is as an oral multi-targeted tyrosine kinase inhibitor (TKI) that inhibits multiple angiogenic factors including the vascular endothelial growth factor receptors 1-3 (VEGFR), the stem-cell factor [SCF] receptor, and the platelet-derived growth factor receptors [193]. Initially in a two-cohort phase II study, sunitinib, administered 50 mg/day orally for 4 weeks followed by 2 weeks off treatment, reported poor response rates in PNETs and carcinoids (16.7% and 2.4%, respectively) [194]. In contrast, a placebo-controlled phase III study in progressive PNETs (SUN 1111 trial) reported increased PFS in the sunitinib group (11.4 months, hazard ratio for progression or death, 0.42; 95% CI 0.26–0.66) compared to placebo (5.5 months), and resulted in study discontinuation due to a higher incidence of adverse events and deaths in the placebo group [80]. Recently a retrospective imaging analysis of 171 patients with well-differentiated metastatic and/or progressive PNETs was performed. Patients received 37.5 mg sunitinib daily or placebo. PFS was increased in the sunitinib group (12.6 months) compared to placebo (5.8 months). 5 years after the study closure, median OS was 38.6 (25.6–56.4) months for sunitinib and 29.1 (16.4–36.8) months for placebo (HR, 0.73; 95% CI 0.50–1.06; p = 0.094). In this study 69% of patients randomized for placebo crossed over to sunitinib, probably affecting the significance of the OS [195], as has been reported in previous studies [196]. As for everolimus, significant improvement in OS has not been reported yet.
Sunitinib is associated with several adverse events including hypertension, diarrhea, nausea, vomiting, asthenia, skin toxicity, and fatigue [80]. Importantly, drug-related adverse events with sunitinib are increased threefold as compared with placebo [193], but fewer grade 3–4 toxicities are observed when compared with chemotherapeutic agents [197]. Dose reduction or temporary interruption of therapy may resolve the adverse clinical situation [193]. Importantly, no differences in the quality-of-life index have been reported when compared to placebo [193].
Sunitinib dose reduction should be considered when co-administered with strong CYP3A4 inhibitors, including ketoconazole, ritonavir, itraconazole, erythromycin, or clarithromycin. In contrast, dose should be increased when administered with CYP3A4 inducers, such as dexamethasone, phenytoin, carbamazepine, or phenobarbital [193].
Sunitinib is currently being compared to other treatment options in NETs including everolimus and PRRT. Current therapeutic options and a possible therapeutic sequence are depicted in Fig. 4.
Fig. 4.
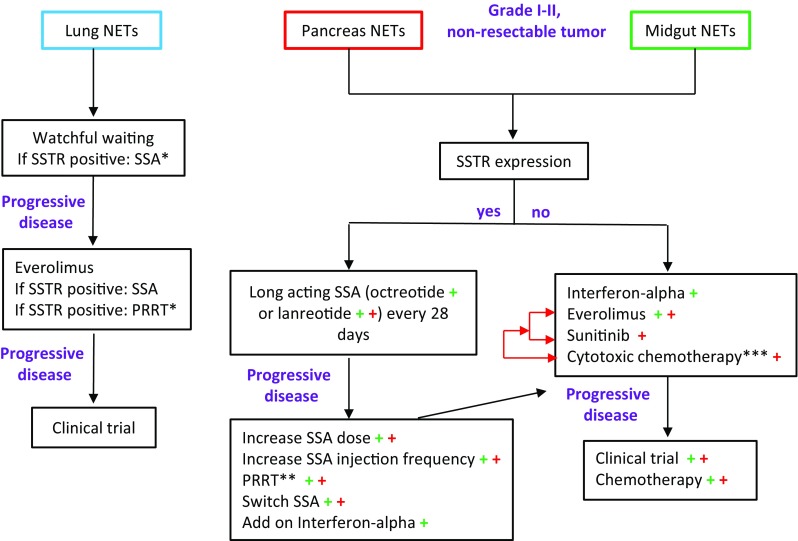
Treatment algorithms for tumor control in neuroendocrine tumors (NETs). Summary of current medical strategies for tumor control in NETs according to the primary tumor site. Legend: Blue, red and green colors represent lung, pancreas and midgut NETs respectively. SSTR somatostatin receptor expression, SSA somatostatin analog, PRRT peptide receptor radionuclide therapy. *Not registered for this indication, **PRRT has been approved in Europe for midgut NETs and in the USA for midgut and pancreatic NETs, ***streptozocin/5-fluorouracil or streptozocin/doxorubicin; temozolomide/capecitabine as an alternative regimen
Other Treatments
Other TKIs and antiangiogenic drugs have been evaluated in NET patients in open-label phase II studies (Table 2). Importantly, these therapies have been considered as promising options in NETs, but further studies are still required. In this sense, despite promising phase II data of bevacizumab administration in patients with gastrointestinal NETs, a phase III trial failed to show superior efficacy of bevacizumab compared to interferon alfa-2b [147]. The combination therapy of sorafenib and bevacizumab can induce antitumoral response, but was accompanied by an unfavorable safety outcome in a phase II study [198]. In contrast, the combination of bevacizumab with everolimus showed improved tumor response rates but no significant effect on PFS [199]; even more, the combination of everolimus with sorafenib reported dose-limiting toxicities [200]. Biochemical and radiological response has been described in PNETs treated with pazopanib [201], but further evaluation is still required; importantly, results of randomized trials are still pending and its combination with temozolomide is currently under evaluation (NCT01465659).
Systemic chemotherapy is indicated for the poorly differentiated grade 3 neuroendocrine carcinomas. Chemotherapy is not discussed in this review, but some publications describe the significant advances in this field in the last years [16, 202–204].
Future Directions
The comprehensive evaluation of signaling pathways regulating cell proliferation involved in NET development and progression has opened new perspectives for the medical treatment of these tumors. Current clinical trials for tumor growth control including novel drugs, treatment comparisons, and combinations are summarized in Table 2.
mTOR inhibitors and TKIs are the most representative examples, but other novel pathway-directed therapeutic compounds are also currently being evaluated in (pre-)clinical studies [205]. Current clinical trials include head-to-head comparisons between novel therapies (PRRT vs. everolimus/sunitinib/capecitabine-temozolomide) and combination therapies (everolimus with bevacizumab/cisplatinum/temozolomide; PRRT with capecitabine-temozolomide). Additionally, trials that compare treatment sequences are also recruiting patients (sequencing of streptozocin-5-FU followed by everolimus vs. the reverse order of treatment, NCT02246127). These results may help to improve the sequence of therapy in NET patients.
Other examples of potential new treatment options include immunotherapy and somatostatin-dopamine multi-receptor chimeras. The mechanisms of action of immune checkpoint inhibitors and multi-receptor chimeras are also depicted in Fig. 2.
Immune checkpoint inhibitors have rapidly advanced and improved the management of several tumors in the last years [206, 207]. Programmed death-ligand 1 (PD-L1) is expressed on several cancer cells and interacts with PD1, which is expressed on T cells. This ligand-receptor interaction inhibits T cells and blocks their antitumor immune response [208, 209]. PD1/PDL1 expression has been demonstrated in several cancers [210–212]. Studies testing anti-PD-L1 or anti-PD1 agents have shown promising results in, for instance, small-cell lung cancer (SCLC) and melanoma [209]. The expression of PD-L1 was demonstrated in GEP- and lung (large cell neuroendocrine carcinoma) NETs using immunohistochemistry and qPCR [208, 213, 214]. Additionally, an inverse relation between PD-L1 and angiogenesis/hypoxia factors (vascular endotelial growth factor-A, hypoxia inducible factor 1a) has been described [215]. Importantly, PD-L1 has been associated with clinical variables in lung and GEP-NETs, including histological type, tumor grade, and survival [208, 216], which have not been described in midgut NETs [214]. Observations that the expression levels of PD-1 and PD-L1 were an independent survival prognostic factor in NETs [216] should be subjected to further study [214].
