

Abstract
Heterosis can increase the yield of many crops and has been extensively applied in agriculture. In maize, female inflorescence architecture directly determines grain yield. Thus, exploring the relationship between early maize ear inflorescence development and heterosis regarding yield-related traits may be helpful for characterizing the molecular mechanisms underlying heterotic performance. In this study, we fine mapped the overdominant heterotic locus (hlEW2b), associated with ear width, in an approximately 1.98-Mb region based on analyses of chromosome segment substitution lines and the corresponding testcross population. Maize ear inflorescences at the floral meristem stage were collected from two inbred lines, one chromosome segment substitution line that carried hlEW2b (sub-CSSL16), the receptor parent lx9801, and the Zheng58 × sub-CSSL16 and Zheng58 × lx9801 hybrid lines. A total of 256 metabolites were identified, including 31 and 24 metabolites that were differentially accumulated between the two hybrid lines and between the two inbred lines, respectively. Most of these metabolites are involved in complex regulatory mechanisms important for maize ear development. For example, nucleotides are basic metabolites affecting cell composition and carbohydrate synthesis. Additionally, nicotinate and nicotinamide metabolism is important for photosynthesis, plant stress responses, and cell expansion. Moreover, flavonoid and phenolic metabolites regulate auxin transport and cell apoptosis. Meanwhile, phytohormone biosynthesis and distribution influence the cell cycle and cell proliferation. Our results revealed that changes in metabolite contents may affect the heterotic performance related to ear width and yield in maize hybrid lines. This study provides new clues in heterosis at the metabolomics level and implies that differentially accumulated metabolites made distinct contributions to the heterosis at an early stage of ear inflorescences development.
Introduction
Heterosis refers to the phenomenon that hybrids perform better than their parents regarding phenotypic traits, including biomass, development rate, fertility, nutrient quality, disease resistance, and crop yield1,2. Heterosis can increase grain yield and has been applied to improve agricultural production for a few hundred years. Three classical genetic hypotheses (i.e., dominance hypothesis, overdominance hypothesis, and epistasis hypothesis) were proposed early last century to explain the genetic basis of heterosis. The overdominance hypothesis has been validated using the SINGLE FLOWER TRUSS (SFT) locus of tomato3. However, these hypotheses did not end the century-long debate regarding whether they fully explain the genetic basis of heterosis because they represent largely conceptual models and do not describe the underlying molecular mechanism. Advances in high-throughput technologies have enabled researchers to develop new strategies for dissecting the genetic or molecular mechanism regulating heterosis. However, multiple omics-based researches (i.e., genomics, transcriptomics and epigenetics) in plants for hybrids and their parents, have been conducted to explore the cause of heterosis, the genetic mechanism mediating heterosis remains unclear up to now4–6.
Genomics-based studies revealed that the complementation of two parental genomes makes an important contribution to heterosis7,8. Moreover, based on transcriptomics and proteomics, additive and non-additive effects have been estimated between parents and hybrids9,10. Due to a large amount of redundant information was generated along with the high-throughput data obtained by genomics, transcriptomics and proteomics technologies, making it even more difficult to elucidate the genetic mechanism of heterosis at one or multiple loci. As so far, at the single-locus level, SFT is the first identified overdominance gene associated with heterosis, has been cloned in tomato3. Although considerable efforts have been made to dissect the genetic basis of heterosis in plants, there is no appropriate method for analyzing heterotic performance because of the complexity of genetic backgrounds and controversies regarding the underlying mechanism.
To eliminate the influence of genetic backgrounds, backcross populations of chromosome segment substitution lines (CSSLs) were developed and used for characterizing the relationship between heterosis and grain yield as well as the associated components in tomato and rice11,12. Compared with other populations, such as recombinant inbred lines (RIL) or doubled haploid populations, CSSLs enable the identification of the chromosomal segment responsible for heterotic effects under the same genetic background in multiple environments. Testcross populations have been widely applied to detect quantitative trait loci associated with heterosis13,14. For example, Wang et al.12 used 66 testcross F1 lines developed from the corresponding CSSLs to demonstrate that partial dominance and overdominance represent the genetic basis of the heterosis of yield and yield-related traits in rice hybrids. Using a set of 202 CSSLs and the corresponding testcross population, 15 heterotic loci (HLs) for plant height were detected in rice13. Integrating the backcross and testcross populations of CSSLs may clarify the genetic and biological basis of heterosis.
Recent metabolomics-based investigations have more thoroughly clarified the metabolic activities of many crops, with potential implications for breeding new high-yielding varieties with improved nutritional composition. Metabolites are useful for efficiently predicting the hybrid performance of several crops, including maize and rice15,16. However, relatively few studies comparing the concentrations of secondary metabolites in hybrids and their parents have been conducted in maize, especially involving two inbred lines (one origin inbred line and its progeny with a single chromosome segment from a backcross), with the corresponding hybrids crossed with a common test parent.
Heterosis of ear traits has significant positive correlations with maize yield17. Ear traits including ear length, ear width, kernel row number and kernel number per row, are determined in developing immature maize ears18,19. During ear inflorescence development, the floret meristem (FM) stage is critical for ear development and heterosis20. Consequently, this stage is ideal for studying heterosis. Furthermore, metabolites represent the basic compounds accumulating in plant organs that influence the phenotype. However, there is very little or no available information regarding the relationship between secondary metabolites and heterosis. Thus, analyzing the secondary metabolites in hybrids and their parents may provide new insights and clarify the genetic mechanism underlying heterosis. In this study, we collected immature maize ears during the FM stage from two inbred lines [i.e., sub-CSSL16 containing an heterotic locus (HL) associated with ear width and its receptor parent lx9801] and their corresponding hybrids crossed with a common tester, Zheng58. We integrated the results of our targeted metabolomics approach with relevant gene expression data to (1) explore the metabolic processes involved in early maize ear development; (2) identify metabolites associated with heterosis; (3) construct a metabolites interaction network explaining heterotic performance. These results may help to elucidate the mechanism underlying heterotic performance at the metabolomics level.
Results
Fine mapping the heterotic locus hlEW2b
We previously mapped hlEW2b on chromosome 2 between markers umc1555 and umc1465, and determined that it accounts for 8.5% of the ear width heterosis over the standard control (Zheng58 × lx9801)21. To more precisely locate hlEW2b, 152 homozygous sub-CSSLs were identified among 1,652 F2 individuals derived from the CSSL125 × lx9801 cross by genotyping with 12 new polymorphism markers within the hlEW2b region. Meanwhile, a test population was generated by crossing 152 homozygous sub-CSSLs with the test parent Zheng58 (Fig. 1). We compared the ear width of Zheng58 × lx9801 with that of the test population. A total of 37 homozygous recombinant lines carrying the hlEW2b-containing donor genomic segment between SSR markers 7HY-304 and umc1024 had ears that were wider than those of Zheng58 × lx9801 (P < 0.01). Screening with molecular markers resulted in the mapping of hlEW2b to a 1.98-Mb region between 7HY-304 and 1HY-100 (see Supplementary Fig. S1).
Figure 1.

Development of a CSSL testcross population and identification of heterotic loci. The Zheng58 × CSSL125 population exhibited significant heterosis for maize ear width. The sub-CSSL population was constructed from 152 homozygous sub-CSSL F2 plants, which were derived from a cross between CSSL125 and lx9801. The corresponding test population was constructed by crossing the test parent Zheng58 with sub-CSSLs. Red, green, and blue represent the genomes of lx9801 (receptor parent), Chang7-2 (donor parent), and Zheng58 (test parent), respectively.
A total of 42 candidate genes, including 30 functional genes coding proteins, 3 transcription factors and 8 unknown functional genes, were found in this mapping region (Supplementary Table S1). Three genes, Zm00001d002896, Zm00001d002929 and Zm00001d002934 encodes TUB-transcription factor, auxin response factor and NAC domain-transcription factor, respectively. Additionally, Zm00001d002882 encodes SKP1-interacting partner and Zm00001d002933 encodes DDB1-, CUL4-associated factor has the same functions, all of which are component of the E3 ubiquitin-protein ligase complex22. One genes Zm00001d002888 [encoding Medicago truncatula NODULIN 21 (MtN21)] was a homolog of Zm00001d002893 [encoding Walls-Are-Thin 1 (WAT1) related protein], which could disrupt auxin transport to regulating cell elongation23. Moreover, several candidate genes encode some important enzymes, for example, Zm00001d002880 encodes an isopropylmalate dehydrogenase, Zm00001d002889 encodes a soluble inorganic pyrophosphatase, Zm00001d002922 encode an inositol polyphosphate multi kinase beta, Zm00001d002912, Zm00001d002916 encode the glyoxal oxidase-related protein and five candidate genes (Zm00001d002897, Zm00001d002898, Zm00001d002899, Zm00001d002901 and Zm00001d002902) encode the peroxidase. These candidate genes maybe contribute to heterosis, more work needed to narrow down the mapping interval and confirm the functional gene. Furthermore, according to the heterosis for ear width and the genotype of the homozygous recombinant lines, one sub-CSSL16 line with the shortest donor chromosomal segment (1.98 Mb) harboring hlEW2b was selected for a metabolomics analysis.
