

Abstract
Many oxidation-reduction (redox) enzymes, particularly oxygenases, have roles in reactions that make and break C-C bonds. The list includes cytochrome P450 and other heme-based monooxygenases, heme-based dioxygenases, non-heme iron mono- and dioxygenases, flavoproteins, radical S-adenosylmethionine enzymes, copper enzymes, and peroxidases. Reactions involve steroids, intermediary metabolism, secondary natural products, drugs, and industrial and agricultural chemicals. Many C-C bonds are formed via either (i) coupling of diradicals or (ii) generation of unstable products that rearrange. C-C cleavage reactions involve several themes: (i) Rearrangement of unstable oxidized products produced by the enzymes, (ii) oxidation and collapse of radicals or cations via rearrangement, (iii) oxygenation to yield products that are readily hydrolyzed by other enzymes, and (iv) activation of O2 in systems in which the binding of a substrate facilitates O2 activation. Many of the enzymes involve metals, but of these iron is clearly predominant.
Graphical Abstract
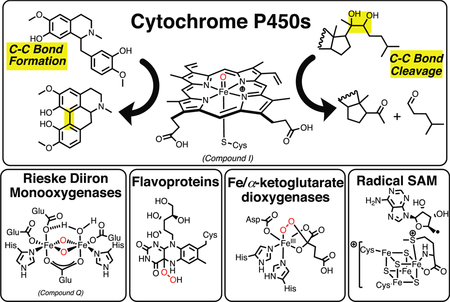
1. Introduction.
The making and breaking of bonds is the basis of metabolism, enzymology, and of biochemistry as a whole. Reviewing all such reactions would not be possible in this article, and we have chosen to restrict the scope of the review to C-C bonds. C-C bonds are inherently difficult to break, if the atoms are not activated, and we will see that a fairly common approach in nature involves first oxidizing one of the carbons in the process. Thus, a rather common paradigm is to couple the breaking of C-C bonds to oxidation-reduction reactions, e.g. oxygenating one of the carbons. In part because of our own research interests (and the original submission invitation), we have restricted our review to the making and breaking of C-C bonds (not including C-O and C-N bonds) by redox processes. The processes are divided into (i) C-C bond-making reactions and (ii) C-C bond-breaking reactions. As we will see, many enzymatic reactions we will consider involve both C-C bond breaking and forming and may not be exclusively classified in one group or the other.
At the outset, we should emphasize that we do not claim to be comprehensive in covering all C-C bond-forming and -breaking reactions catalyzed by redox-active enzymes, but we have attempted to provide many representative enzymes and examples. We have also restricted the definition of redox-active enzymes to those using prosthetic groups to do oxidation-reduction chemistry. For instance, even though there are aspects of pyridoxal chemistry that involve some formal oxidation and reduction processes to generate an electron sink in decarboxylation,1 pyridoxal is not generally considered a redox group and has not been included here. (However, see a recent addition to the literature.2)
Iron has a key role in many of the reactions we will discuss and is the dominant metal ion in the prosthetic groups. (Note: we will refer to “prosthetic groups” as parts of catalysts that do not show up in the overall reaction stoichiometry (e.g., heme, flavins). In a strict sense, “cofactors” is a term meaning co-substrates, which do show up in the overall reaction stoichiometry, e.g. NAD(P)+/NAD(P)H.
1.1. General Aspects of Oxygen Activation.
Description of some fundamental aspects of oxygen activation is in order prior to detailed consideration of mechanisms. Molecular oxygen (O2) is in the triplet state and does not normally react with carbon molecules, which are in the singlet state. (The state refers to the quantum state of the molecules and the energy levels seen in an applied magnetic field.) Singlet oxygen does exist, even in some rare biological situations, but is 22 kcal mol−1 higher in energy and difficult to control in enzymatic reactions that require activated oxygen but also great regio- and stereoselectivity. So how does O2 undergo formally spin-forbidden reactions with biological molecules?
Our understanding of redox enzymes has not always been so complete as today. Before the pioneering work of Mason,3–6 Hayaishi,7–13 and others in the 1950s, the situation was still unclear. Indeed, Wieland was of the opinion that the addition of oxygen to carbon molecules was the result of activation of the carbons or hydrogens, followed by addition of oxygen from water, as exemplified by the fatty acid β-oxidation process (i.e., desaturation followed by hydration, see section 3.3 below). This is in contrast to the concept of activation of the oxygen molecule. For more on the theories of Wieland and the opposing ideas of Warburg, see refs.4,11,12,14–16 Even later there were serious proposals of the activation of mobile species of O2 being generated and then moving to react with carbon atoms,17,18 and some of these have recently surfaced again.19,20 However, any reactive species such as a superoxide anion (O2– •), singlet oxygen, or hydroxyl radical would be extremely hard to control in delicate regioselective hydroxylations and other oxidations.
One major solution to the spin-forbidden dilemma is to bind O2 to a transition metal, most commonly iron or copper, and then consider the electronic state of the metal-oxygen complex in its reactivity with organic molecules (Figure 1). Examples with iron are termed Compound 0 (FeIII–O2¯), Compound I (formally FeO3+), and Compound II (FeOH3+). These species vary in their stability and ease of detection in enzymes and in biomimetic models. In some cases, these have been trapped and studied spectroscopically but in many other cases their roles have been inferred but never proven. As we will see, this has led to some differences in opinion about catalytic mechanisms.
Figure 1.

The issue of reaction of triplet O2 with singlet (carbon) atoms (R). Two solutions are shown, one involving a metal complex M (usually Fe or Cu) and another with a π-stabilized radical (e.g., reduced flavin) that can react with O2. (The reactions are not intended to indicate stoichiometry.)
The other major approach to activating and harnessing O2 to do useful oxidation chemistry is to stabilize superoxide anion (Figure 1). This stabilization can be done with the use of a prosthetic group in which the electronic charge associated with a radical (created in the 1-electron reduction of O2) is stabilized through an extended π system, as in the case of flavins and pterins. The stabilization leads to the reaction of the flavin semiquinone with superoxide to generate a 4a-hydroperoxy flavin, which is the active oxidant in many cases.21 (Flavin N5-oxides are also now recognized as intermediates, possibly arising from initial N5-hydroperoxide products of the reaction of reduced flavins with O2.22) Some recent cases of oxygen activation without prosthetic groups or cofactors have been reported.23,24 These cases involve π-rich substrates that appear to act in the same way as flavins to stabilize O2 and lead to oxygenated species that break down to yield the observed products.25
Among the oxygenases, there is an important distinction between monooxygenases and dioxygenases.4 Monooxygenases incorporate only one of the two atoms of O2 into substrate(s). The other atom is reduced to the level of H2O, i.e. a 2-electron reduction of O2 (Equation 1). These are called mixed-function oxidases and require the input of two electrons, which usually come from NAD(P)H as the donor,4 or occasionally ascorbic acid. An accessory protein, generally a flavoprotein, is required to couple a 2-electron process (NAD(P)H hydride transfer)1 with a 1-electron process (metal center, e.g., Fe, Cu) (Equation 1).
| (Eq. 1) |
Dioxygenases incorporate both atoms of O2 into organic substrates (Equations 2, 3)
| (Eq. 2) |
| (Eq. 3) |
Distinguishing between a dioxygenase that inserts two atoms of oxygen together (Equation 2) from a monooxygenase that does two monooxygenation steps (Equation 1) sequentially may not be easy, especially if cofactor requirements have not been rigorously characterized. The most definitive way is with the use of 18O labeling, particularly with a mixture of 18O2 and 16O2 gas. In a dioxygenase (Equation 2), the product will contain either two atoms of 18O or two atoms of 16O, but not a mixture of 18O and 16O atoms. Mass spectrometry provides a simple answer, as exemplified in the work of Samuelsson with prostaglandin E.26 If a monooxygenase is catalyzing two sequential steps (Equation 1), then the first product will contain a mixture of 18O and 16O. The second product (RO2) will contain a mixture of 18O-18O, 18O-16O, and 16O-16O, which again can be readily distinguished by mass spectrometry.26
Some dioxygenases utilize the stoichiometry shown in Equation 3, where the two atoms of oxygen are incorporated but into separate products. An example is the case of iron/α-ketoglutarate (α-KG) dependent dioxygenases, which will be discussed later (Section 3.10). One oxygen is incorporated in the substrate (R), and the other is used to oxidize α-KG to succinate (with the O label appearing in the carboxylic acid group of succinate).
Monooxygenases generally utilize heme, non-heme iron, copper, or flavins as prosthetic groups. Dioxygenases generally use heme or non-heme iron; copper-based dioxygenases are rare (e.g., quercetinase, which we will deal with later in Section 3.12.1). A few of the enzymes to be discussed use other metals, e.g. nickel (some quercetinases and acireductone dioxygenase, Section 3.12).27 As we will discuss later, no flavin-dependent dioxygenases are known.
2. C-C Bond Forming Reactions.
C-C bonds have to be formed in order to generate both relatively simple and complex molecules. There are a number of ways of doing this, including the use of prosthetic groups, e.g. thiamine pyrophosphate.1 We have restricted the scope of this review to enzymes that normally work by using oxidation and reduction (redox enzymes).
2.1. General Considerations.
Many of the C-C bond-forming reactions catalyzed by redox enzymes fall into one of two major classes, either coupling of diradical systems or the rearrangement of unstable enzyme products, either within or outside of the enzyme. The former are homolytic reactions and the latter are heterolytic, or “ionic.” Some of the diradical systems may involve one carbon radical and the equivalent of a second radical in the form of an iron-oxygen complex until it is used, e.g. some P450 reactions.
The list of enzymes to be addressed for C-C bond formation includes P450s, other heme proteins, non-heme iron proteins, flavoproteins, radical S-adenosylmethionine (SAM) enzymes, and cobalamins.
2.2. P450 Reactions.
Many of the reactions discussed here, both C-C bond forming and C-C bond breaking, are catalyzed by P450 enzymes. Humans have 57 P450 (CYP) genes and most mammals have 40–120, but P450s are also found in plants, most bacteria, fungi, and other forms of life and the total number of known sequences is now ≥ 206,000 (http://www.uniprot.org/uniprot/?query=cytochrome+P450&sort=score). Some species of fungi have >300 P450s.28 Plants each contain hundreds of CYP genes (142–412 among several plant sequences)29 and as many as 1,476 in wheat (drnelson.uthsc.edu/cytochromeP450.html). Collectively the P450s catalyze ≥95% of the reactions known to occur via redox chemistry;30 this value is driven by the literature showing roles of P450s in not only natural product biochemistry but also the dominant role of P450s in the oxidation of drugs, drug candidates, and industrial and agricultural chemicals.
2.2.1. P450 Mechanisms.
It is useful to review general aspects of P450 catalysis before considering individual applications in both C-C bond forming and C-C bond breaking reactions (Figure 2).
Figure 2.

General catalytic cycle for P450 reactions. RH indicates a substrate. The electrons are provided by either NADPH-P450 reductase (microsomal P450s, as shown) or a ferredoxin reductase/ferredoxin system (mitochondrial and some bacterial P450s). For some other electron delivery modes see Guengerich and Munro.31 Note the Fe3+–O2− (ferric peroxide, Compound 0) and FeO3+ (Compound I) forms discussed in the text. The electron transfers from the reductase are simplifications in that the course of electron flow is probably from FMNH2/FADH• to FMNH•/FADH• in the first reduction (step 2) and (assuming that the reductase contributes the second electron to the P450) from FMNH•/FAD• to FMNH•/FAD in the second reduction step (4). In many bacterial systems and in mammalian mitochondria the electrons are donated by ferredoxins (e.g., adrenodoxin, putidaredoxin). In some cases with mammalian P450s, the second electron (step 4) is donated by cytochrome b5 (b5).32 In the literature there exists different nomenclature for the same iron intermediates in this P450 catalytic cycle.33,34 For clarity, throughout the text of this manuscript Compound I is referred to interchangeably with FeO3+, and ferric peroxide is referred to interchangeably with FeIII–O2− or Compound 0 (protonated form FeIII–OOH).
The cycle shown in Figure 2 is for a microsomal P450 utilizing the diflavin NADPH-P450 reductase as a source of electrons. In many cases this reductant is replaced by a flavoprotein/ferredoxin system, particularly in mitochondria and bacteria. There are some other cases involving alternate electron sources, and a few P450s even use H2O2 as an active oxygen source, circumventing the need for electrons.31
The typical reaction cycle contains at least nine discrete electronic steps (Figure 2). It should be noted that at least several of these are accompanied by conformational protein changes,35 but complete roles of such changes are still undefined. An overall point to make is that, although substrate binding may facilitate oxygen activation in some cases,36 the activation of molecular oxygen proceeds by a pathway independent of the substrate. This pattern is shared by flavin-based monooxygenases21 and differs from many dioxygenases, which have iron liganded to both parts of the substrate and to the oxygen, leading to oxygen activation.37,38
The catalytic cycle (Figure 2) begins with binding of the substrate (RH) in Step 1, near the iron but not attached to it. Step 2 is a 1-electron reduction by either the diflavin reductase or a ferredoxin, which in turn has been reduced by a flavoprotein. The rate of reduction may or may not be facilitated by substrate binding.36,39,40 The possibility exists that substrate binding can occur after reduction, and this has been shown.41 Further, a population of ferrous P450 is found in bacterial or liver cells, apparently in the absence of substrates.42 Regardless of the route to the substrate-bound ferrous enzyme, it reacts with O2 to form a ferrous-O2 complex (Step 3), electronically analogous to oxyhemoglobin but much less stable. The stability varies considerably36,43,44 and for several P450s this species has not been detected yet.45 The next step (Step 4) involves the input of another electron. This electron can be provided by the flavoprotein reductase. In some cases this second electron can come from another hemoprotein, cytochrome b5 (b5),46,47 as originally proposed by Hildebrandt and Estabrook,48 although electron transfer has been ruled out in a number of the cases in which stimulation of catalysis by b5 is observed.32,49–51
Following the input of the second electron, the next step (Step 5) is protonation (of Fe3+O2−) to yield an iron hydroperoxide complex (formally FeIII-OOH) (one electron is transferred from the FeIII and one from O2− to form the Fe–O bond, leaving the iron in a formal 3+ (III) state. This complex loses water (Step 6) to generate a formal FeO3+ complex, generally thought to be in the form of FeIV=O with the additional charge and radical (·+) in the porphyrin ring.52–54 This entity is termed Compound I, based on electronic similarity to the entity first characterized in peroxidase chemistry.55 Compound I is highly unstable in P450s, in contrast to some peroxidases and catalase, and to date has only been characterized in the case of a few bacterial P450s.56–58 We will also see elements of this type of chemistry with some other iron-based enzymes (heme oxygenase, dioxygenases).
Compound I abstracts a hydrogen atom in Step 7 (generating Compound II, formally FeIV-OH). The resulting pair of radicals can usually be considered as a caged reaction, i.e. FeOH3+ plus R·, and the Fe-O bond splits homolytically for “oxygen rebound” to the incipient substrate radical (R·), yielding the product ROH (Step 8), which is released in Step 9 to complete the basic cycle. As we will discuss later, Compound I has a high redox potential and can also abstract an electron from an amine or even some hydrocarbons.
Compound I is generally considered to be the active oxoiron (ferryl) species in most P450 reactions.54 Compound I has been trapped through rapid-freeze quenching by reacting a P450 enzyme with a stoichiometric amount of m-chloroperbenzoic acid (m-CPBA), an oxygen surrogate, and has been subsequently characterized by spectroscopic techniques (i.e., Mössbauer, EPR).56 The resulting ferryl species has been clearly shown to hydroxylate alkanes.33 As an electrophilic oxygen species, Compound I abstracts hydrogen atoms of C-H bonds and, in an analogous manner, can abstract a hydrogen atom from an O-H bond to yield an oxygen radical intermediate. One of the precursors to Compound I is formally an iron (III) bound to peroxide (Fe3+-O2− in Figure 2), which is known as ferric peroxide (Compound 0, vide supra) and has been proposed to be an active iron species that performs some of the various other reactions that P450s catalyze (e.g., epoxidation and C-C bond cleavage).59 Compound I has electrophilic properties while ferric peroxide (unprotonated) is nucleophilic.
Two points about this catalytic cycle are relevant here. One is related to both C-C bond- forming and -breaking. In some cases Step 8 (Figure 2) appears to be slow enough to allow the R· (substrate radical) to undergo some rearrangement, moving the site of the radical on the molecule.60 In some cases the FeOH3+ species, which is still a very reactive molecule, can also abstract a hydrogen atom, e.g. in desaturation reactions.45,54,61,62 In the case of the C-C bond-forming reactions, as we will see, a dominant aspect is the formation of diradical systems that can couple together (to form new C-C bonds).
Another major issue that will be discussed regarding several C-C bond-breaking reactions is whether they use the FeO3+ species (Compound I) or Fe3+O2¯ (Compound 0). These are both unstable species and, except in a few cases,34,56,57 cannot be generated in the laboratory. Even if Compound 0 were generated directly,34 it could proceed to yield Compound I before reacting (Figure 2). We will see that both Compound I and Compound 0 mechanisms have been proposed for some of the P450 C-C bond-cleavage reactions (e.g., P450s 19A1, 17A1, 51A1, and 1A2). Mechanistically these are quite different in that Compound I is an electrophile and Compound 0 is a nucleophile, as already mentioned. Distinguishing between the mechanisms has not been trivial in some cases.
2.2.2. P450 Diradical Coupling Reactions.
Many of the P450-dependent C-C bond forming reactions involve diradicals. Direct evidence for their existence has been difficult to obtain even when such rationalizations seem very reasonable, e.g. there is little if any EPR evidence for their existence in almost all of the examples we will cover, presumably due to short half-lives, and such reactions are generally insensitive to the addition of radical scavengers.
It is possible to propose mechanisms with actual diradicals in some cases. In others, the mechanisms have been written with a combination of a carbon radical plus FeOH3+, an entity capable of pulling another hydrogen atom from a C-H bond.
2.2.2.1. Benzyl Isoquinoline Alkaloids.
The benzyl isoquinoline alkaloids are a group of > 2,500 known structures found in nature (mainly in plants), some with medicinal properties that have been known since ancient times.63 These compounds are derived by a number of C-C and other coupling reactions, yielding entities that can be further decorated by various enzymes (Figure 3). The biological advantages of making these alkaloids in the plants are, in general, unknown.29 Many are probably for protection, although in many cases little is known about the natural enemies of the organism.
Figure 3.

Coupling reactions involved in benzylisoquinoline alkaloid biosynthesis.29,63 The colors of the rings (black, red, blue) trace the origins in 1-benzylisoquinoline.
Details of the C-C coupling reactions are usually not known. Some of the reactions probably involve C· diradical coupling, as proposed in one step of morphine synthesis (Figure 4).
Figure 4.

Proposed C-C diradical coupling in the synthesis of salutaridine from reticuline by plant and mammalian P450s.64
Some other examples are shown in Figure 5, and roles of two specific P450s with the enantiomers of reticuline are shown in Figure 6.
Figure 5.

Products of more coupling reactions in the synthesis of isoquinoline alkaloids from (S)-reticuline by plant P450s.65
Figure 6.

C-C bond coupling with reticuline by plant P450s 80G2 and 719B1.29,63,66
2.2.2.2. Morphine Biosynthesis.
The synthesis of morphine begins with tyrosine. Morphine is synthesized in mammals as well as in plants, in very low concentrations, and most of the reaction steps are the same.64,67–69 A key step is the C-C bond coupling (C12-C13) of two phenyl rings in the conversion of (R)-reticuline to generate the important intermediate salutaridine (Figures 4, 6), which goes on to form thebaine, having an additional ether linkage of the two rings. The regiochemistry of the reaction with (R)-reticuline is extremely important (in generating salutaridine), in that alternate coupling products include (+)-pallidine, (−)-isoboldine, and (−)-corytuberine.64 A P450 in opioid poppies catalyzes the coupling to give salutaridine (Figure 6),70 Zenk and his associates also identified a porcine liver P450 enzyme that formed salutaridine.69,71 Human P450s 2D6 and 3A4 can oxidize (R)-reticuline to multiple C-C cyclized products, including salutaridine.64 Subsequent studies showed that human P450s 3A4 and 3A5 can catalyze the O6-demethylation of thebaine (derived from salutaridine) and that P450 2D6 catalyzes the O3-demethylation of codeine, and thus all of the steps in morphine biosynthesis are now established in humans.67 However, the extent of contribution of this pathway to endogenous control of pain remains to be determined.
A mechanism for the oxidative (C12-C13) coupling of (R)-reticuline to form salutaridine has been proposed (Figure 4).64 As in most P450-catalyzed C-C bond-forming reactions, the mechanism involves generation of a diradical system, followed by coupling of two radicals.64
2.2.2.3. P450 3A4 and Raloxifene Dimer.
Another coupling is the formation of a dimer of the drug raloxifene by P450 3A4 (Figure 7).62,72
Figure 7.

Because of the coupling of the phenolic rings, the reaction can easily be rationalized by the coupling of two 1-electron oxidation species generated from the parent drug raloxifene. The authors concluded that the active site of P450 3A4 is sufficiently large enough to accommodate two molecules of (P450 3A4) substrate, a view supported by X-ray crystallography.73,74 P450 3A4 can also form a dimer from 17β-estradiol62,75 but this is an O-linked product and not within the scope of this review.
2.2.2.4. P450 121 and Dicyclotyrosine.
Mycobacterium tuberculosis causes more deaths each year than any other known pathogen, and there is wide interest in finding better drugs to kill the organism. M. tuberculosis has 20 P450 genes, and the functions of only some are known.76 A knockout strain with deletion of rv2276, the gene coding for P450 121, showed the physiological significance of this P450 in the bacterium and suggests it as a drug target. An X-ray crystal structure of M. tuberculosis P450 121 has been published.77
A key to understanding the function of the enzyme came with the identification of cyclization of tyrosine as a reaction catalyzed by the enzyme (Figure 8).78
Figure 8.

C-C diradical coupling in the synthesis of dicyclotyrosine by P450 121, supported by peracetic acid in a model system.79,80 PCET: proton-coupled electron transfer.
The mechanism involves a Compound I intermediate (Figure 2) and, most probably, some type of diradical cyclization (Figure 8).79 More detailed spectroscopic characterization of the enzyme has been reported,80 but the exact biological role of the cyclized product is yet unknown.
2.2.2.5. P450 158A2 and Flaviolin Coupling.
P450 158A2 is one of 18 P450s found in the soil bacterium Streptomyces coelicolor.81 Defining the functions of the P450s of this complex prokaryotic organism has been challenging. The organism uses a Type III polyketide synthase to make hydroxylated naphthalene molecules.82 Some of these polymerize to melanins to contribute to virulence83 and are also UV-protective.82 One compound in this naphthalene group, flaviolin, was found to be oxidized to dimers and trimers by S. coelicolor P450 158A2.84 A crystal structure of the ferric protein contained two molecules of flaviolin, and a diradical pathway for synthesis of dimers is proposed (Figure 9).84
Figure 9.

Proposed C-C diradical coupling in the synthesis of flaviolin by S. coelicolor P450 158A2.84,85
Further X-ray crystallography structures implicated water molecules and the hydroxyl groups of flaviolin in assisting in the oxygen activation process.85 A structure of the Fe2+O2 complex (with two flaviolins bound) was reported.85 The closely related S. coelicolor P450 158A1 catalyzes similar flaviolin dimerization reactions but does not form the trimer seen with P450 158A2.86 Two flaviolin molecules are also seen in the X-ray crystal structure of P450 158A1 but are in different positions than in P450 158A2.86 A lysine group (Lys-90) in P450 158A1 is important in disallowing the binding of the third flaviolin needed to form trimers; this residue is substituted with an isoleucine (Ile-87) in P450 158A2.87 The conclusion was reached that the (diradical) coupling reactions occur within the enzyme, in that P450 158A1 generates only two of the three diflaviolin products that P450 158A2 does, and the molar ratios differ compared with the 158A2 products. Thus each enzyme seems to maintain its own stereo- and regioselectivity.86 This result would not be expected if diradical coupling was thermodynamically controlled and occurred outside of the active site.
2.2.2.6. P450 245A1 and Indolocarbazole Alkaloid Biosynthesis.
Another example of diaryl coupling is the intramolecular cross-linking of two indole moieties in the biosynthesis of indolocarbazole alkaloids by P450 245A1 (Figure 10).62,88,89 The nature of the indole ring argues for a mechanism involving 1-electron abstractions and coupling of the two rings (Figure 10).
Figure 10.

P450 245A1 and the synthesis of indolocarbazole alkaloids.62,88,89
2.2.2.7. P450 107B and Himastatin Biosynthesis.
Another example of apparent diradical coupling is seen with P450 107B in the synthesis of the natural product himastatin (Figure 11). Again, diaryl coupling is seen, probably the result of the proximity of two radicals.
Figure 11.

C-C coupling by bacterial P450 107B1 (HmtS) in the biosynthesis of himastatin.76,90,91
2.2.3. P450 Rearrangements Involving Diradical Coupling.
In some cases bonds are broken and new ones are formed after rearrangement, with the postulated intermediacy of radicals.
2.2.3.1. P450 93C2 and Rearrangement of Flavanone.
P450 93C2 oxidizes flavanone, resulting in 3-hydroxyflavanone and 2-hydroxyisoflavanone (Figure 12). The mechanism can be rationalized by the formation of a carbon radical and an unusual migration of a phenoxy group to the adjacent carbon.92–94
Figure 12.

Proposed mechanism for migration of an aryl group in flavanone by P450 93C2.92–94
2.2.3.2. Oxidation of Littorine by P450 80F1.
Another migration occurs in the oxidation of (R)-littorine, in which a carboalkyl ester migrates to the adjacent carbon. The mechanism can be rationalized in the context of a radical intermediate (Figure 13).95,96
Figure 13.

Oxidation of (R)-littorine to (S)-hyoscyamine aldehyde by Hyoscamus niger (nightshade plant) P450 80F1.95,96
2.2.4. P450s 5A1 and 8A1 and Rearrangements of Prostaglandins.
These mammalian reactions can be considered as both C-C bond-forming and breaking but will be discussed here. The endoperoxide prostaglandin H2 (see Section 2.4) is unstable and reacts with ferric hemeproteins. These reactions are different than those generally catalyzed by P450s, in that the “active oxygen” is supplied by the substrate and the roles of the enzymes are to direct the rearrangements.
The major reactions are catalyzed by P450s 5A1 and 8A1, thromboxane synthase and prostacyclin synthase, respectively (Figure 14).
Figure 14.

Reactions of P450s 5A1 and 8A1 with prostaglandin H2 to form thromboxane A2 (TXA2) and prostacyclin (PGI2).97,98
In general, thromboxane A2 is considered harmful in that it activates blood platelet aggregation and is a vasoconstrictor. Prostacyclin (PGI2) is generally considered a good actor in that it inhibits blood platelet aggregation and is a vasodilator.99 Mechanisms for the rearrangements are shown in Figure 14.97,98,100
In addition to forming thromboxane A2, a side reaction is the conversion to hydroxyheptatrienoic acid (HHT) and malondialdehyde (Figure 15).98
Figure 15.

Conversion of prostaglandin H2 (PGH2) to thromboxane A2 (TxA2) and to hydroxyheptatrienoic acid (HHT) and malondialdehyde by P450s.98,101
This reaction can be catalyzed by a physiologically irrelevant bacterial P45098 or by mammalian hepatic P450s (1A2, 3A4).101 The physiological significance of these latter reactions is not clear.
2.2.5. P450 and Other Natural Products–Coupling of Unstable Products.
In some cases diradical coupling is not involved. Products are generated that appear to leave the active site and then, due to chemical instability, rearrange to form stable products. Two examples with natural products are presented below. (More will be seen with synthetic substrates later, Section 2.2.6.)
2.2.5.1. Mammalian P450s and Indole Oxidation.
The 3-hydroxylation of indole yields indoxyl, which is an inherently unstable molecule. Indoxyl and substituted indoxyls form colored dimers, including indigo, indirubin, and other products (Figure 16).
Figure 16.

P450-catalyzed coupling of indoles to yield dimeric products. (A) Indigo and indirubin;102 (B) oxazole product;102 (C) product of 4-OBn indole.103 Bn: benzyl.
3-Hydroxylation of indoles is catalyzed by several P450s, particularly human P450s 2A6 and 2E1,102,103 and was first detected by the blue color (indigo) that accumulates in bacterial cultures of these enzymes when heterologously expressed in bacteria.102,104,105 The (non-enzymatic) coupling is enhanced in the presence of oxygen and is base-catalyzed.106
2.2.5.2. P450s and Trichothecene Biosynthesis.
P450s are also involved in key steps in the biosynthesis of trichothecenes, mycotoxins produced in several fungi (Figure 17).107
Figure 17.

P450 reactions involved in trichothecene biosynthesis in Trichoderma species.107
Key steps include an allylic hydroxylation and epoxidation to yield the unstable molecule isotrichodiol, which rearranges non-enzymatically to a product that is then hydroxylated to yield trichodermol.
2.2.6. P450 Reactions in Drug Metabolism–Coupling of Unstable Products.
As with natural products (vide infra), drug metabolism by P450s can result in the formation of unstable molecules that couple by chemical means. A few examples are presented.
2.2.6.1. YH3945.
An example of the formation of a new ring structure is shown with the drug candidate YH3945 in Figure 18.108 This path commences with N-demethylation and hydroxylation of a methylene adjacent to a thiourea moiety followed by an O-demethylation step, which facilitates some of the subsequent transformation with the conjugation in the pyridine ring, ultimately generating a new ring.
Figure 18.

Ring formation reaction (to metabolite M14) catalyzed by P450s with the drug candidate YH3945, 1-{3-[3-(4-cyanobenzyl]-3H-imidazol-4-yl]propyl}−3-(6-methoxypyridin-3-yl)-1-(2-trifluoromethylbenzyl)thiourea. The first two reactions are C-hydroxylation and loss of the cyanobenzyl group through N-dealkylation.108
2.2.6.2. Thiophene S-Oxide Coupling.
Another case of C-C bond coupling is seen with thiophene S-oxides (Figure 19). S-Oxygenation is associated with covalent binding to glutathione (GSH) and proteins (Figure 19B), possibly related to toxicity. Both ticlopidine and the simple model compound 2-phenylthiophene have been shown to form S-oxides, which can undergo non-enzymatic Diels-Alder condensation to form dimeric products (Figure 19). These Diels-Alder reactions should show second-order kinetics, and whether they occur at low concentrations in cells is unknown.
Figure 19.

Dimer formation via Diels-Alder reactions of products. (A) Dimerization of thiophene S-oxide formed from ticlodipine by P450 2C19 or 2D6.109 (B) Dimerization of an S-oxide formed from 2-phenylthiophene by P450 1A1 and conjugation with glutathione (GSH).110
2.2.7. P450 Non-redox Reactions.
Essentially all P450 reactions involve either iron redox chemistry or at least the use of iron to make rearrangements of molecules (e.g., prostaglandins or hydroperoxides) via high-valent iron intermediates (e.g. P450s 5A1, 8A1).111 Only a few P450s have been reported to catalyze non-redox chemistry, and two of those cases involve hydrolyses.112,113 The other case, involving S. coelicolor P450 154A1, is one of C-C bond-breaking and reformation to synthesize an unusual Paternò-Büchi-like oxetane product (Figure 20).114
Figure 20.