The use of cytotoxic T-lymphocyte antigen-4 (CTL4) blockers has also been reported in SCLC [217]. CTL4 is a critical negative regulator of the antitumor T-cell response, and its inhibition has encouraging effects in SCLC and melanoma [217, 218]. Additionally, the efficacy of immune checkpoint inhibitors may be affected by the presence of tumor infiltrating lymphocytes (TILs) [219]. TILs have been suggested as a survival predictor for intermediate grade (Ki 67 2–20%) PNETs, since increased recurrence-free survival was observed in patients with higher TIL density [219]. The modulation of TILs density may also be a promising therapeutic option in the future. Despite immunotherapy having an important role in the management of other types of cancer, the effect on well-differentiated NETs according to preliminary data seems to be limited, although it may represent an option for G3NETs/NECs, which needs further investigation [220].
Multi-receptor interaction has been suggested as an efficacious and selective therapeutic strategy for enhancing the effects of somatostatin [221]. The presence of hetero-dimers has been described among SSTRs and between SSTRs and other receptor families, including dopamine receptors, especially the dopamine receptor subtype 2 (D2R) [222, 223]. Based on this, some structural chimeric molecules that combine elements of SSAs and dopamine analogs (DA) have been developed [221]. In vitro studies using a pancreatic NET cell model revealed inhibitory properties of chimeras on hormone secretion without affecting cell proliferation [224]. Importantly, BIM-23A760, a chimeric compound that activates SSTR2 and D2R, acutely decreased growth hormone and prolactin secretion in pituitary tumors, but long-term effects disappeared due to a dopaminergic metabolite that may interfere with the activity of the parent molecule [221]. An open-label, multicenter clinical trial in patients with carcinoid syndrome was started for evaluating the efficacy of BIM-23A760. Unfortunately, this study was prematurely terminated and primary/secondary outcomes were not analyzed (NCT01018953). Currently research is focused on the improvement of chimeric molecules that could keep a long-term effect.
Conclusions
The number of systemic treatment options for NETs aiming to control either hormone production and/or tumor growth has significantly increased and improved in the last years. A higher number of clinical trials and approved therapeutic agents have further facilitated the management of NET patients. SSAs were found to have antiproliferative effects, next to their inhibitory action on hormone secretion, and can induce sustained stabilization in grade 1–2 NETs. If progression occurs under SSAs therapy, PRRT seems to be the most rational second-line treatment, considering its efficacy and side effects, with everolimus and sunitinib (for PNET) as the next options. To further improve PFS and hopefully OS, further randomized studies are needed to establish the optimal role, sequence, and/or combination(s) of the currently available and emerging treatment options. In addition, future studies should further characterize (epi)genetic aspects and regulatory pathways of NETs to identify new targets for medical therapy.
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Justo P. Castaño has received travel or speaker fees from Novartis/Ipsen and research funds from Ipsen/Novartis. Johannes Hofland has received travel or speaker fees from Ipsen, Novartis, and Advanced Accelerator Applications, research funds from Ipsen, and is on the Advisory Boards of Novartis. Wouter W. de Herder has received travel or speaker fees from Novartis and Ipsen, and research funds from Ipsen. Leo J. Hofland has received research funds from Ipsen and Novartis. Aura D. Herrera-Martínez, Tessa Brabander, Ferry A.L.M. Eskens, María A. Gálvez Moreno, Raúl M. Luque, and Richard A. Feelders declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
References
- 1.Ameri P, Ferone D. Diffuse endocrine system, neuroendocrine tumors and immunity: what’s new? Neuroendocrinology. 2012;95(4):267–276. doi: 10.1159/000334612. [DOI] [PubMed] [Google Scholar]
- 2.Rosai J. The origin of neuroendocrine tumors and the neural crest saga. Mod Pathol. 2011;24(Suppl 2):S53–S57. doi: 10.1038/modpathol.2010.166. [DOI] [PubMed] [Google Scholar]
- 3.Fraenkel M, et al. Incidence of gastroenteropancreatic neuroendocrine tumours: a systematic review of the literature. Endocr Relat Cancer. 2014;21(3):R153–R163. doi: 10.1530/ERC-13-0125. [DOI] [PubMed] [Google Scholar]
- 4.Kunz PL. Carcinoid and neuroendocrine tumors: building on success. J Clin Oncol. 2015;33(16):1855–1863. doi: 10.1200/JCO.2014.60.2532. [DOI] [PubMed] [Google Scholar]
- 5.Kim JY, Hong SM, Ro JY. Recent updates on grading and classification of neuroendocrine tumors. Ann Diagn Pathol. 2017;29:11–16. doi: 10.1016/j.anndiagpath.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Dasari A, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017;3(10):1335–1342. doi: 10.1001/jamaoncol.2017.0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofland J, et al. Recent developments in the diagnosis and therapy of well-differentiated neuroendocrine tumours. Neth J Med. 2018;76(3):100–108. [PubMed] [Google Scholar]
- 8.Colao A, Faggiano A, Pivonello R. Somatostatin analogues: treatment of pituitary and neuroendocrine tumors. Prog Brain Res. 2010;182:281–294. doi: 10.1016/S0079-6123(10)82012-6. [DOI] [PubMed] [Google Scholar]
- 9.Baldelli R, et al. Somatostatin analogs therapy in gastroenteropancreatic neuroendocrine tumors: current aspects and new perspectives. Front Endocrinol (Lausanne) 2014;5:7. doi: 10.3389/fendo.2014.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cakir M, Dworakowska D, Grossman A. Somatostatin receptor biology in neuroendocrine and pituitary tumours: part 2–clinical implications. J Cell Mol Med. 2010;14(11):2585–2591. doi: 10.1111/j.1582-4934.2010.01125_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falconi M, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology. 2016;103(2):153–171. doi: 10.1159/000443171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veenstra MJ, et al. Targeting the somatostatin receptor in pituitary and neuroendocrine tumors. Expert Opin Ther Targets. 2013;17(11):1329–1343. doi: 10.1517/14728222.2013.830711. [DOI] [PubMed] [Google Scholar]
- 13.van der Zwan WA, et al. GEPNETs update: radionuclide therapy in neuroendocrine tumors. Eur J Endocrinol. 2015;172(1):R1–R8. doi: 10.1530/EJE-14-0488. [DOI] [PubMed] [Google Scholar]
- 14.Raymond E, et al. Therapy innovations: tyrosine kinase inhibitors for the treatment of pancreatic neuroendocrine tumors. Cancer Metastasis Rev. 2011;30(Suppl 1):19–26. doi: 10.1007/s10555-011-9291-2. [DOI] [PubMed] [Google Scholar]
- 15.Chan J, Kulke M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr Treat Options Oncol. 2014;15(3):365–379. doi: 10.1007/s11864-014-0294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Carbonero R, et al. ENETS consensus guidelines for the standards of care in neuroendocrine neoplasms. Systemic therapy 2: chemotherapy. Neuroendocrinology. 2017;105(3):281–294. doi: 10.1159/000473892. [DOI] [PubMed] [Google Scholar]
- 17.Pavel M, et al. Telotristat ethyl in carcinoid syndrome: safety and efficacy in the TELECAST phase III trial. Endocr Relat Cancer. 2018;25(3):309–322. doi: 10.1530/ERC-17-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavel M, et al. Telotristat etiprate for carcinoid syndrome: a single-arm, multicenter trial. J Clin Endocrinol Metab. 2015;100(4):1511–1519. doi: 10.1210/jc.2014-2247. [DOI] [PubMed] [Google Scholar]
