

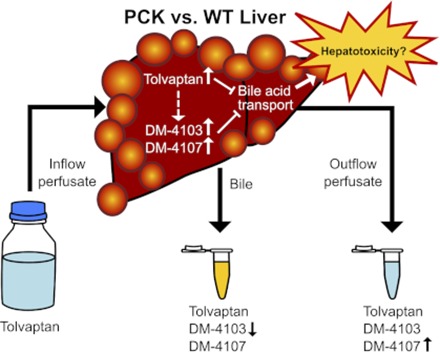
Abstract
Tolvaptan, a vasopressin V2-receptor antagonist, has demonstrated efficacy in slowing kidney function decline in patients with autosomal dominant polycystic kidney disease (ADPKD). In the pivotal clinical trial, the incidence of elevated liver enzymes was higher in patients receiving tolvaptan compared with placebo. Adjudication by a panel of expert hepatologists concluded a causal link of tolvaptan to liver injury in patients with ADPKD. An ex situ isolated perfused liver (IPL) study of tolvaptan disposition was undertaken in a rodent model of ADPKD, the polycystic kidney (PCK) rat (n = 5), and compared with wild-type (WT) Sprague-Dawley rats (n = 6). Livers were perfused with tolvaptan, followed by a tolvaptan-free washout phase. Total recovery (mean ± S.D. percentage of dose; PCK vs. WT) of tolvaptan and two metabolites, DM-4103 and DM-4107, quantified by liquid chromatography–tandem mass spectroscopy, was 58.14% ± 24.72% vs. 43.40% ± 18.11% in liver, 20.10% ± 9.15% vs. 21.17% ± 12.51% in outflow perfusate, and 0.08% ± 0.01% vs. 0.39% ± 0.32% in bile. DM-4103 recovery (mean ± S.D. percentage of dose) was decreased in PCK vs. WT bile (<0.01% ± <0.01% vs. 0.02% ± 0.01%; P = 0.0037), and DM-4107 recovery was increased in PCK vs. WT outflow perfusate (1.60% ± 0.57% vs. 0.43% ± 0.29%; P = 0.0017). A pharmacokinetic compartmental model assuming first-order processes was developed to describe the rate vs. time profiles of tolvaptan and DM-4103 + DM-4107 in rat IPLs. The model-derived estimate of tolvaptan’s biliary clearance was significantly decreased in PCK compared with WT IPLs. The model predicted greater hepatocellular concentrations of tolvaptan and DM-4103 + DM-4107 in PCK compared with WT IPLs. Increased hepatocellular exposure to tolvaptan and metabolites may contribute to the hepatotoxicity in patients with ADPKD treated with tolvaptan.
Introduction
Polycystic kidney disease (PKD) is a class of ciliopathies arising from primary cilium defects in epithelial cells of various organs, resulting in the development of fluid-filled cysts (Igarashi and Somlo, 2007). Autosomal dominant polycystic kidney disease (ADPKD) is the most common type of PKD and hereditary form of kidney disease (Torres et al., 2007), and a significant health concern that leads to kidney failure in approximately one-half of affected individuals (Grantham, 2008). ADPKD is primarily characterized by the progressive development of renal cysts and declining kidney function, but extrarenal cysts are common in this multiorgan disorder. The liver is affected in more than 80% of patients with ADPKD, leading to polycystic liver disease (Grantham, 2008). Despite complications arising from hepatic cysts such as cyst rupture, infection, and obstruction of bile ducts, liver function is thought to be preserved in polycystic liver disease (Cnossen and Drenth, 2014).
ADPKD arises from mutations in the PKD1 or PKD2 gene, encoding polycystin 1 and 2, respectively, which together form a receptor-channel complex (Kaimori and Germino, 2008). The illness has similarities to the less prevalent autosomal recessive PKD, which is caused by mutations in PKHD1, encoding the membrane-associated receptor-like protein fibrocystin/polyductin (Kim et al., 2008). The polycystic kidney (PCK) rat, a rodent model of human PKD that spontaneously arose in a colony of Sprague-Dawley rats, harbors a mutation in the Pkhd1 gene, similar to autosomal recessive PKD (Katsuyama et al., 2000; Masyuk et al., 2004). Since the natural history and the hepatorenal abnormalities in PCK disease and ADPKD are similar, this rodent model has been used to study ADPKD (Lager et al., 2001; Sabbatini et al., 2014).
Tolvaptan (TVP), an oral, selective vasopressin V2-receptor antagonist, has demonstrated efficacy in slowing kidney function decline in patients with ADPKD. A concerning finding in the pivotal clinical trial Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes (TEMPO) 3:4 was the higher incidence of elevated alanine aminotransferase (ALT) in patients receiving TVP (4.9%) compared with placebo (1.2%), with ALT > 2.5× the upper limit of normal (ULN) at any trial visit (Torres et al., 2012). Adjudication by a panel of hepatologists concluded a causal link between TVP and liver injury in patients with ADPKD, and in TEMPO 3:4 and an open-label extension trial, TEMPO 4:4, three patients met Hy’s law criteria (Watkins et al., 2015). Similar percentages (5.6% vs. 1.2%, TVP vs. placebo, respectively) in ALT elevations (>3× the ULN) but no additional Hy’s law cases were observed in a second pivotal trial, Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptan Safety and Efficacy in ADPKD with monthly monitoring of liver enzymes (Torres et al., 2017).
TVP is primarily metabolized by cytochrome P450 3A4 (CYP3A4) (Shoaf et al., 2011); CYP3A5, CYP2D6, uridine 5’-diphospho-glucuronosyltransferase 2B7 (UGT2B7), and UGT2B17 also contribute to TVP metabolite formation (Mazzarino et al., 2017). Numerous TVP metabolites have been identified, but most studies focus on the two major metabolites, DM-4103 and DM-4107 (Jiang et al., 2016; Lu et al., 2016). In humans, the main circulating metabolite is DM-4103, while a substantial portion of TVP is excreted in urine and feces as DM-4107 (Sorbera et al., 2002). Interactions between TVP, DM-4103, and DM-4107 and various human hepatic transporters have been investigated. DM-4103 is a relatively strong inhibitor of Na+-taurocholate cotransporting polypeptide [SLC10A1; half-maximal inhibitory concentration (IC50 value) = 16.3 μM], bile salt export pump (ABCB11; IC50 = 4.15 μM), and multidrug resistance-associated protein 4 (ABCC4; IC50 = 4.26 μM), while TVP is a moderate inhibitor of bile salt export pump (IC50 = 31.6 μM), and DM-4107 is a moderate inhibitor of multidrug resistance-associated protein 4 (IC50 = 37.9 μM) (Slizgi et al., 2016). Furthermore, TVP is a substrate and competitive inhibitor of P-glycoprotein (MDR1/ABCB1) (Shoaf et al., 2011). Currently, it remains unclear whether TVP, DM-4103, DM-4107, or other metabolites are responsible for the elevated liver enzymes observed in the ADPKD clinical trials.