A non-redox rearrangement to yield C-C coupling catalyzed by S. coelicolor P450 154A1.114
2.2.8. Cyclopropanaiton by Engineered P450 Derivatives.
Arnold and associates have engineered bacterial P450s to do some unusual reactions, utilizing the approach of directed evolution (or, perhaps more properly termed “molecular breeding”).115,116 Of particular interest, bacterial P450 102A1 (P450BM-3) has been developed to insert carbenes into olefins, thus accomplishing cyclopropanation reactions (Figure 21).116–119 The same strategy can be applied to generate cyclopropanes, in peptides, beginning with the readily available dehydroalanine.120
Figure 21.

Cyclopropanation reactions catalyzed by unnatural P450 derivatives (P411).118
The mechanism of this reaction is not very similar to that of the usual P450s, in that the enzyme is acting as a carbene transfer reagent. The heme-binding cysteine is not needed, and a serine residue is more useful.121 The engineered P450 is termed P411 due to its altered spectral properties,118 and other proteins and metals can also be utilized in developing catalysts such as these.
2.3. Flavoproteins.
Flavins contain an isoalloxazine ring system that facilitates both electron transfer and reaction with O2 (Figure 1).1,21 Because they (along with pterins) can function in both 1- and 2-electron transfer reactions, they serve as biological “transformers” in coupling 2-electron chemistry (e.g., hydride transfers from NAD(P)H) with 1-electron accepting metal centers (e.g., iron). The roles of 4a-hydroperoxides in monooxygenase reactions have been known for some time1,21,122,123 and will be discussed later under C-C cleavage reactions. Recently a number of examples have been reported of how flavins are used to catalyze many other types of reactions,124,125 including C-C bond-making reactions.
2.3.1. Berberine Bridge Enzyme.
One unusual reaction, the formation of the protoberberine ring system (Figure 3), is catalyzed by a flavoprotein termed berberine bridge enzyme (BBE). The crystal structure of Eschscholzia californica (California poppy) BBE has been published, as well as the rates of the oxidative and reductive half-reactions.126 The FAD prosthetic group has an unusual bicovalent linkage to the protein (Figure 22). The enzymatic mechanism is postulated to involve an unusual oxidation of an N-methyl group concurrent with coupling to a neighboring aryl moiety (Figure 22), followed by rearrangement to give (S)-scoulerine and reoxidation of the reduced FAD.126
Figure 22.

Mechanism of berberine bridge enzyme (BBE).126–128 R indicates the phosphoribitol side chain.
More examples of BBE-related flavoproteins are now known, including the enzyme involved in nicotine formation (Figure 23).129 Other flavoproteins in plants show sequence similarity and catalyze dehydrogenations of benzyl isoquinoline alkaloids but do not appear to catalyze such C-C bond formations.130,131
Figure 23.

More BBE-like flavoprotein-catalyzed C-C bond coupling reactions.129 (A) Cannabigerolic acid (CGBA) is oxidized by tetrahydrocannabinolic acid (THCA) synthase (THCS) to THCA and by cannabidiolic acid (CBDA) synthase (CBDAS) to CBDA).132,133 (B) Synthesis of nicotine. (C) Synthesis of anabasine.129
2.3.1.2. Cannabidiolic Acid Synthase.
BBE is not the only flavoprotein that can use its oxidation/reduction capability to catalyze C-C bond coupling reactions (Figure 23). For example, tetrahydrocannabinolic acid synthase catalyzes an oxidation to form tetrahydrocannabinolic acid, and cannabidiolic acid synthase uses the same substrate (cannabigerolic acid) to form cannabidiolic acid.132,133 In the process, the flavin is reduced; to recycle the flavin, the enzyme reacts with O2 and produces H2O2 (Figure 23A). Other examples involve the synthesis of nicotine (Figure 23B) and its homologue anabasine (Figure 23C). In the latter two processes CO2 is released, a new C-C bond is formed, and H2O2 is produced.
2.3.2. Other Flavoproteins.
Flavins can also act in an auxiliary role. Flavoproteins deliver electrons to P450s, either directly or indirectly (via iron-sulfur proteins) (Figure 2). In the example shown in Figure 24 a flavoprotein delivers electrons to a radical SAM-based transferase, an iron sulfur cluster protein involved in the conversion of tRNA N1-methyl guanosine to wyosine, an etheno derivative.134,135 In this case, the source of the additional carbons is pyruvate.135
Figure 24.

Formation of the modified base wyosine in m1G37-tRNA by Tyw1.134,135 Ado-Met: S-adenosylmethionine (SAM). (See also Figure 40, vide infra.)
2.3.2.1. UDP Galactose Pyranose Mutase.
Another “new” role of a flavin involves the protein UDP galactose pyranose mutase (UGM) (Figure 25).136 A reduced flavin acts as a nucleophile (N5 atom), and the enzyme uses its redox capacity to process through an intermediate in which a UDP sugar hydroxyl acts as a nucleophile, generating a rearrangement leading to a new sugar ring.
Figure 25.

Rearrangement of UDP-galactopyranose to UDP-galactofuranose catalyzed by the flavoprotein UGM.136 R indicates the phosphoribitol side chain.
2.3.2.2. Uridine Methylation in tRNA.
Flavoproteins also work with other proteins in other ways, as exemplified by the post-transcriptional conversion of uridine to thymidine in tRNA, specifically m5U54 (Figure 26A).135
Figure 26.

Reductive methylation of tRNA and rRNA by flavin methyltransferases.135 R indicates the phosphoribitol side chain. (A) Overall reaction catalyzed by TrmFO and RlmFO. (B) Proposed mechanism. N5,N10-CH2-THF: N5,N10-methylene tetrahydrofolate. The N5-substituted flavin is derived by transfer from a methylene (or other one-carbon unit) from the substituted folate.
Covalent bonding of the RNA to Cys-223 of the enyme is required (Figure 26B). The 1-carbon unit is transferred from N5,N10-methylenetetrahydrofolate to the N5 atom of the reduced flavin of TrmFO. The carbon attached to the N5 atom of the flavin is in an electrophilic state and can be attacked by the double bond of the uracil moiety to form a covalent intermediate. Collapse of the intermediate, followed by the addition of a hydride ion, yields the thymine moiety and the oxidized flavin (Figure 26B). The flavin needs to be reduced again to reinitiate the process. TrmFO flavoproteins are found in many bacteria, but the only one for which a 3-dimensional structure is known is the Thermus thermophilus enzyme.137
One aspect of redox enzymes that we are not discussing in depth is electron transfer through the amino acids of proteins. However, tyrosine and tryptophan radicals can be involved in some of the hemeprotein and flavin reactions, and in many protein systems tyrosine can play a functional role as a redox intermediate.138 For instance, the role of a tyrosine radical cation (Tyr-343) in T. thermophilus TrmFO has been studied.139 This tyrosine is very close to the flavin, in an apolar environment and shielded from the cleft forming the substrate binding site by the isoalloxazine ring. Tyr-343 has been identified as the electron donor responsible for quenching the excited state of FAD.139
2.3.2.3. OxaD, NotB, and Epoxidation.
Flavins catalyze epoxidation reactions, although not as commonly as P450s (e.g., squalene epoxidation140). In the case of the biotransformation of notoamide S, the enzyme OxaD forms an epoxide on a 5-hydroxytryptophan entity (Figure 27). Rearrangement leads to the transfer of a pentenyl unit to form a new C-C bond.141
Figure 27.

Rearrangement of notoamide E following epoxidation by the flavoprotein NotB. 141
2.4. Prostaglandin Synthesis.
Arachidonic acid is converted to prostaglandin G2 by prostaglandin synthases, or cyclooxygenases. Prostaglandin G2 is reduced to prostaglandin H2, which then serves as a source of a number of prostaglandins (Figure 28).142
Figure 28.

Prostaglandins synthesized from prostaglandin H2 (PGH2).142 PG: prostaglandin; TX: thromboxane. See also Figure 14.
Mammals, including humans, have two prostaglandin synthases (PTGS1 and PTGS2, sometimes referred to as COX-1 and COX-2, not to be confused with cytochrome oxidase, a more critical enzyme for life). These enzymes are the main targets of almost all non-steroidal anti-inflammatory drugs. Several structures of mammalian prostaglandin synthases are now known.142,143 The enzymes are rather rigid, a property that has facilitated modeling studies and drug discovery efforts.
The conversion of arachidonic acid to prostaglandin H2 involves two functions of the enzyme, the cyclooxygenase and the peroxidase functions. The prosthetic group is heme and the enzyme behaves as a dioxygenase in the cyclooxygenase step. The cyclooxygenase function is of relevance in terms of this review, in that an internal C-C bond is formed. The peroxidase function must also be discussed briefly in light of its role in activation of the system.143 It is of note that the enzyme containing Mn-substituted protophyrin IX (PPIX) has cyclooxygenase activity but not the peroxidase function.142,144
The catalytic mechanism involves initial abstraction of the 13-pro-(S) hydrogen atom of arachidonic acid (Figure 29).142
Figure 29.

Mechanism of oxidation of arachidonic acid to prostaglandin (PG) G2.142
The resulting radical reacts with O2 to form a peroxy radical, which then reacts with the 8,9-double bond to form a 9,11-endoperoxide and move the radical to carbon 8. C-C bond formation them places the radical at carbon 13. Isomerization and reaction with another molecule of O2 yields the 15-peroxy radical, which abstracts a hydrogen atom to form prostaglandin G2 (Figure 29). The hydroperoxide is reduced to the alcohol prostaglandin H2 via the peroxidase function of the enzyme.142 Further transformations of prostaglandin H2 do not involve C-C bond breaking and formation except for the transformations to thromboxane and prostacyclin, which are catalyzed by P450s and are discussed elsewhere (Section 2.2.3).
The coupling of the C-C bond-making cyclooxygenation reaction with the peroxidase function is complex and will only be treated briefly here. However, it is an important part of the action of the enzyme mechanism and merits some discussion (Figure 30). The heme iron uses chemistry similar to other peroxidases and P450s, plus a tyrosyl radical whose role was elusive for many years.145
Figure 30.

Coupling of cyclooxygenation and peroxidation functions in the prostaglandin synthase reaction.143 AA: arachidonic acid..
Initially a peroxide reacts with the heme iron, yielding Compound I (FeO3+ or FeIV=O PPIX+•) and an alcohol. Compound I oxidizes Tyr-385 to a phenoxy radical, apparently via electron transfer involving the proximal heme ligand His-388. The Tyr-385 radical of Intermediate II, a Compound II-like species (Figure 30) abstracts the 13-pro-(S) hydrogen of arachidonic acid to initiate the cyclooxygenation process (Figure 30). The first product is prostaglandin G2, a hydroperoxide that can then be reduced to an alcohol (prostaglandin H2) by the peroxidase cycle, regenerating Compound I to continue the cycle (Figure 30).
Adding an enzyme (e.g., glutathione peroxidase) that scavenges the hydroperoxide will quench the reaction. As might be expected, the initial reaction needs “priming” and there is a lag phase.142,146 One might expect the reaction to continue indefinitely, in that it is a propagative radical reaction (Figure 30). However, the high-valent intermediates are destructive and lead to crosslinking and other destructive processes (Figure 30).142,143
2.5. Lipoxygenase-coupled Reactions.
Lipoxygenases are iron-dependent dioxygenases that catalyze the addition of O2 to allylic or otherwise activated carbons.1 They do not form or cleave C-C bonds by themselves, insofar as we know. However, the hydroperoxide products can be substrates for some unusual P450s and other heme-based enzymes, and a wide variety of natural products result from these processes.
2.5.1. Dicyclobutane Formation.
A highly unusual example of C-C bond forming is seen with a cyanobacterial hemoprotein-lipoxygenase fusion protein (Figure 31).147
Figure 31.

Enzymatic synthesis of a bicyclobutane fatty acid by a cyanobacterial hemoprotein-lipoxygenase fusion protein.147
The reaction begins with a fatty acid hydroperoxide, which can be produced by a dioxygenase reaction, i.e. the lipoxygenase domain of the fusion protein. Two products are observed. One is a leukotriene A-type epoxide. More relevant here is the other product, involving C-C bond formation catalyzed by the hemoprotein. The putative epoxide-carbocation rearranges to create a cyclopropane ring and a new carbocationic center. This center reacts to generate a highly unusual bicyclobutane ring system.
2.5.2. P450 74 Rearrangements.
The rearrangement of oxidized lipids to new biological products was already discussed with P450s 5A1 and 8A1 (Section 2.2.4, Figure 14). The P450 74 Family was first discovered in plants, specifically flaxseed.148 Allene oxides are unstable and form a variety of products, many of which include new C-C bonds (Figure 32).61,99
Figure 32.

Some general product pathways for P450 74 reactions.
The mechanism for formation of these products is shown (Figure 33).99
Figure 33.

Proposed mechanism of radical and ionic pathways for P450 74 reactions.99
Homolytic scission of the hydroperoxide (produced by a lipoxygenase) initiates the reaction. This process is considered to be very physiological, in that many of the products that emanate from the pathway (Figure 34) have important biological roles in plants and other species, e.g. signaling.
Figure 34.

Proposed role of epoxy allylic carbocation in the formation of marine oxylipins.149
Although alkyl hydroperoxides have been utilized with mammalian P450s in oxygen surrogate studies in the laboratory,150 there is no strong evidence to support the view that these play physiological roles. Most studies with these have been done with high concentrations of alkyl hydroperoxides, which are unlikely to be relevant.151–153 Another issue is that homolytic scission of alkyl hydroperoxides generates alkoxy radicals, which can initiate their own non-enzymatic chemistry and may be unrelated to the normal enzyme chemistry.154
Although radical pathways can explain some of the products that have been isolated, ionic intermediates (e.g., carbocations) are preferred to rationalize several products (Figure 33).99 The process of a sequential electron transfer from a carbon radical to Compound II (FeOH3+) is generally well-accepted and, as discussed later, a very rational explanation for events related to steroid aromatization (see Section 3.1.1.1.3, Figures 58, 59, vide infra) and hydride and alkyl group transfers in P450 reactions, as well as some other redox enzymes.99,149 (See also Figure 82, vide infra.)
Figure 58.

Use of 18O labeling studies in defining the mechanism of P450 19A1.177,220,222,223 Steps 1 and 2 are generally agreed to involve the P450 FeO3+ entity and hydrogen atom abstraction/oxygen rebound.224 Two possibilities are shown for Step 3 in the presence of 18O2. In Step 3a, the FeIII–O2− entity participates in a nucleophilic attack on the 19-aldehyde. In Step 3b, the FeO3+ form of the P450 19A1 abstracts the 1β-hydrogen atom of the gem-diol. Electron transfer yields the carbocation, which collapses to yield the estrogen product. In Step 3a, the HCO2H must contain label (18O) but not in Step 3b. “*O” denotes 18O. The step 3(b) pathway is supported by recent evidence.177
Figure 59.

Oxidation of androstenedione by P450 19A1: aromatization and formation of a carboxylic acid by oxidation of the 19-formyl intermediate. The reaction begins with abstraction of the 1β-hydrogen atom by FeO3+. Further electron transfer, as proposed by Hackett et al.,226 yields the C-1 carbocation, and proton abstraction from the gem-diol by Fe–OH and rearrangement yields HCO2H and subsequently estrone. Alternatively oxygen rebound can occur to the A ring to yield a hydroxy derivative of the 19-aldehyde detected by LC–MS (not shown in Figure 59),177 which might be the 2β-hydroxy 19-aldehyde reported by Fishman and associates.209,211 In an alternative initial reaction, FeO3+ abstracts the C-19 hydrogen atom. Oxygen rebound yields a gem-triol, which dehydrates to the 19-carboxylic acid.177 Red color is used to track oxygen atoms.
Figure 82.

Proposed mechanisms for some reactions catalyzed by P450 74 enzymes. Figure 33 is expanded to include C-C cleavage products (divinyl ether and hemiacetal).99
Several examples of the formation of unusual transformations by these pathways are presented. In green algae, the movement of a carbon outside of the chain to form a formyl group is shown (Figure 35).
Figure 35.

Formation of a branched-chain (Acrosiphonia coalita) trienal.149
The formation of a cyclopropyl group and a lactone ring is observed in the synthesis of constanolactones in the red alga Constantinea simplex (Figure 36).149 The red alga Laurencia hybrida synthesizes hybridalactone, a macrocyclic lactone with a cyclopropyl group (Figure 37).149 Finally, the marine diatom Nitzschia pungens forms an unusual bicyclic lactone (Figure 38).149
Figure 36.

Formation of constanolactone (C. simplex).149
Figure 37.

Formation of hybridalactone (L. hybrida).149
Figure 38.

Formation of bacillariolides I and II (N. pungens).149 HPETE: hydroperoxypentatetraenoic acid.
2.6. Rieske Oxygenases RedG and PR4B68 and Prodiginine Synthesis.
Prodiginines are red tripyrrolic alkaloids with a variety of biological activities,155 formed by oxidation reactions (Figure 39).
Figure 39.

Biosynthesis of prodiginines by Rieske/diiron monooxygenases.155 (A) Two coupling reactions. (B) Postulated mechanism of the reaction with prodigiosin, as proposed by de Rond et al.155
In the complex bacterium S. coelicolor, the process involves a family of Rieske non-heme iron monooxygenases, exemplified by RedG. (Rieske iron sulfur clusters were first discovered in 1964 in mitochondrial electron transfer chain proteins156 and have also been implicated in both monooxgenases and dioxygenases, vide infra.) In the marine bacterium Pseudoalteromonas rubin, the reaction is catalyzed by PR4B680, an integral membrane diiron oxygenase that is unrelated to RedG but a member of the fatty acid hydroxylase family and closely related to metazoan alkyl glycerol monooxygenase.155
2.7. Radical SAM and Cobalamin Reactions.
We have already seen the utility of hypervalent iron-oxygen complexes in creating carbon radicals that lead to C-C bond cleavage and rearrangements to yield new C-C bonds (e.g., Figures 4, 8, 9, 12, 13). Complex rearrangements have been observed with both radical SAM and cobalamin (vitamin B12) enzymes, with redox reactions driving the initiation of radical chemistry.1,157
The methyltransferases in the radical SAM enzyme superfamily have been divided into four classes, which differ in their requirements for prosthetic groups and cofactors.158 Class A includes RNA base methylases (vide infra). Class B consists of cobalamin-dependent methyltransferases, e.g. TsrM, Fom3 (vide infra), GenK, CysS, ThuK, Sven0516, PoyC.158 Class C enzymes are cobalamin-independent. Class D enzymes use N5,N10-methylene tetrahydrofolate.
Formation of the hypermodified tRNA base wyosine is considered as an example.134,159 Electron transfer from a flavoprotein (Figure 24) is involved in activation of the iron-sulfur cluster needed to generate a 5´-deoxyadenosyl radical, which is utilized to abstract a hydrogen atom from a substrate (Figure 40), akin to the process in cobalamin chemistry.1
Figure 40.

Mechanistic proposals for synthesis of wyosine by TYW1. See also Figure 24. (A) Mechanism involving covalent catalysis.159 (B) Alternative mechanism.160
At least two mechanistic proposals have been made for the formation of wyosine by TYW1. The first (Figure 40A) is based on covalent catalysis involving a conserved lysine residue in TYW1, which holds the substrate pyruvate via a Schiff base linkage. Hydrogen atom abstraction from the guanine N1-methyl group leaves a methylene radical, which reacts with the C-2 carbon of the pyruvate that in turn is liganded to the iron-sulfur cluster. HCO2H is released, and nucleophilic attack on the Schiff base by the guanyl N2 atom, followed by a series of rearrangements, yields wyosine. An alternate mechanism (Figure 40B)160 invokes a role for the iron-sulfur cluster in the later steps.
Another example of a C-C bond forming reaction involving radical SAM chemistry is the biosynthesis of the antibiotic fosfomycin (Figure 40).161–164 This [FeS] cluster-containing enzyme requires SAM and a methylcorrinoid as substrates.
Cobalamin reactions (vitamin B12) are also known and use similar mechanisms involving adenosine CH2• radicals. A classic case is glutamate mutase (Figure 42A), which uses the general radical mechanism shown for 1,2-alkyl migrations (Figure 42B).1,165–167
Figure 42.

Glutamate mutase and cobalamin chemistry.165–167 (A) Overall reaction; (B) general cobalamin 1,2-migration mechanism. In Part A the carbon atoms are labeled (*, #) to indicate the rearrangement.
An interesting example of a cobalamin-dependent reaction is that of Bacillus megaterium OxsB, involved in a ring contraction in the synthesis of OXT-A (Figure 43).168 A proposed mechanism is shown in Figure 44.168
Figure 43.

Conversion of dAMP to OXT-A.168
Figure 44.

Proposed mechanism for oxsB.168The circled P denotes a phosphate group.
Radical SAM reactions are relatively common, and >113,000 proteins have been annotated as such.157,161 Many are found in bacteria. One important mammalian example of a radical SAM enzyme participating in C-C bond formation is in molybdopterin biosynthesis, involving the protein MOCS1A with its two [4Fe-4S] clusters (Figure 45).169 Molybdopterin deficiency is a pleiotropic genetic disorder. The human enzymes sulfite oxidase, xanthine oxidoreductase, and aldehyde oxidase require this prosthetic group, and administration of Moco precursor Z (Figure 45) rescues Z-deficient knock-out mice displaying the human deficiency phenotype.170,171
Figure 45.

Biosynthesis of Moco precursor Z by MoaA.161
Another example of Class B is TbtI, which involves the methylation of a thiazole ring during posttranslational modification in the biosynthesis of the peptide antibiotic thiomuracin (Figure 46).158 A proposed mechanism is shown in Figure 47.158
Figure 46.

Methyl transfer in the synthesis of thiomuracin by TbtI.158 The course of hydrogens is shown in color (red).
Figure 47.

Proposed mechanism for TbtI.158 The course of hydrogens is shown in color (red).
2.8. Vitamin K-mediated Carboxylation.
Vitamin K has long been known to be important in blood coagulation, but its mechanism remained enigmatic for many years. The reaction of interest is the formation of γ-carboxyglutamate moities in protein side chains, and a redox cycle is involved in the C-C bond formation. The reaction is driven by generation of a strong anion that can deprotonate the γ-carbon of a glutamate residue and cause it to react with CO2 (Figure 48).
Figure 48.

The vitamin K and γ-glutamate carboxylation.
The carboxylase is a non-heme single-iron dioxygenase, with some sequence identity with lipoxygenases.172,173 18O2 incorporation studies showed one 18O in the epoxide oxygen and 5–20% in one of the quinone oxygens.174,175 The epoxidation mechanism proposed by Dowd et al.174 (Figure 49) is realistic when one considers the kinetics of carbonyl oxygen exchange with H2O.176–178
Figure 49.

Mechanism of vitamin K epoxidation.174
The vitamin K epoxide gem-diol is considered to be a strong base and abstracts a proton from a γ-glutamate residue,179 allowing the formal carbanion to attack CO2 and form the γ-carboxyglutamate products. To complete the catalytic cycle, the epoxide must be reduced to the hydroquinone (Figure 50). The oxidative and reductive steps of the cycle are accomplished by the enzyme vitamin K oxidoreductase (VKOR) (Figure 49).
Figure 50.

Proposed mechanism of reduction of vitamin K epoxide.183,184
Along with P450 2C9 (the major enzyme involved in warfarin metabolism),180,181 genetic differences in VKORC1 contribute to dose-response patterns and safety with the anti-coagulant drug warfarin. (There are two forms of VKOR in different tissues, VKORC1 and VKORL1.182) Warfarin (also used as a rodenticide) acts by inhibiting VKOR. The electrons used to reduce VKOR do not come directly from pyridine nucleotides but can be supplied from the simple reagent dithiothreitol in vitro. Two (membrane-imbedded) thiols in VKOR (Cys-132, Cys-135) are apparently involved in a disulfide linkage, which is reduced by Cys-43 and Cys-51.183 Rishavy et al.183,184 have shown that a thioredoxin/thioredoxin reductase system is capable of supporting the reaction in vitro. A plausible mechanism is presented in Figure 50. VKOR is capable of reducing vitamin K quinone to hydroquinone but other enzymes are postulated to also contribute.185
2.9. Lignin Synthesis.
Our discussion of C-C bond-forming reactions would not be complete without mention of what may be the most plentiful natural substance on the earth aside from cellulose, namely lignin. Lignin is of interest in that it is needed for plants to stand erect, but degradation of lignin is also an environmental necessity (see Section 3.12).
2.9.1. Structural Lignins.
Lignin is formed by the polymerization of phenolic substances derived from phenylalanine (Figure 51A). The P450 enzymes cinnamate 4-hydroxylase (P450 73A), p-coumarate 3-hydroxylase (P450 98A), and ferulate 5-hydroxylase (P450 84A) are involved in generating mixtures of the phenols, which yield different types of lignins and mixtures, resulting in polymers with different textures.
Figure 51.

Lignin synthesis catalyzed by peroxidases. (A) Building blocks for lignin synthesis. The three phenols are derived from phenylalanine. The nature of lignin (H, S, G) depends in part upon the balance of the monomeric units utilized. (B) Dimerization of two coniferyl alcohol monomers by plant peroxidases to initiate lignin synthesis.186
Lignin polymerization is the result of oxidative coupling of phenolic radicals, i.e. the diradical process discussed earlier for P450s and some other redox enzymes. Many different peroxidases (and some laccases)187 are involved, in that most plants have large gene families involved (and gene-knockouts are often ineffective due to the extensive gene redundancy).186 An example of the polymerization process is shown for the dimerization of coniferyl alcohol in Figure 51B. Peroxidases are able to abstract electrons from aryl rings, especially when electron-donating groups (e.g., methoxy) are attached (Compound I abstracts an electron, forming Compound II). For instance, rabbit P450 1A2 was demonstrated to be able to abstract an electron from 1,2,4,5-tetramethoxybenzene (E1/2 0.81 V vs. SCE, or 1.05 V vs. NHE)188, yielding a stable radical.189 The resonance forms are shown (Figure 51B), and diradical couplings yield various products that impart different properties to lignins in various plants (and individual tissues).
C-C bond-breaking of lignin is done by similar enzymes, presented in Section 3.13.
2.9.2. Dietary Lignins.
Not all lignans are structural in plants. Some are metabolites that do not have known functions in plants but have biological properties, or at least flavors, when consumed by mammals. An example is sesame. In the biosynthesis of sesame, two molecules of coniferyl alcohol are coupled by a dirigent protein, in a radical process, to form (+)-pinoresinol (Figure 52). Two oxidations by P450 81Q1 yield the di-methylenedioxyphenyl structure (+)-sesamin (Figure 52).190 Further oxidation (of (+)-sesamin) by P450 92B14 yields both C-O and C-C coupled products, i.e. (+)-sesamolin and (+)-sesaminol (Figure 53).190
Figure 52.

Biosynthesis of (+)-sesamin.190
Figure 53.

A proposed mechanism of oxidation of (+)-sesamin to (+)-sesamolin and (+)-sesaminol by P450 92B14.190
2.10. MauG and Tryptophan Crosslinking.
We now return to diradical crosslinking by heme proteins. Bacteria have a number of unsual modifiations of amino acids to generate new prosthetic groups. One is tryptophan tryptophylquinone (TTQ), which functions in some redox reactions. The biosynthesis involves the diheme enzyme MauG (Figure 54), which utilizes long-range electron transfer to form two tryptophan radicals that link, as shown earlier for some of the isoquinoline alkaloids (Figure 4) and indolocarbazole alkaloids (Figure 10).191
Figure 54.

Formation of TTQ in methylamine dehydrogenase by MauG.191
3. C-C Bond Breaking Reactions.
C-C bond cleavage does not occur with unfunctionalized methylene carbons, olefins, and aromatic molecules and requires functionalization before cleavage. In some cases this functionalization is an inherent part of the cleavage enzyme.
3.1. P450 Reactions.
A previous section (Section 2.2) dealt with P450-catalyzed C-C bond-forming reactions. The list of P450 C-C bond-breaking reactions is probably just as long or longer. However, the cleavage of C-C bonds by P450s is not as facile as the cleave of C-N or C-O bonds, which is readily done by C-hydroxylation to form unstable carbinolamines or hemiacetals.61 Cleavage of C-C bonds generally requires oxidation reactions at two adjacent carbon atoms, i.e. a multi-step reaction. For instance, all of the C-C bond-breaking steps observed in mammalian steroid metabolism involve successive reactions (Figures 55, 56).
Figure 55.

Mammalian P450 reactions involved in steroid biosynthesis. The uncircled steps are catalyzed by dehydrogenases.
Figure 56.

C-C bond breaking in steroid biosynthetic reactions catalyzed by P450s 19A1, 17A1, 11A1, and 51A1. C-C bond scission occurs in the last reaction in each sequence.
P450 C-C cleavage reactions involve steroids, drugs, model and industrial chemicals, and many natural products. These fall into a general mechanistic pattern in the formation of unstable products that break C-C bonds during decomposition, either within or outside of the enzyme.
3.1.1. Steroids.
The biosynthesis of steroids in mammals involves four P450 C-C cleavage reactions—lanosterol 14α-demethylation (P450 51A1), cholesterol side chain cleavage (P450 11A1), the 17α-hydroxylation/lyase reactions with pregnenolone and progesterone (P450 17A1), and the aromatization of androgens to form estrogens (P450 19A1) (Figure 56).111,192 (4-Demethylation of sterols is catalyzed by a non-heme diiron protein, to be discussed later, Section 3.9.) Genetic deficiencies in any of these four enzymes are clinically problematic.192,193 On the other hand, the 17α-hydroxylation/lyase (P450 17A1) and aromatization (P450 19A1) reactions are targets for inhibition by drugs in cancer therapy, because of the roles of androgens and estrogens in growth of certain tumors.194–198 14α-Demethylation is critical in formation of ergosterol for membrane synthesis in yeast and fungi, and the enzymes catalyzing this reaction are the most common targets in the development of anti-fungal therapies.199,200 Some P450 enzymes that catalyze sterol oxidative cleavages are also targets for drugs to treat bacterial infections, e.g. tuberculosis.201,202
This section will focus on the mechanism of the C-C bond cleavage process by P450 enzymes. The C-C bond cleavage substrates of P450s 19A1 and 51A1 are both aldehydes, which are cleaved to form the alkene and HCO2H products, while P450s 17A1 and 11A1 require an α-hydroxy ketone (e.g. 17α-hydroxypregnenolone) and a vicinal diol (20R,22R-dihydroxycholesterol), respectively, as their substrates to form ketone products (e.g., dehydroepiandrosterone (DHEA) and pregnenolone). All of these P450s (i.e. P450s 11A1, 17A1, 19A1, 51A1) cleave C-C bonds in a multi-step process from the initial substrate, with varying degrees of processivity.203–205
3.1.1.1. P450 19A1.
P450 19A1 (also known as steroid aromatase) is the enzyme responsible for the formation of 18-carbon estrogens and formic acid from 19-carbon androgens.206 Inhibition of this enzyme is a mechanism for treating estrogen-dependent cancers, and several third-generation selective inhibitors are in wide clinical use today (e.g., exemestane, anastrazole, letrozole).207,208 The C-C bond cleavage process of P450 19A1 occurs in three steps (Figure 56). The first two steps involve hydroxylation of the C19-methyl of the androgen substrate (i.e., testosterone or androstenedione), and the third step is the C-C bond cleavage reaction to afford HCO2H and the estrogen product. The mechanism of the C-C bond cleavage step has been controversial since the 1970s, and various mechanisms had been proposed by experimentalists and theoreticians (Figure 57).177,209–227
Figure 57.