- 19.Halperin DM, et al. Frequency of carcinoid syndrome at neuroendocrine tumour diagnosis: a population-based study. Lancet Oncol. 2017;18(4):525–534. doi: 10.1016/S1470-2045(17)30110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zandee WT, de Herder WW, Jann H. Incidence and prognosis of carcinoid syndrome: hormones or tumour burden? Lancet Oncol. 2017;18(6):e299. doi: 10.1016/S1470-2045(17)30335-2. [DOI] [PubMed] [Google Scholar]
- 21.Mougey AM, Adler DG. Neuroendocrine tumors: review and clinical update. Hosp Physician. 2007;51:12–20. [Google Scholar]
- 22.Niederle B, et al. ENETS consensus guidelines update for neuroendocrine neoplasms of the jejunum and ileum. Neuroendocrinology. 2016;103(2):125–138. doi: 10.1159/000443170. [DOI] [PubMed] [Google Scholar]
- 23.Pavel M, et al. ENETS consensus guidelines update for the management of distant metastatic disease of intestinal, pancreatic, bronchial neuroendocrine neoplasms (NEN) and NEN of unknown primary site. Neuroendocrinology. 2016;103(2):172–185. doi: 10.1159/000443167. [DOI] [PubMed] [Google Scholar]
- 24.Ito T, Lee L, Jensen RT. Carcinoid-syndrome: recent advances, current status and controversies. Curr Opin Endocrinol Diabetes Obes. 2018;25(1):22–35. doi: 10.1097/MED.0000000000000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis GR, et al. Effect of somatostatin infusion on jejunal water and electrolyte transport in a patient with secretory diarrhea due to malignant carcinoid syndrome. Gastroenterology. 1980;78(2):346–349. [PubMed] [Google Scholar]
- 26.Dharmsathaphorn K, et al. Somatostatin inhibits diarrhea in the carcinoid syndrome. Ann Intern Med. 1980;92(1):68–69. doi: 10.7326/0003-4819-92-1-68. [DOI] [PubMed] [Google Scholar]
- 27.Kvols LK, et al. Treatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogue. N Engl J Med. 1986;315(11):663–666. doi: 10.1056/NEJM198609113151102. [DOI] [PubMed] [Google Scholar]
- 28.Rubin J, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol. 1999;17(2):600–606. doi: 10.1200/JCO.1999.17.2.600. [DOI] [PubMed] [Google Scholar]
- 29.O’Toole D, et al. Treatment of carcinoid syndrome: a prospective crossover evaluation of lanreotide versus octreotide in terms of efficacy, patient acceptability, and tolerance. Cancer. 2000;88(4):770–776. doi: 10.1002/(sici)1097-0142(20000215)88:4<770::aid-cncr6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 30.Ruszniewski P, et al. Rapid and sustained relief from the symptoms of carcinoid syndrome: results from an open 6-month study of the 28-day prolonged-release formulation of lanreotide. Neuroendocrinology. 2004;80(4):244–251. doi: 10.1159/000082875. [DOI] [PubMed] [Google Scholar]
- 31.Plockinger U, Wiedenmann B. Neuroendocrine tumors. Biotherapy. Best Pract Res Clin Endocrinol Metab. 2007;21(1):145–162. doi: 10.1016/j.beem.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Janson ET, Oberg K. Long-term management of the carcinoid syndrome. Treatment with octreotide alone and in combination with alpha-interferon. Acta Oncol. 1993;32(2):225–229. doi: 10.3109/02841869309083916. [DOI] [PubMed] [Google Scholar]
- 33.Hofland LJ, Lamberts SW. The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocr Rev. 2003;24(1):28–47. doi: 10.1210/er.2000-0001. [DOI] [PubMed] [Google Scholar]
- 34.Strosberg JR, et al. Clinical benefits of above-standard dose of octreotide LAR in patients with neuroendocrine tumors for control of carcinoid syndrome symptoms: a multicenter retrospective chart review study. Oncologist. 2014;19(9):930–936. doi: 10.1634/theoncologist.2014-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferolla P, et al. Shortened interval of long-acting octreotide administration is effective in patients with well-differentiated neuroendocrine carcinomas in progression on standard doses. J Endocrinol Invest. 2012;35(3):326–331. doi: 10.3275/7869. [DOI] [PubMed] [Google Scholar]
- 36.Wolin EM. The expanding role of somatostatin analogs in the management of neuroendocrine tumors. Gastrointest Cancer Res. 2012;5(5):161–168. [PMC free article] [PubMed] [Google Scholar]
- 37.Sandostatin LAR® depot (octreotide acetate for injectable suspension) [prescribing information]. East Hanover, NJ: Novartis Pharmaceutical Corporation; 2011.
- 38.Teunissen JJ, Kwekkeboom DJ, Krenning EP. Quality of life in patients with gastroenteropancreatic tumors treated with [177Lu-DOTA0, Tyr3]octreotate. J Clin Oncol. 2004;22(13):2724–2729. doi: 10.1200/JCO.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 39.Waldherr C, et al. Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq (90)Y-DOTATOC. J Nucl Med. 2002;43(5):610–6. [PubMed]
- 40.Kiesewetter B, Raderer M. Ondansetron for diarrhea associated with neuroendocrine tumors. N Engl J Med. 2013;368(20):1947–1948. doi: 10.1056/NEJMc1301537. [DOI] [PubMed] [Google Scholar]
- 41.Mota JM, Sousa LG, Riechelmann RP. Complications from carcinoid syndrome: review of the current evidence. Ecancermedicalscience. 2016;10:662. doi: 10.3332/ecancer.2016.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kvols LK, et al. Pasireotide (SOM230) shows efficacy and tolerability in the treatment of patients with advanced neuroendocrine tumors refractory or resistant to octreotide LAR: results from a phase II study. Endocr Relat Cancer. 2012;19(5):657–666. doi: 10.1530/ERC-11-0367. [DOI] [PubMed] [Google Scholar]
- 43.Wolin EM, et al. Phase III study of pasireotide long-acting release in patients with metastatic neuroendocrine tumors and carcinoid symptoms refractory to available somatostatin analogues. Drug Des Dev Ther. 2015;9:5075–5086. doi: 10.2147/DDDT.S84177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frank M, et al. Combination therapy with octreotide and alpha-interferon: effect on tumor growth in metastatic endocrine gastroenteropancreatic tumors. Am J Gastroenterol. 1999;94(5):1381–1387. doi: 10.1111/j.1572-0241.1999.01090.x. [DOI] [PubMed] [Google Scholar]
- 45.Kulke MH, et al. Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome. J Clin Oncol. 2017;35(1):14–23. doi: 10.1200/JCO.2016.69.2780. [DOI] [PubMed] [Google Scholar]
- 46.Kulke MH, et al. A randomized, open-label, phase II study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-differentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309–1315. doi: 10.1093/annonc/mdx078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulke MH, et al. Telotristat etiprate, a novel serotonin synthesis inhibitor, in patients with carcinoid syndrome and diarrhea not adequately controlled by octreotide. Endocr Relat Cancer. 2014;21(5):705–714. doi: 10.1530/ERC-14-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hutcheson JD, et al. Serotonin receptors and heart valve disease–it was meant 2B. Pharmacol Ther. 2011;132(2):146–157. doi: 10.1016/j.pharmthera.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blazevic A, et al. Small intestinal neuroendocrine tumours and fibrosis: an entangled conundrum. Endocr Relat Cancer. 2018;25(3):R115–R130. doi: 10.1530/ERC-17-0380. [DOI] [PubMed] [Google Scholar]
- 50.Grozinsky-Glasberg S, Grossman AB, Gross DJ. Carcinoid heart disease: from pathophysiology to treatment—’something in the way it moves’. Neuroendocrinology. 2015;101(4):263–273. doi: 10.1159/000381930. [DOI] [PubMed] [Google Scholar]
- 51.Hassan SA, et al. Carcinoid heart disease. Heart. 2017;103(19):1488–1495. doi: 10.1136/heartjnl-2017-311261. [DOI] [PubMed] [Google Scholar]
- 52.Davar J, et al. Diagnosing and managing carcinoid heart disease in patients with neuroendocrine tumors: an expert statement. J Am Coll Cardiol. 2017;69(10):1288–1304. doi: 10.1016/j.jacc.2016.12.030. [DOI] [PubMed] [Google Scholar]