Phenotype and heterosis effects analysis
Phenotypic analyses in four environments revealed significant differences in ear width (P < 0.01) between the hybrids of Zheng58 × sub-CSSL16 and the Zheng58 × lx9801, while there were no significant differences in other ear related traits (including ear length, kernel row number and kernel number per row) (Table 1). Additionally, there were no significant differences in all five ear traits between the inbred lines lx9801 and the sub-CSSL16 in two environments (Supplementary Table S2). In order to characterize the genetic effect of HL (hlEW2b), we calculated the additive and dominant value of HL (hlEW2b) based on the following equations: a = (P1–P2)/2 and d = F1−(P1 + P2)/2, here, a is the additive effect, d is the dominant effect, P1 refers to the trait value of Zheng58, P2 stands for the trait value of sub-CSSL16, and F1 refers to the value of the trait for the hybrid Zheng58 × sub-CSSL16). Theoretically, a ratio of estimated dominance to the absolute value of additive effect (d/a) >1, =1, <1 were regarded as overdominance, dominance and partial-dominance, respectively24. Our results revealed that the HL (hlEW2b) of ear width exhibits high overdominance (d/a»1, d = 4.19, a = 0.07) (Supplementary Table S3). Thus, overdominance is the most contributor to heterosis of ear width.
Table 1.
Maize ear traits of Zheng58 × lx9801 and Zheng58 × sub-CSSL16 in four environments.
| Trait | Environments | Hybrids | ||
|---|---|---|---|---|
| Zheng58 × lx9801 | Zheng58 × sub-CSSL16 | P-value | ||
| Ear width(mm) | E1 | 50.71 ± 1.00 | 52.70 ± 1.30** | 1.17E-03 |
| E2 | 46.16 ± 0.96 | 47.64 ± 1.32* | 1.04E-02 | |
| E3 | 51.24 ± 0.65 | 53.08 ± 1.11** | 2.65E-04 | |
| E4 | 50.09 ± 1.43 | 53.12 ± 1.26** | 8.62E-05 | |
| Ear length (cm) | E1 | 19.18 ± 0.77 | 19.25 ± 0.78 | 8.43E-01 |
| E2 | 18.56 ± 1.14 | 19.10 ± 0.75 | 2.27E-01 | |
| E3 | 19.44 ± 0.85 | 18.86 ± 1.30 | 9.14E-02 | |
| E4 | 18.35 ± 1.49 | 17.58 ± 0.93 | 1.83E-01 | |
| Kernel row number | E1 | 13.80 ± 1.48 | 14.40 ± 0.84 | 2.79E-01 |
| E2 | 13.60 ± 1.26 | 13.40 ± 1.65 | 7.64E-01 | |
| E3 | 13.40 ± 1.65 | 13.20 ± 1.40 | 7.73E-01 | |
| E4 | 12.80 ± 1.03 | 13.20 ± 1.03 | 3.98E-01 | |
| Kernel number per row | E1 | 36.00 ± 3.02 | 34.60 ± 1.65 | 2.14E-01 |
| E2 | 38.70 ± 1.16 | 37.20 ± 2.35 | 8.68E-02 | |
| E3 | 38.70 ± 1.89 | 37.30 ± 2.36 | 1.60E-01 | |
| E4 | 37.20 ± 1.99 | 38.40 ± 2.01 | 1.96E-01 | |
| Ear weight (g) | E1 | 181.00 ± 22.35 | 204.00 ± 10.75** | 8.87E-03 |
| E2 | 118.93 ± 5.79 | 125.45 ± 6.38* | 2.78E-02 | |
| E3 | 211.00 ± 13.70 | 225.00 ± 14.34* | 3.85E-02 | |
| E4 | 195.00 ± 13.54 | 216.00 ± 24.13* | 2.74E-02 | |
E1, E2, E3, and E4 represent Xinxiang and Hebi in 2015 and Xinxiang and Hebi in 2016, respectively.
*, ** indicate significant differences at the 0.05 and 0.01 probability levels, respectively.
Global metabolomics
Considerable differences in the secondary metabolite profiles of ears collected at the FM stage were observed (see Supplementary Fig. S2). A total of 256 metabolites were identified in each hybrid and inbred line. A PCA was completed to objectively interpret the metabolomics data for the analyzed hybrids and inbred lines. All samples could be divided into four groups based on the 256 metabolites (Fig. 2). Moreover, 42.4% of the variance in the total data was explained by the first two principal components. An obvious separation between the inbred lines and their corresponding hybrids was detected for PC1, which explained 28.6% of the variance. A clear separation between hybrids was also observed for PC2, which explained 13.8% of the variance. Moreover, two inbred lines, sub-CSSL16 and lx9801, were distributed together along the Y-axis. The PCA model also revealed a generally good biological reproducibility within groups because the three biological replicates of each sample exhibited satisfactory clustering. The general trends for Zheng58 × sub-CSSL16 and Zheng58 × lx9801 indicated their metabolite profiles were similar. This observation was supported by the fact Zheng58 × sub-CSSL16 and Zheng58 × lx9801 were clustered in one subclass (see Supplementary Fig. S2). Moreover, two inbred lines, lx9801 and sub-CSSL16, clustered more with each other than with the hybrid lines (see Supplementary Fig. S2). These results implied that the metabolic activities of the hybrids and inbred lines are likely different during the early stage of maize ear development.
Figure 2.
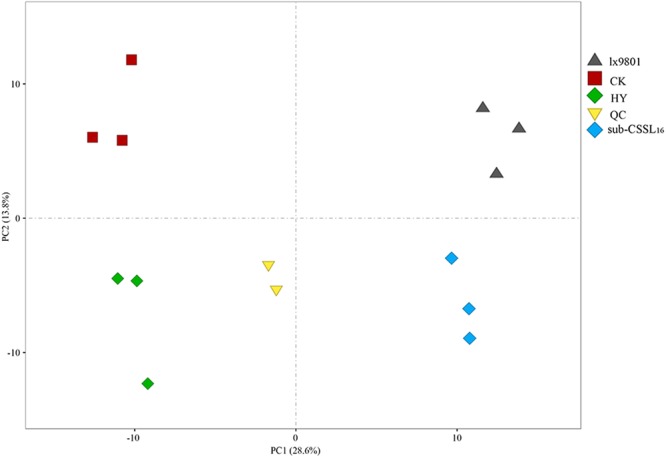
Principal component analysis score plots. The first two principal component (PCs) explained 42.4% of the variation. Additionally, PC1 separated the hybrid lines from the inbred lines, and explained 28.6% of the variation. Meanwhile, PC2 explained 13.8% of the variation, indicating the hybrid lines could be discriminated from the inbred lines. Three biological replicates of each sample were clustered in the same quadrant. HY: Zheng58 × sub-CSSL16; CK: Zheng58 × lx9801.
Metabolite-specific accumulation
Partial least-squares-discriminant analysis (PLS-DA) model is a statistical method for supervised classification that can be exploited to predict the group among new and unlabeled samples25. Thus, the PLS-DA supervised multivariate analysis was used to maximize the variance among groups and identify specific metabolites. We applied this model to identify the metabolites that were differentially abundant in the hybrid lines. On the basis of the PLS-DA score plots, we observed an obvious separation between the hybrids regarding the first component (R2X = 0.478, R2Y = 0.997, Q2 = 0.749) (Fig. 3A). This observation indicated the inter-group differences were significant. Additionally, 31 metabolites were differentially abundant between Zheng58 × sub-CSSL16 and Zheng58 × lx9801 (Table 2). Moreover, the PLS-DA results for the two inbred lines revealed that sub-CSSL16 was markedly different from lx9801 (R2X = 0.702, R2Y = 0.999, and Q2 = 0.974) (Fig. 3B). A total of 24 differentially abundant metabolites were identified based on the VIP and fold-change ratio thresholds (Table 3).
Figure 3.