Five proposed mechanisms (A-E) for P450 19A1 steroid aromatization.177,209–215,217–220,222,223,228–233
Arguments against mechanisms A, B, and C (Figure 57) have been advanced, and these three have generally been dismissed. One controversial aspect has been the proposal of the active iron species directly involved in cleaving the C10-C19 bond in its androgen substrate to form the estrogen product.
Because the direct C-C bond cleavage substrate in the aldehyde form (e.g. 19-oxoandrostenedione) is inherently electrophilic at C19, it has been proposed that a nucleophilic ferric peroxide can attack the aldehyde substrate to form a tetrahedral peroxohemiacetal intermediate, which can break down to form the estrogen product (e.g., estrone) and HCO2H (Figure 57E). In contrast, Compound I has been proposed to abstract the C1β-H bond of the hydrated form of 19-oxoandrostenedione, a 19,19-gem-diol intermediate, to form a C1-radical androgen intermediate. This intermediate can transfer its electron to the iron center of Compound II, forming a carbocation intermediate at C1 (Figure 57D).177,226 The resulting carbocation can break down to form HCO2H and estrone.
3.1.1.1.1. 18O-Isotopic Labeling Studies.
The strongest evidence supporting the active oxoiron species responsible for cleaving the C-C bond has come from oxygen isotope-labeling experiments (Figures 58, 59).220,222,223 The difference between the Compound I mechanism and the ferric peroxide mechanism to form estrogens from 19-oxo androgen precursors is the source of one of the oxygens in the side-product HCO2H. One of the oxygen atoms in HCO2H comes from molecular oxygen in the ferric peroxide mechanism while oxygen comes from water in the Compound I mechanism (Figures 58, 59).
The incorporation of an 18O atom from molecular oxygen (18O2) into the HCO2H product from the incubation of P450 19A1 with its 19-oxo androgen substrate would support a ferric peroxide (Compound 0) mechanism, while the lack of 18O in HCO2H derived from the lyase substrate would support a role for Compound I (Figure 58).177
Analysis of the formic acid product from the incubation of P450 19A1 with the 19-oxo androgen substrate is not trivial. The main challenges with detecting derivatized formate compounds by mass spectrometry are related to the abilities (i) to distinguish between the formic acid from the enzyme incubation and endogenous formic acid and (ii) to detect small amounts of compound (~100 ng to 1 μg scale). Formic acid is ubiquitous and is present in solvents, air, and enzyme preparations. In the analysis of formic acid produced by P450 19A1, differentiating between the background source of formic acid from the enzymatic incubation becomes a major problem. Previous studies had used phenyldiazomethane to derivatize the formic acid product from the enzyme incubation and also used low-resolution mass spectrometry (1000 resolving power with an AEI MS 30 mass spectrometer234) to detect the formic acid produced from placental microsomes (i.e., the source of P450 19A1) as benzyl formate with a minimum of 250 ng quantities.222,223 The strategy in this study used a 19-monodeuterated 19-oxo androgen substrate, which would be aromatized by the enzyme to generate a deuterated formic acid product. The deuterated formic acid should be distinguished from background formic acid through mass spectrometry; however, the natural isotopic abundance of carbon is 98.9% for 12C and 1.1% for 13C. Therefore, the benzyl formate isotopologues that would need to be distinguished by the mass spectrometer are the monodeuterated form resulting from the incubation (m/z 137.0582) and the 13C-labeled form derived from endogenous formic acid (m/z 137.0552), which would require a minimum resolving power of ~46,000 (i.e., M/ΔM = 137.0582/(0.003)) (Figure 60A). These isotopologues were not resolvable with the technology used in 1982.222,223,234
Figure 60.

Comparison of the work done to detect formic acid produced from P450 19A1 steroid aromatase reported in (A) 1976–1982222,223,234 and (B) 2014.177
A more recent study in 2014177 used a new diazo reagent bearing a pyridine moiety to ionize in the electrospray positive mode and high resolution mass spectrometry (HRMS) (ThermoFisher LTQ Orbitrap with resolving power of 100,000) to distinguish between the formic acid produced by P450 19A1 and the endogenous formic acid (i.e., M/ΔM = 58,000) (Figure 52B). With the newer technology (i.e. sensitive derivatization method, high resolution mass spectrometry, and purified enzyme source), it was concluded that P450 19A1 did not incorporate an oxygen atom from molecular oxygen (18O2) into the formic acid product, and therefore only a Compound I active iron species could be responsible for cleaving the C10-C19 bond of androgens to form estrogens.
Spectral studies using resonance Raman measurements have also been interpreted as being consistent with a Compound I intermediate.225 No evidence was seen for the accumulation of a Compound 0 intermediate in EPR work.235 However, the formation of multiple products (e.g., 19-oic acid, vide infra) makes assignments of spectral intermediates ambiguous, in that one intermediate may give rise to only some products. Nevertheless, in this case the spectral conclusions are consistent with our own isotope labeling work.
3.1.1.1.2. Hydration of the 19-Aldehyde to Form the gem-Diol and Catalytic Competency.
The 19-carbon androgen intermediate that is directly converted to the estrone product is 19-oxoandrostenedione. This 19-aldehyde intermediate can potentially exist in equilibrium with its 19,19-gem-diol form in water. In the ferric peroxide mechanism of C10-C19 bond cleavage of P450 19A1 (Figure 57E), the only substrate that can react with the nucleophilic ferric peroxide intermediate is the 19-aldehyde form, which is electrophilic, and not the 19,19-gem-diol. Therefore, determination of the ratio of the 19-aldehyde and the 19,19-gem-diol in water (pH 7.4) is mechanistically informative. If the 19-oxo androgen intermediate exists only as the 19,19-gem-diol in water, then the enzyme will have to catalyze a dehydration of the 19,19-gem-diol to the 19-aldehyde intermediate before ferric peroxide can attack the substrate. On the other hand, if only the 19-aldehyde exists, the enzyme may readily react with the 19-aldehyde in the ferric peroxide mechanism. In addition, the second step of P450 19A1 is the second hydroxylation of its androgen substrate (i.e. 19-hydroxyandrostenedione) to form 19,19-dihydroxyandrostenedione (i.e., 19,19-gem-diol), which must dehydrate to 19-oxoandrostenedione (i.e. 19-aldehyde) at a faster rate than the conversion of 19-hydroxyandrostenedione to estrone by P450 19A1 (kcat 0.13 s−1) in the ferric peroxide-based mechanism. If this non-enzymatic dehydration rate is < 0.13 s−1, then the enzyme must dehydrate the 19,19-gem-diol to the 19-aldehyde to ensure that ferric peroxide can attack the electrophilic 19-oxo intermediate. In other words, the non-enzymatic rate of dehydration of the 19,19-gem-diol must be catalytically competent.177
Conversely, in the Compound I mechanism (Figure 57D) either the 19,19-gem-diol or the 19-oxo intermediate can react with the enzyme. When Compound I abstracts the C1β-H atom of the 19-oxo intermediate, a C1-radical intermediate is generated. This C1-radical intermediate would have to be hydrated at a rate faster than the rate catalyzed by P450 19A1 to convert 19-hydroxyandrostenedione to estrone (kcat or rate of [P450 19A1•19-hydroxyandrostenedione] to [P450 19A1] + [estrone]), which has been measured to be 0.13 s−1. If the non-enzymatic hydration of the 19-aldehyde to the 19,19-gem-diol is < 0.13 s−1, then the hydration of the 19-aldehyde must be catalyzed by the enzyme. In order to experimentally determine the hydration equilibrium, a solution of the 19-oxo compound in D2O (pD 7.8) was analyzed by NMR spectroscopy, and the integrations of the C19-proton of the 19-aldehyde and the 19,19-gem-diol were compared.177
Considering the details supporting either the ferric peroxide or Compound I mechanism from knowledge of the hydration equilibrium of the 19-oxo androgen intermediate, it became imperative to also determine the non-enzymatic rates of hydration and dehydration of the 19-oxo and 19,19-gem-dihydroxy androgen intermediates (Figure 58).177 In order to measure the rates of hydration/dehydration of the 19-aldehyde and 19,19-gem-diol, 19-oxoandrostenedione was initially mixed with 18O-labeled water (H218O) and analyzed by mass spectrometry. However, because 19-oxoandrostenedione contains three carbonyls (i.e. two ketones at C3 and C17, and one aldehyde at C19), the 18O-exchange experiments in H218O resulted in the incorporation of three 18O-atoms into the steroid. Therefore, it was necessary to synthesize 19-[18O]-labeled 19-oxoandrostenedione, which could be exposed to non-labeled water to directly measure the rate of oxygen exchange of the C19-oxygen belonging to the aldehyde. This compound is ideal in that the oxygen isotopes (i.e. 16O) on the C3- and the C17- positions are identical to the medium (i.e. non-labeled water). Hence, even if the C3- and C17- keto groups exchanged with water, this exchange should not interfere with the resulting analysis of the C19-oxygen exchange by mass spectrometry.
Furthermore, because the analysis of the steroid exchange was performed by UPLC-MS using a reversed-phase column, the mobile phase required the use of H2O. In order to avoid the exchange of the 18O-label during the LC-MS analysis, the extracted steroid was derivatized with sodium borohydride to convert 19-oxoandrostenedione to 19-hydroxyandrostenedione. (Because aldehydes are more reactive than ketones, the C3- and C17- keto groups remain intact during these derivatization conditions. Moreover, in the synthesis of 19-deutero-19-oxo-androstenedione, sodium borodeuteride was reacted with 19-oxoandrostenedione in one of the intermediate steps, only reducing the aldehyde to afford 19-deutero-19-hydroxyandrostenedione.)
In a slightly different but related mechanism, the initial hydrogen abstraction can be from an alcohol of the gem-diol, followed by rearrangement to similar intermediates as shown here. Multiscale QM/MM calculations provided some support for this hypothesis,232 which is also entirely consistent with the Compound I mechanism.
3.1.1.1.3. Determination of Catalytic Competency of 19,19-gem-Diol Dehydration.
The t1/2 of 19-[18O]-19-oxoandrostenedione in unlabeled water (H216O) was < 1 s (determined by UPLC-MS).177 Using the first-order relationship t1/2 = 0.693/kobs, kobs was estimated to be ~1 s−1 (or faster). This information combined with the hydration equilibrium constant (Keq) of ~1 between the 19,19-gem-diol and the 19-aldehyde in D2O (determined by NMR)177 allowed for the calculation of the rates of dehydration and hydration of the gem-diol and the aldehyde intermediates by solving for khydration from the following set of equations:
| (Eq. 4) |
| (Eq. 5)236 |
| (Eq. 6) |
Therefore, the rates of hydration and dehydration were both estimated to be ~0.5 s−1 (or faster). Because these rates are faster than the P450 19A1-catalyzed conversion of 19-oxoandrostenedione to estrone (i.e., kcat 0.42 s−1),185 the rates of hydration and dehydration of the 19-aldehyde and the 19,19-gem-diol were determined to be catalytically competent.177
3.1.1.1.4. 19-Carboxylic Acid Androgen as an Alternative Product.
An alternative Compound I product could be formed if the 19-oxoandrogen ligand is positioned so that the C19-position is still in proximity for C-H abstraction to result in a 19-carboxylic acid product. The 19-carboxylic acids were formed in reactions with the 19-formyl derivatives of both testosterone and androstenedione (Figure 59).177 More recent unpublished studies indicate lower catalytic efficiency for the reaction than for estrogen formation (M. J. Reddish and F. P. Guengerich).
3.1.1.2. P450 17A1.
P450 17A1 is the P450 that converts 21-carbon steroids (i.e., pregnenolone and progesterone) to 19-carbon androgens (i.e., DHEA and androstenedione) and CH3CO2H. In addition to its lyase activity (i.e., its second step), P450 17A1 regioselectively hydroxylates the C17-position of 21-carbon steroid precursors, pregnenolone and progesterone (Figures 56, 61).
Figure 61.

The C17-hydroxylated steroids are important precursors of glucocorticoids (e.g., cortisone). Selective inhibition of the C17-C20 lyase reaction is a goal in treating androgen-dependent cancers (e.g., prostate).240–242 The drug abiraterone is currently used in conjunction with prednisolone (a glucocorticoid) to treat these cancers, because of the inhibition of production of glucocorticoids (Figure 55). The C-C bond cleavage step involves a 17α-hydroxy 21-carbon steroid product as an intermediate in forming CH3CO2H and the 17-keto 19-carbon steroid products. The identity of the active iron species responsible for C-C bond cleavage of this enzyme has also been controversial and, similar to the case of P450 19A1, both Compound 0 (ferric peroxide) and Compound I reactions have been proposed (Figure 62).
Figure 62.

(A) Conversion of progesterone to testosterone by a P450 17A1 ferric peroxide and a proposed hemiketal intermediate. (B) Extrapolation to lyase reaction.243,244
3.1.1.2.1. 18O-Isotopic Labeling Experiments.
Unlike the case of P450 19A1, the use of 18O2 in the incubation of P450 17A1 and its lyase substrate does not in itself provide a definitive method to support Compound I or Compound 0 as the active iron species. There have been multiple C-C bond cleavage mechanisms proposed involving both Compound I and Compound 0 that can all incorporate the oxygen atom into the product CH3CO2H from O2.237,238 However, the 18O2 labeling experiments can rule out some of the proposals that involve Compound I, which do not lead to the incorporation of an 18O atom from molecular oxygen.238
3.1.1.2.2. Proposals for a Ferric Peroxide (Compound 0) Mechanism.
The Akhtar group published the results of 18O incorporation experiments demonstrating the incorporation of 18O label from O2 into the product acetic acid,245 which they interpreted as evidence for a ferric peroxide mechanism. This experiment is less difficult than formic acid analysis (vide supra, P450 19A1, Section 3.1.1.1.1) due to the ability to use a 3 a.m.u. difference in the deuterated substrate. We repeated the experiment and obtained similar results (incorporation of one 18O atom from 18O2 into acetic acid).238 However, the result does not rule out some Compound I mechanisms.237–239
Swinney and Mak243,244 reported that either porcine testicular microsomes or purified porcine P450 17A1 could convert progesterone to 17-O-acetyl testosterone and interpreted the results in the context of a ferric peroxide Baeyer-Villiger mechanism (Figure 62). The yield was very low, and we have been unable to detect this product with human P450 17A1 at a level of ≥0.15% of the 17α-hydroxyprogesterone formed (i.e., rate of formation <0.0013 min−1, corresponding to less than one turnover/13 hours).239 Porcine P450 17A1 was not available for direct comparison, but this reaction cannot be universal in its occurrence among P450 17A1 enzymes. Moreover, a Compound I mechanism can be proposed to generate the 17-O-acetyl testosterone product if one of the C20-hydroxy groups of the 20,20-gem-diol form of progesterone were to nucleophilically attack Compound I to form a ferric peroxohemiketal intermediate that rearranges to 17-O-acetyl testosterone. Thus, the previous studies with 17-O-acetyl testosterone cannot be used to support a general FeO2¯-based Baeyer-Villiger mechanism.243,244
Sligar and associates reported a high inverse kinetic deuterium solvent isotope effect (kH/kD 0.39) for the 17α-hydroxypregnenolone lyase reaction with human P450 17A1 and interpreted this as evidence for a ferric peroxide (Compound 0) mechanism, stated as being consistent with inhibition of protonation of the peroxo-ferric species in a manner that serves to facilitate productive rather than unproductive oxidation.246 In contrast, Swinney and Mak244 reported that (30%) D2O attenuated androgen formation from 17α-hydroxyprogesterone using pig testes microsomes as the enzyme source (kH/kD 1.25), suggesting that the 17α,20-lyase reaction is dependent on Compound I formation either through the protonation of the distal oxygen of ferric peroxide (cf. P450 catalytic cycle, Figure 2) or deuterium atom abstraction from the 17-hydroxy group of the substrate. The interpretation of solvent kinetic hydrogen isotope effect is not as straightforward as for C–H bonds, in which rates of bond-breaking are clearly involved.247 We also found a small but repeatable solvent deuterium isotope effect for the lyase reaction using the substrate 17α-hydroxypregnenolone (kH/kD 0.83 ± 0.05, mean ± SD, n = 4) but none for 17α-hydroxyprogesterone (kH/kD 1.05 ± 0.09, n = 4).238 Although we do not have a definite explanation for the small inverse kinetic solvent isotope effect we saw with 17α-hydroxypregnenolone, it seems unlikely the P450 17A1 would use different lyase mechanisms with two different but inherently similar substrates. We are not of the opinion that the inverse kinetic solvent isotope effect,246 at least in itself, provides convincing evidence for a ferric peroxide mechanism. Alternate reasons can be proposed for a kinetic solvent isotope effect.238 One possibility is that the Δ5 substrate (17α-hydroxypregnenolone) 3-hydroxy group exchanges with deuterium and that this has a small effect on the juxtaposition of the substrate in the active site. The hydroxyl moiety was shown by Scott and co-workers248,249 to be in hydrogen bonding distance to Asn-202 of human P450 17A1. A substitution of the 3-hydroxyl group by deuteration (i.e., −OD) could shift the substrate to favor the lyase reaction versus 16-hydroxylation. However, the lack of solvent isotope effects does not allow any definite conclusions about the rate-limiting nature of the abstraction of a proton or hydrogen atom from the 17-OH group, due to the multiple complex influences from solvent deuterium on enzyme function.
The Kincaid and Sligar groups reported that they prepared the ferric peroxide form of P450 17A1 at low temperature (77 K) and published resonance Raman spectra attributed to that form bound to 17α-hydroxypregnenolone in a form indicative of a productive complex.34,225,250 The assignment of the 791 cm−1 band of the hemoprotein complex as a hemiacetal (actually a hemiketal) was based on a biomimetic model for a non-heme iron protein (which was an acylperoxoiron(III) complex, not a hemiketal).251 This (Raman) spectrum differed from that observed with 17α-hydroxyprogesterone and was therefore concluded to be important in the lyase reaction.250 However 17α-hydroxyprogesterone is also a substrate for P450 17A1, although the catalytic specificity value (kcat/Km) for the lyase reaction is less than that for 17α-hydroxypregnenolone.205 The Raman spectrum changed upon warming of the sample, which was interpreted to mean that (a lyase) reaction was occurring.34 However, under the conditions that the experiment was done (60% glycerol and 4 MRad of 60Co γ-radiation), it is not known if the enzyme is catalytically active. Even if the FeO2− complex did form the normal products (androstenedione and DHEA, plus the 16-hydroxylation products, which is unlikely) in these experiments, the simultaneous or subsequent intermediacy of an FeO3+ species could not be ruled out. Measurement of product (i.e., DHEA) was not reported, and whether the system is catalytically competent is unknown. The presence of b5 is rather critical for the lyase reaction205 and no b5 was present in the Raman experiments, so the production of DHEA would be unexpected. If the experiments could be done and the lyase product DHEA recovered in high yield, as in the case of P450 11A1 Compound I and pregnenolone formation,252 then the evidence would be strengthed, although even then the intermediacy of a Compound I intermediate would still be possible (Figure 2).
3.1.1.2.3. Oxygen Surrogate Experiments with Iodosylbenzene.
Oxygen surrogate experiments have been performed to identify the active iron species in the C-C bond cleavage step of P450 17A1. Both H2O2 and iodosylbenzene have been employed to generate either ferric peroxide or Compound I in situ for P450 17A1 in the presence of its C-C lyase substrate, 17α-hydroxypregnenolone. In two separate reports, H2O2 failed to yield the C-C bond cleavage product of P450 17A1.238,253 However, the use of iodosylbenzene resulted in the formation of the C-C bond cleavage product for P450 17A1 (Figure 63).238
Figure 63.

16α-Hydroxylation and 17α,20-lyase action of P450 17A1 in the presence of the oxygen surrogate iodosylbenzene.238 (Reprinted with permission from J. Biol. Chem., 2016, 291, 17143–17164, Figure 6.) Retention times (tR) and integration units are indicated on the chromatograms. (A) Standard compounds. (B) Reaction (0.5 µM P450 17A1) supported by NADPH-P450 reductase (2.0 µM), b5 (0.5 µM), and NADPH (30 s incubation). (C) Reaction (0.5 µM P450 17A1 and b5 (0.5 µM)) with 0.30 mM iodosylbenzene (30 s incubation).
Iodosylbenzene does not support the formation of an iron hydroperoxide species but can produce Compound I or a similar species,238,254 in that it contains a single oxygen atom (Figure 2). The iodosylbenzene-supported reaction was, like the normal NADPH-supported reaction, stimulated by b5.238
3.1.1.2.4. Other Support for a Compound I Active Species in the Lyase Reaction.
In recent experimental mechanistic studies of both human and zebrafish P450 17A1 with its lyase substrates (i.e. 17α-hydroxypregnenolone and 17α-hydroxyprogesterone), other hydroxylation products were identified in addition to the expected lyase products (Figure 61).238,239 The hydroxylation products correspond to 16α-hydroxylation to yield the 16,17-dihydroxy 21-carbon steroid products as well as a further hydroxylation, in the case of the 3-keto-Δ4,5-steroid substrates, at the 6β-position (B-ring of the steroid). P450 17A1 is classically known to catalyze the C17-C20 bond cleavage reaction of 17α-hydroxyprogesterone to yield androstenedione and acetic acid (Figure 62A).
Moreover, P450 17A1 was also found to hydroxylate the 16α-position of the same substrate to yield 16α,17α-dihydroxyprogesterone (Figure 64A).238 The introduction of the 16α-hydroxy group likely resulted in a switch in steroid binding in the active site of P450 17A1 (Figure 62C). In the crystal structures of P450 17A1 with its hydroxylase substrates (pregnenolone and progesterone),249 Asn-202 of the protein hydrogen bonds to the 3β-hydroxy group or the 3-keto group of pregnenolone or progesterone. We hypothesize that the 16α-hydroxy group of the newly identified product of P450 17A1 hydrogen bonds to Asn-202 to position the steroid for 6β-hydroxylation by Compound I (Figure 62C). The introduction of the 16α-hydroxy group must result in two conformations of the steroid in P450 17A1: (i) one that results from hydrogen bonding between the 3-keto group and Asn-202 (Figure 62B) and (ii) a second that arises from the hydrogen bonding between 16α-hydroxy group of the steroid and Asn-202 (Figure 62C). These two conformations lead to the formation of the two products (i.e. 16α-hydroxyandrostenedione and 6β,16,17-trihydroxyprogesterone) when P450 17A1 was incubated with 16α-,17α-dihydroxyprogesterone (Figures 62B, 62C).
Figure 64.

New oxidation products of P450 17A1 detected with 17α-hydroxyprogesterone and 16α-,17α-dihydroxyprogesterone. (A) P450 17A1 oxidizes 17α-hydroxyprogesterone to 16α,17α-dihydroxyprogesterone and androstenedione. (B) P450 17A1 cleaves the C17,C20-bond of 16α-,17α-dihydroxyprogesterone to yield 16α-hydroxyandrostenedione. (C) P450 17A1 hydroxylates 16α-,17α-dihydroxyprogesterone to afford 6β-,16α-,17α-trihydroxyprogesterone.238 In Parts A and B, Compound I is shown to abstract the hydrogen atom of the 17-hydroxy group of the substrate to yield the 19-carbon C17-C20 lyase products, but this C17-C20 lyase reaction can occur also from a nucleophilic attack of the 17-hydroxy group onto Compound I (see text for discussion). (C) A different orientation of the steroid in the enzyme active site is due to the hydrogen bonding interaction between the 16-hydroxy group with Asn-202.
The lyase reactions shown in Figures 62A and62B could process through a hydrogen abstraction mechanism (from the –OH group) that results in the C17-C20 bond cleavage products (androstenedione and 16α-hydroxyandrostenedione). Compound I has been shown to abstract a hydrogen atom from an –OH group in other P450 systems. In particular, Green and co-workers57 have experimentally shown that S. coelicolor P450 158A2 performed a hydrogen abstraction (form the –OH group) of a nearby tyrosyl residue (Tyr-352) when Compound I was formed through the rapid mixing of m-CPBA and the enzyme, generating a tyrosyl radical and Compound II. Compound II was characterized by Mössbauer and UV-visible spectroscopy techniques, and the tyrosyl radical was observed by EPR spectroscopy. When the tyrosine residue was changed to phenylalanine (Y352F) and the resulting protein was subjected to the same conditions with m-CPBA, 80% of the iron-oxo species formed was Compound I, with residual Compound II formation (~3%) and a protein-centered radical (~5%). When two particular tyrosine residues were changed to phenylalanines (Y318F/Y352F) and the enzyme was subjected to the same experimental conditions, Compound II and the tyrosyl radical were not detected and Compound I formation was enhanced (~90% yield). Therefore, in the case of the P450 17A1-catalyzed C17-C20 bond cleavage reaction of 17α-hydroxy 21-carbon steroids to form 19-carbon androgens, abstraction of the 17α-hydroxy group by Compound I is a viable mechanism. This abstraction of the hydrogen from the 17α-hydroxy group results in the homolytic cleavage of the C17-C20 bond to yield the 17-keto androgen and an acyl radical that can recombine with Compound II to form acetic acid. Xu et al.232 have also presented calculations consistent with an initial O–H hydrogen abstraction in the P450 19A1 mechanism, as mentioned earlier (Section 3.1.1.1.2.).
However, an alternative mechanism that involves the nucleophilic attack of the 17-hydroxy group of the steroid onto Compound I can also explain the formation of the C17-C20 bond cleavage products. The attack of the 17-hydroxy group onto Compound I can result in the formation of a 17-peroxide intermediate, which rearranges to a dioxetane that can break down to yield the C17-C20 lyase products.
In order to examine the possible existence of 21-carbon 17-peroxo-type steroid intermediates for the C17-C20 bond scission reaction, 17α-hydroperoxy (OOH) derivatives of the steroids were used as oxygen surrogates (Figure 65).239
Figure 65.

Proposed mechanism of conversion of 17α-hydroperoxypregnenolone to DHEA and relevance to normal P450 17A1 reaction.239,255,256
Physiologically, these 17α-peroxy-type steroids could potentially be formed transiently if the 17α-hydroxy group of the 21-carbon 17α-hydroxy steroid (e.g. 17α-hydroxyprogesterone) could nucleophilically attack Compound I of P450 17A. Human P450 17A1 and both zebrafish P450s 17A1 and P450 17A2 readily converted these hydroperoxy species to the lyase products in the absence of other proteins or cofactors (with catalytically competent kinetics), plus hydroxylated 17α-hydroxy steroids (Figure 66).239
Figure 66.

Conversion of 17α-hydroperoxypregnenolone to lyase products by human P450 17A1.239,256 (Reprinted with permission from J. Biol. Chem. 2018, 293, 541–556, Figure 8) (A) HPLC of standard 17α-hydroperoxypregnenolone, 17α-hydroxypregnenolone, and DHEA, showing tR for elution. (B) Chromatograms of reaction of 25 µM 17α-hydroperoxypregnenolone without (green) or with (blue) 60 nM human P450 17A1 after 150 s. Absorbance (relative units) was integrated over the UV range (200–350 nm). See ref. 239 for other details regarding chromatography. (C) Time course of conversion of 17α-hydroperoxypregnenolone to DHEA, ± human b5. (D) Time course of conversion of 17α-hydroperoxyprogesterone to androstenedione.
In conclusion, there is indirect evidence that P450 17A1 can use a Compound I mechanism in the lyase reaction. However, the use of a ferric peroxide (Compound 0) mechanism in the normal NADPH-supported system (Figure 67A) cannot be ruled out.
Figure 67.

Possible mechanisms of P450 17A1 17α,20-lyase reactions consistent with observed incorporation of 18O from O2 into acetic acid and solvent kinetic isotope results.237–239,245 Mechanism C is favored based on the oxygen surrogate results.238,239
Two viable Compound I mechanisms are presented in Figure 67B and67C. Olsen257 has argued against the mechanism shown in Figure 67B based on theoretical calculations but did not consider the mechanism in Figure 67C, which we presently favor in light of the 17α-hydroperoxide results (Figure 64).239 However, which of these mechanisms is operative in the normal reaction is still not unambiguous.
3.1.1.2.5. The Question of Why Some P450 17A Enzymes Lack Lyase Activity.
Zebrafish P450 17A2 catalyzes only the steroid 17α-hydroxylations, not lyase activity.204,239 Initially, combining the information gathered from (i) the sequence alignment of human P450 17A1, zebrafish P450 17A1, and zebrafish P450 17A2, with (ii) the knowledge that the clinical human P450 17A1 mutations that result in isolated 17,20-lyase deficiency (R358Q, R347H),258 led to the idea that the 17,20-lyase deficient zebrafish P450 17A2 could be related to the clinical mutants. Indeed, alignment between the three enzymes shows a cysteine residue in zebrafish P450 17A2, which corresponds to the human and zebrafish P450 17A1 Arg-358 residues (Figure 68).239 One hypothesis to be tested was the inability of either the clinical R358Q human P450 17A1 variant or zebrafish P450 17A2 to interact with b5, which is known to possess negative charges on the surface of the protein to stimulate 17,20-lyase activity,259 is the reason for the lack of lyase activity. However, the altered proteins (in particular, zebrafish P450 17A2 C358R) have even less lyase activity than the wild-type enzyme in the absence of b5.239 The crystal structures of zebrafish P450 17A1 and 17A2 and human P450 17A1 all have very similar active sites.204,239,248 Five residues near the heme, which differed between zebrafish P450 17A1 and 17A2, were changed but the active site of the modified P450 17A2 still resembled the structure in the other proteins, with some important differences.239 These altered P450 17A1 and 17A2 proteins had catalytic profiles more similar to each other than did the wild-type proteins. Docking with these structures could explain several minor products, which require multiple enzyme conformations.
Figure 68.

Sequence alignment of human P450 17A1 and zebrafish P450s 17A1 and 17A2. Arg-358 (K-helix) in human P450 17A1 and zebrafish P450 17A2 and Cys-369 (K-helix) are marked. The gene nomenclature corresponding to the human and zebrafish P450s is indicated in the last section of the sequences. Red amino acids are identical and blue vary between the sequences.
Even the deficient mutants show lyase activity with the 17α-hydroperoxy steroids.239 The exact reason for the deficiency in the NADPH-supported system remains unclear and is the focus of further studies in this laboratory (F.P.G.). The answer is relevant not only to issues with individuals devoid of lyase activity192,248,258,260,261 but also an important consideration in any development of reaction-selective inhibitors for treatment of prostate cancer.198,205,262–264
3.1.1.3. P450 11A1.
P450 11A1 converts cholesterol to the first 21-carbon steroid hormone, pregnenolone, and isocaproaldehyde through cleavage of the side chain at carbons C20 and C22 (Figures 56, 69).
Figure 69.