- 53.Modlin IM, et al. Therapeutic options for gastrointestinal carcinoids. Clin Gastroenterol Hepatol. 2006;4(5):526–547. doi: 10.1016/j.cgh.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Moller JE, et al. Factors associated with progression of carcinoid heart disease. N Engl J Med. 2003;348(11):1005–1015. doi: 10.1056/NEJMoa021451. [DOI] [PubMed] [Google Scholar]
- 55.Fox DJ, Khattar RS. Carcinoid heart disease: presentation, diagnosis, and management. Heart. 2004;90(10):1224–1228. doi: 10.1136/hrt.2004.040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaltsas G, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: pre- and perioperative therapy in patients with neuroendocrine tumors. Neuroendocrinology. 2017;105(3):245–254. doi: 10.1159/000461583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castillo JG, et al. Management of patients undergoing multivalvular surgery for carcinoid heart disease: the role of the anaesthetist. Br J Anaesth. 2008;101(5):618–626. doi: 10.1093/bja/aen237. [DOI] [PubMed] [Google Scholar]
- 58.Goode PN, et al. Diazoxide in the management of patients with insulinoma. World J Surg. 1986;10(4):586–592. doi: 10.1007/BF01655532. [DOI] [PubMed] [Google Scholar]
- 59.Gill GV, Rauf O, MacFarlane IA. Diazoxide treatment for insulinoma: a national UK survey. Postgrad Med J. 1997;73(864):640–641. doi: 10.1136/pgmj.73.864.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Herder WW, et al. New therapeutic options for metastatic malignant insulinomas. Clin Endocrinol (Oxf) 2011;75(3):277–284. doi: 10.1111/j.1365-2265.2011.04145.x. [DOI] [PubMed] [Google Scholar]
- 61.Baudin E, et al. Malignant insulinoma: recommendations for characterisation and treatment. Ann Endocrinol (Paris) 2013;74(5–6):523–533. doi: 10.1016/j.ando.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 62.Altszuler N, Moraru E, Hampshire J. On the mechanism of diazoxide-induced hyperglycemia. Diabetes. 1977;26(10):931–935. doi: 10.2337/diab.26.10.931. [DOI] [PubMed] [Google Scholar]
- 63.Matej A, Bujwid H, Wronski J. Glycemic control in patients with insulinoma. Hormones (Athens) 2016;15(4):489–499. doi: 10.14310/horm.2002.1706. [DOI] [PubMed] [Google Scholar]
- 64.Oberg K. Pancreatic endocrine tumors. Semin Oncol. 2010;37(6):594–618. doi: 10.1053/j.seminoncol.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 65.Ferrer-Garcia JC, et al. Management of malignant insulinoma. Clin Transl Oncol. 2013;15(9):725–731. doi: 10.1007/s12094-012-0996-7. [DOI] [PubMed] [Google Scholar]
- 66.de Herder WW, et al. Well-differentiated pancreatic tumor/carcinoma: insulinoma. Neuroendocrinology. 2006;84(3):183–188. doi: 10.1159/000098010. [DOI] [PubMed] [Google Scholar]
- 67.Singh V, et al. Characterization of somatostatin receptor subtype-specific regulation of insulin and glucagon secretion: an in vitro study on isolated human pancreatic islets. J Clin Endocrinol Metab. 2007;92(2):673–680. doi: 10.1210/jc.2006-1578. [DOI] [PubMed] [Google Scholar]
- 68.Bruns C, et al. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146(5):707–716. doi: 10.1530/eje.0.1460707. [DOI] [PubMed] [Google Scholar]
- 69.Hendren NS, et al. Pasireotide for the treatment of refractory hypoglycaemia from malignant insulinoma. Clin Endocrinol (Oxf). 2018;88(2):341–343. doi: 10.1111/cen.13503. [DOI] [PubMed] [Google Scholar]
- 70.Tirosh A, et al. Pasireotide for malignant insulinoma. Hormones (Athens) 2016;15(2):271–276. doi: 10.14310/horm.2002.1639. [DOI] [PubMed] [Google Scholar]
- 71.Kulke MH, Bergsland EK, Yao JC. Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med. 2009;360(2):195–197. doi: 10.1056/NEJMc0806740. [DOI] [PubMed] [Google Scholar]
- 72.Bourcier ME, et al. Successful control of intractable hypoglycemia using rapamycin in an 86-year-old man with a pancreatic insulin-secreting islet cell tumor and metastases. J Clin Endocrinol Metab. 2009;94(9):3157–3162. doi: 10.1210/jc.2009-0788. [DOI] [PubMed] [Google Scholar]
- 73.Fuhrer DK, Kobayashi M, Jiang H. Insulin release and suppression by tacrolimus, rapamycin and cyclosporin A are through regulation of the ATP-sensitive potassium channel. Diabetes Obes Metab. 2001;3(6):393–402. doi: 10.1046/j.1463-1326.2001.00150.x. [DOI] [PubMed] [Google Scholar]
- 74.Di Paolo S, et al. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: a crossroad between cancer and diabetes? J Am Soc Nephrol. 2006;17(8):2236–2244. doi: 10.1681/ASN.2006030196. [DOI] [PubMed] [Google Scholar]
- 75.Fiebrich HB, et al. Everolimus induces rapid plasma glucose normalization in insulinoma patients by effects on tumor as well as normal tissues. Oncologist. 2011;16(6):783–787. doi: 10.1634/theoncologist.2010-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Schaik E, et al. Improved control of severe hypoglycemia in patients with malignant insulinomas by peptide receptor radionuclide therapy. J Clin Endocrinol Metab. 2011;96(11):3381–3389. doi: 10.1210/jc.2011-1563. [DOI] [PubMed] [Google Scholar]
- 77.John AM, Schwartz RA. Glucagonoma syndrome: a review and update on treatment. J Eur Acad Dermatol Venereol. 2016;30(12):2016–2022. doi: 10.1111/jdv.13752. [DOI] [PubMed] [Google Scholar]
- 78.Santangelo WC, et al. Somatostatin analog-induced remission of necrolytic migratory erythema without changes in plasma glucagon concentration. Pancreas. 1986;1(5):464–469. doi: 10.1097/00006676-198609000-00013. [DOI] [PubMed] [Google Scholar]
- 79.Schmid HA. Pasireotide (SOM230): development, mechanism of action and potential applications. Mol Cell Endocrinol. 2008;286(1–2):69–74. doi: 10.1016/j.mce.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 80.Raymond E, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 81.Yao JC, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kwekkeboom DJ, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26(13):2124–2130. doi: 10.1200/JCO.2007.15.2553. [DOI] [PubMed] [Google Scholar]
- 83.Jensen RT. Gastrinomas: advances in diagnosis and management. Neuroendocrinology. 2004;80(Suppl 1):23–27. doi: 10.1159/000080736. [DOI] [PubMed] [Google Scholar]
- 84.Brandi ML, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 85.Auernhammer CJ, Goke B. Medical treatment of gastrinomas. Wien Klin Wochenschr. 2007;119(19–20):609–615. doi: 10.1007/s00508-007-0877-1. [DOI] [PubMed] [Google Scholar]
- 86.Termanini B, et al. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger–Ellison syndrome. Am J Med. 1998;104(5):422–430. doi: 10.1016/s0002-9343(98)00087-4. [DOI] [PubMed] [Google Scholar]
- 87.Jensen RT. Consequences of long-term proton pump blockade: insights from studies of patients with gastrinomas. Basic Clin Pharmacol Toxicol. 2006;98(1):4–19. doi: 10.1111/j.1742-7843.2006.pto_378.x. [DOI] [PubMed] [Google Scholar]
- 88.Oberg K, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15(6):966–973. doi: 10.1093/annonc/mdh216. [DOI] [PubMed] [Google Scholar]
- 89.Burgess JR, et al. Octreotide improves biochemical, radiologic, and symptomatic indices of gastroenteropancreatic neoplasia in patients with multiple endocrine neoplasia type 1 (MEN-1). Implications for an integrated model of MEN-1 tumorigenesis. Cancer. 1999;86(10):2154–2159. [PubMed] [Google Scholar]
- 90.Ricci S, et al. Long-acting depot lanreotide in the treatment of patients with advanced neuroendocrine tumors. Am J Clin Oncol. 2000;23(4):412–415. doi: 10.1097/00000421-200008000-00020. [DOI] [PubMed] [Google Scholar]
- 91.Wymenga AN, et al. Efficacy and safety of prolonged-release lanreotide in patients with gastrointestinal neuroendocrine tumors and hormone-related symptoms. J Clin Oncol. 1999;17(4):1111. doi: 10.1200/JCO.1999.17.4.1111. [DOI] [PubMed] [Google Scholar]