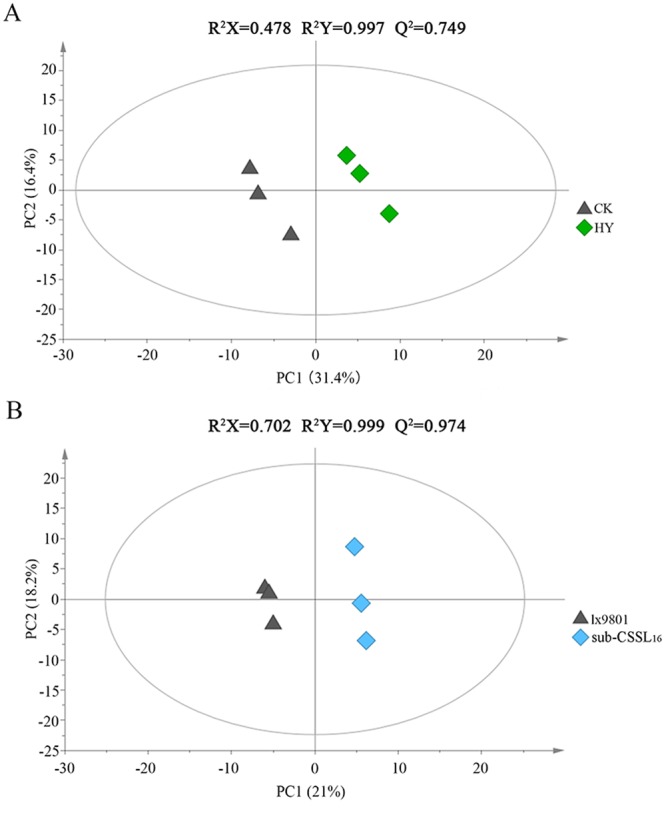
Partial least squares discriminant scores plot. (A) Principal component 1 (PC1) and PC2 explained 31.4% and 16.4% of the variation, respectively, indicating Zheng58 × sub-CSSL16 (HY) could be discriminated from Zheng58 × lx9801 (CK). (B) PC1 and PC2 explained 21% and 18.2% of the variation, illustrating the separation between sub-CSSL16 and lx9801. R2X, R2Y, and Q2 refer to the interpretation and predictability values of the partial least squares discriminant analysis models.
Table 2.
Identified metabolites significantly differentially accumulated in Zheng58 × lx9801 and Zheng58 × sub-CSSL16.
| Compound name | Zheng58 × lx9801 | Zheng58 × sub-CSSL16 | Fold change | VIPa | ||
|---|---|---|---|---|---|---|
| ALKALOIDS | Papaverine | 4.50E + 04 | 2.88E + 04 | 0.64 | 1.38 | down |
| Trigonelline | 3.80E + 05 | 6.42E + 05 | 1.69 | 1.84 | up | |
| AMINO ACID DERIVATIVES | Aspartic acid O-rutinoside | 7.12E + 05 | 4.41E + 05 | 0.62 | 1.73 | down |
| Phenylglycine | 1.28E + 06 | 3.21E + 06 | 2.52 | 2.17 | up | |
| 3-(2-Naphthyl)-D-alanine | 3.81E + 04 | 1.65E + 05 | 4.33 | 2.79 | up | |
| Homocystine | 2.49E + 06 | 4.24E + 06 | 1.70 | 1.61 | up | |
| CARBOHYDRATES | D(-)-Threose | 1.07E + 04 | 1.67E + 04 | 1.56 | 1.29 | up |
| Sucrose | 2.31E + 05 | 4.99E + 05 | 2.16 | 2.29 | up | |
| CHOLINES | Carbachol | 1.41E + 04 | 2.95E + 04 | 2.09 | 2.13 | up |
| COUMARINS AND ITS DERIVATIVES | 4-Methylumbelliferone | 4.74E + 04 | 3.00E + 04 | 0.63 | 1.39 | down |
| FLAVONOID | Tricetin | 1.42E + 05 | 3.16E + 03 | 0.02 | 2.40 | down |
| C-hexosyl-apigenin C-pentoside | 3.76E + 03 | 2.43E + 03 | 0.65 | 1.53 | down | |
| Quercetin O-rutinoside | 2.74E + 03 | 4.24E + 03 | 1.55 | 1.52 | up | |
| LIPIDS-GLYCEROPHOSPHOLIPID | LysoPC 20:1 | 4.08E + 05 | 6.40E + 05 | 1.57 | 1.66 | up |
| LysoPC 20:2 | 1.40E + 05 | 2.34E + 05 | 1.67 | 1.64 | up | |
| NUCLEOTIDE AND ITS DERIVATES | beta-Nicotinamide adenine dinucleotide | 3.53E + 05 | 2.29E + 05 | 0.65 | 1.41 | down |
| Inosine | 2.01E + 05 | 1.33E + 05 | 0.66 | 1.48 | down | |
| Guanine | 1.09E + 05 | 1.77E + 05 | 1.62 | 1.58 | up | |
| 2′-Deoxyadenosine monohydrate | 8.56E + 05 | 1.67E + 06 | 1.96 | 1.82 | up | |
| Guanosine 5′-monophosphate | 5.23E + 04 | 1.04E + 05 | 1.99 | 1.84 | up | |
| Thymine | 1.57E + 05 | 3.48E + 05 | 2.22 | 1.93 | up | |
| PHENYLPROPANOIDS | Sinapoyl O-hexoside | 8.90E + 04 | 2.00E + 05 | 2.25 | 1.96 | up |
| Sinapic acid | 2.04E + 04 | 4.81E + 04 | 2.36 | 2.25 | up | |
| Geranyl acetate | 1.32E + 04 | 4.74E + 04 | 3.60 | 2.95 | up | |
| PHYTOHORMONES | Kinetin riboside | 4.68E + 04 | 7.64E + 04 | 1.63 | 1.67 | up |
| trans-Zeatin riboside (tZR) | 4.02E + 03 | 6.98E + 03 | 1.74 | 1.82 | up | |
| TERPENOIDS | Phytocassane C | 9.95E + 03 | 1.72E + 04 | 1.73 | 1.96 | up |
| Gentiopicrin | 9.36E + 03 | 2.37E + 04 | 2.53 | 2.35 | up | |
| TRYPTAMINES AND ITS DERIVATIVES | N-acetyltryptamine | 1.71E + 03 | 2.56E + 03 | 1.50 | 1.33 | up |
| VITAMINE RELATED | 4-Pyridoxic acid O-hexoside | 1.16E + 04 | 1.84E + 04 | 1.59 | 1.75 | up |
| Nicotinamide | 3.18E + 03 | 5.43E + 03 | 1.71 | 1.46 | up |
Variable importance in the projection (VIP) values were obtained from PLS-DA with the threshold of 1.00.
Table 3.
Identified metabolites significantly differentially accumulated in lx9801 and sub-CSSL16.
| Compound name | lx9801 | sub-CSSL16 | Fold change | VIPa | ||
|---|---|---|---|---|---|---|
| AMINO ACID | N,N-Dimethylglycine | 3.78E + 04 | 2.31E + 04 | 0.61 | 1.89 | down |
| Glu-Glu | 3.80E + 04 | 2.40E + 04 | 0.63 | 1.78 | down | |
| Aspartic acid O-rutinoside | 5.40E + 05 | 1.03E + 06 | 1.91 | 1.97 | up | |
| L-(−)-Threonine | 6.53E + 05 | 1.00E + 06 | 1.53 | 1.51 | up | |
| CARBOHYDRATES | D(−)-Threose | 8.02E + 03 | 1.68E + 04 | 2.09 | 1.95 | up |
| CHOLINES | sn-Glycero-3-phosphocholine | 4.60E + 05 | 7.07E + 05 | 1.54 | 1.02 | up |
| COUMARINS | 4-Methylumbelliferone | 1.43E + 04 | 2.38E + 04 | 1.66 | 1.79 | up |
| FLAVONOID | Cyanidin 3-O-glucoside | 5.78E + 04 | 7.53E + 03 | 0.13 | 3.77 | down |
| Tangeretin | 1.82E + 04 | 7.40E + 03 | 0.41 | 1.81 | down | |
| Tricetin | 4.75E + 03 | 2.63E + 03 | 0.55 | 1.03 | down | |
| LIPIDS-GLYCEROPHOSPHOLIPID | LysoPC 20:1 | 4.53E + 05 | 7.14E + 05 | 1.58 | 1.42 | up |
| NUCLEOTIDE | 5′-Deoxy-5′-Methylthioadenosine | 5.90E + 03 | 8.86E + 03 | 1.50 | 1.53 | up |
| ORGANIC ACID | Ergothioneine | 4.72E + 04 | 2.42E + 04 | 0.51 | 2.29 | down |
| PHENOLAMIDES | N-Feruloyl spermidine | 5.17E + 03 | 9.45E + 03 | 1.83 | 2.15 | up |
| PHENYLPROPANOIDS | Sinapoyl O-hexoside | 1.58E + 05 | 2.41E + 05 | 1.52 | 1.45 | up |
| Coumarin | 3.67E + 04 | 5.96E + 04 | 1.63 | 1.63 | up | |
| Quinic acid | 6.39E + 03 | 1.07E + 04 | 1.68 | 1.65 | up | |
| PHYTOHORMONES | N-[(−)-Jasmonoyl]-(L)-Isoleucine (JA-Ile) | 3.20E + 03 | 1.04E + 03 | 0.33 | 2.52 | down |
| (+)-Jasmonic acid (JA) | 2.14E + 04 | 1.10E + 04 | 0.51 | 2.00 | down | |
| (+)-cis,trans-Abscisic acid (ABA) | 2.57E + 05 | 4.29E + 05 | 1.67 | 1.87 | up | |
| trans-Zeatin (tZ) | 1.88E + 04 | 3.66E + 04 | 1.95 | 2.31 | up | |
| TERPENOIDS | Diosgenin | 1.75E + 06 | 2.91E + 06 | 1.66 | 2.10 | up |
| VITAMINE | Nicotinate ribonucleoside | 1.79E + 05 | 9.55E + 04 | 0.53 | 2.09 | down |
| (S)-(+)-2-(anilinomethyl)pyrrolidine | 1.56E + 04 | 2.48E + 04 | 1.59 | up |
(a) Variable importance in the projection (VIP) values were obtained from PLS-DA with the threshold of 1.00.