Alternative mechanisms of P450 11A1 for the third step of cholesterol side chain cleavage.62,254,265–267 Four Compound I mechanisms are grouped into two categories depicting the C–C bond cleavage step of P450 11A1. (A) Electrophilic Compound I species. (B) HO hydrogen atom abstraction by Compound I. Mechanism A shows no incorporation of an oxygen into the products (5 and 4) from molecular oxygen (*O2), while mechanism B indicates oxygen incorporation from molecular oxygen into products [*O: 18O].
The C-C bond cleavage process involves two successive regio- and stereoselective hydroxylations of cholesterol to afford 22R-hydroxycholesterol followed by 20R,22R-dihydroxycholesterol, which is then cleaved. P450 11A1 is the only one of the four C-C bond-cleaving steroidogenic P450s (P450s 11A1, 17A1, 19A1, and 51A1) located in the mitochondria. The electron transfer process requires both the iron-sulfur protein adrenodoxin and the flavoprotein NADPH-adrenodoxin reductase to transfer the electrons from NADPH to the iron center.
3.1.1.3.1. EPR-ENDOR Experiments Support Compound I as the C-C Bond Cleavage Species.
Davydov et al.252 generated Compound I in the presence of substrate through cryo-reduction of P450 11A1 by γ-irradiation of a sample containing the enzyme in the presence of oxygen, sodium dithionite (10% excess), and glycerol at 77 K using a Gammacell 220 60Co source for 20 h (dose rate of 0.1 Mrad/h).268 The frozen samples were annealed in the range of 70 K to 270 K and monitored by EPR/ENDOR spectroscopy. Based on similar spectroscopic measurements between the hydroxylation substrates (cholesterol and 20R-hydroxycholesterol) and the lyase substrate (20R,22R-dihydroxycholesterol) of P450 11A1 and the formation of the product pregnenolone in high yield, it was concluded that the C20-C22 bond cleavage step of P450 11A1 operates via Compound I.
3.1.1.3.2. 18O Isotopic Labeling Studies.
Several Compound I mechanisms can be used to explain the lyase reaction of P450 11A1 (i.e., cleavage of 20R,22R-dihydroxycholesterol to pregnenolone and isocaproaldehyde). The mechanisms can be divided into two types of Compound I reactivity. In one type, one of the hydroxyl groups of the substrate (C20 or C22) participates in a nucleophilic attack on Compound I; in the second type, Compound I abstracts a hydrogen atom from one of the O-H bonds located on C20 or C22. The distinction existed for over 30 years.62,254,265–267 In this P450 C-C bond cleavage reaction, the use of 18O2 can be employed to distinguish between an electrophilic Compound I mechanism vs. an O-H abstraction mechanism, with the explicit assumption that the dehydration of the gem-diol intermediate in the proposed O-H abstraction mechanism does not stereoselectively dehydrate (Figure 69).267
3.1.1.3.3. Determination of the 18O Content in the C20-C22 Bond Cleavage Products.
The detection of the 18O atom in a carbonyl oxygen is not trivial, due to the fast exchange rate of the carbonyl oxygen with water.176–178,269 In order to trap the carbonyl oxygen in isocaproaldehyde, one of the C20-C22 lyase products of P450 11A1, an enzyme-coupled assay using a yeast alcohol dehydrogenase that converted the aldehyde to the alcohol was employed (Figure 70A). The resulting oxygen in the alcohol product would not be able to exchange with water.
Figure 70.

Enzyme-coupled assays to determine the 18O content in the C20-C22 lyase products of P450 11A1. (A) Yeast alcohol dehydrogenase for isocaproaldehyde; (B) P450 17A1 for pregnenolone.
Conversely, in order to determine the 18O-content in the pregnenolone product of P450 11A1, an enzyme-coupled assay using P450 17A1 was employed. In this enzyme-coupled assay, the oxygen content at the C20-position of the steroid product was determined by converting pregnenolone into acetic acid with P450 17A1 (Figure 61) to perform the C17-C20 lyase reaction (Figure 70B). From our previous studies with P450 17A1,267 we had determined that 18O is completely incorporated into the acetic acid product in the C17-C20 lyase product of P450 17A1 in the presence of molecular oxygen (18O2) when 17α-hydroxy-[21,21,21-2H3]-pregnenolone was used as the substrate.267 Therefore, if P450 11A1 had incorporated an 18O atom from molecular oxygen (18O2), the resulting acetic acid product should contain two 18O atoms.
3.1.1.3.4. O-H Hydrogen Atom Abstraction Mechanism, Stereospecific Oxygen Rebound, and Stereospecific Dehydration.
Although the electrophilic Compound I mechanism for the cleavage of the C20-C22 bond was primarily supported from our 18O-labeling studies, the O-H hydrogen atom abstraction mechanism by Compound I following a stereospecific oxygen rebound by Compound II and stereospecific dehydration of the gem-diol intermediate could not be completely dismissed. In order to illustrate the stereospecific dehydration step, one must consider the various scenarios that arise from an O-H hydrogen atom abstraction from the C20-hydroxy group or the C22-hydroxy group of 20R,22R-dihydroxycholesterol to generate an oxygen radical intermediate either at C22 or C20 (Figure 70). These oxygen radical intermediates can also potentially interconvert if the hydrogen atom transfers between the two adjacent oxygen atoms at the C20 and C22 atoms (Figure 71).
Figure 71.

Generation of an oxygen radical intermediate either through a C22 O-H hydrogen atom abstraction or a C20 O-H hydrogen atom abstraction by P450 11A1. The oxygen radical intermediates are interconvertible through an H atom transfer between the adjacent oxygen atoms.
After C20-C22 bond scission of the oxygen radical intermediate in Figure 71, the carbon-centered radical is produced, which in turn can undergo oxygen rebound. If the oxygen rebound step is slow (Figure 72, steps 1 and 4), then a mixture of 18O-incorporated and non-incorporated products could result. The scrambling would arise from a σ-bond rotation of the C17-C20 bond occurring before oxygen rebound (Figure 72A, step 3). Similarly, if P450 11A1 initially abstracts a hydrogen atom from the C20 O-H bond of 20R,22R-dihydroxycholesterol (Figure 71), a C1-isocaproaldehyde radical forms, which could either undergo: (i) a stereospecific oxygen rebound or (ii) a non-stereospecific oxygen rebound through a rotation of the C1-C2 σ-bond of the radical intermediate (Figure 72B, step 3). The non-stereospecific oxygen rebound followed by a stereospecific dehydration of the gem-diol would result in a mixture of 18O-incorporated and non-incorporated lyase products.
Figure 72.

(A) P450 11A1 non-stereospecific oxygen rebound of the C20-pregnenolone radical followed by stereospecific dehydration. 18O2 isotope-labeling experiments do not support this mechanism because there was no 18O incorporation into pregnenolone. The C20-pregnenolone radical is formed from a C22 O-H abstraction of 20R,22R-dihydroxycholesterol. (B) A C1-isocaproaldehyde radical generated by initial abstraction of the C20 O-H bond of 20R,22R-dihydroxycholesterol by P450 11A1. Step 3 shows how a non-stereospecific oxygen rebound arises from the rotation of the C1-C2 σ-bond (i.e., 60°) in the isocaproaldehyde C1-radical intermediate. *O: 18O.
However, the results from the experiments involving the incubation of the lyase substrate with P450 11A1 in the presence of 18O2 showed complete lack of incorporation of the oxygen atom (18O) into any of the products from molecular oxygen.267 These findings are compelling in ruling out alternative mechanisms involving an O-H abstraction process by Compound I (e.g. Figure 65, with non-stereospecific oxygen rebound). One possible O-H hydrogen atom abstraction mechanism ruled out by the lack of 18O incorporation in the 18O2 studies involves a scenario where Compound II undergoes a stereospecific oxygen rebound onto the carbon radical followed by a non-stereospecific dehydration of either gem-diol intermediate. The non-stereospecific dehydration is not possible, because this process would result in a mixture of 18O-incorporated and non-incorporated carbonyl containing products (i.e., pregnenolone or isocaproaldehyde). Therefore, the 18O-labeling experiments only support mechanistic scenarios that involve both a stereospecific oxygen rebound and a stereospecific gem-diol dehydration step.
In the C20-C22 bond cleavage mechanism involving O-H abstraction with P450 11A1, there is either a C20-gem-diol intermediate with one 18O-atom on the pregnenolone product or a gem-diol containing one 18O-atom on the isocaproaldehyde product (Figure 73). The carbon atom containing the 18O-labeled gem-diol after oxygen rebound is a potential stereogenic center (Figure 73, gem-C1-R-diol).
Figure 73.

Generation of enantiomeric gem-diol isocaproaldehyde intermediates derived from either (A) 20R,22R-dihydroxycholesterol or (B) 20R,22S-dihydroxycholesterol. *O: 18O.
3.1.1.3.5. Electrophilic Compound I Mechanism.
Although the O-H hydrogen abstraction mechanism followed by stereospecific oxygen rebound and stereospecific gem-diol dehydration is consistent with the 18O-labeling experiments, the electrophilic Compound I mechanism is also still consistent with the results. In the electrophilic Compound I mechanism, we cannot discern which alcohol reacts first with the FeO center, and a molozonide intermediate is possible (Figure 67).267
3.1.1.4. P450 51A1.
This enzyme is essential in sterol synthesis in yeasts and fungi199,270 and in mammals271,272 (Figure 56), because the sterols are needed for membrane integrity. Two major studies have been done on the mechanism. Fischer et al.,273 working with rat liver microsomes and lanosterol, tentatively characterized an intermediate as the 14-O-formyl product. The result was interpreted as being consistent with a Baeyer-Villiger reaction, with rearrangement to a formyl product (Figure 75B).
Figure 75.

Ferric peroxide mechanisms proposed for P450 51A enzymes. (A) Hemiacetal mechanism proposed by Akhtar and associates.274 In that reference, the substrate analog used had a Δ7,8 double bond instead of the Δ8,9 bond in lanosterol 32-aldehyde. (B) Baeyer-Villiger mechanism with acyl intermediate proposed by Fischer et al.273
A product could be isolated and an 1H-NMR spectrum was recorded (1-dimensional), but the mass spectrum only showed a product without a formyl group (M+ 412, assigned as M-HCO2H). The NMR spectrum showed limited evidence (δ 7.86 ppm peak) for the postulated oxyformyl intermediate. The authors reported that the sample decomposed due to the acidic nature of the CDCl3, and integration of the peaks was not established. Addition of this intermediate to rat liver microsomes (which contain P450 51A1) generated dehydrolanosterol. Although that reaction should not involve redox chemistry, the reaction was stimulated by NADPH, which presents an enigma. The authors proposed that the ferrous P450 was a better catalyst of the (non-redox) decomposition. The limited mass spectral evidence is one criticism of the study (which was done at the advent of electrospray technology), as well as the lack of measurement of rates of decomposition of the intermediate to establish kinetic competency.
In a subsequent study with yeast (Candida albicans) P450 51A and the Δ6,7 analog of lanosterol, Akhtar’s group274 applied the method of using 18O2 as a co-substrate and analyzing the 18O content of the product HCO2H, as was discussed under P450 19A1 (Section 3.1.1.1.1, vide supra). The authors concluded that one 18O atom was incorporated into HCO2H and that a ferric peroxide mechanism is operative (Figure 75A).274 The same technical concerns about the mass spectral analysis raised about the P450 19A1 work (vide supra) apply here.177,220,222,223 The 18O incorporation was less than quantitative and no raw data are provided; this report is more transparent than the P450 19A1 work220,222,223 in acknowledging that only 5–7% of the analyzed HCO2H was derived from the 32-2H Δ6,7 lanosterol analogue substrate and that the isotopic analysis is problematic.274
These experiments bear repeating with newer mass spectrometry technology, in that the electron impact conditions used by Fischer et al.273 were harsh and the issues of formic acid isotopic analysis274 have already been discussed under P450 19A1 in Section 3.1.1.1.1. A mechanism based on Compound I, similar to that found for P450 19A1 (Figures 58, 59) can be proposed (Figure 76). More experiments with newer technology in this system should help establish a more universal understanding of how P450s cleave carbon-carbon bonds.
Figure 76.

A possible Compound I mechanism for P450 51 enzymes (14α-sterol demethylases). The mechanism is identical to that for P450 19A1 (Figures 58, 59). Use of 18O2 in an experiment with lanosterol 32-aldehyde would not yield any 18O label in the recovered HCO2H.
3.1.1.5. P450 125A1.
P450 125 (from M. tuberculosis and Rhodococci) may use a similar mechanism to that proposed for P450 51A (Figure 75) in the oxidation of cholesterol (Fig. 77). An unusual product is the formate ester (M2 in Fig. 77A). The reaction is shown as releasing formate in the proposed mechanism in Fig. 77A, presumably without extrusion into the medium (equilibration with formate added to the medium was not examined.) An alternate concerted Baeyer-Villiger reaction is shown in Figure 77B.
Figure 77.

Proposed mechanism of sterol side chain oxidation by M. tuberculosis P450 125A1.31,202 (A) Proposed steps involve addition of the ferric peroxo anion (FeIII–O2−, Compound 0) of CYP125 to the C26 carbonyl and subsequent radical fragmentation of the peroxyhemiacetal adduct. The reaction step with the peroxyhemiacetal adduct leads to (a) formation of an alkene (M1) or (b) the formation of a one-carbon deficient alcohol (M4). The Compound I-catalyzed oxidation of M1 generates a diol (M5) via the acid-catalyzed ring-opening of an epoxide intermediate. A C25 cation may also derive from the single-electron oxidation of the C25 radical (c). Trapping of the cation by formate or water (d) results in the formation of the C25-oxyformyl (M2) and the one-carbon reduced alcohol (M4), respectively. Loss of a proton from the C25 cation may also generate M1 (e). The Compound I-catalyzed oxidation of M4 produces a gem-diol intermediate that dehydrates to a keto compound (M3). (B) A possible alternate mechanism involving Baeyer-Villiger oxidation with Compound 0 of CYP125 to yield M2.
3.1.1.6. 17-Acetylenic Steroid D-Ring Annulation.
This is an interesting transformation involving the acetylenic group of the oral contraceptive 17α-ethynylestradiol (Figure 78).275
Figure 78.

A proposed mechanism for steroid ring D homoannulation.275 One carbon of the acetylenic group is incorporated into a new D ring and the other is lost as CO2.
It may apply to some of the 17-acetylenic progestin steroids276,277 in contraceptives as well. The reaction has only been reported in monkey liver microsomes and to our knowledge has not been studied with a purified P450 enzyme.275 In this case CO2 was documented as a product using 14C trapping. The steroid-like product D-homoestrone and related compounds have been identified as in vivo metabolites of 17-ethynyl steroids in humans, monkeys, and rodents.278–280 A plausible mechanism has been proposed (Figure 78),275 although a very reactive oxirene intermediate has been invoked. An oxirene intermediate has also been proposed in the formation of a cyclobutene ring from a propargyl cyclopropane ring in an unrelated drug candidate ((S)-6-chloro-4-(cyclopropropylethynyl)-4-(trifluoromethyl)-3,4-dihydro-2(1H)-quinazolinone (DPC 961, an analogue of the drug efavirenz).61,281
One carbon of the acetylenic group is incorporated into an expanded D ring and the other is lost as CO2. A possible mechanism would involve rearrangement of the enol to a formyl group, followed by oxidation of the aldehyde to a carboxylic acid. This β-keto acid might decarboxylate non-enzymatically, as in the case presented later for steroid 4-demethylation (Section 3.9, Figure 142, vide infra).
Figure 142.

4-Demethylation in sterol biosynthesis. The steps and enzymes are shown for yeast.458 The pathway is similar in mammals and plants, with lanosterol and obtusifoliol, respectively, leading to cholesterol and sitosterol/campesterol instead of ergosterol (yeast, fungi).460 In humans the enzyme corresponding to yeast ERG25 is termed MSMO1 or SC4MOL.461 ERG26 is a dehydrogenase.
3.1.1.7. P450 24A1 and Vitamin D Oxidation.
The secosteroid vitamin D3 is derived from a sterol (Δ6,7-dehydrocholesterol). The side chain is oxidized at the C-23 and C-24 carbons by mammalian P450 24A1 to give a mixture of products (Figure 79).
Figure 79.
Postulated mechanism of P450 24A1 cleavage of vitamin D3.62,282–286
The exact course of these steps has not been delineated, but the cleavage reactions can be proposed to resemble those invoked for the P450s that catalyze the C-C bond cleavage observed for the other steroids considered here (Figures 56–59, 62, 65, 67, 68, 74, 75, 77). Although the reduction of the aldehyde to an alcohol by a P450 has not been demonstrated in this case, there is precedent for carbonyl reduction (under aerobic conditions) with other mammalian P450s.287 One of the shortcomings of research in this area is that the smaller cleavage products have not been characterized, e.g. the proposed dimethyl glycolic acid in Figure 79.
Figure 74.

A molozonoide mechanism proposed for the final step of P450 11A1 side chain cleavage.267
3.1.1.8. P450s 85A2 and 85A3.
Two plant P450s, P450s 85A2 (Arabidopsis thaliana) and 85A3 (Solanum lycopersicum, tomato), have been reported to catalyze what is formally an oxygen insertion reaction in the biosynthesis of brassinosteroids, plant steroid hormones that function in regulation of plant growth and development (Figure 80).
Figure 80.

Oxidation of castasterone to brassinolide by A. thaliana P450s 85A2 and 85A3,96,288,289 using a Baeyer-Villiger oxidation mechanism or a Compound I-based mechanism. *O:18O.
The formation of the 6-keto steroid (from 6-deoxocastasterone) is done by P450s 85A1, 85A2, and 85A3, but only P450s 85A2 and 85A3 catalyze the last step. Thus, this overall sequence has elements of the multistep reactions seen in mammalian steroidogenesis (Figure 56).
This oxygen insertion is clearly an overall Baeyer-Villiger reaction, without any rearrangement. We have included it here because the C6-C7 bond is broken. A case for a FeO2¯-driven reaction can be made here, given the nature of the product. We presume that because the 6-keto product is generated by P450s 85A2 and 85A3, both enzymes may be capable of utilizing both FeO3+ and FeO2¯ (Compounds I and 0, respectively) chemistry for different reactions. Alternatively, the peroxohemiketal, which can form from the nucleophilic attack of ferric peroxide onto the 6-keto intermediate, can also be derived from a nucleophilic attack of the 6,6-gem-diol onto an electrophilic Compound I oxoiron species. The Compound I mechanism would result in the absence of 18O-incorporation into the final lactone product by molecular oxygen (18O2).
3.1.2. P450 74 Cleavage Reactions.
The ability of Family 74 P450s to use hydroperoxides (derived from lipoxygenases) to perform C-C bond forming reaction has already been presented (Section 3.1.1.2.3). Some related reactions are presented here, in which C-C cleavage is seen.
One pathway is the formation of divinyl ethers (Figure 81).290,291
Figure 81.

Synthesis of a divinyl ether by P450 74D1.291
These products, and also the formation of some hemiacetals,99,292 can be explained (Figure 82) by an expansion of the mechanistic scheme presented earlier (Figure 32) for formation of allene oxides.99
3.1.3. Simple Models, Industrial Chemicals, and Drugs.
Many P450 reactions have been studied with model compounds that are not found in nature nor have any uses in medicine, agriculture, or industry, e.g. simple cyclopropylamines. Most of these model reactions have been designed to test hypotheses about catalytic mechanisms. These substrates have been successfully used because of the permissive nature (i.e. large size and limited need for bonding) of the active sites of some P450s, particularly some from mammals.74 However, there are some caveats associated with such an approach, in that one may be selecting for substrates that will support a mechanism that may not be applicable with natural substrates.
We have arbitrarily listed some reactions with fatty acids here as well, although these could be grouped with natural products. The same is true for long-chain aldehydes.
Some of the compounds discussed here have large scale industrial uses, e.g. in plastics (vinyl halides, bisphenol). Information about the metabolism of these compounds has been important in issues of toxicology and environmental health.293,294 P450 reactions are also very important in the field of drug metabolism,111 and several examples of C-C bond-breaking will be presented.
3.1.3.1. Vinyl Halides.
One of the major entries of our own laboratory into P450 enzyme chemistry was studies on the metabolism of vinyl halides, which began with trichloroethylene.295,296 In the original publication from this laboratory, we showed that the known reaction products—especially trichloroacetaldehyde—could not be accounted for by the purported reaction product trichloroethylene oxide, contrary to what was previously assumed.296,297 However, the epoxide was shown to be one of the products,296 but its mechanism of hydrolysis was not elucidated for a number of years and was made possible by advances in LC-MS technology.298 Decomposition of trichloroethylene oxide is proposed to derive from two pathways (Figure 83).
Figure 83.

(A) P450-catalyzed oxidation of the solvent trichloroethylene. Alternate prodducts are shown. (B) C-C cleavage of trichloroethylene oxide.296,298
One pathway (a) yields glyoxylic acid, HCO2H, and CO, two major C-C cleavage products also seen in the enzymatic reaction.296,298 Pathway b involves a 1,2-chloride shift, giving dichloroacetic acid.
Similarly, P450 2E1 converts tetrachloroethylene to an epoxide, which also breaks down through 1,2-chloride shift and hydrolytic pathways, leading to the formation of CO and CO2299 (Figure 84).
Figure 84.

C-C cleavage of tetrachloroethylene via epoxidation.299 Pi: inorganic phosphate (from buffer); the circled P indicates phosphate bound to the glyoxalyl chloride moiety.
A summary of the degradation products of several vinyl halide epoxides (halooxiranes) (Figure 85) indicates that all yield CO (except 2-chlorooxirane, top line) plus another 1-carbon molecule whose valence state depends on the number of halogens present.300
Figure 85.

C-C cleavage and other oxidation products of the epoxides of tribromoethylene and other vinyl halides.296,298,300,301
3.1.3.2. Bisphenol A.
The degradation of the environmental contaminants bisphenol A and nonylphenol involves an unusual cleavage of stable carbon-carbon bonds. Some nonylphenol isomers and bisphenol A possess a quaternary α-carbon atom as a common structural feature. The degradation of nonylphenol in Sphingomonas sp. strain TTNP3 is proposed to occur via an ipso-substitution61,302,303 with the presence of a quaternary α-carbon as a prerequisite. Consequent to the hydroxylation at position C4 according to a type II ipso-substitution mechanism, the C-C bond between the phenolic moiety and the isopropyl group of bisphenol A is broken (Figure 86).304
Figure 86.

Ipso mechanism of cleavage of bisphenol A.304
In addition to hydroquinone and 4-(2-hydroxypropan-2-yl)phenol other compounds resulting from molecular rearrangements involving a carbocationic intermediate were identified.304,305 In resting cells or cell-free extracts of the bacterium Sphingomonas sp. strain TTNP3, one atom of 18O2 was incorporated into hydroquinone. The monooxygenase activity was dependent on both NADPH and, surprisingly, FAD. The degradation pathway of bisphenol A and nonylphenol involves the same monooxygenase, which was proposed to lead to ipso-substitution in other xenobiotics containing a phenol with a quaternary α-carbon.
3.1.3.3. Cyclopropylamines and Other Cycloalkyl Heteroatoms.
Another case in which enzymatic oxidation leads to C-C bond cleavage involves cyclopropylamines (Figure 87). One-electron oxidation, due to the low oxidation potential of the substrate (~ 1 V vs. normal hydrogen electrode (NHE)), yields an aminium radical species. In solution, these are known to ring-open, yielding methylene radicals, with a rate constant of ~108 s−1.306,307 Such radical products have been indirectly implicated in heme destruction with P450 enzymes.308
Figure 87.

A related case involves 4-alkyl 1,4-dihydropyridines (Figure 88A). Mechanistically, these can be considered a vinylogous case of the mechanism presented for cyclopropylamines in Figure 87.
Figure 88.

Oxidations of 1,4-dihydropyridines by P450s. (A) Release of alkyl radical from 4-alkyl 1,4-dihydropyridines. (B) Retention of 4-aryl group in 4-aryl-1,4-dihydropyridines.310,312,313
One-electron oxidation of the amine (which should have an E1/2 of ~ 1 V, vs. NHE) yields an aminium radical, which rearranges to release an alkyl radical.310 The alkyl radical can react with heme to destroy it, in a pseudo-mechanism-based inactivation.311 Alternatively, the alkyl radical can react with an added spin-trapping reagent.310 Oxidation (and release of a radical) does not occur when the 4-position is unsubstituted or when an aryl group is attached, in that an aryl radical would not form as easily and be a good leaving group (Figure 88B).312 This is one of the few examples of P450s generating radicals, other than O2∸.
In the case of a cyclobutylamine (Figure 89), the ring expansion product could be characterized.314 O-Ethyl cyclopropanone hydrate was also converted to a ring-opened product (3-hydroxypropionic acid ethyl ester) (Figure 89).309
Figure 89.

Oxidations of strained ring compounds and C-C cleavage.309,314–316
3.1.3.4. Ring-strained Hydrocarbons.
C-C bond breaking can also be observed in strained cycloalkanes, in the absence of heteroatoms, as a result of low potentials for 1-electron oxidation (also ~ 1 V vs. NHE). Such C-C bond breaking has been observed in quadricyclane315 and thujone316 (Figure 89), as well as rearranged products derived from model cyclopropyl fatty acids.317
3.1.3.5. Fatty Acids.
The bacterial P450 107H1 (P450BioI) catalyzes two successive hydroxylations to form a (cis-)glycol (Figure 90).62,318–320
Figure 90.

These hydroxylations are straightforward P450 reactions, probably involving an FeO3+ (Compound I) intermediate. The mechanism of cleavage of the diol to two aldehydes has not been investigated but can be postulated to occur, as in the third step of the P450 11A1 reaction with cholesterol (Figures 56, 68, 74).
Another bacterial P450 reaction in which C-C scission of a fatty acid occurs is with P450 152L1 (P450OleT) (Figure 91).
Figure 91.

Proposed mechanisms for decarboxylation of myristic acid (C14:0) to terminal olefins by P450 152L1 (P450OleT).62,321–323 P450OleT also has the ability to catalyze β-hydroxylation. R: CH3(CH2)10.
This reaction is supported by H2O2 and not the usual NADPH/O2 electron transfer pathway.321 Strong spectral evidence for the involvement of Compound I has been published by the Makris group.322,324 Two reactions occur, β-hydroxylation and decarboxylation to yield a terminal olefin. These are considered to involve the same intermediates, either a β-carbon radical or the carbocation formed from a subsequent electron transfer step. An alternate path, leading only to loss of CO2 via abstraction of a hydrogen atom from the protonated carboxylic acid, can also be considered.62
3.1.3.6. Aldehydes.
Aldehydes are readily oxidized to carboxylic acids by aldehyde dehydrogenases325 or by P450s.61 At least two processes have been shown to cleave aldehydes.
Oxidative cleavage of model aldehydes to generate terminal olefins has been demonstrated for several mammalian P450s (Figure 92).326 The process presumably also yields HCO2H. A Compound 0 (FeIII–O2¯) mechanism has been proposed (Figure 92).
Figure 92.

Proposed mechanism of deformylation of aldehydes to yield olefins.326
Another reaction with long chain aldehydes has been shown with housefly P450 enzymes (e.g. P450 4G, 6A1) (Figure 93).327,328
Figure 93.

Mechanisms proposed for decarbonylation of long-chain aldehydes.62,327,328 (A) Compound I;327 Compound 0.62,329
The mechanism has been proposed to involve Compound 0 (FeIII–O2¯) but can be supported by alkyl hydroperoxide and iodosylbenzene oxygen surrogates, which is not consistent with the proposed mechanism (Figure 93). An interesting feature of the proposed mechanism (Figure 93) is the abstraction of a hydrogen atom from the product HCO2H to produce CO2, based on isotopic labeling studies. In the propsed Compound I mechanism (Figure 93B) there is an imbalance of the electrons in the oxidations of HCO2H to CO2.
It should be noted that the 1-carbon products formed in both Figures 92 and 93 have not actually been identified and are only inferred. One- and two-carbon molecules (e.g., HCO2H, HCHO, CO, CO2, CH3CHO, CH3CO2H) are extremely difficult to analyze at low levels177,298,330,331 due to background contamination (vide supra, Section 3.1.1.1.1) and can generally only be done with the use of isotopically labeled precursors in our experience. The identification of the nature of these products is probably necessary to complete the understanding of the catalytic mechanisms.
3.1.3.7. Drugs.
Two seemingly endless supplies of novel P450 reactions are found in drugs and natural products, fields of research that may well go on forever. In drug metabolism, all significant metabolites must be identified for regulatory purposes, as well as for realizing goals in terms of aspects of pharmacokinetics, safety, and back-up programs. In this section we will only consider drug C-C cleavage. There is some overlap between P450 reactions that cleave C-C bonds and those in which new ones are formed (vide supra).
3.1.3.7.1. Nabumetone.
Nabumetone undergoes (along with other reactions332) an oxidation cleavage of a carbonyl group to shorten the side chain (Figure 94A).333
Figure 94.

Possible Compound I and ferric peroxide mechanisms (of human P450 1A2) for the oxidation of the drug nabumetone. (A) Observed reaction. (B) Ferric peroxide (Compound 0) mechanism.332 (C) Compound I mechanism involving hydrogen atom abstraction.31 (D) Dioxetane mechanism based on the mechanism proposed for P450 17A1 (Figures 65, 67C).239
At least two pathways have been proposed (Figure 94B, 94C). Both begin with C-hydroxylation at the methylene α to the carbonyl. At this point we are at the mechanistic equivalent to the P450 17A1 problem (Figures 32, 41). One pathway (Figure 94B) involves Compound 0 (FeIII–O2¯) and a nucleophilic attack on the ketone, followed by a collapse of the peroxy-glycol to yield an aldehyde. From the available data332 it is also possible to evoke a classical Compound I (FeO3+) mechanism (Figure 94C), yielding the aldehyde.31 Another option is a dioxetane, as proposed for P450 17A1 (Figures 65, 67, 94D). All of these options generate an aldehyde, which could be readily oxidized (by a Compound I mechanism) to generate the observed carboxylic acid product.333 (Acetic acid is the expected side product but its formation has not been investigated.)
Figure 41.

Radical SAM enzyme-catalyzed methylation in the formation of fosfomycin.161,163
3.1.3.7.2. Alpidine.
C-Dealkylation (Figure 95) has been reported, and two reactions have been attributed to P450 2A6 and to P450 3A4.334,335 No mechanism has been proposed.336
Figure 95.

C-Dealkylation of alpidine in incubations with human liver microsomes.334,335 Sites of demethylation are indicated with arrows.
3.1.3.7.3. LC15–0133.
The C-demethylation of a tert-butyl group was reported for a drug candidate (LC15–0133) by an unidentified P450 in rat liver microsomes (Figure 96).336,337
Figure 96.

C-Demethylation of drug candidate LC15–0133 (1-[2-(5-tert-butyl-[1,3,4]oxadiazole-2-carbonyl)-4-fluoropyrrolidin-1-yl]-2-(2-hydroxy-1,1-dimethylethylamino)-ethanone) in rat liver microsomes.336,337
The reaction can be rationalized in terms of oxidation of a methyl group to a carboxylic acid and then loss of CO2 by facilitation of the “electron sink” properties of an adjacent heterocyclic ring system (oxadiazole), similar to the cases of pyridoxal and thiamine chemistry.1
3.1.3.7.4. C-Demethylation of a tert-Butyl Group.
Another example of the C-demethylation of a tert-butyl group is considered in Figure 97.336,338 This reaction can be rationalized in the context of either Compound 0 (FeIII–O2¯) or Compound I (FeO3+) chemistry, in the absence of more data.
Figure 97.

(A) Proposed initial steps in the loss of a carbon from a tert-butyl compound, 3-{[(4-tert-butylbenzyl)-(pyridine-3-sulfonyl)-amino]-methyl}-phenoxy)-acetic acid (CP-533,536). (B) Final step in formation of the alcohol. (C) Final step in formation of desaturation product.336,338
3.1.3.7.5. Isomoxole.
The cleavage of the olefinic bond of the oxazole ring of the drug isomoxole is rationalized in the context of an epoxide, which can collapse in several ways (Figure 98).339–342 In this case a new type of heterocyclic ring can form, namely a hydantoin.
Figure 98.