The isolated perfused liver (IPL) is a physiologically relevant ex situ model used to assess hepatobiliary drug clearance (Brouwer and Thurman, 1996), and evaluate the hepatic transport and metabolic properties of a compound (Booth et al., 1998; Xiong et al., 2000; Chandra et al., 2005; Miranda et al., 2008; Pfeifer et al., 2013). To our knowledge, hepatobiliary function in PCK rats has not been evaluated previously using the IPL model. In the present study, the disposition of TVP and the major metabolites, DM-4103 and DM-4107, was investigated in IPLs from PCK and wild-type (WT) Sprague-Dawley rats to elucidate disease-mediated alterations in hepatobiliary function.
Materials and Methods
Chemicals.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Livers were perfused with freshly prepared, protein-free, modified Krebs-Henseleit buffer supplemented with 0.373 g/l calcium chloride dihydrate and 2.1 g/l sodium bicarbonate (Fisher Scientific, Fair Lawn, NJ); 7.735 mg/l taurocholic acid sodium salt hydrate was added (15 µM) to maintain bile flow. The pH of the buffer was adjusted to 7.4 with sodium hydroxide or hydrochloric acid. TVP, DM-4103, DM-4107, and OPC-41100, which was used as the internal standard (IS), were provided by Otsuka Pharmaceutical Co., Ltd. (Tokushima, Japan).
Animals.
Male, 20- to 21-week-old, WT and PCK (PCK/CrljCrl-Pkhd1pck/Crl) Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were used for the IPL studies. Cyst development in the PCK rat is substantial at this age, evidenced by an increase of approximately 85% in hepatic cyst volume from 12 to 20 weeks, and by significantly increased liver and kidney weights in 20-week-old PCK rats compared with age-matched WT rats (Brock et al., 2018). Rats were housed in an alternating 12-hr light/dark cycle, allowed water and food ad libitum, and acclimated for a minimum of 1 week prior to liver perfusions. All animal procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee (University of North Carolina, Chapel Hill, NC). All surgeries were performed under full anesthesia with ketamine:xylazine (60:12 mg/kg, i.p. injection).
Tolvaptan Disposition in Rat Isolated Perfused Livers.
Following cannulation of the bile duct and portal vein, the livers were perfused ex situ in a single-pass manner with continuously oxygenated, albumin-free Krebs-Henseleit buffer (35 ml/min) as described previously (Brouwer and Thurman, 1996). Albumin was excluded so that more TVP, which is highly protein bound, could be taken up by the hepatocytes, resulting in more quantifiable concentrations of TVP and metabolites in outflow perfusate and bile. After a 15-min equilibration period with blank perfusate, livers were perfused for 30 min with TVP-containing perfusate (∼7 μM; the exact concentration was measured for each experiment), followed by TVP-free perfusion for 30 min. Bile was collected continuously over 5-min intervals for 60 min. Outflow perfusate was collected directly from the perfusate flowing out of the vena cava every 5 min for 60 min. Blank bile was collected during the surgery immediately after bile duct cannulation. After each perfusion, the liver was blotted and weighed. All samples were initially snap frozen, and subsequently stored at −80°C. Liver viability was assessed by monitoring portal perfusion pressure (<15 cm H2O), gross morphology, bile flow (>2 μl/min) (Zhang et al., 2016), and lactate dehydrogenase (LDH) levels in the outflow perfusate measured by a Pierce LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific, Waltham, MA) per the manufacturer’s instructions. As a positive control for the LDH assay, one WT rat liver was perfused with 1% Triton X-100, which resulted in immediate liver toxicity based on gross morphology and major LDH release in outflow perfusate. Using dilutions and a standard curve for the outflow perfusate sample with the most LDH release (defined as 100% cytotoxicity), the percentage of cytotoxicity in the other perfused livers was evaluated; LDH release from these livers was negligible (<1% cytotoxicity, data not shown).
Bioanalysis.
TVP, DM-4103, and DM-4107 were quantified by liquid chromatography/electrospray ionization (ESI)–tandem mass spectroscopy. After thawing, liver tissue was mixed with saline and homogenized with a Shake Master NEO (BioMedical Science, Tokyo, Japan) prior to the pretreatment. Each sample was mixed with the IS and methanol in an ice-water bath. After centrifugation at 20,400g for 5 min at 10°C or below, the supernatant was analyzed by liquid chromatography/ESI–tandem mass spectroscopy. The liquid chromatography–tandem mass spectroscopy system consisted of a Prominence UFLC system (Shimadzu, Kyoto, Japan) and an API4000 triple quadrupole mass spectrometer equipped with an ESI source (AB SCIEX, Framingham, MA) and a switching valve (Valco Instruments, Houston, TX). Analyte separation was achieved on a reversed-phase high-performance liquid chromatography column XBridge C18 (3.5 μm, 2.1 mm i.d. × 50 mm; Waters, Milford, MA) by the binary gradient method with mobile phases of water-formic acid (1000:1, v/v) and acetonitrile-formic acid (1000:1, v/v) at a flow rate of 0.35 ml/min. The mass spectrometer was operated in ESI positive ion mode using multiple reaction monitoring transitions at mass-to-charge ratio (m/z) 449.2/252 for TVP, m/z 479.1/252 for DM-4103, m/z 481.1/252 for DM-4107, and m/z 463.2/266 for IS. Quantification of each analyte was achieved by comparison of the analyte/IS peak area ratios to those of a calibration curve (Analyst 1.5.1; AB SCIEX). Blank perfusate, blank bile, and blank liver homogenate served as matrices for the corresponding samples. TVP and metabolites were quantified in outflow perfusate, bile, and terminal liver samples from the IPL studies; these data were used to assess mass balance and conduct pharmacokinetic analyses. The lower limits of quantitation were 5nM, 50 nM, and 0.6 nmol/g tissue for the outflow perfusate, bile, and liver samples, respectively. Beal’s M5 method (Beal, 2001; Jusko, 2012) was used to manage individual values below the lower limit of quantitation.
Pharmacokinetic Modeling.
Modeling and simulation were used to evaluate the disposition of TVP and the two major metabolites in rat IPLs. A compartmental model assuming first-order processes was developed to describe the rate vs. time profiles using WinNonlin version 8.1 (Phoenix, St. Louis, MO), and using a mixed (combined additive and proportional) residual error model. The model scheme depicting the single-pass IPL system (Fig. 1) consisted of an extrahepatic and hepatic compartment, which was divided into five subcompartments to mimic liver physiology (Watanabe et al., 2009), and a bile compartment. The model was fit simultaneously to the average biliary excretion and outflow perfusate-time data for TVP and DM-4103 + DM-4107, for the WT IPLs and separately for the PCK IPLs. The individual outflow perfusate appearance and biliary excretion rates are presented in Supplemental Table 1. To describe the apparent delay in the biliary detection of TVP and DM-4103 + DM-4107 in PCK IPLs (likely due to the cyst burden in the PCK bile ducts), the bile compartment consisted of five transit compartments. The differential equations used to describe the model scheme in Fig. 1 are as follows, where the initial conditions in all compartments were assumed to be zero:
Fig. 1.