A Venn diagram revealed six common metabolites in both comparison groups (i.e., Zheng58 × sub-CSSL16 vs. Zheng58 × lx9801 and sub-CSSL16 vs. lx9801). Furthermore, 25 metabolites were differentially accumulated between Zheng58 × sub-CSSL16 and Zheng58 × lx9801, while 18 metabolites were only differentially accumulated between sub-CSSL16 and lx9801 (Fig. 4A). Of all the differentially abundant metabolites, 24 and 15 metabolites were upregulated, while seven and nine metabolites were downregulated in the Zheng58 × sub-CSSL16 vs. Zheng58 × lx9801 and sub-CSSL16 vs. lx9801 comparisons, respectively (Fig. 4B). Clearly, many metabolites identified in the current study by metabolomics presenting further evidence to understand metabolic processes that might involve in heterosis.
Figure 4.

Differentially accumulated metabolites between hybrid and inbred lines. (A) Venn diagram of differentially accumulated metabolites in sub-CSSL16 and lx9801 as well as in Zheng58 × sub-CSSL16 (HY) and Zheng58 × lx9801 (CK). (B) Number of differentially accumulated metabolites in hybrids and inbred lines.
Secondary metabolite biosynthetic pathway
To fully characterize the pathways that maybe involve in heterosis during an early stage of ear development, a network was constructed based on differentially abundant metabolites in the two hybrids. In this study, regarding nucleotide metabolism, guanosine 5′-monophosphate (GMP) belongs to purine nucleotide was 1.99-fold higher in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801, and guanine as an important compounds of GMP having the same trend was 1.62-fold higher in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 (Fig. 5A). Additionally, under the catalysis of structural genes, the flux of inosine to tZR monophosphate was enhanced in the trans-zeatin riboside (tZR) biosynthetic pathway and tZR monophosphate can be further hydrolyzed to tZR. Consequently, the tZR abundance was 1.74-fold higher in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 (Fig. 5A). In terms of the nicotinate and nicotinamide metabolic pathway, the abundance of nicotinamide showed 1.71-fold increments in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 and nicotinamide as the precursor for synthesizing trigonelline may be also supported the higher accumulation of trigonelline (1.69-fold increments) in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 (Fig. 5B).
Figure 5.

Integrated metabolite changes and differentially expressed genes for analyzing the secondary metabolite biosynthetic pathways. Differentially accumulated metabolites indicated in red and blue were upregulated and downregulated, respectively. The red-to-blue colors in squares indicate high-to-low gene expression levels, respectively. (A) surE, 5′-nucleotidase; IMPDH, inosine-5′-phosphate dehydrogenase; GMPS, guanosine 5′-monophosphate synthetase; APRT, adenine phosphoribosyl transferase; ADA, adenosine deaminase; ADK, adenosine kinase; CYP735A, cytokinin trans-hydroxylase; (B) NMNAT, nicotinamide mononucleotide adenylyltransferase; SDT1, pyrimidine and pyridine-specific 5′-nucleotidase; URH1, uridine nucleosidase; PNC1, nicotinamidase; NNM, nicotinamide N-methyltransferase.
Gene expression pattern analysis
Most secondary metabolic pathways are regulated at the gene expression level. To investigate the expression of key genes, we completed quantitative real-time PCR (qRT-PCR) analyses. A total of 15 candidate genes associated with major secondary metabolic pathways were selected for an analysis of the secondary metabolism related to heterosis during the early stage of maize ear development. During the nucleotide metabolism, the genes of surE [5′-nucleotidase; one differentially expressed gene (DEG)], IMPDH (inosine-5′-phosphate dehydrogenase; three DEGs) and GMPS (guanosine 5′-monophosphate synthetase; one DEG) that involved in the purine metabolic pathway were expressed at lower levels in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 (Fig. 6). These results implied that the conversion of inosine to GMP was less efficiency in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801. In contrast, the expression levels of several genes were upregulated in Zheng58 × sub-CSSL16, including APRT (adenine phosphoribosyl transferase; two DEGs), ADA (adenosine deaminase; one DEG), and ADK (adenosine kinase; two DEGs) (Fig. 6). Meanwhile, the expression of CYP735A (cytokinin trans-hydroxylase; two DEGs), which is involved in zeatin biosynthetic pathways, was significantly upregulated in Zheng58 × sub-CSSL16 (Fig. 6). These genes expression pattern were responsible for the increased biosynthesis of tZR in Zheng58 × sub-CSSL16.
Figure 6.

Relative expression levels of genes in secondary metabolite biosynthetic pathways as determined by qRT-PCR. Relative expression levels are presented as the mean of three replicates ± standard error. HY: Zheng58 × sub-CSSL16; CK: Zheng58 × lx9801. * and **Indicate significant differences at P < 0.05 and P < 0.01, respectively, as determined by a one-way ANOVA with Student’s t-test.
In the nicotinate and nicotinamide metabolic pathway, the expression of NMNAT (nicotinamide mononucleotide adenylyl transferase; one DEG), which is an important structural gene related to nicotinamide mononucleotide (NaMN) synthesis, was significantly upregulated in Zheng58 × sub-CSSL16 (Fig. 6). Additionally, SDT1(pyrimidine and pyridine-specific 5′-nucleotidase; one DEG) was one of the most significantly upregulated DEGs (P = 5.25E−03) in Zheng58 × sub-CSSL16 vs. Zheng58 × lx9801 group. Meanwhile, URH1(uridine nucleosidase; one DEG) catalyzing nicotinamide riboside (NR) to nicotinamide was upregulated in Zheng58 × sub-CSSL16 (Fig. 6). These results are consistent with our metabolomics analysis that trigonelline contents were higher in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801.
Discussion
Application of a CSSL test population to investigate the genetic basis of heterosis
Heterosis is considerably influenced by the genetic background and environmental conditions. In previous studies, several primary segregated populations, such as BC1, F2, and F2:3, were used to study the genetic basis of heterosis. However, these populations represent temporary genotypes, making it difficult to obtain accurate phenotypic values over multiple years. To overcome the shortcomings of these populations, several permanently segregated populations, such as introgression lines and immortalized F2 (IF2) populations, have been developed for identifying heterotic loci24,26. Hua et al.27 identified 44 HLs for grain yield and yield-related components in rice using IF2 population that were developed from paired crosses of recombinant inbred lines, while Tang et al.28 detected 13 HLs for grain yield and its components in maize using the same genetic population. Compared with the primary segregated population and other permanently segregated population, the CSSLs and receptor parent test hybrids used in this study may be more appropriate for dissecting the genetic basis of heterosis. First, the influence of the genetic background can be eliminated during the identification of HLs in comparisons between the test population and the control hybrid Zheng58 × lx9801. Second, accurate phenotypic values may be obtained for the test hybrids under different environmental conditions during the identification of HLs.
Predicting the generation of high-performance hybrids is an integral part of maize breeding. However, only small subsets of potential crosses have been evaluated under field conditions for selecting elite hybrids. Thus, predicting hybrid performance is important for breeding programs. Advanced molecular technologies enabled the acquisition of system-wide genome data for predicting hybrid performance15. High-throughput technologies were recently applied to accurately measure the accumulation of metabolites, resulting in large amounts of metabolomics data for analyzing rice hybrid performance16. In another study, Lima et al.29 detected more robust metabolite changes (e.g., organic acids and amino acids) in hybrid lines than in inbred lines. They also observed that a few secondary metabolites could be used as biological markers to predict heterosis. In this study, we conducted a metabolomics analysis with one CSSL and its receptor parent as well as test hybrids to identify nucleotides and their derivatives, alkaloids, flavonoids, phenolics, and phytohormones mediating heterosis for maize ear development. Our data suggested that several important metabolic pathways affect the heterosis for maize ear development.