Oxidative transformation of an azole ring to a hydantoin.339–342
3.1.4. More Natural Products.
C-C cleavage is not a particularly unusual P450 reaction in natural product chemistry. Bacteria and plants collectively contain many more and arguably more diversified sets of P450s than mammals do, being involved in the biosynthesis of a broader range of compounds (drnelson.uthsc.educytochromeP450.html). Some of the transformations cited under C-C bond forming reactions earlier also involve C-C cleavages and rearrangement. Some examples of P450 cleavages of natural products follow, although the list is not intended to be comprehensive.
3.1.4.1. P450 88A and ent-Kaurenoic Acid.
P450 88A is involved in the synthesis of gibberellins in plants (Figure 99).62,343,344
Figure 99.

P450 88A-catalyzed ring contraction.62,343,344 The product, GA12, is involved in gibberelin biosynthesis.
A key step is the contraction of one of the rings of the diterpenoid ent-kaurenoic acid. Following an initial hydroxylation, the mechanism is proposed to proceed by hydrogen atom abstraction, a subsequent electron transfer to yield a carbocation, and rearrangement to yield the cyclopentyl ring-aldehyde, with a final oxidation of the aldehyde.
3.1.4.2. P450 82G1 and Nerolidol.
Nerolidol is cleaved by plant P450 82G1 (Figure 100).
Figure 100.

This reaction is easily rationalized by the pathway shown, which consists of hydrogen atom transfer and then electron abstraction to generate a carbocation β to an alcohol, which readily collapses to yield methyl vinyl ketone plus the other conjugated product (Figure 100).62,345
3.1.4.3. P450 71AJ1 and Furanocoumarin Synthesis.
P450s are involved in the biosynthesis of furanocoumarins in plants. P450 71AJ1 oxidizes the substrate marmesin to psoralen (Figure 101).62,346–348
Figure 101.

P450 71AJ1-catalyzed cleavage of an isopropanol group in furanocoumarin synthesis.62,346–348
As in the case of nerolidol (Figure 100), hydrogen abstraction and subsequent transfer of an electron β to an alcohol yield a carbocation that can collapse, in this case yielding acetone and the important natural product psoralen (Figure 101A). A similar mechanism can be proposed for the conversion of columbianetin to angelicin (Figure 101B).
3.1.4.4. P450Fma and Fumagillin Synthesis.
P450Fma catalyzes an unusual rearrangement in the synthesis of fumagillin in the fungus Aspergillus fumigatus (Figure 102).62,349
Figure 102.

C-C bond cleavage by the P450 enzyme Fma (A. fumigatus af510), with a proposed ferric peroxide reaction in the upper pathway and a Compund I mechanism in the lower part.62,349
The substrate is first hydroxylated in a straightforward reaction. The next step can be rationalized by either Compound I (FeO3+) or Compound 0 (FeIII–O2¯) chemistry, with either path yielding an iron peroxide-carbon bond. This intermediate rearranges to yield an epoxide. Finally, the exocyclic methine is oxygenated to yield the final diepoxide product.
3.1.4.5. P450s PenM and PntM and Methyl Migration.
The reactions catalyzed by P450s PenM and PntM in the bacteria Streptomyces exfoliates and Streptomyces arenae, respectively, involve methyl group migrations (Figure 103). Hydrogen atom abstraction and subsequent electron transfer yield a carbocation, which undergoes stereospecific transfer of a methyl group to yield the final pentalenolactone product.62,350
Figure 103.

Methyl group migration catalyzed by P450s PenM and PntM (P450s 161C3 and 161C2, from S. exfoliatus UC5319 and S. arenae TÜ469, respectively).62,350
3.1.4.6. P450 725A4 and Taxavoid Synthesis.
One of the more unusual examples of C-C bond cleavage (and new C-C bond forming in this case) involves P450 725A4 and the synthesis of taxanoids in Pacific yew trees (Figure 104).31,351
Figure 104.

Rearrangement of taxa-4(5),11(12)-diene to 5(12)-oxa-3(11)-cyclotaxane, catalyzed by P450 725A4 from yew trees. (A) Overall pathway. (B) Proposed mechanism.351
As in several other cases, the reaction can be rationalized in terms of the use of Compound I (FeO3+) in hydrogen atom and subsequent electron transfer, followed by C-C bond cleavage and rearrangement of a carbocationic intermediate.
3.1.4.7. P450 72A1 and Loganin.
Another example of C-C bond cleavage, without formation of any new C-C bonds, is seen with Cantharanthus roseus (rose periwinkle flower) P450 72A1 in the conversion of loganin to secologanin (Figure 105).96,352
Figure 105.

C. roseus P450 72A1-catalyzed C-C bond cleavage in the conversion of loganin to secologanin.96,352
The reaction is rationalized in terms of hydrogen atom abstraction, homolytic cleavage of a C-C bond to yield a methine and a new carbinol radical, and oxygen rebound to yield a gem-diol, followed by dehydration to the aldehyde. Interestingly, this reaction occurs with the glucose-linked monoterpenoid.
3.2. Flavoproteins.
We have presented some examples of flavoproteins being involved in C-C bond formation (Section 2.3), but there are also examples of C-C bond cleavage.125 These fall into several categories involving both oxygenated flavins and other reactions. Although these reactions have largely been characterized in microorganisms, some (Baeyer-Villiger oxidations) have now been observed in humans and shown to be involved in drug metabolism.
3.2.1. Flavin Monooxygenase Reactions.
Flavoproteins are well-known for their roles in electron-transfer reactions; they play important roles as biological “transformers” in coupling 2-electron donors (NAD(P)H) with 1-electron acceptors (e.g., Fe3+). They are also oxygenases, acting as mixed-function oxidases. The general mechanism involves a 4a-hydroperoxide species, which is formed by the reaction of a reduced flavin (usually FADH2) with O2. Reactions involve the hydroxylation of activated phenyl groups, e.g. phenols and anilines,353 and the oxygenation of the soft nucleophiles nitrogen and sulfur.354 In these reactions the 4a-hydroperoxide behaves as an electrophile.
3.2.1.1. Electrophilic C-C Bond Cleavage Monooxygenase Reactions.
6-Hydroxy-3-succinoyl-pyridine 3-monooxygenase (HspB), a bacterial enzyme involved in nicotine degradation, is an NADH-dependent flavoprotein (containing FAD) (Figure 106). Several related flavoprotein monooxygenases catalyze oxidations of substituted phenols,355 with several of these fitting into the category of flavin-based C-C bond cleaving reactions (Figure 106). These reactions utilize the 4a-hydroperoxide form of the flavin with the protonated hydroperoxide behaving as an electrophile (Figure 107). As shown in Figure 107, the phenolic oxygen and a carbonyl at the p-benzylic position are used to produce C-C bond cleavage,
Figure 106.

C-C bond cleavage reaction of HspB and several structurally related flavoprotein monooxygenases.355
Figure 107.

Proposed mechanism of HspB.355
In the case of 4HPA1M (Figure 103a), the breakdown of an intermediate analogous to that shown for HspB in Figure 103b can be rationalized with a 1,2-alkyl migration (Figure 108).
Figure 108.

A proposed mechanism for the 1,2-acetyl migration catalyzed by the flavoprotein 4HPA1M (Figure 106).
3.2.1.2. Baeyer-Villiger Reactions.
Flavin 4a-hydroperoxides (unprotonated) can also act as nucleophiles, catalyzing Baeyer-Villiger reactions with carbonyl substrates (Figure 109).356
Figure 109.

Baeyer-Villiger oxidation of cyclohexanone by the human flavoprotein monooxygenase FMO5. Uncoupling also occurs to generate H2O2.1,357 R indicates the phosphoribitol side chain.
This is an important reaction in some bacteria, allowing the breaking of a (ketone) ring structure to generate lactones, which are readily hydrolyzed to acidic products that can be degraded (e.g., fatty acid oxidation enzymes, Section 3.3) for use as a carbon source.1 Another example is EpnF, which converts a ketone into a carbonate in the biosynthesis of precytochalasin E in the fungus Aspergilllus clavatus.358 An example of a mammalian enzyme that catalyzes Baeyer-Villiger reations is human liver microsomal flavin-containing monooxygenase 5 (FMO5). Other FMOs have been characterized in regard to electrophilic roles in the N- and S-oxygenation of drugs,354,359 but FMO5 appears to be adapted for nucleophilic Baeyer-Villiger chemistry. Examples of reactions attributed to FMO5 are presented in Figure 110, including at least four drugs.
Figure 110.

Some Baeyer-Villiger C-C oxidations of drugs catalyzed by the flavoprotein monooxygenase FMO5, a “BVMO” (Baeyer-Villiger monooxygenase) enzyme, that lead to C-C cleavage.357,360–362
Thus, a C-C oxygen insertion reaction can be utilized to cleave a C-C bond. Recently an alternate flavin mechanism involved in some oxygenations has been shown to involve a flavin N5-oxide,22 but it is unknown whether this intermediate can also catalyze Baeyer-Villiger oxidations.
3.2.2. Flavin-based Pseudodioxygenases.
In his classic 1979 text, Walsh1 discussed flavin-based dioxygenases and presented an enzyme involved in pyridoxol degradation as an example (Figure 111).363
Figure 111.

Bacterial reactions involved in oxidative catabolism of pyridoxol (from vitamin B6) and the model substrate 2-methyl-3-hydroxypyridine-5-carboxylic acid.364
The classification as a dioxygenase was based upon some 18O incorporation mass spectral data that are probably equivocal in retrospect, in that some 18O label derived from H218O was also detected.363 The proposed mechanism involved hydride addition to the aromatic ring, with the addition of O2 and an oxetane intermediate. A second enzyme was isolated that catalyzes the same reaction.364
More extensive mass spectrometry studies showed that one oxygen atom of the product was derived from O2 and one from H2O.365 The proposed mechanism (Figure 112) uses a 4a-hydroperoxide and resembles that of anthranilic acid hydroxylase366 and other flavin-containing enzymes that hydroxylate activated aromatic rings.367
Figure 112.

Mechanism of 2-methyl-3-hydroxypyridine-5-carboxylic acid monooxygenase.365 R indicates the phosphoribitol side chain.
At this time, based on the literature and on mechanistic considerations regarding the roles of flavin 4a-hydroperoxides367,368 and N5-oxides, there do not appear to be any flavin-based dioxygenases, only monooxygenases.
3.2.3. Prenyl FMN.
One of the more interesting recent developments in flavin chemistry is the discovery of prenylated flavins in some bacteria (Figure 113).369,370,371
Figure 113.

Interconversions of prenylated FMN species (prFMN).369 The carbons added to the isoalloxazine ring system are derived form dimethylallyl phosphate. R indicates the phosphoribitol side chain.
Prenylated flavins are capable of redox chemistry (Figure 113). One unusual reaction is 1,3-dipolar cycloaddition (Figure 113). One example (Figure 114) involves the reaction with acrylic acid, releasing CO2. The same mechanism is used to decarboxylate cinnamic acid to generate styrene.124
Figure 114.

Decarboxylation of acrylic acid by a prenyl FMN enzyme.369 R indicates the phosphoribitol side chain.
3.2.4. N5 Nucleophlic Reactions.
In classic enzymology, many decarboxylations are catalyzed in processes that involve pyridoxal and thiamine as electron sinks, to avoid the localization of a negative charge on a carbon atom.1 In some cases flavins can catalyze decarboxylations, as demonstrated with CndG (Figure 115).372
Figure 115.

Oxidative decarboxylation of an N-acyl tyrosine derivative by the flavoprotein CndG.372 (A) Overall reaction; (B) proposed mechanism. R indicates the phosphoribitol side chain.
Two flavoproteins can catalyze the oxidative decarboxylation of N-substituted tyrosine derivatives (Figure 115A), CndG from the myxobacterium Chondromyces crocatus and FeeG, from an unidentified soil microbe. The enzymes are part of the 4PO (4-phenyl oxidizing) subgroup of the VAO/PCMH (vanillyl alcohol oxidase/p-cresol methyl hydroxylase) flavoprotein family.372–374 A mechanism is shown in Figure 115B, with the involvement of a flavin N5-adduct and the generation of a p-quinone methide intermediate, which decomposes to yield CO2.
A bond is formed between the benzylic carbon of the tyrosine derivative and an oxidized flavin, effectively transferring a hydride ion to the flavin. The phenolic oxygen of the tyrosine assists in the process, generating a quinoid species that collapses, with electrons moving in from the carboxylic acid to release CO2. The reduced flavin reacts with O2 to (generate H2O2 and) complete the process.
3.3. Desaturation and Cleavage of Fatty Acids.
Fatty acids undergo redox reactions, both C-C bond forming and cleaving. There are two systems for introducing double bonds into fatty acids. Stearoyl CoA and palmitoyl CoA both undergo desaturation, as well as their products.375 Eventually arachidonic acid is produced, the important C20:4 fatty acid. Humans have two fatty acid desaturases and mice have four. The electron transfer pathway involves the flavoprotein NADH-b5 reductase, the hemoprotein b5, and a diiron non-heme oxygenase (Figure 116).
Figure 116.

Oxidation of stearoyl CoA (18:0-SCoA) to oleyl CoA (18:1-SCoA) by liver microsomal fatty acid desaturase (non-heme iron protein) and the associated electron transfer pathway.
A crystal structure of a mammalian stearoyl CoA desaturase has been published.376 Mechanistically, desaturation is related to hydroxylation, and some P450s can do both.45,377,378 The initial step in both reactions is hydrogen atom abstraction; this is followed by either oxygen rebound (hydroxylation) or the abstraction of a second hydrogen atom (or the equivalent).45,379 The balance between hydroxylation and desaturation can be fragile, and changing only one or a few amino acids can shift this ratio.380–382
Another mechanism of desaturation of fatty acids involves hydride transfer to flavoproteins, i.e. FAD-acyl CoA dehydrogenase, and in turn the electrons are transferred to another flavoprotein, electron-transferring flavoprotein (ETFP), and then into the mitochondrial electron transport chain.375 The resulting trans-enoyl CoA products are enzymatically hydrated and then oxidized by a dehydrogenase to β-keto fatty acyl CoA esters, which are cleaved by a thiol cleavage enzyme to liberate acetyl CoA plus a shortened fatty acyl CoA (Fig. 117).
Figure 117.

β-Oxidation pathway for mammalian metabolism of fatty acids. The electrons transferred to FADH2 are utilized in the mitochondrial electron transport chain.
Both of the desaturase reactions can be considered in the context of this review, in that the enzyme called the desaturase (Figure 116) is forming a C-C (π) bond, and the other desaturase (dehydrogenase, Figure 117) is part of a sequence of events leading to C-C cleavage.
3.4. Oxidations of Heme.
C-C bond cleavage occurs during the synthesis of heme, in that PPIX formation requires the conversion of two side-chain propionic acid residues to ethylenes. Heme is not recycled (at least not completely) but is degraded, and there are several means of opening the ring system. In mammals, the major pathway involves the use of heme oxygenase(s) to convert heme to biliverdin and CO. Biliverdin is reduced to bilirubin, which is conjugated with glucuronic acid and excreted. The overall pathways of heme and iron homeostasis can be complex in both microorganisms and mammals.383–385
3.4.1. Non-enzymatic Heme Degradation.
The heme of P450s is degraded during incubations with NADPH-P450 reductase and NADPH.386,387 This process is exacerbated under conditions in which lipid peroxidation is enhanced.388 In addition, NADPH-P450 reductase (with NADPH) can destroy free heme or the heme in several other heme proteins, including hemoglobin and methemalbumin.389 The process is related to the production of H2O2 and is more pronounced with ferrous heme than ferric.389 The process can be demonstrated in liver microsomes, although these are usually contaminated with enough catalase to prevent the destruction (the reaction can be demonstrated by inhibiting the catalase with NaN3).389–391 (Catalase is localized in the peroxisomes, which contaminate the microsomal fraction obtained in differential centrifugation.)
The products of this type of heme destruction are dipyrrolic propentdyopents and their cleavage products, maleimides.389,392 The pentdyopent products have unique spectral properties and were already known to be formed in model systems involving heme, ascorbate, and H2O2.393 (The name propentdyopent derives from the pink spectrum (λmax 525 nm; i.e., Greek “pent-dyo-pent”) upon reduction.) Mechanisms for the formation of pentdyopents and maleimides are shown in Figure 115.392
This process has been documented in vitro389,392 and what appear to be such dipyrrolic adducts have been shown to be crosslinked to P450 3A4 following treatment with alkyl hydroperoxides.394 However, the relevance of the process in vivo is unknown. There is some in vivo evidence for heme degradation that does not form CO (i.e., heme oxygenase, vide infra).395
3.4.2. Heme Oxygenases.
Heme oxygenases catalyze the first steps in the degradation of heme. This is an important process in the homeostasis of heme and iron itself. In some organisms, high concentrations of heme are lethal (e.g., this is very important in the parasite that causes malaria396). As already discussed, reduced heme can react with H2O2 to destroy itself (Figure 118).389,392 However, compared to heme oxygenase, these reactions are not selective regarding which of the meso bridges (Figure 119) is broken.
Figure 118.

Proposed mechanisms for non-specific oxidative degradation of heme by peroxides.392 (A) Degradation of heme methine linkages to yield propentdyopents. (B) Proposed mechanism of cleavage of a formyl group from a 2-formylpyrrolonone to generate a maleimide.
Figure 119.

Heme degradation by heme oxygenase.397
Although some of the heme catabolism in mammals is not accounted for,395 the major pathway involves the heme oxygenase enzymes and the formation of biliverdin and bilirubin (Figure 114). There are two heme oxygenases in mammals, including humans, a constitutive form and an inducible form.398 Specific enzymes reduce biliverdin to bilirubin (biliverdin reductase)399 and conjugate bilirubin with UDP-glucuronic acid.400 Although bilirubin formation can be seen in a positive light due to its antioxidant properties,401 high concentrations cause jaundice.402 The heme oxygenase cleavage reaction yields CO, which can have signaling properties of its own.403–405
The mechanism of heme oxygenase is complex.397,406 The substrate, heme, is used in the oxygen activation process that acts on it, and the role of the enzyme is to set the substrate up for electron delivery to the iron atom and also set the correct positioning of the substrate for cleavage. In mammals, electrons come from NADPH-P450 reductase, the same enzyme that delivers electrons to microsomal P450s.407 The intermediates in the reaction of the heme with O2, as might be expected, resemble those with P450s (Figures 2, 65, 67, 68, 74) and other hemeprotein and non-heme iron oxygenases (Figure 39). The two forms of mammalian heme oxygenase have some differences but the basic mechanisms are believed to be the same for both.408
The overall reaction catalyzed by heme oxygenase has at least three distinguishable steps.397,409 Although the kinetics of the overall reaction have been studied using stopped-flow spectroscopy,410 the on and off rates were not considered and it is not known if the reaction is processive or distributive. The rates of all of the individual steps were < 1 s−1, so using the expression Kd = koff/kon (and a typical kon of 107 M−1s−1) then binding affinities of < 0.1 µM would be necessary to make the reaction processive. The rate-limiting step in steady-state turnover was concluded to be the release of biliverdin from the enzyme, although the kinetics were enhanced in the presence of added biliverdin reductase.410 The release of the product biliverdin should be a first-order process and insensitive to the inclusion of a trapping reaction, however, and the authors suggest an allosteric effect of the biliverdin reductase as a possible explanation.410
The first reaction step is meso-hydroxylation, at the α-methine carbon (Figure 119). A (protonated) ferric hydroperoxy species (FeIII-OOH) is believed to be involved (Figure 120).397
Figure 120.

Three of the pathways proposed for oxidation of heme to α-meso-hydroxyheme by heme oxygenase.397
The first step can be supported by H2O2 but not the nominal single-oxygen donor m-CPBA or an alkyl hydroperoxide. The FeIII-OOH intermediate has been observed at low temperature411 and is stabilized by the enzyme. The distal aspartate residue has been proposed to be a hydrogen bond acceptor to prevent protonation of the FeIII-OOH species by H2O in the region,412 but Ikeda-Saito and his associates have interpreted FeIII-OOH activation by proton donation397,411 (see also ref.413 for discussion). Heme hydroxylation is proposed to occur by one of two mechanisms: (i) electrophilic addition of the terminal oxygen of the ferric hydroperoxo species (FeIII–OO−) to the α-meso carbon or (ii) nucleophilic addition of the terminal oxygen of the protonated (FeIII-OOH) species.413 The FeIII–OOH heme hydroxylation reaction outweighs constraints required for an alternate pathway to the FeIV=O species.413
The second step differs from the first and the third (Figure 114) in that it is not inhibited by CO ligation and cannot be supported by exogenous H2O2 (in place of O2 and an electron source).397 Hydroxyheme (in the heme oxygenase complex) is stable under anaerobic conditions but aerobically, even in the absence of the protein, it reacts (with O2) to form verdoheme (Figure 119). There are two ways to form verdoheme (Figure 121).397
Figure 121.

Resonance structures and reaction pathways for meso-hydroxyheme conversion to verdoheme.397 See Figure 119 for conversion of verdoheme to biliverdin and CO.
The scheme (Figure 121) is an oversimplification in that the process is a multistep reaction that includes O2 binding, O-O bond cleavage, and reorganization of the macrocycle. O2 binding is proposed to occur at the α-pyrrole carbon (FeIII)-hydroxyheme on the basis of the apparent lack of CO inhibition.397
The third step (conversion of verdoheme to biliverdin, Figures 119, 122) is probably still the least understood of the three steps.
Figure 122.

Mechanisms proposed for opening of verdoheme to biliverdin in the heme oxygenase reaction. 397,406
Verdoheme can be non-enzymatically converted to biliverdin by hydrolysis or by oxidation using either O2 or H2O2.397 The hydrolytic reaction is relatively straightforward, with nucleophilic attack of a hydroxide ion at the positively charged α-pyrrole carbon of the verdoheme ring (Figure 122). However, heme oxygenase is known to incorporate an oxygen atom from O2, rather than H2O, into biliverdin.414 FeIII-verdoheme does not react with H2O but FeII-verdoheme does.415 Some possible mechanisms are shown in Figure 122.397 (See also ref.413.)
The enzyme HutZ, a crucial protein of the iron uptake system in the bacterium Vibrio cholerae, is a heme oxygenase and appears to work in the same manner as the mammalian heme oxygenases, but it uses ascorbic acid as a reductant.416
3.4.3. Coproheme Decarboxylation.
The “tailoring” of the porphyrin for the synthesis of heme b in Gram-positive bacteria involves decarboxylation of two of the propionic acid moieties of iron-bound coproheme (FeIII) to produce the vinyl groups (Figure 123).417
Figure 123.

Possible coproheme decarboxylase pathways.418 (A) Oxidase; (B) oxygenase; (C) peroxide-dependent decarboxylation. P indicates a propionic acid side chain.
The enzyme involved in decarboxylation is HemQ (also called ChdC).417–419 Several mechanisms are possible, including oxidase, oxygenase, and peroxide-dependent schemes (Figure 123).418 A mechanism for H2O2-dependent decarboxylation is shown in Figure 124.418
Figure 124.

Proposed decarboxylation pathways for oxidative decarboxylation of coproheme.418 P indicates propionic acid side chains.
This process involves a Compound I intermediate, akin to P450s and heme oxygenase. Possible mechanisms include decarboxylation driven by a radical β to the carboxylic acid or by a hydroxyl group β to the carboxylic acid, generating the vinyl group(s).
3.4.4. HemF.
In Escherichia coli the radical SAM enzyme HemF catalyzes the oxidative decarboxylation of coprophyrinogen III to yield the vinyl groups, in this case without iron being present in the porphyrin (Figure 125).420 The reaction is metal-dependent, and Mn2+ appears to be the favored metal. A manganese peroxide mechanism is proposed (Figure 125B).420
Figure 125.

HemF-catalyzed decarboxylation of coproporphrinogen III. (A) Overall reaction; (B) Mn-dependent reaction.420
3.4.5. Mycobilin and Staphylobilin Formation.
A variant on the heme oxygenase mechanism to degrade heme is used in M. tuberculosis, in which CO is not released (Figure 126).421
Figure 126.

Comparison of heme oxidation pathways for heme oxygenase and MhuD (leading to mycobilin). (Side chains are not showon, for simplicity.)
The enzyme involved is MhuD and the product is mycobilin. Depending upon the cleavage pattern, there are two forms of mycobilin, with a formyl group still attached to the tetrapyrrole at either of the two α-sites.422 The first steps (up to hydroxyheme, Figure 126) are the same as in heme oxygenase and the mechanism shown in Figure 127 is supported by mass spectral and other spectroscopic evidence.422 “Ruffling” of the heme plane has been proposed to play a role in radical localization during the mechanism and directing the site of cleavage (Figure 127).422
Figure 127.

Proposed mechanism of heme degradation by MhuD.422
Another means of cleaving a C-C bond is seen in the bacterium Staphylococcus aureus.423 Both the paralogues IsdG and IsdI are part of a heme uptake process called the Isd system. These both cleave heme to form HCHO and a tetrapyrrole termed staphylobilin (Figure 128).423 The source of the electrons is unknown, as are intermediates in the reaction.
Figure 128.

Heme degradation to staphylobilin by IsdG and IsdI.423
3.5. Radical SAM Reactions.
Radical SAM enzymes have already been discussed in the context of C-C bond forming reactions (Figures 24, 40, 45). Enzymes in this family are also involved in C-C cleavage as well.
3.5.1. Repair of Photodimers in DNA.
One example is the repair of DNA T-T 6,4-photoproduct dimers (5-thymidyl-5,6-dihydrothymidine) in Bacillus subtilis spores (Figure 129).424 The process restores the original two thymidines in DNA.
Figure 129.

Radical SAM-mediated repair of a thymine-thymine 6,4-photoproduct dimer in DNA.424
3.5.2. Coproporphyrinogen Oxidase.
Another radical SAM C-C bond-cleaving reaction is the decarboxylation of the two propionic acid groups of coproporphyrinogen by coproporphyrinogen oxidase (HemN) (Figure 130A).
Figure 130.

Anaerobic decarboxylation of coproporphyrinogen III by HemN.161 (A) Overall reaction; (B) proposed mechanism.
This enzyme reaction resembles that seen with coproheme (Figure 123), which works with the iron-substituted porphyrin and uses the iron atom for oxidative chemistry (Figures 123, 124), but in this case no iron is present. HemN is utilized in anaerobes, in which no oxygen is present for use. The electron acceptors are SAM and an unidentified biological acceptor (Figure 130B).161
In a somewhat related reaction, the radical SAM enzyme NirJ removes two propionic acid groups from 12,18-didecarboxy-siroheme to form dihydro-heme d1 (which is desaturated to form heme d1).425
3.5.3. Mycofactin-biosynthetic Protein.
Another interesting radical SAM enzyme is involved in post-translational modification of a peptide, catalyzing the oxidative decarboxylation of the C-terminal tyrosine (Tyr-30) of the mycofactin precursor peptide MftA.426,427 Mycofactin is proposed to serve as a redox cofactor for other nicotinamide-dependent proteins in Mycobacterium ulcerans, although its exact function in unknown. A radical SAM mechanism has been proposed for the enzyme MftC and is shown in Figure 131.426,427
Figure 131.

Proposed mechanism of C-terminal tyrosine modification by MftC.426,427 R denotes the rest of the peptide (28 N-terminal residues).
3.6. β-Carotene Dioxygenases.
Although the mammalian conversion of β-carotene to retinal (Figure 132) had been known since 1930, the enzyme systems were not characterized for another 70 years.428
Figure 132.

Monooxygenase vs. dioxygenase reaction possibilities for cleavage of carotenoids.429
Although initial cloning and expression studies had described the enzyme as a dioxygenase,430 several subsequent papers in the field referred to the enzyme as a monooxygenase.431,432 The difference between monooxygenase and dioxygenase pathways,1,4 leading to the same carbonyl products (Figure 132), was considered429,433 using chemical 18O labeling approaches, and the results clearly demonstrate that the enzyme is a dioxygenase.
Mammals have two related enzymes catalyzing the reaction, BCO1 and BCO2.434 The enzymes use non-heme iron bound to several histidines and possibly a glutamate residue.431 A viable mechanism has been proposed by Harrison and Bugg (Figure 133).435 As with many dioxygenases, oxygen activation is linked to the presence of the substrate.
Figure 133.

A proposed mechanism of cleavage of carotenoids, based on corn (maize, Zea mays) Vp14 (9´-cis-epoxycarotenoid dioxygenase 1 (NED1)).435 R indicates the rest of the carotenoid.
The CCO (carotenoid cleaving oxygenase) family now includes multiple members. Further, the enzymes in this family are widespread and have even been characterized in marine bacteria.436 The members cleave carotenoids with varying specificity. For instance, Synechocystis (cyanobacter) ACO cleaves all-trans-8´-apocarotenal to generate all-trans-8´-retinal and C10-apocarotenol (Figure 134).433
Figure 134.

Reactions catalyzed by CCO family dioxygenases.433
Bacterial Novosphingobium NOV1 cleaves the stilbene resveratrol (Figure 134),437 as does the (fungal) Ustilago maydis enzyme Rco1.438 A mechanism similar to that proposed by Harrison and Bugg (Figure 133)435 has been proposed for NOV1.437 The atypical CCO member RPE65, which has an important role in the visual cycle, has esterase activity but is not an oxygenase.433 Even the two classic human, chicken, and plant CCO dioxygenases can cleave different retinoids at varying positions (Figure 135).439–441
Figure 135.

Cleavage of a variety of carotenoids by chicken dioxygenase BCO2.441
3.7. Tryptophan and Indoleamine 2,3-Dioxygenases.
Hayaishi’s group discovered the conversion of tryptophan to N-formylkynurenine in 1957 (Figure 136).10
Figure 136.

Working model for tryptophan oxidation.443 The intermediates are proposed to be the same for indole dioxygenase and tryptophan dioxygenase. The rate-limiting step(s) is proposed to be the addition (electrophilic or radical) of an oxygen atom for tryptophan dioxygenase and in the reaction of the epoxide with Compound II in the case of indole dioxygenase.
Although this was one of the first recognized dioxygenase reactions, the mechanism is still not totally understood today.16,442–444 Indoleamine-1,2-dioxygenases have broad substrate specificity, including L-tryptophan. Tryptophan-2,3-dioxygenases use only L-tryptophan as a substrate.442,443
A number of catalytic mechanisms for this heme-based dioxygenase have been proposed over the years,16,444 including a dioxetane intermediate.1,10 The enzyme is a heme protein and accordingly uses its iron atom (in the heme) to activate O2. Both radical and electrophilic reactions can be considered (Figure 136).
The most recent proposal by Raven and associates has one atom of the activated O2 used to form a 2,3-epoxide in the presence of a Compound II-like intermediate (FeIV=O, without another positive charge in the porphyrin like Compound I) (Figure 136).16,443 There is spectral evidence for this Compound II entity.443 The oxygen atom of the Compound II species reacts with the C2 atom of the indole ring to yield a diol-like species, with one of the oxygens liganded to the iron atom. Rearrangement of the species yields the dicarbonyl product (e.g. N-formylkynurenine).443
Indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase differ in two aspects: (i) catalytic specificity (i.e., features of the active site), and (ii) which step is rate-limiting in the overall reaction (Figure 136).445
3.8. Mononuclear Non-heme Iron Dioxygenases.
The cleavage of aromatic rings is a very important process for many bacteria, allowing them to generate alicyclic hydrocarbons that can be processed by typical enzymatic processes such as fatty acid β-oxidation. Monooxygenation yields phenols, the phenyl rings of which can be further hydroxylated by flavoproteins or P450 enzymes.1 Thus bacteria can utilize some aromatic ring compounds as sole carbon sources.
3.8.1. Examples of Mononuclear Non-heme Iron Dioxygenases.
One approach used in nature is that of the Rieske dioxygenases, which catalyze the addition of O2 to aromatic molecules to generate cis-dihydrodiols (Figure 137).
Figure 137.