Model scheme describing the hepatobiliary disposition of TVP and metabolites in rat isolated perfused livers. The rate of change in the amount of TVP in perfusate flowing into the first sinusoidal compartment with respect to time,  , was determined by multiplying the inflow concentration (Cin) by the perfusate flow rate (Q). The rate of change in the amount of TVP in the outflow perfusate from the final sinusoidal compartment with respect to time,
, was determined by multiplying the inflow concentration (Cin) by the perfusate flow rate (Q). The rate of change in the amount of TVP in the outflow perfusate from the final sinusoidal compartment with respect to time, 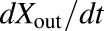 , was determined by multiplying the outflow concentration (Cout) by Q. Based on the well-stirred model (Pang and Rowland, 1977a,b,c), Cout was assumed to be equal to the concentration in the final sinusoidal compartment, CEC,5. The uptake clearance of TVP from the sinusoidal space to the hepatocellular space was described by CLUP, whereas CLBL described the efflux clearance from the hepatocellular space to the sinusoidal space and CLBile described the clearance of TVP from the hepatocellular space to bile. The biliary excretion rate,
, was determined by multiplying the outflow concentration (Cout) by Q. Based on the well-stirred model (Pang and Rowland, 1977a,b,c), Cout was assumed to be equal to the concentration in the final sinusoidal compartment, CEC,5. The uptake clearance of TVP from the sinusoidal space to the hepatocellular space was described by CLUP, whereas CLBL described the efflux clearance from the hepatocellular space to the sinusoidal space and CLBile described the clearance of TVP from the hepatocellular space to bile. The biliary excretion rate, 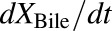 , was determined by multiplying the intracellular concentration of TVP (CIC) by the biliary clearance CLBile, as detailed in the differential equations. In the case of polycystic kidney rats, the biliary excretion rates of TVP and DM-4103 + DM-4107 were best described by including transit compartments (XBile,1–5 and X03,07,Bile,1–5, respectively) and transit rate constants (KLag,Bile and KLag,Bile,03,07, respectively) following the excretion of these compounds into bile (denoted by the gray shading). Cytochrome P450–mediated biotransformation of TVP into DM-4103 + DM-4107 and other metabolites occurs in the hepatocellular space, and the formation of these metabolites was described by the clearance parameters CL03,07 and CLMet, respectively. Once DM-4103 and DM-4107 were formed in the hepatocellular space, the hepatobiliary disposition of these metabolites was described with similar clearance parameters as those for TVP, while an uptake clearance parameter for DM-4103 + DM-4107 was not necessary to describe the metabolite profiles. The hepatobiliary disposition of other TVP metabolites could not be assessed directly because only TVP, DM-4103, and DM-4107 concentrations were quantified. Here, XEC and XIC denote the amount of TVP in the extracellular and intracellular space, respectively, and X03,07,EC and X03,07,IC denote the amount of DM-4103 + DM-4107 in the extracellular and intracellular space, respectively. Likewise, VEC, VIC, V03,07,EC, and V03,07,IC denote the volumes of the extracellular and intracellular space for TVP and metabolites, respectively. VEC and V03,07,EC were assumed to be equal; likewise, VIC and V03,07,IC were assumed to be equal. The term CIC,n represents the concentration in the nth liver compartment.
, was determined by multiplying the intracellular concentration of TVP (CIC) by the biliary clearance CLBile, as detailed in the differential equations. In the case of polycystic kidney rats, the biliary excretion rates of TVP and DM-4103 + DM-4107 were best described by including transit compartments (XBile,1–5 and X03,07,Bile,1–5, respectively) and transit rate constants (KLag,Bile and KLag,Bile,03,07, respectively) following the excretion of these compounds into bile (denoted by the gray shading). Cytochrome P450–mediated biotransformation of TVP into DM-4103 + DM-4107 and other metabolites occurs in the hepatocellular space, and the formation of these metabolites was described by the clearance parameters CL03,07 and CLMet, respectively. Once DM-4103 and DM-4107 were formed in the hepatocellular space, the hepatobiliary disposition of these metabolites was described with similar clearance parameters as those for TVP, while an uptake clearance parameter for DM-4103 + DM-4107 was not necessary to describe the metabolite profiles. The hepatobiliary disposition of other TVP metabolites could not be assessed directly because only TVP, DM-4103, and DM-4107 concentrations were quantified. Here, XEC and XIC denote the amount of TVP in the extracellular and intracellular space, respectively, and X03,07,EC and X03,07,IC denote the amount of DM-4103 + DM-4107 in the extracellular and intracellular space, respectively. Likewise, VEC, VIC, V03,07,EC, and V03,07,IC denote the volumes of the extracellular and intracellular space for TVP and metabolites, respectively. VEC and V03,07,EC were assumed to be equal; likewise, VIC and V03,07,IC were assumed to be equal. The term CIC,n represents the concentration in the nth liver compartment.