- 92.Shojamanesh H, et al. Prospective study of the antitumor efficacy of long-term octreotide treatment in patients with progressive metastatic gastrinoma. Cancer. 2002;94(2):331–343. doi: 10.1002/cncr.10195. [DOI] [PubMed] [Google Scholar]
- 93.Hiraide S, Ono S, Kato S. Long-term efficacy of S-1 chemotherapy plus administration of octreotide for a patient with metastatic neuroendocrine tumor (Gastrinoma) Case Rep Oncol. 2017;10(2):420–427. doi: 10.1159/000475463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peghini PL, et al. Effect of chronic hypergastrinemia on human enterochromaffin-like cells: insights from patients with sporadic gastrinomas. Gastroenterology. 2002;123(1):68–85. doi: 10.1053/gast.2002.34231. [DOI] [PubMed] [Google Scholar]
- 95.Krenning EP, et al. Somatostatin receptor scintigraphy in carcinoids, gastrinomas and Cushing’s syndrome. Digestion. 1994;55(Suppl 3):54–59. doi: 10.1159/000201202. [DOI] [PubMed] [Google Scholar]
- 96.O’Dorisio TM, Mekhjian HS, Gaginella TS. Medical therapy of VIPomas. Endocrinol Metab Clin N Am. 1989;18(2):545–556. [PubMed] [Google Scholar]
- 97.Williamson JM, et al. Pancreatic and peripancreatic somatostatinomas. Ann R Coll Surg Engl. 2011;93(5):356–360. doi: 10.1308/003588411X582681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Friesen SR. Update on the diagnosis and treatment of rare neuroendocrine tumors. Surg Clin N Am. 1987;67(2):379–393. doi: 10.1016/s0039-6109(16)44190-3. [DOI] [PubMed] [Google Scholar]
- 99.Oberg K, et al. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii124–vii130. doi: 10.1093/annonc/mds295. [DOI] [PubMed] [Google Scholar]
- 100.Eriksson B, et al. High-dose treatment with lanreotide of patients with advanced neuroendocrine gastrointestinal tumors: clinical and biological effects. Ann Oncol. 1997;8(10):1041–1044. doi: 10.1023/a:1008205415035. [DOI] [PubMed] [Google Scholar]
- 101.Delbaldo C, et al. Sunitinib in advanced pancreatic neuroendocrine tumors: latest evidence and clinical potential. Ther Adv Med Oncol. 2012;4(1):9–18. doi: 10.1177/1758834011428147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Faivre S, et al. Sunitinib in pancreatic neuroendocrine tumors: updated progression-free survival and final overall survival from a phase III randomized study. Ann Oncol. 2017;28(2):339–343. doi: 10.1093/annonc/mdw561. [DOI] [PubMed] [Google Scholar]
- 103.Hubold C, Brabant G. [Ectopic hormone secretion by neuroendocrine tumors] Ektope Hormonsekretion bei neuroendokrinen Tumoren. Internist (Berl). 2012;53(2):145–51. [DOI] [PubMed]
- 104.Kamp K, et al. Parathyroid hormone-related peptide (PTHrP) secretion by gastroenteropancreatic neuroendocrine tumors (GEP-NETs): clinical features, diagnosis, management, and follow-up. J Clin Endocrinol Metab. 2014;99(9):3060–3069. doi: 10.1210/jc.2014-1315. [DOI] [PubMed] [Google Scholar]
- 105.Herrera-Martinez AD, Padillo Cuenca JC, Bahamondes Opazo R, Barrera Martín A, Rebollo Roman A, Díaz Iglesia C, Gálvez Moreno MA. ACTH producing pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1. J Pancreas. 2017;02:175–80.
- 106.Ilias I, et al. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90(8):4955–4962. doi: 10.1210/jc.2004-2527. [DOI] [PubMed] [Google Scholar]
- 107.Kamp K, et al. Prevalence and clinical features of the ectopic ACTH syndrome in patients with gastroenteropancreatic and thoracic neuroendocrine tumors. Eur J Endocrinol. 2016;174(3):271–280. doi: 10.1530/EJE-15-0968. [DOI] [PubMed] [Google Scholar]
- 108.van der Pas R, et al. New developments in the medical treatment of Cushing’s syndrome. Endocr Relat Cancer. 2012;19(6):R205–R223. doi: 10.1530/ERC-12-0191. [DOI] [PubMed] [Google Scholar]
- 109.Aniszewski JP, et al. Cushing syndrome due to ectopic adrenocorticotropic hormone secretion. World J Surg. 2001;25(7):934–940. doi: 10.1007/s00268-001-0032-5. [DOI] [PubMed] [Google Scholar]
- 110.Lamberts SW, et al. A role of (labeled) somatostatin analogs in the differential diagnosis and treatment of Cushing’s syndrome. J Clin Endocrinol Metab. 1994;78(1):17–19. doi: 10.1210/jcem.78.1.7904612. [DOI] [PubMed] [Google Scholar]
- 111.Phlipponneau M, et al. Somatostatin analogs for the localization and preoperative treatment of an adrenocorticotropin-secreting bronchial carcinoid tumor. J Clin Endocrinol Metab. 1994;78(1):20–24. doi: 10.1210/jcem.78.1.7904613. [DOI] [PubMed] [Google Scholar]
- 112.Pivonello R, et al. Dopamine receptor expression and function in corticotroph ectopic tumors. J Clin Endocrinol Metab. 2007;92(1):65–69. doi: 10.1210/jc.2006-0728. [DOI] [PubMed] [Google Scholar]
- 113.Pivonello R, et al. Cabergoline plus lanreotide for ectopic Cushing’s syndrome. N Engl J Med. 2005;352(23):2457–2458. doi: 10.1056/NEJM200506093522322. [DOI] [PubMed] [Google Scholar]
- 114.de Herder WW, et al. Somatostatin receptor scintigraphy: its value in tumor localization in patients with Cushing’s syndrome caused by ectopic corticotropin or corticotropin-releasing hormone secretion. Am J Med. 1994;96(4):305–312. doi: 10.1016/0002-9343(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 115.Baudry C, Paepegaey AC, Groussin L. Reversal of Cushing’s syndrome by vandetanib in medullary thyroid carcinoma. N Engl J Med. 2013;369(6):584–586. doi: 10.1056/NEJMc1301428. [DOI] [PubMed] [Google Scholar]
- 116.Barroso-Sousa R, et al. Complete resolution of hypercortisolism with sorafenib in a patient with advanced medullary thyroid carcinoma and ectopic ACTH (adrenocorticotropic hormone) syndrome. Thyroid. 2014;24(6):1062–1066. doi: 10.1089/thy.2013.0571. [DOI] [PubMed] [Google Scholar]
- 117.Kamenicky P, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2011;96(9):2796–2804. doi: 10.1210/jc.2011-0536. [DOI] [PubMed] [Google Scholar]
- 118.Schulte HM, et al. Infusion of low dose etomidate: correction of hypercortisolemia in patients with Cushing’s syndrome and dose-response relationship in normal subjects. J Clin Endocrinol Metab. 1990;70(5):1426–1430. doi: 10.1210/jcem-70-5-1426. [DOI] [PubMed] [Google Scholar]
- 119.Nieman LK, et al. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(8):2807–2831. doi: 10.1210/jc.2015-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Athanassiadi K, et al. Acromegaly caused by ectopic growth hormone-releasing hormone secretion by a carcinoid bronchial tumor: a rare entity. J Thorac Cardiovasc Surg. 2004;128(4):631–632. doi: 10.1016/j.jtcvs.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 121.Dayal Y, et al. Immunocytochemical demonstration of growth hormone-releasing factor in gastrointestinal and pancreatic endocrine tumors. Am J Clin Pathol. 1986;85(1):13–20. doi: 10.1093/ajcp/85.1.13. [DOI] [PubMed] [Google Scholar]
- 122.Gola M, et al. Neuroendocrine tumors secreting growth hormone-releasing hormone: pathophysiological and clinical aspects. Pituitary. 2006;9(3):221–229. doi: 10.1007/s11102-006-0267-0. [DOI] [PubMed] [Google Scholar]
- 123.Giustina A, et al. Diagnosis and treatment of acromegaly complications. J Endocrinol Invest. 2003;26(12):1242–1247. doi: 10.1007/BF03349164. [DOI] [PubMed] [Google Scholar]
- 124.van Hoek M, et al. Effects of somatostatin analogs on a growth hormone-releasing hormone secreting bronchial carcinoid, in vivo and in vitro studies. J Clin Endocrinol Metab. 2009;94(2):428–433. doi: 10.1210/jc.2008-1712. [DOI] [PubMed] [Google Scholar]
- 125.Van den Bruel A, et al. Hormonal and volumetric long term control of a growth hormone-releasing hormone-producing carcinoid tumor. J Clin Endocrinol Metab. 1999;84(9):3162–3169. doi: 10.1210/jcem.84.9.6001. [DOI] [PubMed] [Google Scholar]