Dioxygenases involved in cleavage of aromatic C-C bonds. Rieske dioxygenases add two oxygen atoms to an aromatic ring. The resulting diol can be oxidized to the catechol, which can be cleaved by an extradiol dioxygenase (FeII) or an intradiol dioxygenase (FeIII).37 O2 positional labeling is indicated in red. R indicates a variable constituent.
These cis-dihydrodiols can be oxidized to o-catechols by dehydrogenases. P450s can also generate o-catechols by two successive monooxygenations or can make dihydrodiols (through the formation of an epoxide followed by hydrolysis (e.g., Figure 132)), but the stereochemistry will be trans in the latter case due to the mode of addition of H2O, either with or without the use of the enzyme epoxide hydrolase.446 The Rieske dioxygenases require auxiliary proteins for the delivery of two electrons, which are derived from NAD(P)H through a ferredoxin with the utilization of a flavoprotein.37
The difference between these three types of non-heme dioxygenases, which incorporate both atoms of oxygen into the same substrate, is shown in Figure 137. A list of some Rieske dioxygenases and intradiol and extradiol dioxygenases is shown in Figure 138.
Figure 138.

Some typical mononuclear non-heme iron and Rieske center-containing dioxygenases.37 O2 positional labeling is indicated in red.
Studies of these dioxygeanses have a long history, and they were among the first oxygenases characterized.7,9–11,13,447,448 Both the intradiol and extradiol dioxygeanases use a non-heme iron center to facilitate the reaction of triplet oxygen with singlet substrates.
3.8.2. Mechanisms of Mononuclear Non-heme Iron Dioxygenases.
The patterns of oxygen incorporation are shown in Figure 137. All three types of dioxygenases (Figure 137) have complex mechanisms. In all cases the non-heme iron serves as the core and, in contrast to what is usually seen in P450 monooxygenases (Figure 2), forms ligands with the substrate and intermediates (Figures 139–141).37 The other ligands to the iron are key tyrosine, histidine, and acidic amino acids, as opposed to the cysteine in P450s.
Figure 139.

Proposed catalytic mechanisms of Rieske dioxygenases.37 (A) Homolytic; (B) heterolytic.
Figure 141.

Proposed catalytic mechanism of extradiol ring-cleaving dioxygenases.37 R indicates a variable constituent.
3.8.2.1. Rieske Dioxygenases.
We have included these here, even though it may not be obvious that a C-C bond is being broken (Figure 137). However, closer inspection reveals that a C-C π bond is being broken. Mechanisms of these enzymes have been reviewed by Wang et al.37 and are shown in Figure 139 (see also ref.449). The mechanism begins with the reaction of ferrous iron with O2 to form a complex. A homolytic mechanism is shown in Figure 139A and a heterolytic mechanism in Figure 139B. Reaction of the Fe-O2 complex occurs with the aryl substrate, with both oxygens being incorporated. Regardless of the mechanism (Figure 139A or 139B), the reaction proceeds to yield a dihydrodiol product that chelates the iron. Two H2O molecules bind as the product is released. Note that, in contrast to the ring-cleaving dioxygenases (Figures 140, 141), this dioxygenase requires the input of two electrons to break a C-C π bond.
Figure 140.

Proposed catalytic mechanism of intradiol ring-cleaving dioxygenases.37 R indicates a variable constituent.
3.8.2.2. Intradiol Ring-cleaving Dioxygenases.
The mechanism of the intradiol dioxygenases (Figure 140) involves FeIII serving to activate the substrate to allow formation of an alkyl peroxy intermediate, polarizing the substrate and building up the charge on one of the hydroxyl-bearing carbons.37 The oxygen accepts one electron from the substrate and the other from the (d-orbital of the) iron (different from the one accepting an electron from the substrate). Thus, two electrons of different spin are transferred to the incoming alkylperoxo intermediate. O-O bond cleavage occurs via a Criegee rearrangement, forming a cyclic anhydride intermediate and FeIII-OH. Hydrolysis of the anhydride by FeII-OH yields the product, muconic acid.37
3.8.2.3. Extradiol Ring-cleaving Dioxygenases.
The extradiol dioxygenases use FeII and feature nucleophilic attack by an iron-bound oxygen on an organic substrate to yield an alkyl peroxo intermediate (Figure 141). Three types of extradiol dioxygenases have been characterized, namely Types I, II, and III.37 The three enzymes appear to have similar mechanisms. The mechanism described in Figure 136 is based largely on work with the Type III enzyme protocatechuate 4,5-dioxygenase. The iron-bound superoxo, alkylperoxo, and gem-diol intermediates and the product complex have all been observed using X-ray crystallography450,451 and spectroscopy.452–454 The binding of the organic substrate leads to reorganization of the iron center to promote O2 binding and subsequent reactions. O2 binds to ferrous iron and may form a transient FeIII-O2∸ species.37 The FeIII iron then activates the substrate, accepting an electron to generate a substrate semiquinone radical-FeII-O2∸ species. Radical-radical bond forming yields an FeII-bound alkylperoxo intermediate (Figure 141). Further support for the proposed mechanism comes from the trapping of an FeIII-superoxide radical in a variant of homoprotocatechuate 2,3-dioxygenase (Figure 138).37
3.9. Sterol 4-Demethylation.
An early step in the synthesis of cholesterol in mammals and the analogous sterols in plants and fungi is 14α-demethylation to yield HCO2H, which is catalyzed by P450 51 enzymes (section 3.1.1.4). A later step in sterol biosynthesis is removal of the two 4-methyl groups retained from the isoprenoid pathway. These are both lost as CO2.455–457 The pathway has now been determined in rat liver and yeast and is inherently conserved in most species.457,458 Rahier, 2011, 59021,459 The yeast pathway is outlined in Figure 142.458 In yeast and fungi, the pathway generates zymosterol and the final product ergosterol, in plants 24-methylene-cycloartanol is used to make sitosterol and campesterol, and in mammals lanosterol is used to synthesize cholesterol.457,458,460
Based largely on sequence comparisons,460 the enzyme appears to be a non-heme diiron protein. It receives electrons from b5 (via NADH- b5 reductase).457,461
The fungus A. fumigatus has two genes (Erg25). In plants the two 4-demethylation steps are separated by several intermediate steps instead of being sequential.460 There are five SMO (sterol 4α-methyl oxidase) cDNAs in the plant A. thaliana, belonging to two gene families.460 In humans, the protein now has the names MSMO1 and SC4MOL.461 Deficiencies in the gene have been reported to cause dermatitis, as well as microencephaly, congenital cataracts, and growth retardation.462
The mechanism (Figure 142) involves 3-step oxidations of the methyl groups to carboxylic acids, analogous to P450 reactions. The β-keto carboxylic acids are inherently unstable and are known to decarboxylate readily.463 It is unclear why nature has used multiple enzyme systems to do similar (C-demethylation) tasks in these pathways. A recent report indicates that a similar β-keto acid pathway is used in the synthesis of helvoic acid in the fungus Aspergillus oryzae but that the enzyme catalyzing the 3-step oxidation of a single C4 methyl group is a P450 (HePB1),464 which is surprising in light of the system in A. fumigatus (vide supra).
3.10. Iron/α-KG (Fe/α-KG) Dioxygenases.
This group of enzymes appears to be even more ubiquitous than the P450s, at least in the number of individual genes (although P450s collectively catalyze the oxidation of more substrates due to their less restricted catalytic selectivity).30 The reactions that Fe/α-KG enzymes catalyze have similarity to those catalyzed by P450s, if the cofactors are not considered. In some cases, the same reaction can be catalyzed by an Fe/α-KG dioxygenase in one species and by a P450 in another (e.g. thebaine O6-demethylation by an Fe/α-KG dioxygenase in plants465 and by P450s in mammals67).
3.10.1. Mechanisms of Iron/α-KG Dioxygenases.
The Fe/α-KG dioxygenases are inherently very different than P450s and use the following stoichiometry, where R is a substrate (Equation 7):
| 7 |
One of the two atoms of O2 is incorporated into succinate and the other into the substrate (R). Another feature of these dioxygenases is that they generally ligand the co-substrate α-KG to the catalytic iron center and then use other coordination sites of the iron center to bind O2, so that the activation of the iron is coupled to the presence of substrate.466 In contrast, P450s and flavin-based monooxygenases activate oxygen in a manner in which it is uncoupled from any linkage with a substrate (although in some cases substrates do facilitate the delivery of electrons to P450s, through indirect mechanisms).
3.10.2. Examples of Iron/α-KG Dioxygenases.
Fe/α-KG enzymes are involved in many hydroxylations and dealkylations, including many involving proteins and DNA. They appear to have relatively fewer roles in C-C bond formation and cleavage (aside from the C-C bond in α-KG), although there are at least several cases of C-C bond cleavage that will be considered here.
3.10.2.1. Ethylene-forming Enzyme.
The first example is the ethylene-forming enzyme EFE (Figure 143).467
Figure 143.

Proposed reaction mechanism for ethylene-forming enzyme (EFE).467
Ethylene is an important ripening agent in plants, and its production is under regulatory control. With EFE, α-KG is converted to ethylene and possibly oxalate, with release of CO2. Some potential mechanisms for the conversion of oxalate to CO2 are shown (Figure 144).467
Figure 144.

Some proposed mechanisms for decomposition of oxalate intermediates by EFE.467
3.10.2.2. AusE and Spiro Ring Formation.
Another example of a C-C bond cleavage reaction is the AusE reaction (Figure 145), in which a spiro center is formed.468 The mechanism is reminiscent of P450 chemistry, with a hydroxylation triggering the rearrangement leading to the spiro linkage.
Figure 145.

Proposed mechanism of spiro center formation by the Fe/α-KG dioxygenase AusE.468
3.10.2.3. TropC Ring Expansion.
TropC (Figure 141) is a case that can be considered both as a C-C bond cleavage and new C-C bond formation.469 An exocyclic methyl group is utilized in the expansion of a 6-membered ring to a 7-membered one.
3.10.2.4. EasH and Cycloclavine Biosynthesis.
An Fe/α-KG enzyme can also catalyze a ring contraction, as in the case of EasH involved in cycloclavine biosynthesis (Figure 147).470 In pathway a it is not clear why the hydroxylated product would be unstable. In pathway b the authors470 propose that EasH might transfer the (benzylic) hydride to NADP+, but there is no evidence that this enzyme uses pyridine nucleotides. Pathway c involves an unusual rearrangement initiated by hydrogen atom abstraction, although it may be most likely in light of the caveats about the other two possible pathways.
Figure 147.

Proposed mechanisms for an Fe/α-KG dioxygenase (EasH) involved in cycloclavine biosynthesis.470
3.10.2.5. An Iron/α-KG Dioxygenase in Gibberellin Biosynthesis.
Giberellin synthesis in plants involves a number of complex rearrangements, which we have seen with a P450 in this review already (Figure 99). In the case of an Fe/α-KG enzyme in the reaction shown in Figure 110, CO2 is removed in the addition of a new lactone ring.471 The authors propose an unusual mechanism involving a protein thiol and thioester intermediate.471
3.10.2.6. AmdA-mediated Transformation.
The Fe/α-KG dioxygenase AmdA has been shown to catalyze a very complex rearrangement (Figure 149).472 In the overall process, there is a ring contraction, the addition of a new ring, and a major change in another ring.
Figure 149.

Proposed mechanism of a transformation catalyzed by the Fe/α-KG dioxygenase AndA.472
3.10.2.7. AsqJ-mediated Reaction.
The Fe/α-KG dioxygenase AsqJ catalyzes two steps leading to a ring contraction (Figure 150).473,474 A nitrogen and two carbons are lost in the process. The other product has not been identified but might be methylisocyanate (CH3-N=C=O).
Figure 150.

Proposed mechanism for the Fe/α-KG dioxygenase AsqJ.473
3.10.2.8. Oxidative Decarboxylation Catalyzed by IsnB and AmbI3.
An oxidative decarboxylation by an Fe/α-KG-dependent dioxygenase has been characterized recently.475 A proposed mechanism is presented in Figure 151.
Figure 151.

Proposed mechaims for decarboxylation-assisted desaturation catalyzed by IsnB, AmbI3, and other Fe/α-KG enzymes.475
3.11. 1-Aminocyclopropane-1-carboxylic Acid Oxidase.
1-Aminocyclopropane-1-carboxylic acid oxidase (Figure 152) is a member of the 2-histidine/1-carboxylate family of non-heme FeII oxidases, which contains the Fe/α-KG dioxygenases.
Figure 152.

1-Aminocyclopropane-1-carboxylic acid oxidase. (A) Overall reaction. (B) Proposed mechanism for 1-aminocyclopropane-1-carboxylic acid oxidase.476
This enzyme and another in the family, isopenicillin N synthase, are unusual in that they do not use α-KG.476 Ascorbic acid plays a role as an electron donor (Figure 152).476 (Ascorbate also functions as the electron donor in some mixed-function oxidases, e.g., the copper enzyme dopamine β-hydroxylase.)
This particular enzyme is very important because the simple molecule ethylene acts in several physiological roles in plants, particularly in fruit ripening (vide supra). The unusual cyclopropane amino acid substrate is the product of a pyridoxal-dependent enzyme, aminocyclopropane-1-carboxylic acid synthase, which makes this compound from SAM. The enzyme mechanism is unusual in that CO2 is used as a ligand, stimulating the reaction, and is also a product (Figure 152).
The generation of the FeO complex with ascorbate is apparently rate-limiting.477 The mechanism (Figure 152)476 has some similarity to the P450 reactions described earlier (Figure 87). An aminium radical is produced and cyclopropane ring opening follows. In Figure 89 a hydroxylated product was characterized in a P450 reaction but, with 1-aminocyclopropane-1-carboxylic acid oxidase, homolytic scission generates ethylene. The other product is cyanoformic acid, which decomposes to CO2 and cyanide, regenerating the FeII/CO2 complex (Figure 152).476
3.12. Dioxygenases with Variable Metal Groups.
Some dioxygenase enzymes function with different metals. In these cases there appears to be no redox function of the metals. Rather, the metal serves as a ligand to bridge the substrate and O2 to facilitate the oxygenation.
3.12.1. Quercetinase.
Quercetinase is a dioxygenase that cleaves the heterocyclic ring of the flavonoid quercetin to yield CO and 2-protocatechuoylphloroglucinol carboxylic acid (Figure 153).
Figure 153.

(A) Reaction catalyzed by quercetinase (flavonol 2,4-dioxygenase).478 (B) Proposed mechanism for Ni2+-quercetinase.479 The electron needed to reduce molecular oxygen to superoxide anion may be derived from quercetin (as shown) or Ni2+.
This dioxygenase was reported to contain copper and to be the first copper-containing dioxygenase.480 It appears to be a true dioxygeanse in that 18O label from O2 is incorporated into both the products 2-protocatechuoylphloroglucinol carboxylic acid and CO.27,481 The peroxy-type intermediate shown in Figure 153B is based on an X-ray crystal structure but exactly how 18O-labeled CO would evolve is not clear.
Several quercetinases have now been identified in bacteria and fungi, members of the cupin family.478 The quercetinases isolated to date contain copper (Aspergillus) or are rather promiscuous with regard to metal selectivity (Bacillus).478 The bacterial Streptomyces sp. FLA quercetinase QueD was active with Ni2+ or Co2+ but not with Cu2+, Mn2+, Fe2+, or Zn2+.478 Because the metal can be substituted in this way, no electron transfer appears to be involved.27
The mechanism proposed for the enzyme is reminiscent of the non-heme iron dioxygenases and is based on an X-ray crystal structure.479 No change in the valence state of the metal appears to be required (Figure 147). An alternative mechanism, also involving a peroxide bridge, has been presented.27
3.12.2. Acireductone Dioxygenase.
An enzyme with some mechanistic resemblance to quercetinase is bacterial acireductone dioxygenase, which operates with Ni2+, Fe2+, or Co2+, also yielding CO in some cases (Figure 154).27,482 Acireductone (1,2-dihydroxy-3-keto-5-(methylthio)pentene, Figure 154) is an intermediate in a bacterial methionine salvage pathway.
Figure 154.

Cleavage of acireductone by acireductone dioxygenases.27,482 The red color is used to indicate the sites of O2 incorporation.
With the iron (Fe2+) enzyme, no CO is formed. The enzymes both feature 3-His/1-Glu coordination, and apparently the valence states of the metals do not change. O2 is bound to a metal-bound substrate dianion (Figure 155).
Figure 155.

Proposed mechanisms of acireductone dioxygenases. (A) Ni2+ enzyme. (B) Fe2+ enzyme.482
Thus, it is proposed that these are examples of substrate activation by the metalloenzyme, not of oxygen activation. This so-called chelate hypothesis explains the selectivity of these enzymes related to distinct coordination modes of the substrate in the Fe2+ and Ni2+ enzymes, perhaps harkening back to the original thoughts of Wieland on the subject (Section 1.1). The selectivity of the two metals has not been reproduced in synthetic models.27
3.13. Lignin Degradation.
In Section 2.9 the roles of peroxidases and laccases were discussed in regard to the synthesis of lignin in plants. Similar enzyme systems are also involved in lignin degradation. These enzymes are of practical interest in the processing of forest products. Structures of several of these peroxidases have been reviewed.483
3.13.1. Lignin-degrading Peroxidases.
Several groups of peroxidases are involved in lignin peroxidation.483,484 These include lignin peroxidases, manganese peroxidases, versatile peroxidases, and “dye-decolorizing peroxidase.”483,484 These peroxidases are abundant in fungi (e.g., Phanerochaete chrysosporium, or “white rot” fungi) that degrade dead wood. All of these peroxidases use H2O2, and the general principle is 1-electron oxidation of the phenolic components of lignin (Figure 156).483 The radical (and possibly a carbocation) are used to break linkages.
Figure 156.

A C-C cleavage reaction catalyzed by lignin peroxidase.483 (A) Mechanistic features (B) Proposed role of veratryl alcohol (VA). R indicates a variable constituent.
One of the issues is that degradation of a solid polymer like lignin is difficult because the enzyme can only have contact with the surface. Roles for diffusible mediators have been proposed,485,486 including veratryl alcohol (VA) (Figure 156).
3.13.1.1. Lignin Peroxidase.
This is a heme-based glycoprotein peroxidase with a high redox potential (E1/2 1.2 V at pH 3.0). The general mechanism is that shown in Figure 150, which is basically that for most peroxidases.487 The VA cation radical is proposed to protect the peroxidase from inactivation as a result of reaction of Compound I (cf. prostaglandin synthase, Figure 30, Section 2.4). The exact role of a VA cation radical as a mobile species is controversial,485,486,488–491 and recent work with fungal lignin peroxidase now argues strongly against a role for the VA cation radical.492
Efforts have been made to utilize the P. chrysosporium enzyme in biotechnology applications involving lignin degradation. However, there is an inherent problem when one considers that the peroxidase mechanisms for lignin synthesis and degradation are very similar, even if the actual enzymes are not the same (Figures 51, 156). Thus, polymerization occurs when the phenolic products are not removed, and the preference of lignin peroxidase for phenolic lignin units can explain the propensity to polymerize rather than depolymerize lignin under in vitro conditions.483,493
Another interesting aspect of lignin peroxidase is its ability to oxidize small aromatic molecules, unrelated to lignin. Thus, lignin peroxidase can degrade a number of persistent environmental pollutants.494 One example is benzo[a]pyrene,495 known to be accessible to 1-electron oxidation by other peroxidases.496,497 Other polycyclic hydrocarbons can be oxidized in this way, even by P450s.498 Efforts have been made to utilize the oxidation properties of lignin peroxidase and other peroxidases in bioremediation.499
3.13.1.2. Manganese Peroxidase.
This is another heme-based peroxidase found in wood-colorizing basidiomycetes (fungi), including P. chrysosporium. The peroxidase catalytic cycle is similar to that of lignin peroxidase (Figure 151) except that it oxidizes Mn2+ to Mn3+, which is then used as a “mobile” oxidant in the same way that VA has been proposed with lignin peroxidase. The redox potential (~ 0.8 V at pH 4.5, NHE) is reported not to be as high as for lignin peroxidase.483
3.13.1.3. Versatile Peroxidase.
This is another broad specificity heme-based peroxidase found in white-rot fungi. It has a different, broader specificity; like lignin peroxidase it will oxidize VA but will also oxidize other dyes that lignin peroxidase does and, unlike manganese peroxidase, it does not require Mn2+.483,484
3.13.1.4. Dye-decolorizing Peroxidase.
This is a relatively new class of peroxidases (DyP) found in fungi and bacteria, unrelated to other peroxidases.483,500,501 These enzymes can oxidize VA and some dyes,501 but their actual roles in lignin degradation have not been studied.
3.13.2. Laccases.
Laccases are 4-copper enzymes with moderately low redox potentials (0.42–0.79 V), 483 which are found in plants, bacteria, and fungi. They reduce O2 through two consecutive (Figure 158), as depicted in the scheme for a “trinuclear copper” (TNC) site.502
Figure 158.

As do the heme-containing peroxidases, laccases abstract an electron from phenolic (or other suitable aryl) groups, leading to Cα oxidation, alkyl-aryl cleavage and Cα-Cβ cleavage (Figure 159).483 They can oxidize non-phenolic lignin model compounds but only in the presence of redox mediators.484
Figure 159.

Products of cleavage of a lignin unit by laccase.483 (A) Phenolic model; (B) non-phenolic model.
4. Summary and Conclusions.
Redox enzymes are found everywhere, from mammals to anaerobic bacteria. They enable a number of aspects of life, and we have reviewed the roles of many of these in the formation and cleavage of C-C bonds.
The list of examples continues to grow and will continue to expand. Although the diversity of P450 reactions has been appreciated for several decades now, the discovery of many new reactions with flavins has been more recent.124 The broad contribution of the P450s is apparent,30 and even with more than 180,000 genes already identified, the list of these P450s will continue to expand. A major issue is annotation of the functions of these genes, which (somewhat embarrassingly) are largely unknown in all forms of life. The diversity of radical SAM and iron/α-KG reactions continues to expand.38,503,504 One point of interest in the C-C bond forming and cleaving reactions is the limited number of examples with copper, with only quercetinase and laccases (and the copper in quercetinase is not specific for the enzyme).
There are some common features of these steps (e.g., diradical formation for many of the C-C bond-forming steps) but also a diversity of both prosthetic groups, cofactors, and reaction mechanisms. In returning to the concepts of Wieland and Warburg (Section 1.1), we note that in many enzymes oxygen is activated, with a requirement for energy, in the absence of substrate (e.g., the FMO flavoproteins and many P450s and peroxidases) but that in other systems binding of a substrate or co-substrate to a metal lattice is what facilitates oxygen activation and, in some cases, the substrate itself is activated for the reaction (e.g., mononuclear dioxygenases, quercetinase, acireductone dioxygenases). Nevertheless, in most of the cases in which oxygen incorporation occurs in the C-C bond making and breaking reactions we have discussed, the source of the oxygen is O2, not water.
The nature of the reactions discussed here involves unstable intermediates, which of course are difficult to study and characterize. Thus, it is not surprising that there have been controversies about what mechanisms are involved, and there have been some paradigm shifts in dogma about some of the enzymes. Nevertheless, we return to the basic concepts about oxygenation in Section 1.1 (Figure 1), which are generally settled, even with the discovery of “cofactor-less” reactions.23,24 There are still many details of many of these reactions to understand, and the discovery of new natural products and drugs every day leads to more opportunities in the future. We have elaborated on some controversies in the P450 field and have even suggested experiments to do.
As pointed out above, despite the enormous scientific progress that has been made in the last few decades, we have a very limited understanding of what most enzymes (and genes) even do. We are of the opinion that this is a major need in biology in general today and that the answers will come not only from predictions but more from experimental approaches. With the discovery of new reactions, some will undoubtedly involve C-C bond breaking and forming, and there will be not only the systems discussed here but even more new ones to be discovered.
Figure 146.

Ring expansion catalyzed by the Fe/α-KG dioxygenase TropC.469 The asterisk (*) is used to track the fate of the methyl carbon atom.
Figure 148.

A proposed mechanism for an iron/α-KG dioxygenase involved in gibberellin biosynthesis.471
Figure 157.