- WT and PCK extracellular compartment 1 (TVP)
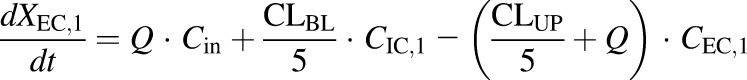
(1) - WT and PCK extracellular compartments 2–5 (TVP)

(2) - WT and PCK intracellular compartments 1–5 (TVP)

(3) - WT and PCK concentration data in outflow perfusate expressed as dX/dt (TVP)
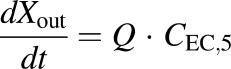
(4) - WT concentration data in bile expressed as dX/dt (TVP)
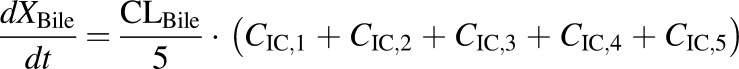
(5) - PCK transit compartment 1 following biliary excretion (TVP)

(6) - PCK transit compartments 2–5 following biliary excretion (TVP)
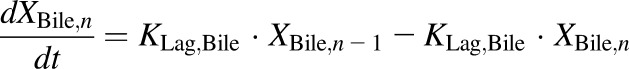
(7) - PCK concentration data in bile expressed as dX/dt (TVP)
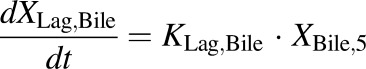
(8) - WT and PCK extracellular compartment 1 (DM-4103 + DM-4107)
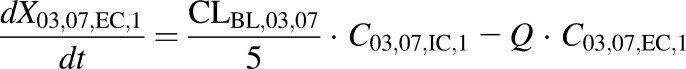
(9) - WT and PCK extracellular compartments 2–5 (DM-4103 + DM-4107)
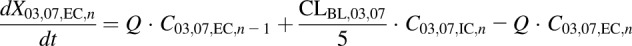
(10) - WT and PCK intracellular compartments 1–5 (DM-4103 + DM-4107)
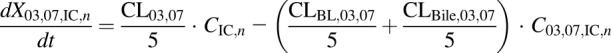
(11) - WT and PCK concentration data in outflow perfusate expressed as dX/dt (DM-4103 + DM-4107)
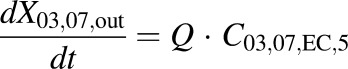
(12) - WT concentration data in bile expressed as dX/dt (DM-4103 + DM-4107)
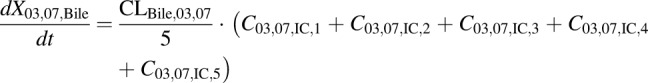
(13) - PCK transit compartment 1 following biliary excretion (DM-4103 + DM-4107)
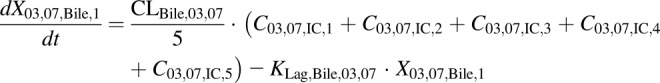
(14) - PCK transit compartments 2–5 following biliary excretion (DM-4103 + DM-4107)

(15) - PCK concentration data in bile expressed as dX/dt (DM-4103 + DM-4107)

(16)
In eqs. 1–16, the variables and parameters are defined as in Fig. 1, with further explanation as follows: EC denotes extracellular; IC denotes intracellular; CL03,07 denotes the formation clearance of DM-4103 + DM-4107 from TVP; CLBile denotes biliary clearance of TVP; CLBile,03,07 denotes biliary clearance of DM-4103 + DM-4107; CLBL denotes basolateral clearance of TVP; CLBL,03,07 denotes basolateral clearance of DM-4103 + DM-4107; CLMet denotes formation clearance of other metabolites from TVP; CLUP denotes uptake clearance of TVP; KLag,Bile denotes the transit rate constant for the biliary excretion of TVP; KLag,Bile,03,07 denotes the transit rate constant for the biliary excretion of DM-4103 + DM-4107; CEC,n represents the extracellular concentration in the corresponding sinusoidal compartment, calculated as  ; and VEC is the extracellular volume of the liver, which was assumed to be in equilibrium with the sinusoidal space and estimated as 20% of the total liver mass (Watanabe et al., 2009; Hobbs et al., 2012). Total liver mass was converted to total liver volume (VL) using the average density of rat liver (1.084 g/ml) (Pretlow and Pretlow, 1987). Here, Q represents the perfusate flow rate of 35 ml/min, which was assumed to be constant throughout the sinusoidal space of the liver; Cin represents the concentration of TVP in the inflow perfusate, measured for each preparation; binding of TVP to the perfusion tubing and apparatus was negligible (data not shown); and CIC,n and C03,07,IC,n represent the total intracellular concentrations of TVP and DM-4103+DM-4107, respectively, in the corresponding hepatocellular compartment. The total liver concentration was calculated as the sum of the mass in intra- and extracellular liver subcompartments 1–5 divided by VL. The total intracellular liver concentration was calculated as the sum of the mass in hepatocellular subcompartments 1–5, divided by the intracellular volume (VIC), calculated as
; and VEC is the extracellular volume of the liver, which was assumed to be in equilibrium with the sinusoidal space and estimated as 20% of the total liver mass (Watanabe et al., 2009; Hobbs et al., 2012). Total liver mass was converted to total liver volume (VL) using the average density of rat liver (1.084 g/ml) (Pretlow and Pretlow, 1987). Here, Q represents the perfusate flow rate of 35 ml/min, which was assumed to be constant throughout the sinusoidal space of the liver; Cin represents the concentration of TVP in the inflow perfusate, measured for each preparation; binding of TVP to the perfusion tubing and apparatus was negligible (data not shown); and CIC,n and C03,07,IC,n represent the total intracellular concentrations of TVP and DM-4103+DM-4107, respectively, in the corresponding hepatocellular compartment. The total liver concentration was calculated as the sum of the mass in intra- and extracellular liver subcompartments 1–5 divided by VL. The total intracellular liver concentration was calculated as the sum of the mass in hepatocellular subcompartments 1–5, divided by the intracellular volume (VIC), calculated as  . The perfusate flow rate multiplied by the concentration in the final extracellular liver compartment (
. The perfusate flow rate multiplied by the concentration in the final extracellular liver compartment (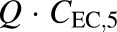 ) was fit to the observed appearance rate in outflow perfusate, while the biliary excretion rate (
) was fit to the observed appearance rate in outflow perfusate, while the biliary excretion rate ( and
and 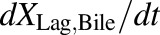 for WT and PCK rats, respectively) was fit to the observed biliary excretion rate data. The hepatocellular concentrations of TVP and DM-4103 + DM-4107 were simulated using the final parameter estimates based on the extensive sampling of outflow perfusate and bile.
for WT and PCK rats, respectively) was fit to the observed biliary excretion rate data. The hepatocellular concentrations of TVP and DM-4103 + DM-4107 were simulated using the final parameter estimates based on the extensive sampling of outflow perfusate and bile.
Data Analysis.
The majority of the data sets passed the Shapiro-Wilk normality test. All raw data are presented as mean ± S.D. from n = 5 livers in the PCK group, and n = 6 in the WT group. Statistical tests [t tests and Bonferroni-Dunn multiple comparisons tests (α = 0.05)] were performed using GraphPad Prism 7.02 for Windows (GraphPad Software, La Jolla, CA) to assess differences in: 1) concentrations of TVP, DM-4103, and DM-4107 in outflow perfusate at 30 and 60 min; 2) cumulative amounts of TVP, DM-4103, and DM-4107 in bile at the end of the perfusion; and 3) recovery (in terms of percentage of dose) of TVP, DM-4103, and DM-4107 in the outflow perfusate, bile, and liver. Final parameter estimates were considered significantly different if the 95% confidence intervals determined in Phoenix WinNonlin version 8.1 between the WT and PCK groups did not overlap. This method of judging the significance of differences is more conservative than the “standard” method (Schenker and Gentleman, 2001), which assumes consistency, asymptotic normality, and asymptotic independence of the estimates.
Results
At the end of liver perfusion, larger amounts of TVP (1.29-fold), DM-4103 (1.42-fold), and DM-4107 (1.45-fold) were recovered in PCK compared with WT rat livers, but differences were not statistically significant (Fig. 2). PCK rat livers weighed significantly more than those of WT rats (P = 0.0002; Supplemental Table 2), consistent with previous reports (Katsuyama et al., 2000; Lager et al., 2001; Shimomura et al., 2015).
Fig. 2.

Liver recovery of tolvaptan, DM-4103, and DM-4107. Amounts (nmol) of TVP, DM-4103, and DM-4107 recovered at 60 min in isolated perfused livers from WT and PCK rats. Data are presented as mean ± S.D. (n = 6 for WT; n = 5 for PCK).