- 126.Rindi G, et al. TNM staging of neoplasms of the endocrine pancreas: results from a large international cohort study. J Natl Cancer Inst. 2012;104(10):764–777. doi: 10.1093/jnci/djs208. [DOI] [PubMed] [Google Scholar]
- 127.Green JR. Bisphosphonates: preclinical review. Oncologist. 2004;9(Suppl 4):3–13. doi: 10.1634/theoncologist.9-90004-3. [DOI] [PubMed] [Google Scholar]
- 128.Loftus LS, Edwards-Bennett S, Sokol GH. Systemic therapy for bone metastases. Cancer Control. 2012;19(2):145–153. doi: 10.1177/107327481201900209. [DOI] [PubMed] [Google Scholar]
- 129.Jensen RT. Carcinoid and pancreatic endocrine tumors: recent advances in molecular pathogenesis, localization, and treatment. Curr Opin Oncol. 2000;12(4):368–377. doi: 10.1097/00001622-200007000-00015. [DOI] [PubMed] [Google Scholar]
- 130.Wangberg B, et al. Somatostatin receptors. A new way to diagnosis and therapy of neuroendocrine tumors. Lakartidningen. 1997;94(10):829–30, 835–8. [PubMed]
- 131.Delle Fave G, et al. ENETS consensus guidelines update for gastroduodenal neuroendocrine neoplasms. Neuroendocrinology. 2016;103(2):119–124. doi: 10.1159/000443168. [DOI] [PubMed] [Google Scholar]
- 132.Rinke A, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 133.Rinke A, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (PROMID): results of long-term survival. Neuroendocrinology. 2017;104(1):26–32. doi: 10.1159/000443612. [DOI] [PubMed] [Google Scholar]
- 134.Caplin ME, Pavel M, Ruszniewski P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(16):1556–1557. doi: 10.1056/NEJMc1409757. [DOI] [PubMed] [Google Scholar]
- 135.Zomerhuis MT, et al. Octreotide exerts only acute, but no sustained, effects on MRI enhancement of liver metastases in carcinoid syndrome. Neuroendocrinology. 2005;82(1):41–48. doi: 10.1159/000090636. [DOI] [PubMed] [Google Scholar]
- 136.Imam H, et al. Induction of apoptosis in neuroendocrine tumors of the digestive system during treatment with somatostatin analogs. Acta Oncol. 1997;36(6):607–614. doi: 10.3109/02841869709001323. [DOI] [PubMed] [Google Scholar]
- 137.Welin SV, et al. High-dose treatment with a long-acting somatostatin analogue in patients with advanced midgut carcinoid tumours. Eur J Endocrinol. 2004;151(1):107–112. doi: 10.1530/eje.0.1510107. [DOI] [PubMed] [Google Scholar]
- 138.Yao JC, et al. Phase I dose-escalation study of long-acting pasireotide in patients with neuroendocrine tumors. Onco Targets Ther. 2017;10:3177–3186. doi: 10.2147/OTT.S128547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cives M, et al. Phase II clinical trial of pasireotide long-acting repeatable in patients with metastatic neuroendocrine tumors. Endocr Relat Cancer. 2015;22(1):1–9. doi: 10.1530/ERC-14-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ferolla P, et al. Efficacy and safety of long-acting pasireotide or everolimus alone or in combination in patients with advanced carcinoids of the lung and thymus (LUNA): an open-label, multicentre, randomised, phase II trial. Lancet Oncol. 2017;18(12):1652–1664. doi: 10.1016/S1470-2045(17)30681-2. [DOI] [PubMed] [Google Scholar]
- 141.Gillis JC, Noble S, Goa KL. Octreotide long-acting release (LAR). A review of its pharmacological properties and therapeutic use in the management of acromegaly. Drugs. 1997;53(4):681–99. [DOI] [PubMed]
- 142.Oberg K, Eriksson B. The role of interferons in the management of carcinoid tumours. Br J Haematol. 1991;79(Suppl 1):74–77. doi: 10.1111/j.1365-2141.1991.tb08125.x. [DOI] [PubMed] [Google Scholar]
- 143.Oberg K, Funa K, Alm G. Effects of leukocyte interferon on clinical symptoms and hormone levels in patients with mid-gut carcinoid tumors and carcinoid syndrome. N Engl J Med. 1983;309(3):129–133. doi: 10.1056/NEJM198307213090301. [DOI] [PubMed] [Google Scholar]
- 144.Plockinger U, et al. Guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumours. A consensus statement on behalf of the European Neuroendocrine Tumour Society (ENETS). Neuroendocrinology. 2004;80(6):394–424. [DOI] [PubMed]
- 145.Kolby L, et al. Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. Br J Surg. 2003;90(6):687–693. doi: 10.1002/bjs.4149. [DOI] [PubMed] [Google Scholar]
- 146.Oberg K. Interferon-alpha versus somatostatin or the combination of both in gastro-enteropancreatic tumours. Digestion. 1996;57(Suppl 1):81–83. doi: 10.1159/000201403. [DOI] [PubMed] [Google Scholar]
- 147.Yao JC, et al. Phase III prospective randomized comparison trial of depot octreotide plus interferon Alfa-2b versus depot octreotide plus bevacizumab in patients with advanced carcinoid tumors: SWOG S0518. J Clin Oncol. 2017;35(15):1695–1703. doi: 10.1200/JCO.2016.70.4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pavel ME, et al. Efficacy and tolerability of pegylated IFN-alpha in patients with neuroendocrine gastroenteropancreatic carcinomas. J Interferon Cytokine Res. 2006;26(1):8–13. doi: 10.1089/jir.2006.26.8. [DOI] [PubMed] [Google Scholar]
- 149.Zandee WT, de Herder WW. The evolution of neuroendocrine tumor treatment reflected by ENETS guidelines. Neuroendocrinology. 2018;106(4):357–365. doi: 10.1159/000486096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cives M, Strosberg J. Radionuclide therapy for neuroendocrine tumors. Curr Oncol Rep. 2017;19(2):9. doi: 10.1007/s11912-017-0567-8. [DOI] [PubMed] [Google Scholar]
- 151.Pool SE, et al. Preclinical and clinical studies of peptide receptor radionuclide therapy. Semin Nucl Med. 2010;40(3):209–218. doi: 10.1053/j.semnuclmed.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 152.Kwekkeboom DJ, et al. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17(1):R53–R73. doi: 10.1677/ERC-09-0078. [DOI] [PubMed] [Google Scholar]
- 153.van Adrichem RC, et al. Is there an additional value of using somatostatin receptor subtype 2a immunohistochemistry compared to somatostatin receptor scintigraphy uptake in predicting gastroenteropancreatic neuroendocrine tumor response? Neuroendocrinology. 2016;103(5):560–566. doi: 10.1159/000441604. [DOI] [PubMed] [Google Scholar]
- 154.Severi S, et al. Peptide receptor radionuclide therapy in the management of gastrointestinal neuroendocrine tumors: efficacy profile, safety, and quality of life. Onco Targets Ther. 2017;10:551–557. doi: 10.2147/OTT.S97584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brabander T, et al. Long-term efficacy, survival, and safety of [(177)Lu-DOTA(0), Tyr(3)]octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin Cancer Res. 2017;23(16):4617–4624. doi: 10.1158/1078-0432.CCR-16-2743. [DOI] [PubMed] [Google Scholar]
- 156.Kwekkeboom DJ, et al. Radiolabeled somatostatin analog [177Lu-DOTA0, Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23(12):2754–2762. doi: 10.1200/JCO.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 157.Ezziddin S, et al. Predictors of long-term outcome in patients with well-differentiated gastroenteropancreatic neuroendocrine tumors after peptide receptor radionuclide therapy with 177Lu-octreotate. J Nucl Med. 2014;55(2):183–190. doi: 10.2967/jnumed.113.125336. [DOI] [PubMed] [Google Scholar]
- 158.Valkema R, et al. Survival and response after peptide receptor radionuclide therapy with [90Y-DOTA0, Tyr3]octreotide in patients with advanced gastroenteropancreatic neuroendocrine tumors. Semin Nucl Med. 2006;36(2):147–156. doi: 10.1053/j.semnuclmed.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 159.Bushnell DL, Jr, et al. 90Y-edotreotide for metastatic carcinoid refractory to octreotide. J Clin Oncol. 2010;28(10):1652–1659. doi: 10.1200/JCO.2009.22.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Imhof A, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29(17):2416–2423. doi: 10.1200/JCO.2010.33.7873. [DOI] [PubMed] [Google Scholar]