Catalytic cycle of lignin-degrading manganese peroxidase.483 R indicates a substrate.
Acknowledgments.
Work on the subject in this laboratory (F.P.G.) is supported by National Institutes of Health grant R01 GM118122. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank K. Trisler for assistance in preparation of the manuscript. We also thank Prof. R. P. Hausinger (Michigan State University) for input on iron/α-KG dioxygenases and Profs. A. R. Brash and C. Schneider (Vanderbilt University School of Medicine) for comments on the manuscript. We are also indebted to the reviewers for their suggestions which were very helpful.
ABBREVIATIONS
- b5
cytochhrome b5
- BBE
berberine bridge enzyme
- m-CPBA
m-chloroperbenzoic acid
- CYP
cytochrome P450
- DHEA
dehydroepiandrosterone
- ENDOR
electron nuclear double resonance
- EPR
electron paramagnetic resonance
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- FMO
(microsomal) flavin-containing monooxygenase
- α-KG
α-ketoglutarate
- NHE
normal hydrogen electrode (or standard hydrogen electrode)
- P450
cytochrome P450
- PCET
proton-coupled electron transfer
- PPIX
protoporphyrin IX
- SAM
S-adenosylmethionine
- SCE
saturated calomel electrode
- UV-vis
ultraviolet-visible
- VA
veratryl alcohol
- VKOR
vitamin K oxidoreductase
Biographies
Dr. F. Peter Guengerich graduated (B.S., Agricultural Sciences) from the University of Illinois with University Honors (Bronze Tablet) in 1970. He received a Ph.D. in Biochemistry in 1973 from Vanderbilt University under the mentorship of the late Prof. Harry P. Broquist, where he studied alkaloid biosynthesis in fungi. From 1973–1975 he trained under the direction of Prof. Minor J. Coon at the University of Michigan Medical School, where he first developed his interests in cytochrome P450 enzymes. In 1975 he started his own laboratory in the Department of Biochemistry at Vanderbilt University School of Medicine, being promoted to the rank of associate professor in 1980 and to (full) professor in 1983. From 1980–2011 he served as Director of the Center in Molecular Toxicology at Vanderbilt. His research has been in the areas of the enzymology of cytochromes P450, DNA polymerases, and other enzymes involved in the biotransformation of xenobiotic chemicals, steroids, vitamins, and fatty acids. In 2009 Prof. Guengerich was one of the inaugural class of Fellows of the American Chemical Society (ACS) and in 2011 was the recipient of the Founders Award of the ACS Division of Chemical Toxicology. He chaired the Division from 2007–2008. Since 2006 he has been an Associate Editor of The Journal of Biological Chemistry and has been Deputy Editor since 2013, also serving as Editor-in-Chief in 2015–2016. In addition, Prof. Guengerich served as an Associate Editor of the ACS journal Chemical Research in Toxicology from 1989–2017. He has held an endowed chair at Vanderbilt since 2007 and is currently the Tadashi Inagami Professor of Biochemistry. Prof. Guengerich has published >700 primary research papers and >250 review articles and book chapters. He has also received two awards at Vanderbilt for training postdoctoral fellows; more than 20 graduate students and 135 postdoctoral fellows and visiting scientists have trained in his group.
Dr. Francis K. Yoshimoto received his bachelor’s degrees (B.S. in Chemistry and B.A. in Linguistics) from the University of California at Berkeley in 2005, where he was first introduced to research in synthetic organic chemistry in the laboratory of Prof. Richmond Sarpong. He completed his Ph.D. in Biochemistry (2012), first at the University of Texas Southwestern Medical Center and then at the University of Michigan Medical School in the research laboratory of Prof. Richard Auchus, where Dr. Yoshimoto elucidated the mechanisms of two steroidogenic cytochrome P450 enzymes, P450s 17A1 and 21A2. He conducted postdoctoral studies (2012–2016) in the research laboratory of Prof. F. Peter Guengerich at Vanderbilt University School of Medicine, where he cultivated his expertise in the P450 field, exploring the mechanisms of various P450s (including P450s 19A1, 17A1, and 11A1). In 2016 Dr. Yoshimoto established his independent research laboratory at the University of Texas at San Antonio in the Department of Chemistry where, as an Assistant Professor, he continues to work at the interface of chemistry and biology with the goal of training students to discover chemical and biochemical phenomena that will benefit human health.
Footnotes
Dedication. This review is dedicated to Professor Minor J. Coon, University of Michigan Medical School, a major figure in the field of cytochrome P450 research who introduced one of us (F.P.G.) to the fields of cytochrome P450 and enzyme mechanisms.
Conflict of Interest Disclosure. The authors declare no competing financial interests.
References
- (1).Walsh C Enzymatic Reaction Mechanisms; W. H. Freeman Co.: San Francisco, 1979. [Google Scholar]
- (2).Huang Y; Liu X; Cui Z; Wiegmann D; Niro G; Ducho C; Song Y; Yang Z; Van Lanen SG Pyridoxal-5´phosphate as an oxygenase cofactor: Discovery of a Carboxamide-forming, α-Amino Acid Monooxygenase-decarboxylase. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 974–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mason HS; Fowlks WB; Peterson EW Oxygen transfer and electron transport by the phenolase complex. J. Am. Chem. Soc 1955, 77, 2914–2915. [Google Scholar]
- (4).Mason HS Mechanisms of oxygen metabolism. Science 1957, 125, 1185–1190. [DOI] [PubMed] [Google Scholar]
- (5).Murakami K; Mason HS An Electron Spin Resonance Study of Microsomal Fex. J. Biol. Chem 1967, 242, 1102–1110. [PubMed] [Google Scholar]
- (6).Keevil T; Mason HS Moleular Oxygen in Biological Oxidations–An Overview. Methods Enzymol 1978; 52, 3–40. [DOI] [PubMed] [Google Scholar]
- (7).Hayaishi O; Hashimoto K Pyrocatecase: A New Enzyme Catalizing Oxidative Breakdown of Pyrocatechin. J. Biochem 1950, 37, 371–374. [Google Scholar]
- (8).Hayaishi O Enzymic Studies on the Mechanism of Double Hydroxylation. Pharmacol. Rev 1966, 18, 71–75. [PubMed] [Google Scholar]
- (9).Hayaishi O; Katagiri M; Rothberg S Studies on Oxygenases: Pyrocatechase. J. Biol. Chem 1957, 229, 905–920. [PubMed] [Google Scholar]
- (10).Hayaishi O; Rothberg S; Mehler AH; Saito Y Studies on Oxygenases: Enzymatic Formation of Kynurenine from Tryptophan. J. Biol. Chem 1957, 229, 889–896. [PubMed] [Google Scholar]
- (11).Hayaishi O From Oxygenase to Sleep. J. Biol. Chem 2008, 283, 19165–19175. [DOI] [PubMed] [Google Scholar]
- (12).Hayaishi O An Odyssey with Oxygen. Biochem. Biophys. Res. Commun 2005, 338, 2–6. [DOI] [PubMed] [Google Scholar]
- (13).Hayaishi O; Katagiri M; Rothberg S Mechanism of the Pyrocatechase Reaction. J. Am. Chem. Soc 1955, 77, 5450–5451. [Google Scholar]
- (14).Witkop B Remembering Heinrich Wieland (1877–1957): Portrait of an Organic Chemist and Founder of Modern Biochemistry. Med. Res. Rev 1992, 12, 195–274. [DOI] [PubMed] [Google Scholar]
- (15).Dixon M The History of Enzymes and of Biological Oxidations. In The Chemistry of Life: Eight Lectures on the History of Biochemistry; Needham J, Ed.; Cambridge Univeristy Press: Cambridge, UK, 1971; pp. 15–37. [Google Scholar]
- (16).Raven EL A Short History of Heme Dioxygenases: Rise, Fall and Rise Again. J. Biol. Inorg. Chem 2017, 22, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Strobel HW; Coon MJ Effect of Superoxide Generation and Dismutation on Hydroxylation Reactions Catalyzed by Liver Microsomal Cytochrome P-450. J. Biol. Chem 1971, 246, 7826–7829. [PubMed] [Google Scholar]
- (18).Aust SD; Roerig DL; Pederson TC Evidence for Superoxide Generation by NADPH-Cytochrome c Reductase of Rat Liver Microsomes. Biochem. Biophys. Res. Commun 1972, 47, 1133–1137. [DOI] [PubMed] [Google Scholar]
- (19).Manoj KM; Parashar A; Gade SK; Venkatachalam A Functioning of Microsomal Cytochrome P450s: Murburn Concept Explains the Metabolism of Xenobiotics in Hepatocytes. Front. Pharmacol 2016, 7, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Manoj KM; Parashar A; Venkatachalam A; Goyal S; Satyalipsu; Singh PG; Gade SK; Periyasami K; Jacob RS; Sardar D et al. Atypical Profiles and Modulations of Heme-enzymes Catalyzed Outcomes by Low Amounts of Diverse Additives Suggest Diffusible Radicals’ Obligatory Involvement in Such Redox Reactions. Biochimie 2016, 125, 91–111. [DOI] [PubMed] [Google Scholar]
- (21).Massey V The Chemical and Biological Versatility of Riboflavin. Biochem. Soc. Trans 2000, 28, 283–296. [PubMed] [Google Scholar]
- (22).Teufel R; Stull F; Meehan MJ; Michaudel Q; Dorrestein PC; Palfey B; Moore BS Biochemical Establishment and Characterization of EncM’s Flavin-N5-oxide Cofactor. J. Am. Chem. Soc 2015, 137, 8078–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Widboom PF; Fielding EN; Liu Y; Bruner SD Structural Basis for Cofactor-independent Dioxygenation in Vancomycin Biosynthesis. Nature 2007, 447, 342–345. [DOI] [PubMed] [Google Scholar]
- (24).Fielding EN; Widboom PF; Bruner SD Substrate Recognition and Catalysis by the Cofactor-independent Dioxygenase DpgC. Biochemistry 2007, 46, 13994–14000. [DOI] [PubMed] [Google Scholar]
- (25).Machovina MM; Usselman RJ; DuBois JL Monooxygenase Substrates Mimic Flavin to Catalyze Cofactorless Oxygenations. J. Biol. Chem 2016, 291, 17816–17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Samuelsson B On the Incorporation of Oxygen in the Conversion of 8,11,14-Eicosatrienoic Acid to Prostaglandin E1. J. Am. Chem. Soc 1965, 87, 3011–3013. [DOI] [PubMed] [Google Scholar]
- (27).Fiedler AT; Fischer AA Oxygen Activation by Mononuclear Mn, Co, and Ni Centers in Biology and Synthetic Complexes. J. Biol. Inorg. Chem 2017, 22, 407–424. [DOI] [PubMed] [Google Scholar]
- (28).Moktali V; Park J; Fedorova-Abrams ND; Park B; Choi J; Lee Y-H; Kang S Systematic and Searchable Classification of Cytochrome P450 Proteins Encoded by Fungal and Oomycete Genomes. BMC Genomics 2012, 13, 525–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schuler MA P450s in Plants Insect, and Their Fungal Pathogens. In Cytochrome P450. Structure, Mechanism, and Biochemistry; 4th ed.; Ortiz de Montellano PR, Ed.; Springer: New York, 2015, pp. 409–449. [Google Scholar]
- (30).Rendic S; Guengerich FP Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chem. Res. Toxicol 2015, 28, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Guengerich FP; Munro AW Unusual Cytochrome P450 Enzymes and Reactions. J. Biol. Chem 2013, 288, 17065–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yamazaki H; Nakamura M; Komatsu T; Ohyama K; Hatanaka N; Asahi S; Shimada N; Guengerich FP; Shimada T; Nakajima M et al. Roles of NADPH-P450 Reductase and Apo- and Holo-cytochrome b5 on Xenobiotic Oxidations Catalyzed by 12 Recombinant Human Cytochrome P450s Expressed in Membranes of Escherichia coli. Prot. Express. Purif 2002, 24, 329–337. [DOI] [PubMed] [Google Scholar]
- (33).Krest CM; Onderko EL; Yosca TH; Calixto JC; Karp RF; Livada J; Rittle J; Green MT Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mak PJ; Gregory MC; Denisov IG; Sligar SG; Kincaid JR Unveiling the Crucial Intermediates in Androgen Production. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tripathi S; Li H; Poulos TL Structural Basis for Effector Control and Redox Partner Recognition in Cytochrome P450. Science 2013, 340, 1227–1230. [DOI] [PubMed] [Google Scholar]
- (36).Denisov IG; Sligar SG Activation of Moleular Oxygen in Cytochromes P450. In Cytochrome P450: Structure, Mechanism, and Biochemistry; 4th ed.; Ortiz de Montellano PR, Ed.; Springer: New York, 2015, pp. 69–109. [Google Scholar]
- (37).Wang YF; Li JS; Liu AM Oxygen Activation by Mononuclear Nonheme Iron Dioxygenases Involved in the Degradation of Aromatics. J. Biol. Inorg. Chem 2017, 22, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Martinez S; Hausinger RP Catalytic Mechanisms of FeII- and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Guengerich FP Oxidation-Reduction Properties of Rat Liver Cytochromes P-450 and NADPH-cytochrome P-450 Reductase Related to Catalysis in Reconstituted Systems. Biochemistry 1983, 22, 2811–2820. [DOI] [PubMed] [Google Scholar]
- (40).Guengerich FP; Johnson WW Kinetics of Ferric Cytochrome P450 Reduction by NADPH-Cytochrome P450 Reductase: Rapid Reduction in the Absence of Substrate and Variations among Cytochrome P450 Systems. Biochemistry 1997, 36, 14741–14750. [DOI] [PubMed] [Google Scholar]
- (41).Yun CH; Kim KH; Calcutt MW; Guengerich FP Kinetic Analysis of Oxidation of Coumarins by Human Cytochrome P450 2A6. J. Biol. Chem 2005, 280, 12279–12291. [DOI] [PubMed] [Google Scholar]
- (42).Johnston WA; Hunter DJ; Noble CJ; Hanson GR; Stok JE; Hayes MA; De Voss JJ; Gillam EM Cytochrome P450 Is Present in Both Ferrous and Ferric Forms in the Resting State within Intact Escherichia coli and Hepatocytes. J. Biol. Chem 2011, 286, 40750–40759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Denisov IG; Grinkova YV; McLean MA; Sligar SG The One-Electron Autoxidation of Human Cytochrome P450 3A4. J. Biol. Chem 2007, 282, 26865–26873. [DOI] [PubMed] [Google Scholar]
- (44).Guengerich FP; Krauser JA; Johnson WW Rate-Limiting Steps in Oxidations Catalyzed by Rabbit Cytochrome P450 1A2. Biochemistry 2004, 43, 10775–10788. [DOI] [PubMed] [Google Scholar]
- (45).Johnson KM; Phan TTN; Albertolle ME; Guengerich FP Human Mitochondrial Cytochrome P450 27C1 is Localized in Skin and Preferentially Desaturates trans-Retinol to 3,4-Dehydroretinol. J. Biol. Chem 2017, 292, 13672–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhang H; Im SC; Waskell L Cytochrome b5 Increases the Rate of Product Formation by Cytochrome P450 2B4 and Competes with Cytochrome P450 Reductase for a Binding Site on Cytochrome P450 2B4. J. Biol. Chem 2007, 282, 29766–29776. [DOI] [PubMed] [Google Scholar]
- (47).Pearl NM; Wilcoxen J; Im S; Kunz R; Darty J; Britt RD; Ragsdale SW; Waskell L Protonation of the Hydroperoxo Intermediate of Cytochrome P450 2B4 Is Slower in the Presence of Cytochrome P450 Reductase Than in the Presence of Cytochrome b5. Biochemistry 2016, 55, 6558–6567. [DOI] [PubMed] [Google Scholar]
- (48).Hildebrandt A; Estabrook RW Evidence for the Participation of Cytochrome b5 in Hepatic Microsomal Mixed-Function Oxidation Reactions. Arch. Biochem. Biophys 1971, 143, 66–79. [DOI] [PubMed] [Google Scholar]
- (49).Yamazaki H; Johnson WW; Ueng YF; Shimada T; Guengerich FP Lack of Electron Transfer from Cytochrome b5 in Stimulation of Catalytic Activities of Cytochrome P450 3A4. Characterization of a Reconstituted Cytochrome P450 3A4/NADPH-Cytochrome P450 Reductase System and Studies with Apo-Cytochrome b5. J. Biol. Chem 1996, 271, 27438–27444. [DOI] [PubMed] [Google Scholar]
- (50).Yamazaki H; Shimada T; Martin MV; Guengerich FP Stimulation of Cytochrome P450 Reactions by Apo-Cytochrome b5: Evidence Against Transfer of Heme from Cytochrome P450 3A4 to Apo-Cytochrome b5 or Heme Oxygenase. J. Biol. Chem 2001, 276, 30885–30891. [DOI] [PubMed] [Google Scholar]
- (51).Auchus RJ; Lee TC; Miller WL Cytochrome b5 Augments the 17,20-lyase activity of Human P450c17 Without Direct Electron Transfer. J. Biol. Chem 1998, 273, 3158–3165. [DOI] [PubMed] [Google Scholar]
- (52).Groves JT Models and Mechanisms of Cytochrome P450 Action. In Cytochrome P450: Structure, Mechanism,and Biochemistry; 3rd ed.; Ortiz de Montellano PR Ed.; Kluwer Academic/Plenum Publishers: New York, 2005, pp. 1–43. [Google Scholar]
- (53).Bell SR; Groves JT A Highly Reactive P450 Model Compound I. J. Am. Chem. Soc 2009, 131, 9640–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Theorell H; Ehrenberg A; Chance B Electronic structure of the peroxidase-peroxide complexes. Arch. Biochem. Biophys 1952, 37, 237–239. [DOI] [PubMed] [Google Scholar]
- (56).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (57).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV) Hydroxide pKa and the Role of Thiolate Ligation in C-H bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Erdogan H; Vandemeulebroucke A; Nauser T; Bounds PL; Koppenol WH Jumpstarting the Cytochrome P450 Catalytic Cycle with a Hydrated Electron. J. Biol. Chem 2017, 292, 21481–21489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Newcomb M; Hollenberg PF; Coon MJ Multiple Mechanisms and Multiple Oxidants in P450-Catalyzed Hydroxylations. Arch. Biochem. Biophys 2003, 409, 72–79. [DOI] [PubMed] [Google Scholar]
- (60).Groves JT; McClusky GA; White RE; Coon MJ Aliphatic Hydroxylation by Highly Purified Liver Microsomal Cytochrome P-450: Evidence for a Carbon Radical Intermediate. Biochem. Biophys. Res. Commun 1978, 81, 154–160. [DOI] [PubMed] [Google Scholar]
- (61).Guengerich FP Common and Uncommon Cytochrome P450 Reactions Related to Metabolism and Chemical Toxicity. Chem. Res. Toxicol 2001, 14, 611–650. [DOI] [PubMed] [Google Scholar]
- (62).Ortiz de Montellano PR Substrate Oxidation. In Cytochrome P450: Structure, Mechanism, and Biochemistry; 4th ed.; Ortiz de Montellano PR, Ed.; Springer: New York, 2015, pp. 111–176. [Google Scholar]
- (63).Hagel JM; Facchini PJ Benzylisoquinoline Alkaloid Metabolism: A Century of Discovery and a Brave New World. Plant Cell Physiol 2013, 54, 647–672. [DOI] [PubMed] [Google Scholar]
- (64).Grobe N; Zhang B; Fisinger U; Kutchan TM; Zenk MH; Guengerich FP Mammalian Cytochrome P450 Enzymes Catalyze the Phenol-Coupling Step in Endogenous Morphine Biosynthesis. J. Biol. Chem 2009, 284, 24425–24431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ikezawa N; Iwasa K; Sato F CYP719A Subfamily of Cytochrome P450 Oxygenases and Isoquinoline Alkaloid Biosynthesis in Eschscholzia californica. Plant Cell Rep 2009, 28, 123–133. [DOI] [PubMed] [Google Scholar]
- (66).Gesell A; Rolf M; Ziegler J; Diaz Chavez ML; Huang FC; Kutchan TM CYP719B1 is Salutaridine Synthase, the C-C Phenol-Coupling Enzyme of Morphine Biosynthesis in Opium Poppy. J. Biol. Chem 2009, 284, 24432–24442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kramlinger VM; Alvarado Rojas M; Kanamori T; Guengerich FP Cytochrome P450 3A Enzymes Catalyze the O6-Demethylation of Thebaine, a Key Step in Endogenous Mammalian Morphine Biosynthesis. J. Biol. Chem 2015, 290, 20200–20210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Poeaknapo C; Schmidt J; Brandsch M; Drager B; Zenk MH Endogenous Formation of Morphine in Human Cells. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 14091–14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Amann T; Zenk MH Formation of the Morphine Precursor Salutaridine is Catalyzed by a Cytochrome P-450 Enzyme in Mammalian Liver. Tet. Lett 1991, 32, 3675–3678. [Google Scholar]
- (70).Gerardy R; Zenk MH Formation of Salutaridine from (R)-Reticuline by a Membrane-Bound Cytochrome P-450 Enzyme from Papaver somniferum. Phytochemistry 1992, 32, 79–86. [Google Scholar]
- (71).Amann T; Roos PH; Huh H; Zenk MH Purification and Characterization of a Cytochrome P450 Enzyme from Pig Liver, Catalyzing the Phenol Oxidative Coupling of (R)-Reticuline to Salutaridine, the Critical Step in Morphine Biosynthesis. Heterocycles 1995, 40, 425–440. [Google Scholar]
- (72).Davis JA; Greene RJ; Han S; Rock DA; Wienkers LC Formation of Raloxifene Homo-Dimer in CYP3A4, Evidence for Multi-Substrate Binding in a Single Catalytically Competent P450 Active Site. Arch. Biochem. Biophys 2011, 513, 110–118. [DOI] [PubMed] [Google Scholar]
- (73).Yano JK; Wester MR; Schoch GA; Griffin KJ; Stout CD; Johnson EF The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-ray Crystallography to 2.05 Å Resolution. J. Biol. Chem 2004, 38091–38094. [DOI] [PubMed] [Google Scholar]
- (74).Poulos TL; Johnson EF Structures of Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Function, and Biochemistry; Ortiz de Montellano PR, Ed.; Springer: New York, 2015; pp. 3–32. [Google Scholar]
- (75).Chen AY; Lee AJ; Jiang XR; Zhu BT Chemical Synthesis of Six Novel 17β-Estradiol and Estrone Dimers and Study of Their Formation Catalyzed by Human Cytochrome P450 Isoforms. J. Med. Chem 2007, 50, 5372–5381. [DOI] [PubMed] [Google Scholar]
- (76).McLean KJ; Leys D; Munro AW Microbial Cytochromes P450. iIn Cytochrome P450. Structure, Mechanism, and Biochemistry; 4th ed.; Ortiz de Montellano P, Ed.; Springer: New York, 2015; pp. 261–407. [Google Scholar]
- (77).Leys D; Mowat CG; McLean KJ; Richmond A; Chapman SK; Walkinshaw MD; Munro AW Atomic Structure of Mycobacterium tuberculosis CYP121 to 1.06 Å Reveals Novel Features of Cytochrome P450. J. Biol. Chem 2003, 278, 5141–5147. [DOI] [PubMed] [Google Scholar]
- (78).Belin P; Le Du MH; Fielding A; Lequin O; Jacquet M; Charbonnier JB; Lecoq A; Thai R; Courcon M; Masson C et al. Identification and Structural Basis of the Reaction Catalyzed by CYP121, an Essential Cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Dornevil K; Davis I; Fielding AJ; Terrell JR; Ma L; Liu A Cross-linking of Dicyclotyrosine by the Cytochrome P450 Enzyme CYP121 from Mycobacterium tuberculosis Proceeds Through a Catalytic Shunt Pathway. J. Biol. Chem 2017, 292, 13645–13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Fielding AJ; Dornevil K; Ma L; Davis I; Liu A Probing Ligand Exchange in the P450 Enzyme CYP121 from Mycobacterium tuberculosis: Dynamic Equilibrium of the Distal Heme Ligand as a Function of pH and Temperature. J. Am. Chem. Soc 2017, 139, 17484–17499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Lamb DC; Skaug T; Song H-L; Jackson CJ; Podust LM; Waterman MR; Kell DB; Kelly DE; Kelly SL The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem 2002, 277, 24000–24005. [DOI] [PubMed] [Google Scholar]
- (82).Austin MB; Izumikawa M; Bowman ME; Udwary DW; Ferrer J-L; Moore BS; Noel JP Crystal Structure of a Bacterial Type III Polyketide Synthase and Enzymatic Control of Reactive Polyketide Intermediates. J. Biol. Chem 2004, 279, 45162–45174. [DOI] [PubMed] [Google Scholar]
- (83).Feng B; Wang X; Hauser M; Kaufmann S; Jentsch S; Haase G; Becker JM; Szaniszlo PJ Molecular Cloning and Characterization ofWdPKS1, a Gene Involved in Dihydroxynaphthalene Melanin Biosynthesis and Virulence in Wangiella (Exophiala) dermatitidis. Infect. Immun 2001, 69, 1781–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Zhao B; Guengerich FP; Bellamine A; Lamb DC; Izumikawa M; Lei L; Podust LM; Sundaramoorthy M; Kalaitzis JA; Reddy LM et al. Binding of Two Flaviolin Substrate Molecules, Oxidative Coupling, and Crystal Structure of Streptomyces coelicolor A3(2) Cytochrome P450 158A2. J. Biol. Chem 2005, 280, 11599–11607. [DOI] [PubMed] [Google Scholar]
- (85).Zhao B; Guengerich FP; Voehler M; Waterman MR Role of Active Site Water Molecules and Substrate Hydroxyl Groups in Oxygen Activation by Cytochrome P450 158A2: A New Mechanism of Proton Transfer. J. Biol. Chem 2005, 280, 42188–42197. [DOI] [PubMed] [Google Scholar]
- (86).Zhao B; Lamb DC; Lei L; Kelly SL; Yuan H; Hachey DL; Waterman MR Different Binding Modes of two flaviolin substrate molecules in cytochrome P450 158A1 (CYP158A1) compared to CYP158A2. Biochemistry 2007, 46, 8725–8733. [DOI] [PubMed] [Google Scholar]
- (87).Zhao B; Bellamine A; Lei L; Waterman MR The Role of Ile87 of CYP158A2 in Oxidative Coupling Reaction. Arch. Biochem. Biophys 2012, 518, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Makino J; Sugimoto H; Shiro Y; Asamizu S; Onaka H; Nagano S Crystal Structures and Catalytic Mechanism of Cytochrome P450 StaP that Produces the Indolocarbazole Skeleton. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 11591–11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Wang Y; Hirao H; Chen H; Onaka H; Nagano S; Shaik S Electron Transfer Activation of Chromopyrrolic Acid by Cytochrome P450 En Route to the Formation of an Antitumor Indolocarbazole Derivative: Theory Supports Experiment. J. Am. Chem. Soc 2008, 130, 7170–7171. [DOI] [PubMed] [Google Scholar]
- (90).Ma J; Wang Z; Huang H; Luo M; Zuo D; Wang B; Sun A; Cheng Y-Q; Zhang C; Ju J Biosynthesis of Himastatin: Assembly Line and Characterization of Three Cytochrome P450 Enzymes Involved in the Post-tailoring Oxidative Steps. Angew. Chem. Int. Ed 2011, 50, 7797–7802. [DOI] [PubMed] [Google Scholar]
- (91).Zhang H; Chen J; Wang H; Xie Y; Ju J; Yan Y; Zhang H Structural Analysis of HmtT and HmtN Involved in the Tailoring Steps of Himastatin Biosynthesis. FEBS Lett 2013, 587, 1675–1680. [DOI] [PubMed] [Google Scholar]
- (92).Hashim MF; Hakamatsuka T; Ebizuka Y; Sankawa U Reaction Mechanism of Oxidative Rearrangement of Flavanone in Isoflavone Biosynthesis. FEBS Lett 1990, 271, 219–222. [DOI] [PubMed] [Google Scholar]
- (93).Sawada Y; Kinoshita K; Akashi T; Aoki T; Ayabe S Key Amino Acid Residues Required for Aryl Migration Catalysed by the Cytochrome P450 2-Hydroxyisoflavanone Synthase. Plant J 2002, 31, 555–564. [DOI] [PubMed] [Google Scholar]
- (94).Sawada Y; Ayabe S Multiple Mutagenesis of P450 Isoflavonoid Synthase Reveals a Key Active-Site Residue. Biochem. Biophys. Res. Commun 2005, 330, 907–913. [DOI] [PubMed] [Google Scholar]
- (95).Nasomjai P; Reed DW; Tozer DJ; Peach MJ; Slawin AM; Covello PS; O’Hagan D Mechanistic Insights into the Cytochrome P450-Mediated Oxidation and Rearrangement of Littorine in Tropane Alkaloid Biosynthesis. Chembiochem 2009, 10, 2382–2393. [DOI] [PubMed] [Google Scholar]
- (96).Mizutani M; Sato F Unusual P450 Reactions in Plant Secondary Metabolism. Arch. Biochem. Biophys 2011, 507, 194–203. [DOI] [PubMed] [Google Scholar]
- (97).Ullrich V Thoughts on Thiolate Tethering. Tribute and Thanks to a Teacher. Arch. Biochem. Biophys 2003, 409, 45–51. [DOI] [PubMed] [Google Scholar]
- (98).Hecker M; Ullrich V On the Mechanism of Prostacyclin and Thromboxane A2 Biosynthesis. J. Biol. Chem 1989, 264, 141–150. [PubMed] [Google Scholar]
- (99).Brash AR Mechanistic Aspects of CYP74 Allene Oxide Synthases and Related Cytochrome P450 Enzymes. Phytochemistry 2009, 70, 1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Ullrich V; Brugger R Prostacyclin and Thromboxane Synthase–New Aspects of Hemethiolate Catalysis. Angew. Chem. Int. Ed 1994, 33, 1911–1919. [Google Scholar]
- (101).Plastaras JP; Guengerich FP; Nebert DW; Marnett LJ Xenobiotic-Metabolizing Cytochromes P450 Convert Prostaglandin Endoperoxide to Hydroxyheptadecatrienoic Acid and the Mutagen, Malondialdehyde. J. Biol. Chem 2000, 275, 11784–11790. [DOI] [PubMed] [Google Scholar]
- (102).Gillam EM; Notley LM; Cai H; De Voss JJ; Guengerich FP Oxidation of Indole by Cytochrome P450 Enzymes. Biochemistry 2000, 39, 13817–13824. [DOI] [PubMed] [Google Scholar]
- (103).Wu ZL; Podust LM; Guengerich FP Expansion of Substrate Specificity of Cytochrome P450 2A6 by Random and Site-Directed Mutagenesis. J. Biol. Chem 2005, 280, 41090–41100. [DOI] [PubMed] [Google Scholar]
- (104).Gillam EM; Aguinaldo AM; Notley LM; Kim D; Mundkowski RG; Volkov AA; Arnold FH; Soucek P; DeVoss JJ; Guengerich FP Formation of Indigo by Recombinant Mammalian Cytochrome P450. Biochem. Biophys. Res. Commun 1999, 265, 469–472. [DOI] [PubMed] [Google Scholar]
- (105).Nakamura K; Martin MV; Guengerich FP Random Mutagenesis of Human Cytochrome P450 2A6 and Screening with Indole Oxidation Products. Arch. Biochem. Biophys 2001, 395, 25–31. [DOI] [PubMed] [Google Scholar]
- (106).Russell GA; Kaupp G Oxidation of Carbanions. IV. Oxidation of Indoxyl to Indigo in Basic Solution. J. Am. Chem. Soc 1969, 91, 3851–3859. [Google Scholar]
- (107).Hussain R; Kumari I; Sharma S; Ahmed M; Khan TA; Akhter Y Catalytic Diversity and Homotropic Allostery of Two Cytochrome P450 Monooxygenase Like Proteins from Trichoderma brevicompactum. J. Biol. Inorg. Chem 2017, 22, 1197–1209. [DOI] [PubMed] [Google Scholar]
- (108).Lee J; La S; Ahn BR; Jeong TC; Kim DH Metabolism of 1-(3-[3-(4-Cyanobenzyl)-3H-imidazol-4-yl]-propyl)-3-(6-methoxypyridin-3-yl )-1-(2-trifluoromethylbenzyl)thiourea (YH3945), a Novel Anti-Cancer Drug, in Rats using the 14C-Labeled Compound. Rapid Commun. Mass Spectrom 2004, 18, 1901–1910. [DOI] [PubMed] [Google Scholar]
- (109).Dalvie DK; O’Connell TN Characterization of Novel Dihydrothienopyridinium and Thienopyridinium Metabolites of Ticlopidine in vitro: Role of Peroxidases, Cytochromes P450, and Monoamine Oxidases. Drug Metab. Dispos 2004, 32, 49–57. [DOI] [PubMed] [Google Scholar]
- (110).Dansette PM; Bertho G; Mansuy D First Evidence that Cytochrome P450 may Catalyze both S-Oxidation and Epoxidation of Thiophene Derivatives. Biochem. Biophys. Res. Commun 2005, 338, 450–455. [DOI] [PubMed] [Google Scholar]
- (111).Guengerich FP Human Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; 4th ed.; Ortiz de Montellano PR, Ed.; Springer: New York, 2015; pp. 523–785. [Google Scholar]
- (112).Yun CH; Ahn T; Guengerich FP; Yamazaki H; Shimada T Phospholipase D Activity of Cytochrome P450 in Human Liver Endoplasmic Reticulum. Arch. Biochem. Biophys 1999, 367, 81–88. [DOI] [PubMed] [Google Scholar]
- (113).Zhao B; Lei L; Vassylyev DG; Lin X; Cane DE; Kelly SL; Yuan H; Lamb DC; Waterman MR Crystal Structure of Albaflavenone Monooxygenase Containing a Moonlighting Terpene Synthase Active Site. J. Biol. Chem 2009, 284, 36711–36719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Cheng Q; Lamb DC; Kelly SL; Lei L; Guengerich FP Cyclization of a Cellular Dipentaenone by Streptomyces coelicolor Cytochrome P450 154A1 Without Oxidation/Reduction. J. Am. Chem. Soc 2010, 132, 15173–15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Joo H; Lin ZL; Arnold FH Laboratory Evolution of Peroxide-Mediated Cytochrome P450 Hydroxylation. Nature 1999, 399, 670–673. [DOI] [PubMed] [Google Scholar]