Outflow Perfusate Concentrations of Tolvaptan and Metabolites.
After 30 min of TVP perfusion, when the perfusate was switched from TVP-containing to blank buffer, the TVP concentrations in outflow perfusate did not differ between PCK and WT IPLs (Fig. 3A). However, at the end of the washout phase, the outflow perfusate concentration of TVP was 2.33-fold higher in PCK compared with the WT rat livers (PCK = 522.4 ± 127.6 nM vs. WT = 224.6 ± 162.3 nM, P = 0.0089). DM-4103 concentrations in outflow perfusate trended higher for most time points in PCK compared with WT IPLs, especially toward the end of the perfusion, but these differences were not statistically significant (Fig. 3B). A similar trend was seen for DM-4107, but the concentrations at 30 min (3.94-fold increase in PCK) and at the end of the washout (3.69-fold increase in PCK) were significantly different (PCK = 62.57 ± 25.05 nM vs. WT = 15.89 ± 8.67 nM, P = 0.0020, and PCK = 93.04 ± 30.01 nM vs. WT = 25.23 ± 19.38 nM, P = 0.0014, respectively; Fig. 3C).
Fig. 3.

Outflow perfusate concentration-time profiles. Concentration (nM) vs. time (min) profiles of (A) TVP, (B) DM-4103, and (C) DM-4107 in the outflow perfusate of isolated perfused livers (IPLs) from WT and PCK rats. Statistically significant differences in concentrations between WT and PCK IPLs were assessed at t = 30 and 60 min. Perfusions with tolvaptan (∼7 μM) and subsequently with blank buffer (washout) for 30 min each are represented by the black and gray bars, respectively. Data are presented as mean ± S.D. (n = 6 for WT; n = 5 for PCK). *P < 0.05, PCK vs. WT.
Biliary Excretion of Tolvaptan and Metabolites.
The overall bile flow in PCK rats was reduced ∼1.4-fold (data not shown). The cumulative amount of TVP, DM-4103, and DM-4107 recovered in the bile of PCK compared with WT rat livers tended to be lower throughout the perfusion (Fig. 4). The cumulative biliary recovery at 60 min was significantly lower for DM-4103 (P = 0.0038; Fig. 4B).
Fig. 4.

Cumulative bile amount-time profiles. Cumulative amount (nmol) vs. time (min) profiles of (A) TVP, (B) DM-4103, and (C) DM-4107 in the bile of isolated perfused livers (IPLs) from WT and PCK rats. Statistically significant differences in cumulative biliary excretion between WT and PCK IPLs were assessed at 60 min. Perfusions with tolvaptan (∼7 μM) and subsequently with blank buffer (washout) for 30 min each are represented by the black and gray bars, respectively. Data are presented as mean ± S.D. (n = 6 for WT; n = 5 for PCK). *P < 0.05, PCK vs. WT.
Total Recovery of Tolvaptan and Metabolites.
The total recovery of TVP, expressed as a percentage of dose, in liver tissue (Fig. 5A), outflow perfusate (Fig. 5B), and bile (Fig. 5C) at the end of the 60-min perfusions was similar between PCK and WT IPLs. DM-4107 recovery was significantly increased in the outflow perfusate of PCK (1.60% ± 0.57%) compared with WT (0.43% ± 0.29%) IPLs (P = 0.0017). DM-4103 recovery was significantly decreased in the bile of PCK (<0.01% ± <0.01%) compared with WT (0.02% ± 0.01%) IPLs (P = 0.0037). When assessing the metabolite/parent area under the curve ratios in liver tissue, outflow perfusate, and bile, only DM-4107 in outflow perfusate (PCK = 9.40% ± 2.63%; WT = 2.67% ± 1.98%) reached statistical significance (P = 0.0019), which coincides with Fig. 5B.
Fig. 5.

Total recovery. Total recovery at 60 min of TVP and two TVP metabolites, DM-4103 and DM-4107, expressed as a percentage of dose, in (A) liver, (B) outflow perfusate, and (C) bile in isolated perfused livers from WT and PCK rats. Significant differences between WT and PCK per sample type were assessed for each of the three chemical species. Data are presented as mean ± S.D. (n = 6 for WT; n = 5 for PCK). *P < 0.05, PCK vs. WT.
Pharmacokinetic Modeling.
Differential equations corresponding to that the model described in Fig. 1 were fit to the data to obtain estimates for the seven clearance parameters, and also for the two rate constants in the case of PCK rats (Table 1). The appearance rate in outflow perfusate, and the excretion rate in bile are plotted vs. time in Fig. 6. Additional model diagnostic plots are shown in Supplemental Fig. 1.
TABLE 1.
Estimates of TVP and DM-4103 + DM-4107 clearance values in IPLs from WT and PCK rats based on the model scheme depicted in Fig. 1
Livers were perfused with TVP (∼7 μM) and subsequently with blank buffer (washout) for 30 min each. Statistically significant differences were based on nonoverlapping 95% confidence intervals (CI): *P < 0.05, PCK vs. WT; #P < 0.05, CLBL vs. CLBile within the same rat IPL group; †P < 0.05, compared with CLMet within the same rat IPL group.
| Parameter | Estimate (CV%); [95% CI] |
|
|---|---|---|
| WT | PCK | |
| CLBL (l/h) | 0.20 (20%); [0.12, 0.28]#,† | 0.29 (22%); [0.16, 0.43]#,† |
| CLUP (l/h) | 21 (14%); [15, 26] | 29 (14%); [21, 38] |
| CLBile (l/h) | 0.11 × 10−3 (10%); [0.85 × 10−4, 0.13 × 10−3]*,#,† | 0.49 × 10−4 (16%); [0.33 × 10−4, 0.65 × 10−4]*,#,† |
| CLMet (l/h) | 0.44 × 10−1 (4.2%); [0.40 × 10−1, 0.48 × 10−1] | 0.38 × 10−1 (8.9%); [0.31 × 10−1, 0.45 × 10−1] |
| CL03,07 (l/h) | 0.18 × 10−2 (20%); [0.10 × 10−2, 0.26 × 10−2]† | 0.47 × 10−2 (39%); [0.99 × 10−3, 0.85 × 10−2]† |
| CLBL,03,07 (l/h) | 0.50 × 10−2 (24%); [0.26 × 10−2, 0.74 × 10−2] | 0.91 × 10−2 (43%); [0.11 × 10−2, 0.17 × 10−1] |
| CLBile,03,07 (l/h) | 0.16 × 10−2 (23%); [0.84 × 10−3, 0.24 × 10−2] | 0.11 × 10−2 (76%); [−0.62 × 10−3, 0.28 × 10−2] |
| KLag,Bile (h−1) | 17 (15%); [12, 22] | |
| KLag,Bile,03,07 (h−1) | 4.1 (21%); [2.3, 5.8] | |
CI, confidence interval.
Fig. 6.