- 161.Bodei L, et al. Peptide receptor radionuclide therapy with (1)(7)(7)Lu-DOTATATE: the IEO phase I-II study. Eur J Nucl Med Mol Imaging. 2011;38(12):2125–2135. doi: 10.1007/s00259-011-1902-1. [DOI] [PubMed] [Google Scholar]
- 162.Sabet A, et al. Specific efficacy of peptide receptor radionuclide therapy with (177)Lu-octreotate in advanced neuroendocrine tumours of the small intestine. Eur J Nucl Med Mol Imaging. 2015;42(8):1238–1246. doi: 10.1007/s00259-015-3041-6. [DOI] [PubMed] [Google Scholar]
- 163.Paganelli G, et al. 177 Lu-Dota-octreotate radionuclide therapy of advanced gastrointestinal neuroendocrine tumors: results from a phase II study. Eur J Nucl Med Mol Imaging. 2014;41(10):1845–1851. doi: 10.1007/s00259-014-2735-5. [DOI] [PubMed] [Google Scholar]
- 164.Kim SJ, et al. The efficacy of (177)Lu-labelled peptide receptor radionuclide therapy in patients with neuroendocrine tumours: a meta-analysis. Eur J Nucl Med Mol Imaging. 2015;42(13):1964–1970. doi: 10.1007/s00259-015-3155-x. [DOI] [PubMed] [Google Scholar]
- 165.Khan S, et al. Quality of life in 265 patients with gastroenteropancreatic or bronchial neuroendocrine tumors treated with [177Lu-DOTA0, Tyr3]octreotate. J Nucl Med. 2011;52(9):1361–1368. doi: 10.2967/jnumed.111.087932. [DOI] [PubMed] [Google Scholar]
- 166.Strosberg J, et al. Phase 3 trial of (177)Lu-dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–135. doi: 10.1056/NEJMoa1607427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ianniello A, et al. Peptide receptor radionuclide therapy with (177)Lu-DOTATATE in advanced bronchial carcinoids: prognostic role of thyroid transcription factor 1 and (18)F-FDG PET. Eur J Nucl Med Mol Imaging. 2016;43(6):1040–1046. doi: 10.1007/s00259-015-3262-8. [DOI] [PubMed] [Google Scholar]
- 168.Severi S, et al. Feasibility and utility of re-treatment with (177)Lu-DOTATATE in GEP-NENs relapsed after treatment with (90)Y-DOTATOC. Eur J Nucl Med Mol Imaging. 2015;42(13):1955–1963. doi: 10.1007/s00259-015-3105-7. [DOI] [PubMed] [Google Scholar]
- 169.Forrer F, et al. Treatment with 177Lu-DOTATOC of patients with relapse of neuroendocrine tumors after treatment with 90Y-DOTATOC. J Nucl Med. 2005;46(8):1310–1316. [PubMed] [Google Scholar]
- 170.Claringbold PG, et al. Phase II study of radiopeptide 177Lu-octreotate and capecitabine therapy of progressive disseminated neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2011;38(2):302–311. doi: 10.1007/s00259-010-1631-x. [DOI] [PubMed] [Google Scholar]
- 171.Claringbold PG, Price RA, Turner JH. Phase I-II study of radiopeptide 177Lu-octreotate in combination with capecitabine and temozolomide in advanced low-grade neuroendocrine tumors. Cancer Biother Radiopharm. 2012;27(9):561–569. doi: 10.1089/cbr.2012.1276. [DOI] [PubMed] [Google Scholar]
- 172.Kong G, et al. Assessment of predictors of response and long-term survival of patients with neuroendocrine tumour treated with peptide receptor chemoradionuclide therapy (PRCRT) Eur J Nucl Med Mol Imaging. 2014;41(10):1831–1844. doi: 10.1007/s00259-014-2788-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Claringbold PG, Turner JH. NeuroEndocrine tumor therapy with lutetium-177-octreotate and everolimus (NETTLE): a phase I study. Cancer Biother Radiopharm. 2015;30(6):261–269. doi: 10.1089/cbr.2015.1876. [DOI] [PubMed] [Google Scholar]
- 174.Barber TW, et al. The potential for induction peptide receptor chemoradionuclide therapy to render inoperable pancreatic and duodenal neuroendocrine tumours resectable. Eur J Surg Oncol. 2012;38(1):64–71. doi: 10.1016/j.ejso.2011.08.129. [DOI] [PubMed] [Google Scholar]
- 175.Ezziddin S, et al. Neoadjuvant downsizing by internal radiation: a case for preoperative peptide receptor radionuclide therapy in patients with pancreatic neuroendocrine tumors. Clin Nucl Med. 2012;37(1):102–104. doi: 10.1097/RLU.0b013e318238f111. [DOI] [PubMed] [Google Scholar]
- 176.van Vliet EI, et al. Neoadjuvant treatment of nonfunctioning pancreatic neuroendocrine tumors with [177Lu-DOTA0, Tyr3]octreotate. J Nucl Med. 2015;56(11):1647–1653. doi: 10.2967/jnumed.115.158899. [DOI] [PubMed] [Google Scholar]
- 177.Brabander T, et al. Pitfalls in the response evaluation after peptide receptor radionuclide therapy with [(177)Lu-DOTA(0), Tyr(3)]octreotate. Endocr Relat Cancer. 2017;24(5):243–251. doi: 10.1530/ERC-16-0524. [DOI] [PubMed] [Google Scholar]
- 178.Ginj M, et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci USA. 2006;103(44):16436–16441. doi: 10.1073/pnas.0607761103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nonnekens J, et al. Potentiation of peptide receptor radionuclide therapy by the PARP inhibitor olaparib. Theranostics. 2016;6(11):1821–1832. doi: 10.7150/thno.15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bodei L, et al. Measurement of circulating transcripts and gene cluster analysis predicts and defines therapeutic efficacy of peptide receptor radionuclide therapy (PRRT) in neuroendocrine tumors. Eur J Nucl Med Mol Imaging. 2016;43(5):839–851. doi: 10.1007/s00259-015-3250-z. [DOI] [PubMed] [Google Scholar]
- 181.Kidd M, Modlin IM. Therapy: the role of liquid biopsies to manage and predict PRRT for NETs. Nat Rev Gastroenterol Hepatol. 2017;14(6):331–332. doi: 10.1038/nrgastro.2017.26. [DOI] [PubMed] [Google Scholar]
- 182.Bodei L, et al. PRRT genomic signature in blood for prediction of (177)Lu-octreotate efficacy. Eur J Nucl Med Mol Imaging. 2018;45(7):1155–1169. doi: 10.1007/s00259-018-3967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bodei L, Pepe G, Paganelli G. Peptide receptor radionuclide therapy (PRRT) of neuroendocrine tumors with somatostatin analogues. Eur Rev Med Pharmacol Sci. 2010;14(4):347–351. [PubMed] [Google Scholar]
- 184.Yao JC, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. 2016;34(32):3906–3913. doi: 10.1200/JCO.2016.68.0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lombard-Bohas C, et al. Impact of prior chemotherapy use on the efficacy of everolimus in patients with advanced pancreatic neuroendocrine tumors: a subgroup analysis of the phase III RADIANT-3 trial. Pancreas. 2015;44(2):181–189. doi: 10.1097/MPA.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Anthony LB, et al. Impact of previous somatostatin analogue use on the activity of everolimus in patients with advanced neuroendocrine tumors: analysis from the phase III RADIANT-2 trial. Neuroendocrinology. 2015;102(1–2):18–25. doi: 10.1159/000381715. [DOI] [PubMed] [Google Scholar]
- 187.Yao JC, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase III study. Lancet. 2016;387(10022):968–977. doi: 10.1016/S0140-6736(15)00817-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pavel ME, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase III study. Lancet. 2011;378(9808):2005–2012. doi: 10.1016/S0140-6736(11)61742-X. [DOI] [PubMed] [Google Scholar]
- 189.Fazio N, et al. Everolimus plus octreotide long-acting repeatable in patients with advanced lung neuroendocrine tumors: analysis of the phase III, randomized, placebo-controlled RADIANT-2 study. Chest. 2013;143(4):955–962. doi: 10.1378/chest.12-1108. [DOI] [PubMed] [Google Scholar]
- 190.Castellano D, et al. Everolimus plus octreotide long-acting repeatable in patients with colorectal neuroendocrine tumors: a subgroup analysis of the phase III RADIANT-2 study. Oncologist. 2013;18(1):46–53. doi: 10.1634/theoncologist.2012-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ferolla P. Everolimus and pasireotide LAR alone or in combination significantly improved outcome in patients with advanced lung and thymic carcinoids. In: ESMO conference 2016. Abstract. 2016.