Differentially expressed genes associated with nucleotide metabolism and nicotinate and nicotinamide metabolism
Biosynthetic and catabolic enzymes could regulate the pool size of nucleotides and nucleosides30. In this study, the enzyme of surE participated in the substrate cycles and influence the balance of nucleotides and nucleosides pools at the cellular levels31, which have the lower expressed levels in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801. Additionally, the enzymes of IMPDH and GMPS exist in one branch of the guanine nucleotide synthesis and IMPDH controlling the gateway to guanine nucleotides for most organism32. The conversion of inosine 5′-monophosphate (IMP) to xanthosine 5′-phosphate (XMP) is the first committed and rate-limiting step in guanine nucleotide biosynthesis, which was catalyzed by IMPDH. Subsequently, XMP is converted to GMP through the action of GMPS32. In this study, some genes in the same branch of guanine nucleotide biosynthetic pathway were expressed at lower levels in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801, including surE, IMPDH and GMPS. These observations suggested that the conversion of inosine to GMP was less activity in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801. Another branch of guanine nucleotide regeneration is the recycling of guanine. The APRT belongs to the phosphoribosyl transferases family. Each APRTs can reutilize purine bases (such as guanine), being substrates for the synthesis of high-energy nucleotides, and convert them into mononucleotides (such as GMP), instead of nucleotides de novo synthesis consuming high-energy33. In this branch of guanine nucleotide regeneration, two genes encoding APRT were significantly upregulated in Zheng58 × sub-CSSL16 comparing with in Zheng58 × lx9801. This could largely explain the high accumulation of GMP in the Zheng58 × sub-CSSL16. Moreover, there are two enzymes potentially contribute to the adenosine metabolism in plant, ADA and ADK. One major route for adenosine salvage may be catalyzed by ADA in specific subcellular compartments34, and ADK as the housekeep enzymes phosphorylate adenosine to adenosine monophosphate (AMP) in Arabidopsis thaliana35. Moreover, in many organisms, AMP not only as an essential component in purine nucleotide pools, but also as the substrate be catalyzed to produce isopentenyladenine36,37. Furthermore, hydroxylation of isopentenyladenine was a key step, which was catalyzed by CYP735A in tZR biosynthetic pathway37. In this study, some upregulated expression genes involved in the adenosine metabolism including ADA, ADK and in the trans-zeatin riboside (tZR) biosynthetic pathway including CYP735A were recruited in Zheng58 × sub-CSSL16, implied that expression pattern of these genes might be responsible for the increased biosynthesis of tZR in Zheng58 × sub-CSSL16.
In the nicotinate and nicotinamide metabolic pathway, the expression of NMNAT, as the central enzyme to control this conversion of nicotinamide adenine dinucleotide (NAD) to NaMN38, was significantly upregulated in Zheng58 × sub-CSSL16. In addition, SDT1 are responsible for production of NR in cells and its activity is correlates with reduced cellular NAD39, which was one of the most significantly DEGs between Zheng58 × sub-CSSL16 and Zheng58 × lx9801. These two enzymes have significant impact on the cellular NAD homeostasis, which are the regular steps in NAD metabolism38,39. Moreover, URH1, as an apparent hydrolytic enzyme, makes a greater contribution to the conversion of NR to nicotinamide, and maintains the higher expression levels than other enzymes in this pathway of NR to nicotinamide salvage40. The expression of URH1 was upregulated in Zheng58 × sub-CSSL16, which may result in the higher accumulation of nicotinamide in Zheng58 × sub-CSSL16 than Zheng58 × lx9801, thus, more trigonelline was synthesized from the nicotinamide.
Nucleotide and phytohormone metabolism
Purine and pyrimidine nucleotides are precursors for the synthesis of primary and secondary products as well as the building blocks for nucleic acids. In higher plants, GMP can be used as a feedback signal to terminate the pathway responsible for de novo guanine nucleotide biosynthesis35. Salvage pathways are essential for the biosynthesis of nucleic acids in all organisms. In plants, they also help regenerate nucleotide pools by converting nucleosides to nucleotides, and they contribute to plant cell metabolism and catabolism41. To preserve energy, the purine salvage pathway is activated to regenerate guanine from GMP, while consuming only one adenosine triphosphate (ATP)42. Guanine nucleotides are important components of nucleotide pools. A small increase in nucleotide pools is accompanied by an increase in carbohydrate content43. In the present study, the purine salvage pathway affected the accumulation of GMP and guanine, with higher levels in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801. These results may be explained by the fact that carbohydrates, such as sucrose (2.16-fold change) and D-(−)-threose (1.56-fold change), accumulated more in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 (Table 2). Additionally, carbohydrate synthesis might be limited by the nucleotide supply. Moreover, high sugar concentrations may provide sufficient nutrients for vigorous tissue morphogenesis44.
The conversion of inosine to AMP occurs by sequential reactions catalyzed by ADA and ADK45,46. A previous study reported that the first step of cytokinin biosynthesis was the isopentenylation of AMP47. Thus, AMP is a direct precursor involved in cytokinin synthesis. Cytokinin free bases, such as tZR and isopentenyl adenine, are physiologically active in plant tissues48. The disequilibrium distribution of tZR is a key signal for organ-to-organ communication and positively regulates various growth and developmental processes37. As the cell cycle progresses, tZR seems to activate cell division during the early part of the S phase49. In this study, the tZR differentially accumulated in the two hybrids. Moreover, the upregulated expression of CYP375A, which encodes a cytokinin hydroxylase, enhanced tZR synthesis in Zheng58 × sub-CSSL16 by releasing active cytokinins37, which may be important for cell division and the early stage of maize ear development. These results implied that tZR may contribute to the heterosis of maize ear development.
Alkaloid metabolism
Nicotinamide is a vitamin B3 precursor that functions as a fundamental mediator of diverse biological processes50. In higher plants, including Arabidopsis thaliana and Oryza sativa, nicotinamide is likely converted to nicotinic acid and salvaged for NAD biosynthesis, which is generally active in vigorously growing cells51. Previous studies involving tobacco (Nicotiana sylvestris) provided evidence that NAD is an important coenzyme for photosynthesis and plant stress responses52,53. Additionally, trigonelline synthesis during seed germination may contribute to the detoxification of surplus nicotinamide54. Moreover, trigonelline induces cell expansion, which is an important determinant of grain width55. In this study, the abundance of trigonelline (1.69-fold change) and nicotinamide (1.71-fold change) increased in Zheng58 × sub-CSSL16 (Table 2). Thus, nicotinamide and trigonelline may be involved in the heterotic performance during the early stage of ear development. These findings implied that nicotinate and nicotinamide metabolism was more active in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801.
Flavonoid and phenolic metabolism
Flavonoids are common and widespread plant secondary metabolites that affect numerous physiological activities, including the negative regulation of auxin transport56. Phosphoglycoprotein is an auxin transporter that requires the hydrolysis of ATP57. The PIN expression pattern and the subcellular localization of the encoded protein are consistent with the fact PIN functions in polar auxin transport58. Flavonoids appear to alter PIN cycling, possibly because of induced changes in auxin concentrations via modulated auxin transport directly mediated by phosphoglycoprotein, rather than through an interaction with PIN59. Additionally, C-hexosyl-apigenin C-pentoside was detected as a major flavonoid compound, with lower accumulation in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801. These results indicated that flavonoids were more abundant in Zheng58 × lx9801 than in Zheng58 × sub-CSSL16, thereby inhibiting the transport of indole-3-acetic acid (IAA) and influencing the early stage of maize ear inflorescence development. Moreover, we also detected other flavonoids. For example, tricetin is a flavonoid exhibiting cytostatic properties by inducing the production of endogenous reactive oxygen species involved in apoptosis60. Tricetin concentrations were higher in Zheng58 × lx9801 than in Zheng58 × sub-CSSL16 (Table 2), but were similar between inbred line lx9801 and sub-CSSL16 (Table 3). These results revealed that the elevated levels of intracellular flavonoids adversely affected cell growth and development by regulating apoptosis, with potential implications for heterosis in maize.