Model fits to outflow perfusate appearance and biliary excretion rate-time data. Appearance rate in outflow perfusate (nmol/hr) and biliary excretion rate (nmol/hr) vs. time (min) profiles of [(A and C), respectively] TVP and [(B and D), respectively] DM-4103 + DM-4107 in IPLs from WT and PCK rats. Perfusions with tolvaptan (∼7 μM) and subsequently with blank buffer (washout) for 30 min each are represented by the black and gray bars, respectively. Observed data are presented as mean ± S.D. (n = 6 for WT; n = 5 for PCK). PCKobs and WTobs are the observed outflow perfusate appearance and biliary excretion rates in PCK and WT IPLs, whereas PCKpred and WTpred are the rates predicted by the model scheme depicted in Fig. 1. See Supplemental Fig. 1 for observed vs. predicted outflow perfusate appearance and biliary excretion rate plots.
The biliary clearance value of TVP (CLBile) was significantly decreased in PCK compared with WT IPLs (2.19-fold; Table 1). Based on the confidence intervals of the remaining parameter estimates, no additional differences in clearance parameters were significant between PCK and WT IPLs. For those parameters describing clearance from a compartment of identical volume, additional comparisons were made within the same rat group (indicated by the # or † symbols in Table 1). In both PCK and WT groups, CLBL > CLBile suggesting a higher affinity of TVP for hepatic basolateral efflux transporters than for canalicular transporters. Additionally, in both groups, CLMet > CL03,07 indicating that the formation clearance of other metabolites is greater in rat IPLs than the formation clearance of DM-4103 + DM-4107, assuming that subsequent metabolism of these two chemical species is negligible. In both WT and PCK rat IPLs, CLBL > CLMet > CL03,07 > CLBile suggesting that the formation clearance of metabolites is more substantial than the biliary clearance of TVP, but not as extensive as the basolateral clearance of TVP.
Pharmacokinetic Simulations.
The model described in Fig. 1 also was used to simulate concentrations of TVP and DM-4103 + DM-4107 in the hepatocellular space of PCK and WT IPLs (Fig. 7). The model predicted similar hepatocellular concentrations of TVP at the end of the 30-min perfusion with TVP-containing buffer in the PCK and WT IPLs, whereas TVP concentrations were predicted to be higher in PCK compared with WT IPLs throughout the washout phase and beyond. Based on the simulations, hepatocellular concentrations of DM-4103 + DM-4107 were higher in PCK compared with WT IPLs throughout the 60-min perfusion and beyond.
Fig. 7.

Simulated hepatocelluar concentrations. Total hepatocellular concentration (nM) vs. time (hr) profiles of (A) TVP and (B) DM-4103 + DM-4107 simulated in the hepatocellular space of isolated perfused livers from WT and PCK rats using the model depicted in Fig. 1. Perfusions with tolvaptan (∼7 μM) and subsequently with blank buffer (washout) for 30 min each are represented by the black and gray bars, respectively.
Discussion
The current ex situ IPL study revealed that the hepatobiliary disposition of TVP and two metabolites was altered in a rodent model of ADPKD. Significantly more DM-4107 (percentage of TVP dose) was recovered in the outflow perfusate from PCK compared with WT IPLs. In contrast, significantly less DM-4103 (percentage of TVP dose) was recovered in the bile of PCK compared with WT IPLs. A pharmacokinetic model was developed to describe the disposition of TVP and metabolites in perfusate and bile and to estimate relevant clearance processes.
The estimated biliary clearance value for TVP (CLBile) was significantly decreased in PCK compared with WT IPLs, suggesting that this biliary excretion process is impaired in PCK rat livers. However, in both groups, basolateral efflux was a more prominent clearance mechanism for TVP (CLBL > CLBile). The formation clearance of DM-4103 + DM-4107 and other metabolites from TVP (CL03,07 and CLMet, respectively) was not significantly different between PCK and WT IPLs; however, in both groups CLMet > CL03,07, indicating that a substantial amount of other metabolites is formed. For example, DM-4103 and DM-4107 are formed through separate sequential metabolic pathways (Furukawa et al., 2014; Mazzarino et al., 2017). However, based on our data set, a more complex pharmacokinetic model could not be justified.
Using the final clearance parameter estimates, simulations revealed that TVP and metabolite concentrations at the end of the perfusion were increased in hepatocytes of PCK rats. These findings are significant because TVP, DM-4107, and especially DM-4103 inhibit hepatic efflux transporters (Slizgi et al., 2016). Inhibition of hepatic efflux transporters may be greater in PCK disease compared with healthy controls due to elevated hepatocellular TVP and metabolite concentrations, resulting in accumulation of hepatotoxic endogenous molecules (e.g., lithocholate, glycine-conjugated deoxycholate, and chenodeoxycholate) (Galle et al., 1990; Fickert et al., 2006; Chatterjee et al., 2014) and leading to liver injury. Therefore, increased hepatocellular concentrations of TVP and DM-4103 + DM-4107 in PCK disease could be related to the higher incidence of hepatotoxic events in patients with ADPKD treated with TVP in the pivotal clinical trial. Bile acid concentrations are increased in PCK rat livers (Munoz-Garrido et al., 2015; Brock et al., 2018), and elevated hepatocyte concentrations of TVP and metabolites may exacerbate this imbalance. Although the IC50 values obtained from previous studies with human hepatic transporters (Slizgi et al., 2016) cannot be compared directly to the simulated hepatocellular TVP and DM-4103 + DM-4107 concentrations in rat IPLs due to species differences, the simulations illustrate the possibility that these chemical species, which are capable of inhibiting hepatic transporters, could accumulate to a greater degree in patients with ADPKD compared with healthy controls. Hepatocellular accumulation of TVP and metabolites may take time to reach inhibitory concentrations, which could be related to ADPKD progression, and this could be one explanation for the delay in TVP toxicity in patients with ADPKD (Watkins et al., 2015).
All rats used in the current study were 20- to 21-week-old males. Although liver function is thought to be preserved in polycystic liver disease (Cnossen and Drenth, 2014), hepatic cyst volume in PCK rats increases with age (Brock et al., 2018), potentially resulting in age-dependent changes in hepatobiliary disposition of compounds. A definitive correlation between hepatic cyst involvement and clinically observed TVP-mediated hepatotoxicity cannot be made. However, of those subjects that were adjudicated as highly likely or probably related to study drug, 76% had some level of hepatic cyst burden (Watkins et al., 2015). Furthermore, in TEMPO 3:4 and 4:4, all three Hy’s law cases and 18 out of 30 subjects with hepatotoxicity attributable to TVP with a probable (or higher) likelihood, were women (Watkins et al., 2015). However, sex differences in TVP-mediated hepatotoxicity could not be concluded.