- 192.Bago-Horvath Z, et al. Synergistic effects of erlotinib and everolimus on bronchial carcinoids and large-cell neuroendocrine carcinomas with activated EGFR/AKT/mTOR pathway. Neuroendocrinology. 2012;96(3):228–237. doi: 10.1159/000337257. [DOI] [PubMed] [Google Scholar]
- 193.Wiedmann MW, Mossner J. Safety and efficacy of sunitinib in patients with unresectable pancreatic neuroendocrine tumors. Clin Med Insights Oncol. 2012;6:381–393. doi: 10.4137/CMO.S7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kulke MH, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26(20):3403–3410. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
- 195.Faivre S, et al. Sunitinib in pancreatic neuroendocrine tumors: updated progression-free survival and final overall survival from a phase III randomized study. Ann Oncol. 2017;28(2):339–343. doi: 10.1093/annonc/mdw561. [DOI] [PubMed] [Google Scholar]
- 196.Faivre S, et al. Imaging response in neuroendocrine tumors treated with targeted therapies: the experience of sunitinib. Target Oncol. 2012;7(2):127–133. doi: 10.1007/s11523-012-0216-y. [DOI] [PubMed] [Google Scholar]
- 197.Valle JW, et al. A systematic review of non-surgical treatments for pancreatic neuroendocrine tumours. Cancer Treat Rev. 2014;40(3):376–389. doi: 10.1016/j.ctrv.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 198.Castellano D, et al. Sorafenib and bevacizumab combination targeted therapy in advanced neuroendocrine tumour: a phase II study of Spanish Neuroendocrine Tumour Group (GETNE0801) Eur J Cancer. 2013;49(18):3780–3787. doi: 10.1016/j.ejca.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 199.Kulke MH, Niedzwiecki D, Foster NR, et al. Randomized phase II study of everolimus (E) versus everolimus plus bevacizumab (E + B) in patients (Pts) with locally advanced or metastatic pancreatic neuroendocrine tumors (pNET), CALGB 80701 (Alliance). J Clin Oncol. 2015;33(15) (suppl; abstr 4005).
- 200.Chan JA, et al. Phase I study of sorafenib in combination with everolimus (RAD001) in patients with advanced neuroendocrine tumors. Cancer Chemother Pharmacol. 2013;71(5):1241–1246. doi: 10.1007/s00280-013-2118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Phan AT, et al. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: a multicentre, single-group, phase II study. Lancet Oncol. 2015;16(6):695–703. doi: 10.1016/S1470-2045(15)70136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fazio N, Spada F, Giovannini M. Chemotherapy in gastroenteropancreatic (GEP) neuroendocrine carcinomas (NEC): a critical view. Cancer Treat Rev. 2013;39(3):270–274. doi: 10.1016/j.ctrv.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 203.Auernhammer CJ, et al. Advanced neuroendocrine tumours of the small intestine and pancreas: clinical developments, controversies, and future strategies. Lancet Diabetes Endocrinol. 2018;6(5):404–415. doi: 10.1016/S2213-8587(17)30401-1. [DOI] [PubMed] [Google Scholar]
- 204.Kunz PL, et al. Oxaliplatin-fluoropyrimidine chemotherapy plus bevacizumab in advanced neuroendocrine tumors: an analysis of 2 phase II trials. Pancreas. 2016;45(10):1394–1400. doi: 10.1097/MPA.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 205.Aristizabal Prada ET, Auernhammer CJ. Targeted therapy of gastroenteropancreatic neuroendocrine tumours: preclinical strategies and future targets. Endocr Connect. 2018;7(1):R1–R25. doi: 10.1530/EC-17-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pennock GK, Chow LQ. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist. 2015;20(7):812–822. doi: 10.1634/theoncologist.2014-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chauhan A, et al. Immune checkpoint inhibitors in neuroendocrine tumors: a single institution experience with review of literature. Oncotarget. 2018;9(10):8801–8809. doi: 10.18632/oncotarget.23753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kim ST, et al. The impact of PD-L1 expression in patients with metastatic GEP-NETs. J Cancer. 2016;7(5):484–489. doi: 10.7150/jca.13711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nomi T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13(7):2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 211.Badoual C, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73(1):128–138. doi: 10.1158/0008-5472.CAN-12-2606. [DOI] [PubMed] [Google Scholar]
- 212.Droeser RA, et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur J Cancer. 2013;49(9):2233–2242. doi: 10.1016/j.ejca.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 213.Tsuruoka K, et al. PD-L1 expression in neuroendocrine tumors of the lung. Lung Cancer. 2017;108:115–120. doi: 10.1016/j.lungcan.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 214.Lamarca A, et al. PD-L1 expression and presence of TILs in small intestinal neuroendocrine tumours. Oncotarget. 2018;9(19):14922–14938. doi: 10.18632/oncotarget.24464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pinato D, Brown M, White S, Zhang H, Toloue S, Dina R, Trivedi R, Stebbing J, Mauri F, Sharma R. Programmed cell death (PD-1) ligands expression in gastro-entero-pancreatic neuroendocrine tumours (GEP-NETs): relationship with angiogenesis and clinical outcome. J Clin Oncol. 2016;34(15_suppl):e15658.
- 216.Fan Y, et al. Prognostic value of PD-L1 and PD-1 expression in pulmonary neuroendocrine tumors. Onco Targets Ther. 2016;9:6075–6082. doi: 10.2147/OTT.S115054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Reck M, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II trial. Ann Oncol. 2013;24(1):75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 218.Tarhini A, Lo E, Minor DR. Releasing the brake on the immune system: ipilimumab in melanoma and other tumors. Cancer Biother Radiopharm. 2010;25(6):601–613. doi: 10.1089/cbr.2010.0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Katz SC, et al. T cell infiltrate and outcome following resection of intermediate-grade primary neuroendocrine tumours and liver metastases. HPB (Oxford) 2010;12(10):674–683. doi: 10.1111/j.1477-2574.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cavalcanti E, et al. Role of PD-L1 expression as a biomarker for GEP neuroendocrine neoplasm grading. Cell Death Dis. 2017;8(8):e3004. doi: 10.1038/cddis.2017.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Culler MD. Somatostatin-dopamine chimeras: a novel approach to treatment of neuroendocrine tumors. Horm Metab Res. 2011;43(12):854–857. doi: 10.1055/s-0031-1287769. [DOI] [PubMed] [Google Scholar]
- 222.Rocheville M, et al. Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. J Biol Chem. 2000;275(11):7862–7869. doi: 10.1074/jbc.275.11.7862. [DOI] [PubMed] [Google Scholar]
- 223.Rocheville M, et al. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science. 2000;288(5463):154–157. doi: 10.1126/science.288.5463.154. [DOI] [PubMed] [Google Scholar]
- 224.Couvelard A, et al. Antisecretory effects of chimeric somatostatin/dopamine receptor ligands on gastroenteropancreatic neuroendocrine tumors. Pancreas. 2017;46(5):631–638. doi: 10.1097/MPA.0000000000000813. [DOI] [PubMed] [Google Scholar]