Several known phenolic compounds, such as phenylpropanoids and coumarins, have been classified as natural regulators of plant growth, with phenylpropanoids important for responses to abscisic acid (ABA)61. Abscisic acid is a major regulator of plant development, including seed dormancy and germination62. Sinapic acid is a small natural molecule from the phenylpropanoid family that might be imbibed into seeds, where it is converted into sinapic acid esters that regulate ABA homeostasis and may promote early seed germination in Arabidopsis thaliana63. In this study, sinapic acid concentrations were more than 2-fold higher in Zheng58 × sub-CSSL16 than in Zheng58 × lx9801 (Table 2). Thus, sinapic acid might interfere with ABA homeostasis to induce early maize ear inflorescence growth in Zheng58 × sub-CSSL16.
Auxin biosynthesis and transport promote heterosis in Arabidopsis thaliana64. Regarding auxin distribution, PIN is the crucial factor facilitating polar auxin transport, while SLR/IAA14 is a key mediator of auxin redistribution that is regulated by 4-methylumbelliferone65,66. In this study, 4-methylumbelliferone was detected during the early development of maize ear inflorescences. Additionally, 4-methylumbelliferone accumulated more in Zheng58 × lx9801 than in Zheng58 × sub-CSSL16 (Table 2), implying that it might restrict the IAA redistribution in Zheng58 × lx9801 and interfere with maize ear growth and development in the FM stage. These results provide new evidence that changes to phenolic compounds influence heterosis in maize.
In this study, variations in gene expression patterns and metabolite contents were analyzed in maize ear inflorescences in CSSL, lx9801, and their corresponding hybrid lines. Additionally, a large-scale targeted metabolomics analysis revealed 24 metabolites that were differentially accumulated between inbred lines, while the contents of 31 metabolites were significantly different in the hybrid lines. The variability in the abundance of metabolites, including nucleotides, alkaloids, flavonoids, and phenolics, as well as in gene expression levels were associated with many biological processes, such as carbohydrate synthesis, cell division and apoptosis, and phytohormone transport and distribution. This study offers novel insights into the influence on secondary metabolism related to heterosis and provides additional information on the mechanism of secondary metabolic pathway involving in heterosis at the metabolomics level.
Materials and Methods
Plant materials and the field experiment
Previously, a maize CSSL panel of 184 lines was developed used two maize inbred lines (Chang7-2 as donor parent and lx9801 as the receptor parent), and a corresponding testcross population was constructed by crossing the CSSL panel with a test parent Zheng58. Of the 169 heterotic loci (HL) associated with grain yield were detected in the testcross population of Zheng58 × CSSL after comparing with the control hybrid Ludan9002 (Zheng58 × lx9801)21. A maize CSSL125 line carrying the HL hlEW2b was obtained from one of the CSSL test hybrids (Zheng58 × CSSL125) with an ear width significantly different from that of Ludan900221. In this study, various mapping populations, including backcross and inbred populations, were developed in the winter of 2013 (for backcrossing) in Sanya, Hainan province, China (E18°15′, N109°30′) and in the summer of 2014 (for selfing) in Xinxiang, Henan province, China (E113°31′, N35°11′). The sub-CSSL test population was constructed by crossing sub-CSSLs with Zheng58 in the winter of 2014 in Sanya. Additionally, the phenotypes of the sub-CSSL test population and the control hybrid, Ludan9002, were evaluated at two locations, Xinxiang and Hebi (E114°17′, N35°90′), in 2014 and 2015. Plants were grown with two replicates and a randomized complete block design. The control hybrid was also planted after every five testcross plants. Plots consisted of two rows (4 m long and 0.6 m wide) that were separated by 0.22 m. The final plant density was 67,500 plants per hectare. The plants were grown using standard agricultural practices.
Phenotypic data collection and fine mapping
A maize inbred line, sub-CSSL16, which contained the shortest donor chromosomal segment harboring hlEW2b, was compared with other homozygous lines in the sub-CSSL population. Two inbred lines, sub-CSSL16 and lx9801, along with their corresponding testcrosses, Zheng58 × sub-CSSL16 and Zheng58 × lx9801, were planted in two-row plots in four environments (Xinxiang and Hebi in 2015 and 2016). In each plot, 10 ears were harvested at physiological maturity for phenotypic measurements. Ear length (cm), ear width (mm), ear weight (g), kernel row number, and kernel number per row were measured using standard procedures to confirm the presence of hlEW2b. Raw data underwent a one-way analysis of variance (ANOVA) using the SPSS 17.0 program (SPSS, Inc., Chicago, IL, USA). The HL associated with ear width were considered to exist in sub-CSSL16 when the ear width in Zheng58 × sub-CSSL16 differed significantly from that of Zheng58 × lx9801 (P < 0.05).
In this study, we developed molecular markers for fine mapping. The simple sequence repeat (SSR) markers in the region flanked by umc1555 and umc1465 on chromosome 2 were obtained from the IBM 2008 Neighbors maize linkage map (http://www.maizegdb.org/data_center/map). Additionally, SSR sites were identified with the SSR Hunter program67. Primers were designed using the Primer 5.0 program (Premier Biosoft International, Palo Alto, CA, USA). Details regarding the primers used for mapping are listed in Supplementary Table S4. Furthermore, possible candidate genes were identified for hlEW2b within 1.98-Mb region based on the filtered working gene list of the maize genome downloaded from MaizeGDB (Zea mays. AGPv4.37, http://www.maizegdb.org). Candidate genes were annotated according to InterProScan (http://www.ebi.ac.uk/interpro/scan.html).
Sample preparation
Two hybrids, Zheng58 × sub-CSSL16 and Zheng58 × lx9801, as well as their corresponding inbred lines, sub-CSSL16 and lx9801, were planted at the experimental station of Henan Agricultural University (Zhengzhou, China; E113°65′, N34°76′) on June 20, 2016. The ear development stage was confirmed based on the leaf age index and microscopic observation. Immature ears at the FM development stage were collected. Three biological replicates were collected for each sample, which comprised five randomly collected fresh immature ears. All samples were immediately frozen in liquid nitrogen and stored at −80 °C prior to metabolite analyses.
Metabolite identification and quantification
Samples were freeze-dried in a vacuum and then homogenized using an Retsch MM400 Mixer Mill (RETSCH GmbH, Haan, Germany) with zirconia beads (90 s at 30 Hz). Metabolites were extracted from 100 mg powder with 70% aqueous methanol containing 0.1 mg/L lidocaine. Samples were incubated overnight at 4 °C and then centrifuged at 10,000 × g for 10 min. A 400-µL aliquot of the extract was filtered through an SCAA-104 membrane (0.22 µm pore size; Millipore), and the filtrate was stored in sample bottles for a subsequent analysis by liquid chromatography (LC) and tandem mass spectrometry (MS/MS). The LC-MS/MS analysis was completed with a Shim-pack UFLC Shimadzu CBM30A HPLC system and a 4500 QTRAP ESI–triple quadrupole–linear ion trap mass spectrometer (Applied Biosystems) equipped with an ESI Turbo Ion-Spray interface, which was operated in a positive ion mode and controlled with the Analyst 1.6 program (AB/SCIEX, Framingham, MA, USA).
Metabolites were identified and quantified according to a published protocol68. Briefly, the multiple reaction monitoring model was applied to identify the precursor ion of the target material. Many ion fragments were formed after a collision-induced dissociation, and characteristic ions were filtered from the fragment ions. The retention time and mass-to-charge ratio were measured for all detectable ions. Metabolites were quantified according to integral correction and normalization methods for analyzing mass spectra for metabolites in different samples (i.e., peak area evaluation). Metabolites were analyzed qualitatively and quantitatively.
Metabolite data analysis
Metabolite data were log2-transformed before undergoing a multivariate statistical analysis with the SIMCA-P 13.0 program (Umetrics, Umeå, Sweden). The data then underwent an unsupervised principal component analysis (PCA), which is a widely used unsupervised dimensionality reduction method, to visualize general clustering and trends among the samples. To identify differentially accumulated metabolites for each group, the partial least squares discriminant analysis (PLS-DA) was applied to remove irrelevant variables and maximize the variance among samples25. The efficiency and reliability of the PLS-DA model was assessed by a cross-validation with R2. The predictive performance was described by the Q2 model. The variable importance in the projection (VIP) reflected the contribution of each variable to the model. Variables with a VIP value > 1.0 were considered to influence the separation of samples in the PLS-DA score plots69. In addition to the multivariate approaches, fold-change ratios were used to assess the significance of each metabolite. A VIP value > 1.0 and a fold-change ratio ≥1.5 or ≤0.67 were set as the thresholds for screening potential metabolites with significant differences in abundance between samples70.