PCK rat livers weighed significantly more than WT rat livers, consistent with prior studies (Katsuyama et al., 2000; Lager et al., 2001). These differences may be due to the cysts in PCK rat livers (Brock et al., 2018). Interestingly, bile flow in PCK IPLs was slightly lower than in WT IPLs during perfusion. One possible explanation may be that cystic bile ducts in PCK rat livers are dilated (Masyuk et al., 2004). Similarly, renal tubules in PKD are dilated by cyst formation and proliferation (Igarashi and Somlo, 2007). The resulting structural compression in the tubules hinders urine flow (Grantham et al., 2011). Following this logic, it is conceivable that bile flow in the cystic bile ducts of PCK rats is reduced.
Considerable variability was observed in the TVP and metabolite excretion rates in outflow perfusate and bile between individual perfused livers in each group of rats (Supplemental Table 1). Liver function may be inherently different from rat to rat, especially in the case of PCK rats where hepatic cyst burden can differ markedly at a specific age (Brock et al., 2018). Despite variability between individual IPLs, the average data for WT and PCK rat IPLs revealed distinct differences and trends between the two groups.
In conclusion, the hepatobiliary disposition of TVP, DM-4103, and DM-4107 was modified in PCK compared with WT IPLs. The altered disposition resulted in higher simulated concentrations of these chemical species in PCK rat livers, which could impair transport of endogenous compounds such as hepatotoxic bile acids. We hypothesize that this is a contributing mechanism in relation to the increase in liver injury observed in some patients with ADPKD who are administered TVP.
Acknowledgments
We gratefully acknowledge Certara (Princeton, NJ) for providing academic research licenses for the Phoenix software as part of the Center of Excellence program for educational institutions. We thank Dr. Yang Lu for conducting the pilot study, Dr. Brian C. Cooley for surgical expertise, and Dr. Noora Sjöstedt for support with the visual abstract.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- CLBile
biliary clearance of tolvaptan
- CLBile,03,07
biliary clearance of DM-4103 + DM-4107
- CLBL
basolateral clearance of tolvaptan
- CLBL,03,07
basolateral clearance of DM-4103 + DM-4107
- CLMet
formation clearance of other metabolites from tolvaptan
- CLUP
uptake clearance of tolvaptan
- CL03,07
formation clearance of DM-4103 + DM-4107 from tolvaptan
- EC
extracellular
- ESI
electrospray ionization
- IC
intracellular
- IPL
isolated perfused liver
- IS
internal standard
- KLag,Bile
transit rate constant for the biliary excretion of tolvaptan
- KLag,Bile,03,07
transit rate constant for the biliary excretion of DM-4103 + DM-4107
- LDH
lactate dehydrogenase
- m/z
mass-to-charge ratio
- PCK
polycystic kidney
- PKD
polycystic kidney disease
- TEMPO
Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes
- TVP
tolvaptan
- WT
wild-type
Authorship Contributions
Participated in research design: Beaudoin, Roth, Brock, Brouwer.
Conducted experiments: Beaudoin, Mizuno.
Performed data analysis: Beaudoin, Bezençon, Cao, Roth, Brock, Mizuno, Brouwer.
Wrote or contributed to the writing of the manuscript: Beaudoin, Bezençon, Brouwer.
Footnotes
This work was supported, in part, by Otsuka Pharmaceutical Development & Commercialization, Inc., and the National Institutes of Health National Institute of General Medical Sciences [Grants R01GM041935 and R35GM122576].
This work was presented, in part, at the 21st North American Meeting of the International Society for the Study of Xenobiotics in 2017.
Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the Otsuka Pharmaceutical Group or the National Institutes of Health.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Beal SL. (2001) Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28:481–504. [DOI] [PubMed] [Google Scholar]
- Booth CL, Brouwer KR, Brouwer KLR. (1998) Effect of multidrug resistance modulators on the hepatobiliary disposition of doxorubicin in the isolated perfused rat liver. Cancer Res 58:3641–3648. [PubMed] [Google Scholar]
- Brock WJ, Beaudoin JJ, Slizgi JR, Su M, Jia W, Roth SE, Brouwer KLR. (2018) Bile acids as potential biomarkers to assess liver impairment in polycystic kidney disease. Int J Toxicol 37:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer KLR, Thurman RG. (1996) Isolated perfused liver. Pharm Biotechnol 8:161–192. [DOI] [PubMed] [Google Scholar]
- Chandra P, Johnson BM, Zhang P, Pollack GM, Brouwer KLR. (2005) Modulation of hepatic canalicular or basolateral transport proteins alters hepatobiliary disposition of a model organic anion in the isolated perfused rat liver. Drug Metab Dispos 33:1238–1243. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Bijsmans IT, van Mil SW, Augustijns P, Annaert P. (2014) Toxicity and intracellular accumulation of bile acids in sandwich-cultured rat hepatocytes: role of glycine conjugates. Toxicol In Vitro 28:218–230. [DOI] [PubMed] [Google Scholar]
- Cnossen WR, Drenth JP. (2014) Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis 9:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickert P, Fuchsbichler A, Marschall HU, Wagner M, Zollner G, Krause R, Zatloukal K, Jaeschke H, Denk H, Trauner M. (2006) Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol 168:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa M, Miyata K, Kawasome C, Himeda Y, Takeuchi K, Koga T, Hirao Y, Umehara K. (2014) Liquid chromatography-tandem mass spectrometry method for determining tolvaptan and its nine metabolites in rat serum: application to a pharmacokinetic study. Arch Pharm Res 37:1578–1587. [DOI] [PubMed] [Google Scholar]
- Galle PR, Theilmann L, Raedsch R, Otto G, Stiehl A. (1990) Ursodeoxycholate reduces hepatotoxicity of bile salts in primary human hepatocytes. Hepatology 12:486–491. [DOI] [PubMed] [Google Scholar]
- Grantham JJ. (2008) Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 359:1477–1485. [DOI] [PubMed] [Google Scholar]
- Grantham JJ, Mulamalla S, Swenson-Fields KI. (2011) Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 7:556–566. [DOI] [PubMed] [Google Scholar]
- doi: 10.17226/12910. [DOI] [Google Scholar]
- Hobbs M, Parker C, Birch H, Kenworthy K. (2012) Understanding the interplay of drug transporters involved in the disposition of rosuvastatin in the isolated perfused rat liver using a physiologically-based pharmacokinetic model. Xenobiotica 42:327–338. [DOI] [PubMed] [Google Scholar]
- Igarashi P, Somlo S. (2007) Polycystic kidney disease. J Am Soc Nephrol 18:1371–1373. [DOI] [PubMed] [Google Scholar]
- Jiang J, Tian L, Huang Y, Yan Y, Li Y. (2016) A rapid and sensitive LC-MS/MS-ESI method for the determination of tolvaptan and its two main metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 1027:158–164. [DOI] [PubMed] [Google Scholar]