RNA extraction and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted from each sample using the Trizol® Reagent (Invitrogen, Carlsbad, CA, USA) and then treated with DNase I. The quality of the purified RNA (i.e., degradation and contamination) was characterized by 1% agarose gel electrophoresis and with the Nano LabChip kit (Agilent Technologies, Santa Clara, CA, USA). Purified RNA was quantified at OD260nm by using a NanoDrop™ One (Thermo Fisher Scientific, Wilmington, DE, USA). The RNA was then used as the template for a reverse transcription with the PrimeScript™ RT reagent kit with gDNA Eraser (Takara, Japan). A quantitative real-time polymerase chain reaction (qRT-PCR) assay was conducted with the SYBR Premix Ex Taq Kit (Takara) and the BioRad iQ Cycler (Bio-Rad, Hercules, CA, USA) according to the manufacturer′s instructions. The Primer 5.0 program was used to design the qRT-PCR primers, which are listed in Supplementary Table S5. Each sample was analyzed using three biological replicates and three technical replicates. The 2−ΔΔCt method was applied to estimate relative gene expression levels. Additionally, gene expression data underwent a one-way ANOVA with the SPSS 17.0 program. The significance threshold was set as P < 0.05.
Electronic supplementary material
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (91335205), the Central Plains Scholar of Henan, and the Major Science and Technology Programs of Henan Province (161100110500), the Henan Province Science and Technology Attack Project (152102110062, 182102110349).
Author Contributions
J.T. and W.L. designed the experiments and together with, X.S. and X.Z. carried out data analysis. X.S., D.S. and X.Z. carried out maize morphological investigation, validation of gene expression level. X.S., X.Z. and J.T. was mainly involved in preparation of manuscript. All authors reviewed and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xia Shi and Xuehai Zhang contributed equally.
Contributor Information
Weihua Li, Email: liwh416@163.com.
Jihua Tang, Email: tangjihua1@163.com.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-36446-5.
References
- 1.Shull GH. The composition of a field of maize. Journal of Heredity. 1908;1:296–301. doi: 10.1093/jhered/os-4.1.296. [DOI] [Google Scholar]
- 2.Lippman ZB, Zamir D. Heterosis: revisiting the magic. Trends in Genetics. 2007;23:60–66. doi: 10.1016/j.tig.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Krieger U, Lippman ZB, Zamir D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nature genetics. 2010;42:459–463. doi: 10.1038/ng.550. [DOI] [PubMed] [Google Scholar]
- 4.Yao H, Dogra GA, Auger DL, Birchler JA. Genomic dosage effects on heterosis in triploid maize. Proc. Natl. Acad. Sci. USA. 2013;110:2665–2669. doi: 10.1073/pnas.1221966110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frisch M, et al. Transcriptome-based distance measures for grouping of germplasm and prediction of hybrid performance in maize. Theor. Appl. Genet. 2010;120:441–450. doi: 10.1007/s00122-009-1204-1. [DOI] [PubMed] [Google Scholar]
- 6.Groszmann M, et al. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc. Natl. Acad. Sci. USA. 2011;108:2617–2622. doi: 10.1073/pnas.1019217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu H, Dooner HK. Intraspecific violation of genetic colinearity and its implications in maize. Proc. Natl. Acad. Sci. USA. 2002;99:9573–9578. doi: 10.1073/pnas.132259199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiao Y, et al. Genome-wide genetic changes during modern breeding of maize. Nature genetics. 2012;44:812–815. doi: 10.1038/ng.2312. [DOI] [PubMed] [Google Scholar]
- 9.Swanson-Wagner RA, et al. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA. 2006;103:6805–6810. doi: 10.1073/pnas.0510430103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahal D, Mooney BP, Newton KJ. Specific changes in total and mitochondrial proteomes are associated with higher levels of heterosis in maize hybrids. Plant J. 2012;72:70–83. doi: 10.1111/j.1365-313X.2012.05056.x. [DOI] [PubMed] [Google Scholar]
- 11.Semel Y, et al. Overdominant quantitative trait loci for yield and fitness in tomato. Proc. Natl. Acad. Sci. USA. 2006;103:12981–12986. doi: 10.1073/pnas.0604635103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, et al. Identification of Indica rice chromosome segments for the improvement of Japonica inbreds and hybrids. Theor. Appl. Genet. 2012;124:1351–1364. doi: 10.1007/s00122-012-1792-z. [DOI] [PubMed] [Google Scholar]
- 13.Shen G, Zhan W, Chen H, Xing Y. Dominance and epistasis are the main contributors to heterosis for plant height in rice. Plant Science. 2014;215–216:11–18. doi: 10.1016/j.plantsci.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 14.Wei X, et al. Heterotic loci for various morphological traits of maize detected using a single segment substitution lines test-cross population. Molecular Breeding. 2015;35:1–13. doi: 10.1007/s11032-015-0287-4. [DOI] [Google Scholar]
- 15.Riedelsheimer C, et al. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nature genetics. 2012;44:217–220. doi: 10.1038/ng.1033. [DOI] [PubMed] [Google Scholar]
- 16.Xu S, Xu Y, Gong L, Zhang Q. Metabolomic Prediction of Yield in Hybrid Rice. Plant J. 2016;88:219–227. doi: 10.1111/tpj.13242. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, et al. Relationship between differential gene expression and heterosis during ear development in Maize (Zea mays L.) J Genet Genomics. 2007;34:160–170. doi: 10.1016/S1673-8527(07)60017-4. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Y, et al. Genome-wide analysis of gene expression profiles during ear development of maize. Plant Molecular Biology. 2009;70:63–77. doi: 10.1007/s11103-009-9457-2. [DOI] [PubMed] [Google Scholar]
- 19.Miller ND, et al. A robust, high-throughput method for computing maize ear, cob, and kernel attributes automatically from images. Plant J. 2016;89:169–178. doi: 10.1111/tpj.13320. [DOI] [PubMed] [Google Scholar]
- 20.Hu X, et al. Transcriptome profiling and comparison of maize ear heterosis during the spikelet and floret differentiation stages. Bmc Genomics. 2016;17:959–976. doi: 10.1186/s12864-016-3296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, et al. Identification of heterotic loci associated with grain yield and its components using two CSSL test populations in maize. Scientific Reports. 2016;6:38205–38218. doi: 10.1038/srep38205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, et al. Arabidopsis DDB1-CUL4 ASSOCIATED FACTOR1 forms a nuclear E3 ubiquitin ligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant Cell. 2008;20(6):1437–1455. doi: 10.1105/tpc.108.058891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranocha P, et al. Walls are thin 1 (WAT1), an Arabidopsis homolog of Medicago truncatula NODULIN21, is a tonoplast-localized protein required for secondary wall formation in fibers. Plant J. 2010;63:469–483. doi: 10.1111/j.1365-313X.2010.04256.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhou G, et al. Genetic composition of yield heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA. 2012;109:15847–15852. doi: 10.1073/pnas.1214141109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Worley B, Powers R. Multivariate analysis in metabolomics. Curr Metabolomics. 2013;1:92–107. doi: 10.2174/2213235X11301010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi H, et al. Identification of combining ability loci for five yield-related traits in maize using a set of testcrosses with introgression lines. Theor. Appl. Genet. 2013;126:369–377. doi: 10.1007/s00122-012-1985-5. [DOI] [PubMed] [Google Scholar]
- 27.Hua J, et al. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA. 2003;100:2574–2579. doi: 10.1073/pnas.0437907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang J, et al. Dissection of the genetic basis of heterosis in an elite maize hybrid by QTL mapping in an immortalized F2 population. Theor. Appl. Genet. 2010;120:333–340. doi: 10.1007/s00122-009-1213-0. [DOI] [PubMed] [Google Scholar]
- 29.Lima, F. A. et al. Metabolic robustness in young roots underpins a predictive model of maize hybrid performance in the field. Plant J. 90, 10.1111/tpj.13495 (2017). [DOI] [PubMed]
- 30.Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- 31.Rampazzo C, et al. Mitochondrial deoxyribonucleotides, pool sizes, synthesis, and regulation. J Biol Chem. 2004;279(17):17019–17026. doi: 10.1074/jbc.M313957200. [DOI] [PubMed] [Google Scholar]
- 32.Hedstrom L. IMP dehydrogenase: structure, mechanism and inhibition. Chem Rev. 2009;109(7):2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baranowska-Bosiacka I, et al. Inhibition of erythrocyte phosphoribosyltransferases (APRT and HPRT) by Pb2+: A potential mechanism of lead toxicity. Toxicology. 2009;259:77–83. doi: 10.1016/j.tox.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Schoor S, Moffatt BA. Applying high throughput techniques in the study of adenosine kinase in plant metabolism and development. Front Biosci. 2004;9:1771–1781. doi: 10.2741/1346. [DOI] [PubMed] [Google Scholar]
- 35.Zrenner R, Stitt M, Sonnewald U, Boldt R. Pyrimidine and purine biosynthesis and degradation in plants. Annual Review of Plant Biology. 2006;57:805–836. doi: 10.1146/annurev.arplant.57.032905.105421. [DOI] [PubMed] [Google Scholar]
- 36.Moffatt BA, et al. Adenosine kinase of Arabidopsis. Kinetic properties and gene expression. Plant Physiol. 2000;124:1775–1785. doi: 10.1104/pp.124.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takei K, Yamaya T, Sakakibara H. Arabidopsis CYP735A1 and CYP735A2 encode cytokinin hydroxylases that catalyze the biosynthesis of trans-Zeatin. J Biol Chem. 2004;279:41866–41872. doi: 10.1074/jbc.M406337200. [DOI] [PubMed] [Google Scholar]
- 38.Lau C, Niere M, Ziegler M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front Biosci. 2009;14:410–431. doi: 10.2741/3252. [DOI] [PubMed] [Google Scholar]
- 39.Bogan KL, et al. Identification of Isn1 and Sdt1 as glucose- and vitamin-regulated nicotinamide mononucleotide and nicotinic acid mononucleotide 5′-nucleotidases responsible for production of nicotinamide riboside and nicotinic acid riboside. J Biol Chem. 2009;284:34861–34869. doi: 10.1074/jbc.M109.056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belenky P, Christensen KC, Gazzaniga F, Pletnev AA, Brenner C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J Biol Chem. 2008;284:158–164. doi: 10.1074/jbc.M807976200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moffatt BA, Ashihara H. Purine and Pyrimidine Nucleotide Synthesis and Metabolism. Arabidopsis Book. 2002;1:e0018. doi: 10.1199/tab.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stasolla C, Katahira R, Thorpe TA, Ashihara H. Purine and pyrimidine nucleotide metabolism in higher plants. J Plant Physiol. 2003;160:1271–1295. doi: 10.1078/0176-1617-01169. [DOI] [PubMed] [Google Scholar]
- 43.Schröder M, Giermann N, Zrenner R. Functional analysis of the pyrimidine de novo synthesis pathway in solanaceous species. Plant Physiol. 2005;138:1926–1938. doi: 10.1104/pp.105.063693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogawa A, Ando F, Toyofuku K, Kawashima C. Sucrose metabolism for the development of seminal root in Maize seedlings. Plant Production Science. 2009;12:9–16. doi: 10.1626/pps.12.9. [DOI] [Google Scholar]
- 45.Ginés S, et al. Regulation of epithelial and lymphocyte cell adhesion by adenosine deaminase-CD26 interaction. Biochemical Journal. 2002;361:203–209. doi: 10.1042/0264-6021:3610203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Long MC, Escuyer V, Parker WB. Identification and characterization of a unique adenosine kinase from Mycobacterium tuberculosis. Journal of Bacteriology. 2003;185:6548–6555. doi: 10.1128/JB.185.22.6548-6555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kakimoto T. Biosynthesis of cytokinins. Journal of Plant Research. 2003;116:233–239. doi: 10.1007/s10265-003-0095-5. [DOI] [PubMed] [Google Scholar]
- 48.Frébort I, Kowalska M, Hluska T, Frébortová J, Galuszka P. Evolution of cytokinin biosynthesis and degradation. J. Exp. Bot. 2011;62:2431–2452. doi: 10.1093/jxb/err004. [DOI] [PubMed] [Google Scholar]
- 49.Dobrev P, et al. Transient accumulation of cis - and trans -zeatin type cytokinins and its relation to cytokinin oxidase activity during cell cycle of synchronized tobacco BY-2 cells. Plant Physiology & Biochemistry. 2002;40:333–337. doi: 10.1016/S0981-9428(02)01383-9. [DOI] [Google Scholar]
- 50.Hunt L, Holdsworth MJ, Gray JE. Nicotinamidase activity is important for germination. Plant J. 2007;51:341–351. doi: 10.1111/j.1365-313X.2007.03151.x. [DOI] [PubMed] [Google Scholar]
- 51.Matsui A, Yin Y, Yamanaka K, Iwasaki M, Ashihara H. Metabolic fate of nicotinamide in higher plants. Physiologia Plantarum. 2007;131:191–200. doi: 10.1111/j.1399-3054.2007.00959.x. [DOI] [PubMed] [Google Scholar]
- 52.Dutilleul C, et al. Functional mitochondrial complex I is required by tobacco leaves for optimal photosynthetic performance in photorespiratory conditions and during transients. Plant Physiol. 2003;131:264–275. doi: 10.1104/pp.011155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dutilleul C, et al. Leaf mitochondria modulate whole cell redox homeostasis, set antioxidant capacity, and determine stress resistance through altered signaling and diurnal regulation. Plant Cell. 2003;15:1212–1216. doi: 10.1105/tpc.009464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng XQ, Hayashibe E, Ashihara H. Changes in trigonelline (N-methylnicotinic acid) content and nicotinic acid metabolism during germination of mungbean (Phaseolus aureus) seeds. J. Exp. Bot. 2005;56:1615–1623. doi: 10.1093/jxb/eri156. [DOI] [PubMed] [Google Scholar]
- 55.Chen W, et al. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nature Communications. 2016;7:12767–12776. doi: 10.1038/ncomms12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown DE, et al. Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis. Plant Physiol. 2001;126:524–535. doi: 10.1104/pp.126.2.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geisler M, et al. Cellular efflux of auxin catalyzed by the Arabidopsis MDR/PGP transporter AtPGP1. Plant J. 2005;44:179–194. doi: 10.1111/j.1365-313X.2005.02519.x. [DOI] [PubMed] [Google Scholar]
- 58.Rahman A, et al. Gravitropism of Arabidopsis thaliana roots requires the polarization of PIN2 toward the root tip in meristematic cortical cells. Plant Cell. 2010;22:1762–1776. doi: 10.1105/tpc.110.075317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peer WA, et al. Variation in expression and protein localization of the PIN family of auxin efflux facilitator proteins in flavonoid mutants with altered auxin transport in Arabidopsis thaliana. Plant Cell. 2004;16:1898–1911. doi: 10.1105/tpc.021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chien MH, et al. Tricetin induces apoptosis of human leukemic HL-60 cells through a reactive oxygen species-mediated c-Jun N-terminal kinase activation pathway. International Journal of Molecular Sciences. 2017;18:1667–1680. doi: 10.3390/ijms18081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Medinapuche L, et al. MYB10 plays a major role in the regulation of flavonoid/phenylpropanoid metabolism during ripening of Fragaria × ananassa fruits. J. Exp. Bot. 2014;65:401–417. doi: 10.1093/jxb/ert377. [DOI] [PubMed] [Google Scholar]
- 62.Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR. Abscisic acid: emergence of a core signaling network. Annual Review of Plant Biology. 2010;61:651–679. doi: 10.1146/annurev-arplant-042809-112122. [DOI] [PubMed] [Google Scholar]
- 63.Bi B, Tang J, Shuang H, Guo J, Miao Y. Sinapic acid or its derivatives interfere with abscisic acid homeostasis during Arabidopsis thaliana seed germination. Bmc Plant Biology. 2017;17:99–110. doi: 10.1186/s12870-017-1048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L, et al. PIF4-controlled auxin pathway contributes to hybrid vigor in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 2017;114:E3555–E3562. doi: 10.1073/pnas.1703179114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fukaki H, Tameda S, Masuda H, Tasaka M. Lateral root formation is blocked by a gain-of-function mutation in the SOLITARY-ROOT/IAA14 gene of Arabidopsis. Plant Journal for Cell & Molecular Biology. 2002;29:153–168. doi: 10.1046/j.0960-7412.2001.01201.x. [DOI] [PubMed] [Google Scholar]
- 66.Petrasek J, et al. PIN proteins perform a rate-limiting function in cellular auxin efflux. Science. 2006;312:914–918. doi: 10.1126/science.1123542. [DOI] [PubMed] [Google Scholar]
- 67.Li, Q. & Wan, J.M. SSRHunter: development of a local searching software for SSR sites. Hereditas27, 808–810, 10.3321/j.issn:0253-9772.2005.05.023 (2005). [PubMed]
- 68.Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature Protocols. 2006;1:387–396. doi: 10.1038/nprot0915-1457a. [DOI] [PubMed] [Google Scholar]
- 69.Saccenti E, Hoefsloot HCJ, Smilde AK, Westerhuis JA, Hendriks MMWB. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics. 2014;10:361–374. doi: 10.1007/s11306-013-0598-6. [DOI] [Google Scholar]
- 70.Jin J, et al. Integrated transcriptomics and metabolomics analysis to characterize cold stress responses in Nicotiana tabacum. Bmc Genomics. 2017;18:496–510. doi: 10.1186/s12864-017-3871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.