- Jusko WJ. (2012) Use of pharmacokinetic data below lower limit of quantitation values. Pharm Res 29:2628–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimori JY, Germino GG. (2008) ARPKD and ADPKD: first cousins or more distant relatives? J Am Soc Nephrol 19:416–418. [DOI] [PubMed] [Google Scholar]
- Katsuyama M, Masuyama T, Komura I, Hibino T, Takahashi H. (2000) Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp Anim 49:51–55. [DOI] [PubMed] [Google Scholar]
- Kim I, Fu Y, Hui K, Moeckel G, Mai W, Li C, Liang D, Zhao P, Ma J, Chen XZ, et al. (2008) Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol 19:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager DJ, Qian Q, Bengal RJ, Ishibashi M, Torres VE. (2001) The PCK rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int 59:126–136. [DOI] [PubMed] [Google Scholar]
- Lu Y, Slizgi JR, Brouwer KR, Claire RL, Freeman KM, Pan M, Brock WJ, Brouwer KLR. (2016) Hepatocellular disposition and transporter interactions with tolvaptan and metabolites in sandwich-cultured human hepatocytes. Drug Metab Dispos 44:867–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, Larusso NF. (2004) Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol 165:1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarino M, Buccilli V, de la Torre X, Fiacco I, Palermo A, Ughi D, Botrè F. (2017) Characterization of the phase I and phase II metabolic profile of tolvaptan by in vitro studies and liquid chromatography-mass spectrometry profiling: relevance to doping control analysis. J Pharm Biomed Anal 145:555–568. [DOI] [PubMed] [Google Scholar]
- Miranda SR, Lee JK, Brouwer KLR, Wen Z, Smith PC, Hawke RL. (2008) Hepatic metabolism and biliary excretion of silymarin flavonolignans in isolated perfused rat livers: role of multidrug resistance-associated protein 2 (Abcc2). Drug Metab Dispos 36:2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Garrido P, Marin JJ, Perugorria MJ, Urribarri AD, Erice O, Sáez E, Úriz M, Sarvide S, Portu A, Concepcion AR, et al. (2015) Ursodeoxycholic acid inhibits hepatic cystogenesis in experimental models of polycystic liver disease. J Hepatol 63:952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KS, Rowland M. (1977a) Hepatic clearance of drugs. I. Theoretical considerations of a “well-stirred” model and a “parallel tube” model. Influence of hepatic blood flow, plasma and blood cell binding, and the hepatocellular enzymatic activity on hepatic drug clearance. J Pharmacokinet Biopharm 5:625–653. [DOI] [PubMed] [Google Scholar]
- Pang KS, Rowland M. (1977b) Hepatic clearance of drugs. II. Experimental evidence for acceptance of the “well-stirred” model over the “parallel tube” model using lidocaine in the perfused rat liver in situ preparation. J Pharmacokinet Biopharm 5:655–680. [DOI] [PubMed] [Google Scholar]
- Pang KS, Rowland M. (1977c) Hepatic clearance of drugs. III. Additional experimental evidence supporting the “well-stirred” model, using metabolite (MEGX) generated from lidocaine under varying hepatic blood flow rates and linear conditions in the perfused rat liver in situ preparation. J Pharmacokinet Biopharm 5:681–699. [DOI] [PubMed] [Google Scholar]
- Pfeifer ND, Bridges AS, Ferslew BC, Hardwick RN, Brouwer KLR. (2013) Hepatic basolateral efflux contributes significantly to rosuvastatin disposition II: characterization of hepatic elimination by basolateral, biliary, and metabolic clearance pathways in rat isolated perfused liver. J Pharmacol Exp Ther 347:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretlow TG, II, Pretlow TP. (1987) Cell Separation: Methods and Selected Applications, pp 1–20, Harcourt Brace Jovanovich, New York. [Google Scholar]
- Sabbatini M, Russo L, Cappellaio F, Troncone G, Bellevicine C, De Falco V, Buonocore P, Riccio E, Bisesti V, Federico S, et al. (2014) Effects of combined administration of rapamycin, tolvaptan, and AEZ-131 on the progression of polycystic disease in PCK rats. Am J Physiol Renal Physiol 306:F1243–F1250. [DOI] [PubMed] [Google Scholar]
- Schenker N, Gentleman JF. (2001) On judging the significance of differences by examining the overlap between confidence intervals. Am Stat 55:182–186. [Google Scholar]
- Shimomura Y, Brock WJ, Ito Y, Morishita K. (2015) Age-related alterations in blood biochemical characterization of hepatorenal function in the PCK rat: a model of polycystic kidney disease. Int J Toxicol 34:479–490. [DOI] [PubMed] [Google Scholar]
- Shoaf SE, Ohzone Y, Ninomiya S, Furukawa M, Bricmont P, Kashiyama E, Mallikaarjun S. (2011) In vitro P-glycoprotein interactions and steady-state pharmacokinetic interactions between tolvaptan and digoxin in healthy subjects. J Clin Pharmacol 51:761–769. [DOI] [PubMed] [Google Scholar]
- Slizgi JR, Lu Y, Brouwer KR, St Claire RL, Freeman KM, Pan M, Brock WJ, Brouwer KLR. (2016) Inhibition of human hepatic bile acid transporters by tolvaptan and metabolites: contributing factors to drug-induced liver injury? Toxicol Sci 149:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbera LA, Castañer J, Bayes M, Silvestre J. (2002) Tolvaptan: treatment for heart failure vasopressin V2 antagonist. Drugs Future 27:350–357. [Google Scholar]
- Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, TEMPO 3:4 Trial Investigators (2012) Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367:2407–2418.23121377 [Google Scholar]
- Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, Ouyang J, McQuade RD, Blais JD, Czerwiec FS, et al. REPRISE Trial Investigators (2017) Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med 377:1930–1942. [DOI] [PubMed] [Google Scholar]
- Torres VE, Harris PC, Pirson Y. (2007) Autosomal dominant polycystic kidney disease. Lancet 369:1287–1301. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. (2009) Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther 328:652–662. [DOI] [PubMed] [Google Scholar]
- Watkins PB, Lewis JH, Kaplowitz N, Alpers DH, Blais JD, Smotzer DM, Krasa H, Ouyang J, Torres VE, Czerwiec FS, et al. (2015) Clinical pattern of tolvaptan-associated liver injury in subjects with autosomal dominant polycystic kidney disease: analysis of clinical trials database. Drug Saf 38:1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Turner KC, Ward ES, Jansen PL, Brouwer KLR. (2000) Altered hepatobiliary disposition of acetaminophen glucuronide in isolated perfused livers from multidrug resistance-associated protein 2-deficient TR− rats. J Pharmacol Exp Ther 295:512–518. [PubMed] [Google Scholar]
- Zhang A, Jia Y, Xu Q, Wang C, Liu Q, Meng Q, Peng J, Sun H, Sun P, Huo X, et al. (2016) Dioscin protects against ANIT-induced cholestasis via regulating Oatps, Mrp2 and Bsep expression in rats. Toxicol Appl Pharmacol 305:127–135. [DOI] [PubMed] [Google Scholar]