- (116).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed 2017, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Coelho PS; Brustad EM; Kannan A; Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- (118).Brandenberg OF; Prier CK; Chen K; Knight AM; Wu Z; Arnold FH Stereoselective Enzymatic Synthesis of Heteroatom-Substituted Cyclopropanes. ACS Catalysis 2018, 8, 2629–2634. [Google Scholar]
- (119).Renata H; Lewis RD; Sweredoski MJ; Moradian A; Hess S; Wang ZJ; Arnold FH Identification of Mechanism-Based Inactivation in P450-Catalyzed Cyclopropanation Facilitates Engineering of Improved Enzymes. J. Am. Chem. Soc 2016, 138, 12527–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Gober JG; Ghodge SV; Bogart JW; Wever WJ; Watkins RR; Brustad EM; Bowers AA P450-Mediated Non-natural Cyclopropanation of Dehydroalanine-Containing Thiopeptides. ACS Chem. Biol 2017, 12, 1726–1731. [DOI] [PubMed] [Google Scholar]
- (121).Coelho PS; Wang ZJ; Ener ME; Baril SA; Kannan A; Arnold FH; Brustad EM A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in vivo. Nat. Chem. Biol 2013, 9, 485–U433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Ball S; Bruice TC 4a-Hydroperoxyflavin N-Oxidation of Tertiary Amines. J. Am. Chem. Soc 1979, 101, 4017–4019. [Google Scholar]
- (123).Muto S; Bruice TC Dioxygen Rtransfer from 4a-Hydroperoxyflavin Anion. 3. Oxygen Transfer to the 3-Position of Substituted Indoles. J. Am. Chem. Soc 1980, 102, 7559–7564. [Google Scholar]
- (124).Piano V; Palfey BA; Mattevi A Flavins as Covalent Catalysts: New Mechanisms Emerge. Trends Biochem. Sci 2017, 42, 457–469. [DOI] [PubMed] [Google Scholar]
- (125).Romero E; Gomez Castellanos JR; Gadda G; Fraaije MW; Mattevi A Same Substrate, Many Reactions: Oxygen Activation in Flavoenzymes. Chem. Rev 2018, 118, 1742–1769. [DOI] [PubMed] [Google Scholar]
- (126).Winkler A; Łyskowski A; Riedl S; Puhl M; Kutchan TM; Macheroux P; Gruber K A Concerted Mechanism for Berberine Bridge Enzyme. Nat. Chem. Biol 2008, 4, 739. [DOI] [PubMed] [Google Scholar]
- (127).Kutchan TM; Dittrich H Characterization and Mechanism of the Berberine Bridge Enzyme, a Covalently Flavinylated Oxidase of Benzophenanthridine Alkaloid Biosynthesis in Plants. J. Biol. Chem 1995, 270, 24475–24481. [DOI] [PubMed] [Google Scholar]
- (128).Gaweska HM; Roberts KM; Fitzpatrick PF Isotope Effects Suggest a Stepwise Mechanism for Berberine Bridge Enzyme. Biochemistry 2012, 51, 7342–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Daniel B; Konrad B; Toplak M; Lahham M; Messenlehner J; Winkler A; Macheroux P The Family of Berberine Bridge Enzyme-Like Enzymes: A Treasure-trove of Oxidative Reactions. Arch. Biochem. Biophys 2017, 632, 88–103. [DOI] [PubMed] [Google Scholar]
- (130).Gesell A; Díaz Chávez ML; Kramell R; Piotrowski M; Macheroux P; Kutchan TM Heterologous Expression of Two FAD-Dependent Oxidases with (S)-Tetrahydroprotoberberine Oxidase Activity from Arge mone mexicana and Berberis wilsoniae in Insect Cells. Planta 2011, 233, 1185–1197. [DOI] [PubMed] [Google Scholar]
- (131).Hagel JM; Beaudoin GAW; Fossati E; Ekins A; Martin VJJ; Facchini PJ Characterization of a Flavoprotein Oxidase from Opium Poppy Catalyzing the Final Steps in Sanguinarine and Papaverine Biosynthesis. J. Biol. Chem 2012, 287, 42972–42983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Taura F; Sirikantaramas S; Shoyama Y; Yoshikai K; Shoyama Y; Morimoto S Cannabidiolic-acid Synthase, the Chemotype-Determining Enzyme in the Fiber-Type Cannabis sativa. FEBS Lett 2007, 581, 2929–2934. [DOI] [PubMed] [Google Scholar]
- (133).Sirikantaramas S; Morimoto S; Shoyama Y; Ishikawa Y; Wada Y; Shoyama Y; Taura F The Gene Controlling Marijuana Psychoactivity: Molecular Cloning and Heterologous Expression of Δ1-Tetrahydrocannabinolic Acid Synthase from Cannabis sativa L. J. Biol. Chem 2004, 279, 39767–39774. [DOI] [PubMed] [Google Scholar]
- (134).Young AP; Bandarian V Radical Mediated Ring Formation in the Biosynthesis of the Hypermodified tRNA Base Wybutosine. Curr. Opin. Chem. Biol 2013, 17, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Lombard M; Hamdane D Flavin-dependent Epitranscriptomic World. Arch. Biochem. Biophys 2017, 632, 28–40. [DOI] [PubMed] [Google Scholar]
- (136).Sobrado P; Tanner JJ Multiple Functionalities of Reduced Flavin in the Non-Redox Reaction Catalyzed by UDP-Galactopyranose Mutase. Arch. Biochem. Biophys 2017, 632, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Nishimasu H; Ishitani R; Yamashita K; Iwashita C; Hirata A; Hori H; Nureki O Atomic Structure of a Folate/FAD-dependent tRNA T54 Methyltransferase. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 8180–8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Winkler JR; Gray HB Long-Range Electron Tunneling. J. Am. Chem. Soc 2014, 136, 2930–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Nag L; Sournia P; Myllykallio H; Liebl U; Vos MH Identification of the TyrOH•+ Radical Cation in the Flavoenzyme TrmFO. J. Am. Chem. Soc 2017, 139, 11500–11505. [DOI] [PubMed] [Google Scholar]
- (140).Ono T; Nakazono K; Kosaka H Purification and Partial Characterization of Squalene Epoxidase from Rat Liver Microsomes. Biochim. Biophys. Acta 1982, 709, 84–90. [DOI] [PubMed] [Google Scholar]
- (141).Li S; Finefield JM; Sunderhaus JD; McAfoos TJ; Williams RM; Sherman DH Biochemical Characterization of NotB as an FAD-dependent Oxidase in the Biosynthesis of Notoamide Indole Alkaloids. J. Am. Chem. Soc 2012, 134, 788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Rouzer CA; Marnett LJ Mechanism of Free Radical Oxygenation of Polyunsaturated Fatty Acids by Cyclooxygenases. Chem. Rev 2003, 103, 2239–2304. [DOI] [PubMed] [Google Scholar]
- (143).Smith WL; DeWitt DL; Garavito RM Cyclooxygenases: Structural, Cellular, and Molecular Biology. Annu. Rev. Biochem 2000, 69, 145–182. [DOI] [PubMed] [Google Scholar]
- (144).Ogino N; Ohki S; Yamamoto S; Hayaishi O Prostaglandin Endoperoxide Synthetase from Bovine Vesicular Gland Microsomes. Inactivation and Activation by Heme and Other Metalloporphyrins. J. Biol. Chem 1978, 253, 5061–5068. [PubMed] [Google Scholar]
- (145).Dietz R; Nastainczyk W; Ruf HH Higher Oxidation States of Prostaglandin H Synthase. Rapid Electronic Spectroscopy Detected Two Spectral Intermediates During the Peroxidase Reaction with Prostaglandin G2. Eur. J. Biochem 1988, 171, 321–328. [DOI] [PubMed] [Google Scholar]
- (146).Kulmacz RJ Prostaglandin G2 Levels During Reaction of Prostaglandin H Synthase with Arachidonic Acid. Prostaglandins 1987, 34, 225–240. [DOI] [PubMed] [Google Scholar]
- (147).Schneider C; Niisuke K; Boeglin WE; Voehler M; Stec DF; Porter NA; Brash AR Enzymatic Synthesis of a Bicyclobutane Fatty Acid by a Hemoprotein Lipoxygenase Fusion Protein from the Cyanobacterium Anabaena PCC 7120. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 18941–18945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Song WC; Brash AR Purification of an Allene Oxide Synthase and Identification of the Enzyme as a Cytochrome P-450. Science 1991, 253, 781–784. [DOI] [PubMed] [Google Scholar]
- (149).Gerwick WH Epoxy Allylic Carbocations as Conceptual Intermediates in the Biogenesis of Diverse Marine Oxylipins. Lipids 1996, 31, 1215–1231. [DOI] [PubMed] [Google Scholar]
- (150).Kadlubar FF; Morton KC; Ziegler DM Microsomal-catalyzed Hydroperoxide-Dependent C-Oxidation of Amines. Biochem. Biophys. Res. Commun 1973, 54, 1255–1261. [DOI] [PubMed] [Google Scholar]
- (151).Bui P; Imaizumi S; Beedanagari SR; Reddy ST; Hankinson O Human CYP2S1 Metabolizes Cyclooxygenase- and Lipoxygenase-Derived Eicosanoids. Drug Metab. Dispos 2011, 39, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Bui PH; Hsu EL; Hankinson O Fatty Acid Hydroperoxides Support Cytochrome P450 2S1-Mediated Bioactivation of Benzo[a]pyrene-7,8-dihydrodiol. Mol. Pharmacol 2009, 76, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Weiss RH; Aranold JL; Estabrook RW Transformation of an Arachidonic Acid Hydroperoxide into Epoxyhydroxy and Trihydroxy Fatty Acids by Liver Microsomal Cytochrome P-450. Arch. Biochem. Biophys 1987, 252, 334–338. [DOI] [PubMed] [Google Scholar]
- (154).Mansuy D; Bartoli JF; Momenteau M Alkane Hydroxylation Catalyzed by Metalloporphyrins: Evidence for Different Active Oxygen Species with Alkylhydroperoxides and Iodosobenzene as Oxidants. Tet. Lett 1982, 23, 2781–2784. [Google Scholar]
- (155).de Rond T; Stow P; Eigl I; Johnson RE; Chan LJG; Goyal G; Baidoo EEK; Hillson NJ; Petzold CJ; Sarpong R et al. Oxidative Cyclization of Prodigiosin by an Alkylglycerol Monooxygenase-loike Enzyme. Nat. Chem. Biol 2017, 13, 1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Rieske JS; MacLennan DH; Coleman R Isolation and Properties of an Iron-protein from the (Reduced Coenzyme Q)-Cytochrome c Reductase Complex of the Respiratory Chain. Biochem. Biophys. Res. Commun 1964, 15, 338–344. [Google Scholar]
- (157).Yokoyama K Radical Breakthroughs in Natural Product and Cofactor Biosynthesis. Biochemistry 2018, 57, 390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Zhang ZG; Mahanta N; Hudson GA; Mitchell DA; van der Donk WA Mechanism of a Class C Radical S-Adenosyl-L-methionine Thiazole Methyl Transferase. J. Am. Chem. Soc 2017, 139, 18623–18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Young AP; Bandarian V Pyruvate Is the Source of the Two Carbons That Are Required for Formation of the Imidazoline Ring of 4-Demethylwyosine. Biochemistry 2011, 50, 10573–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Perche-Letuvée P; Kathirvelu V; Berggren G; Clemancey M; Latour J-M; Maurel V; Douki T; Armengaud J; Mulliez E; Fontecave M et al. 4-Demethylwyosine Synthase from Pyrococcus abyssi is a Radical-S-adenosyl-L-methionine Enzyme with an Additional [4Fe-4S]+2 Cluster that Interacts with the Pyruvate Co-substrate. J. Biol. Chem 2012, 287, 41174–41185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Frey PA; Hegeman AD; Ruzicka FJ The Radical SAM Superfamily. Crit. Rev. Biochem. Mol. Biol 2008, 43, 63–88. [DOI] [PubMed] [Google Scholar]
- (162).Kuzuyama T; Hidaka T; Kamigiri K; Imai S; Seto H Studies on the Biosynthesis of Fosfomycin. 4. The Biosynthetic Origin of the Methyl Group of Fosfomycin. J. Antibiotics 1992, 45, 1812–1814. [DOI] [PubMed] [Google Scholar]
- (163).Sato S; Kudo F; Kim SY; Kuzuyama T; Eguchi T Methylcobalamin-Dependent Radical SAM C-Methyltransferase Fom3 Recognizes Cytidylyl-2-hydroxyethylphosphonate and Catalyzes the Nonstereoselective C-Methylation in Fosfomycin Biosynthesis. Biochemistry 2017, 56, 3519–3522. [DOI] [PubMed] [Google Scholar]
- (164).Schweifer A; Hammerschmidt F Stereochemical Course of Methyl Transfer by Cobalamin-Dependent Radical SAM Methyltransferase in Fosfomycin Biosynthesis. Biochemistry 2018, 57, 2069–2073. [DOI] [PubMed] [Google Scholar]
- (165).Banerjee R Radical Carbon Skeleton Rearrangements: Catalysis by Coenzyme B12-Dependent Mutases. Chem. Rev 2003, 103, 2083–2094. [DOI] [PubMed] [Google Scholar]
- (166).Brooks AJ; Fox CC; Marsh ENG; Vlasie M; Banerjee R; Brunold TC Electronic Structure Studies of the Adenosylcobalamin Cofactor in Glutamate Mutase. Biochemistry 2005, 44, 15167–15181. [DOI] [PubMed] [Google Scholar]
- (167).Román-Meléndez GD; von Glehn P; Harvey JN; Mulholland AJ; Marsh ENG Role of Active Site Residues in Promoting Cobalt–Carbon Bond Homolysis in Adenosylcobalamin-Dependent Mutases Revealed through Experiment and Computation. Biochemistry 2014, 53, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Bridwell-Rabb J; Zhong A; Sun HG; Drennan CL; Liu HW A B12-dependent Radical SAM Enzyme Involved in Oxetanocin A Biosynthesis. Nature 2017, 544, 322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Hänzelmann P; Hernández HL; Menzel C; García-Serres R; Huynh BH; Johnson MK; Mendel RR; Schindelin H Characterization of MOCS1A, an Oxygen-sensitive Iron-Sulfur Protein Involved in Human Molybdenum Cofactor Biosynthesis. J. Biol. Chem 2004, 279, 34721–34732. [DOI] [PubMed] [Google Scholar]
- (170).Schwarz G; Santamaria-Araujo JA; Wolf S; Lee HJ; Adham IM; Grone HJ; Schwegler H; Sass JO; Otte T; Hanzelmann P et al. Rescue of Lethal Molybdenum Cofactor Deficiency by a Biosynthetic Precursor from Escherichia coli. Human Mol. Genetics 2004, 13, 1249–1255. [DOI] [PubMed] [Google Scholar]
- (171).Lee HJ; Adham IM; Schwarz G; Kneussel M; Sass JO; Engel W; Reiss J Molybdenum Cofactor-Deficient Mice Resemble the Pphenotype of Human Patients. Human Mol. Genetic 2002, 11, 3309–3317. [DOI] [PubMed] [Google Scholar]
- (172).Wu SM; Cheung WF; Frazier D; Stafford DW Cloning and Expression of the cDNA for Human γ-Glutamyl Carboxylase. Science 1991, 254, 1634–1636. [DOI] [PubMed] [Google Scholar]
- (173).Wu SM; Morris DP; Stafford DW Identification and Purification to Near Homogeneity of the Vitamin K-Dependent Carboxylase. Proc. Natl. Acad. Sci. U. S. A 1991, 88, 2236–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Dowd P; Ham SW; Hershline R Role of Oxygen in the Vitamin K-Dependent Carboxylation Reaction: Incorporation of a Second Atom of 18O from Molecular Oxygen-18O2 into Vitamin K Oxide During Carboxylase Activity. J. Am. Chem. Soc 1992, 114, 7613–7617. [Google Scholar]
- (175).Kuliopulos A; Hubbard BR; Lam Z; Koski IJ; Furie B; Furie BC; Walsh CT Dioxygen Transfer During Vitamin K Dependent Carboxylase Catalysis. Biochemistry 1992, 31, 7722–7728. [DOI] [PubMed] [Google Scholar]
- (176).Bell RP The Reversible Hydration of Carbonyl Compounds. Adv. Phys.Org. Chem 1966, 4, 1–29. [Google Scholar]
- (177).Yoshimoto FK; Guengerich FP Mechanism of the Third Oxidative Step in the Conversion of Androgens to Estrogens by Cytochrome P450 19A1 Steroid Aromatase. J. Am. Chem. Soc 2014, 136, 15016–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Greenzaid P; Luz Z; Samuel D A Nuclear Magnetic Resonance Study of the Reversible Hydration of Aliphatic Aldehydes and Ketones. II. The Acid-Catalyzed Oxygen Exchange of Acetaldehyde. J. Am. Chem. Soc 1967, 89, 756–759. [Google Scholar]
- (179).Rishavy MA; Hallgren KW; Yakubenko AV; Shtofman RL; Runge KW; Berkner KL Brønsted Analysis Reveals Lys218 as the Carboxylase Active Site Base that Deprotonates Vitamin K Hydroquinone to Initiate Vitamin K-Dependent Protein Carboxylation. Biochemistry 2006, 45, 13239–13248. [DOI] [PubMed] [Google Scholar]
- (180).Tatarunas V; Lesauskaite V; Veikutiene A; Grybauskas P; Jakuska P; Jankauskiene L; Bartuseviciute R; Benetis R The Effect of CYP2C9, VKORC1 and CYP4F2 Polymorphism and of Clinical Factors on Warfarin Dosage during Initiation and Long-term Treatment after Heart Valve Surgery. J. Thrombosis Thrombolysis 2014, 37, 177–185. [DOI] [PubMed] [Google Scholar]
- (181).Higashi MK; Veenstra DL; Kondo LM; Wittkowsky AK; Srinouanprachanh SL; Farin FM; Rettie AE Association between CYP2C9 Genetic Variants and Anticoagulation-Related Outcomes during Warfarin Therapy. J. Am. Med. Assoc 2002, 287, 1690–1698. [DOI] [PubMed] [Google Scholar]
- (182).Caspers M; Czogalla KJ; Liphardt K; Muller J; Westhofen P; Watzka M; Oldenburg J Two Enzymes Catalyze Vitamin K 2,3-Epoxide Reductase Activity in Mouse: VKORC1 is Highly Expressed in Exocrine Tissues while VKORC1L1 is Highly Expressed in Brain. Thromb. Res 2015, 135, 977–983. [DOI] [PubMed] [Google Scholar]
- (183).Rishavy MA; Usubalieva A; Hallgren KW; Berkner KL Novel Insight into the Mechanism of the Vitamin K Oxidoreductase (VKOR): Electron Relay Through Cys43 and Cys51 Reduces VKOR to Allow Vitamin K Reduction and Facilitation of Vitamin K-Dependent Protein Carboxylation. J. Biol. Chem 2011, 286, 7267–7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Rishavy MA; Hallgren KW; Wilson LA; Usubalieva A; Runge KW; Berkner KL The Vitamin K Oxidoreductase Is a Multimer That Efficiently Reduces Vitamin K Epoxide to Hydroquinone to Allow Vitamin K-dependent Protein Carboxylation. J. Biol. Chem 2013, 288, 31556–31566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Ingram BO; Turbyfill JL; Bledsoe PJ; Jaiswal AK; Stafford DW Assessment of the Contribution of NAD(P)H-dependent Quinone Oxidoreductase 1 (NQO1) to the Reduction of vitamin K in Wild-type and NQO1-deficient Mice. Biochem. J 2013, 456, 47–54. [DOI] [PubMed] [Google Scholar]
- (186).Vanholme R; Demedts B; Morreel K; Ralph J; Boerjan W Lignin Biosynthesis and Structure. Plant Physiol 2010, 153, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).McCaig BC; Meagher RB; Dean JF Gene Structure and Molecular Analysis of the Laccase-Like Multicopper Oxidase (LMCO) Gene Family in Arabidopsis thaliana. Planta 2005, 221, 619–636. [DOI] [PubMed] [Google Scholar]
- (188).Zweig A; Hodgson WG; Jura WH The Oxidation of Methoxybenzenes. J. Am. Chem. Soc 1964, 86, 4124–4129. [Google Scholar]
- (189).Sato H; Guengerich FP Oxidation of 1,2,4,5-Tetramethoxybenzene to a Cation Radical by Cytochrome P450. J. Am. Chem. Soc 2000, 122, 8099–8100. [Google Scholar]
- (190).Murata J; Ono E; Yoroizuka S; Toyonaga H; Shiraishi A; Mori S; Tera M; Azuma T; Nagano AJ; Nakayasu M et al. Oxidative Rearrangement of (+)-Sesamin by CYP92B14 Co-Generates Twin Dietary Lignans in Sesame. Nat. Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Davidson VL Protein-Derived Cofactors Revisited: Empowering Amino Acid Residues with New Functions. Biochemistry 2018, in press. DOI: 10.1021/acs.biochem.8b0012310.1021/acs.biochem.8b00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Auchus RJ; Miller WL P450 Enzymes in Steroid Processing. In Cytochrome P450: Structure, Mechanism, and Biochemistry; 4th ed.; Ortiz de Montellano PR, Ed.; Springer: New York, 2015; pp. 851–879. [Google Scholar]
- (193).Turcu AF; Rege J; Chomic R; Liu J; Nishimoto HK; Else T; Moraitis AG; Palapattu GS; Rainey WE; Auchus RJ Profiles of 21-Carbon Steroids in 21-hydroxylase Deficiency. J. Clin. Endocrinol. Metab 2015, 100, 2283–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Brodie AMH Aromatase, Its Inhibitors and Their Use in Breast Cancer Treatment. Pharmacol. Therapeut 1993, 60, 501–515. [DOI] [PubMed] [Google Scholar]
- (195).Haidar S; Ehmer PB; Barassin S; Batzl-Hartmann C; Hartmann RW Effects of Novel 17α-Hydroxylase/C17, 20-lyase (P450 17, CYP 17) Inhibitors on Androgen Biosynthesis in vitro and in vivo. J. Steroid Biochem. Mol. Biol 2003, 84, 555–562. [DOI] [PubMed] [Google Scholar]
- (196).Njar VCO; Brodie AMH Discovery and Development of Galeterone (TOK-001 or VN/124–1) for the Treatment of All Stages of Prostate Cancer. J. Med. Chem 2015, 58, 2077–2087. [DOI] [PubMed] [Google Scholar]
- (197).Gomez L; Kovac JR; Lamb DJ CYP17A1 Inhibitors in Castration-resistant Prostate Cancer. Steroids 2015, 95, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Alyamani M; Li ZF; Berk M; Li JN; Tang JJ; Upadhyay S; Auchus RJ; Sharifi N Steroidogenic Metabolism of Galeterone Reveals a Diversity of Biochemical Activities. Cell Chem. Biol 2017, 24, 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Bard M; Lees ND; Turi T; Craft D; Cofrin L; Barbuch R; Koegel C; Loper JC Sterol Synthesis and Viability of Erg11 (Cytochrome P450 Lanosterol Demethylase) Mutations in Saccharomyces cerevisiae and Candida albicans. Lipids 1993, 28, 963–967. [DOI] [PubMed] [Google Scholar]
- (200).Hargrove TY; Friggeri L; Wawrzak Z; Qi A; Hoekstra WJ; Schotzinger RJ; York JD; Guengerich FP; Lepesheva GI Structural Analyses of Candida albicans Sterol 14α-Demethylase Complexed with Azole Drugs Address the Molecular Basis of Azole-Mediated Inhibition of Fungal Sterol Biosynthesis. J. Biol. Chem 2017, 292, 6728–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).McLean KJ; Dunford AJ; Neeli R; Driscoll MD; Munro AW Structure, Function and Drug Targeting in Mycobacterium tuberculosis Cytochrome P450 Systems. Arch. Biochem. Biophys 2007, 464, 228–240. [DOI] [PubMed] [Google Scholar]
- (202).Sivaramakrishnan S; Ouellet H; Matsumura H; Guan S; Moenne-Loccoz P; Burlingame AL; Ortiz de Montellano PR Proximal Ligand Electron Donation and Rreactivity of the Cytochrome P450 Ferric-Peroxo Anion. J. Am. Chem. Soc 2012, 134, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Sohl CD; Guengerich FP Kinetic Analysis of the Three-Step Steroid Aromatase Reaction of Human Cytochrome P450 19A1. J. Biol. Chem 2010, 285, 17734–17743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Pallan PS; Nagy LD; Lei L; Gonzalez E; Kramlinger VM; Azumaya CM; Wawrzak Z; Waterman MR; Guengerich FP; Egli M Structural and Kinetic Basis of Steroid 17α,20-Lyase Activity in Teleost Fish Cytochrome P450 17A1 and Its Absence in Cytochrome P450 17A2. J. Biol. Chem 2015, 290, 3248–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Gonzalez E; Guengerich FP Kinetic Processivity of the Two-Step Oxidations of Progesterone and Pregnenolone to Androgens by Human Cytochrome P450 17A1. J. Biol. Chem 2017, 292, 13168–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Ryan KJ Biological Aromatization of Steroids. J. Biol. Chem 1959, 234, 268–272. [PubMed] [Google Scholar]
- (207).Brodie AMH Aromatase Inhibition and Its Pharmacologic Implications. Biochem. Pharmacol 1985, 34, 3213–3219. [DOI] [PubMed] [Google Scholar]
- (208).Chumsri S; Howes T; Bao T; Sabnis G; Brodie A Aromatase, Aromatase Inhibitors, and Breast Cancer. J. Steroid Biochem. Mol. Biol 2011, 125, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Goto J; Fishman J Participation of a Nonenzymatic Transformation in the Biosynthesis of Estrogens from Androgens. Science 1977, 195, 80–81. [DOI] [PubMed] [Google Scholar]
- (210).Hosoda H; Fishman J Unusually Facile Aromatization of 2β-Hydroxy-19-oxo-4-androstene-3,17-dione to Estrone. Implications in Estrogen Biosynthesis. J. Am. Chem. Soc 1974, 96, 7325–7329. [DOI] [PubMed] [Google Scholar]
- (211).Hahn EF; Fishman J Immunological Probe of Estrogen Biosynthesis. Evidence for the 2β-Hydroxylative Pathway in Aromatization of Androgens. J. Biol. Chem 1984, 259, 1689–1694. [PubMed] [Google Scholar]
- (212).Fishman J Biochemical Mechanism of Aromatization. Cancer Res 1982, 42, 3277s–3280s. [PubMed] [Google Scholar]
- (213).Cole PA; Robinson CH Synthesis of and Reactivity Studies with 19-Peroxide-Androstenedione Derivatives: Analogues of a Proposed Aromatase Intermediate. J. Chem. Soc., Perk. Trans 1 1990, 2119–2125. [Google Scholar]
- (214).Cole PA; Robinson CH Conversion of 19-Oxo[2β−2H]androgens into Estrogens by Human Placental Aromatase. An Unexpected Stereochemical Outcome. Biochem. J 1990, 268, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Cole PA; Robinson CH Mechanistic Studies on a Placental Aromatase Model Reaction. J. Am. Chem. Soc 1991, 113, 8130–8137. [Google Scholar]
- (216).Cole PA; Robinson CH Mechanism and Inhibition of Cytochrome P-450 Aromatase. J. Med. Chem 1990, 33, 2933–2942. [DOI] [PubMed] [Google Scholar]
- (217).Cole PA; Robinson CH A Peroxide Model Reaction for Placental Aromatase. J. Am. Chem. Soc 1988, 110, 1284–1285. [Google Scholar]
- (218).Caspi E; Njar VC Concerning the Pathway from 19-Ooxoandrost-4-ene-3,17-dione to Estrone. Steroids 1987, 50, 347–362. [DOI] [PubMed] [Google Scholar]
- (219).Caspi E; Wicha J; Arunachalam T; Nelson P; Spiteller G Estrogen Biosynthesis: Concerning the Obligatory Intermediacy of 2β-Hydroxy-10β-formyl Androst-4-ene-3,17-dione. J. Am. Chem. Soc 1984, 106, 7282–7283. [Google Scholar]
- (220).Caspi E; Arunachalam T; Nelson PA Biosynthesis of Estrogens: Aromatization of (19R)-,(19S)-, and (19S)-[19–3H,2H,1H]-3β-Hydroxyandrost-5-en-17-ones by Human Placental Aromatase. J. Am. Chem. Soc 1986, 108, 1847–1852. [Google Scholar]
- (221).Akhtar M; Lee-Robichaud P; Akhtar ME; Wright JN The Impact of Aromatase Mechanism on Other P450s. J. Steroid Biochem. Mol. Biol 1997, 61, 127–132. [PubMed] [Google Scholar]
- (222).Akhtar M; Corina D; Pratt J; Smith T Studies on the Removal of C-19 in Oestrogen Biosynthesis using 18O2. J. Chem. Soc., Chem. Commun 1976, 854–856. [Google Scholar]
- (223).Akhtar M; Calder MR; Corina DL; Wright JN Mechanistic Studies on C-19 Demethylation in Oestrogen Biosynthesis. Biochem. J 1982, 201, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Khatri Y; Luthra A; Duggal R; Sligar SG Kinetic Solvent Isotope Effect in Steady-State Turnover by CYP19A1 Suggests Involvement of Compound 1 for Both Hydroxylation and Aromatization steps. FEBS Lett 2014, 588, 3117–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Mak PJ; Luthra A; Sligar SG; Kincaid JR Resonance Raman Spectroscopy of the Oxygenated Intermediates of Human CYP19A1 Implicates a Compound I Intermediate in the Final Lyase Step. J. Am. Chem. Soc 2014, 136, 4825–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Hackett JC; Brueggemeier RW; Hadad CM The Final Catalytic Step of Cytochrome P450 Aromatase: A Density Functional Theory Study. J. Am. Chem. Soc 2005, 127, 5224–5237. [DOI] [PubMed] [Google Scholar]
- (227).Sen K; Hackett JC Coupled Electron Transfer and Proton Hopping in the Final Step of CYP19-catalyzed Androgen Aromatization. Biochemistry 2012, 51, 3039–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Guengerich FP; Sohl CD; Chowdhury G Multi-step Oxidations Catalyzed by Cytochrome P450 Enzymes: Processive vs. Distributive Kinetics and the Issue of Carbonyl Oxidation in Chemical Mechanisms. Arch. Biochem. Biophys 2011, 507, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Akhtar M; Wright JN; Lee-Robichaud P A Review of Mechanistic Studies on Aromatase (CYP19) and 17α-Hydroxylase-17,20-lyase (CYP17). J. Steroid Biochem. Mol. Biol 2011, 125, 2–12. [DOI] [PubMed] [Google Scholar]
- (230).Korzekwa KR; Trager WF; Mancewicz J; Osawa Y Studies on the Mechanism of Aromatase and Other Cytochrome P450 Mediated Deformylation Reactions. J. Steroid Biochem. Mol. Biol 1993, 44, 367–373. [DOI] [PubMed] [Google Scholar]
- (231).Oh SS; Robinson CH Mechanism of Human Placental Aromatase: A New Active Site Model. J. Steroid Biochem. Mol. Biol 1993, 44, 389–397. [DOI] [PubMed] [Google Scholar]
- (232).Xu K; Wang Y; Hirao H Estrogen Formation via H-Abstraction from the O–H Bond of gem-Diol by Compound I in the Reaction of CYP19A1: Mechanistic Scenario Derived from Multiscale QM/MM Calculations. ACS Catalysis 2015, 5, 4175–4179. [Google Scholar]
- (233).Oh SS; Robinson CH Synthesis of and Chemical Model Reaction Studies with 3-Deoxyandrogens: Evidence Supporting a 2,3-Enolization Hypothesis in Human Placental Aromatase Catalyasis. J. Chem. Soc. Perk. Tran 1 1994, 2237–2243. [Google Scholar]
- (234).Corina DL Determination of the 18O Content of Formic Acid as Benzyl Formate. Anal. Biochem 1977, 80, 639–642. [DOI] [PubMed] [Google Scholar]
- (235).Gantt SL; Denisov IG; Grinkova YV; Sligar SG The Critical Iron-Oxygen Intermediate in Human Aromatase. Biochem. Biophys. Res. Commun 2009, 387, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Johnson KA INtroduction to Kinetic Analysis of Enzyme Systems. In Kinetic Analysis of Macromolecules. A Practical Approach; Johnson KA, Ed.; Oxford University Press: Oxford, UK, 2003; pp. 1–18. [Google Scholar]
- (237).Yoshimoto FK; Auchus RJ The Diverse Chemistry of Cytochrome P450 17A1 (P450c17, CYP17A1). J. Steroid Biochem. Mol. Biol 2014, 151, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Yoshimoto FK; Gonzalez E; Auchus RJ; Guengerich FP Mechanism of 17α,20-Lyase and New Hydroxylation Reactions of Human Cytochrome P450 17A1: 18O Labeling and Oxygen Surrogate Evidence for a Role of a Perferryl Oxygen. J. Biol. Chem 2016, 291, 17143–17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Gonzalez E; Johnson KM; Pallan PS; Phan TTN; Zhang W; Lei L; Wawrzak Z; Yoshimoto FK; Egli M; Guengerich FP Inherent Steroid 17α,20-Lyase Activity in Defunct Cytochrome P450 17A Enzymes. J. Biol. Chem 2018, 293, 541–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Scott LJ Abiraterone Acetate: A Review in Metastatic Castration-Resistant Prostrate Cancer. Drugs 2017, 77, 1565–1576. [DOI] [PubMed] [Google Scholar]
- (241).Stein MN; Goodin S; Dipaola RS Abiraterone in Prostate Cancer: A New Angle to an Old Problem. Clin. Cancer Res 2012, 18, 1848–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Yang LP Abiraterone Acetate: In Metastatic Castration-Resistant Prostate Cancer. Drugs 2011, 71, 2067–2077. [DOI] [PubMed] [Google Scholar]
- (243).Mak AY; Swinney DC 17-O-Acetyltestosterone Formation from Progesterone in Microsomes from Pig Testes.Evidence for the Baeyer-Villiger Rearrangement in Androgen Formation Catalyzed by Cyp17. J. Am. Chem. Soc 1992, 114, 8309–8310. [Google Scholar]
- (244).Swinney DC; Mak AY Androgen Formation by Cytochrome P450 CYP17. Solvent Isotope Effect and pL Studies Suggest a Role for Protons in the Regulation of Oxene versus Peroxide Chemistry. Biochemistry 1994, 33, 2185–2190. [DOI] [PubMed] [Google Scholar]
- (245).Miller SL; Wright JN; Corina DL; Akhtar M Mechanistic Studies on Pregnene Side-Chain Cleavage Enzyme (17α-Hydroxylase-17,20-Lyase) Using 18O. J. Chem. Soc., Chem. Commun 1991, 157–159. [Google Scholar]
- (246).Gregory MC; Denisov IG; Grinkova YV; Khatri Y; Sligar SG Kinetic Solvent Isotope Effect in Human P450 CYP17A1-Mediated Androgen Formation: Evidence for a Reactive Peroxoanion Intermediate. J. Am. Chem. Soc 2013, 135, 16245–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Guengerich FP Kinetic Deuterium Isotope Effects in Cytochrome P450 Enzyme Reactions. Methods Enzymol 2017, 596, 217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).DeVore NM; Scott EE Structures of Cytochrome P450 17A1 with Prostate Cancer Drugs Abiraterone and TOK-001. Nature 2012, 482, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Petrunak EM; DeVore NM; Porubsky PR; Scott EE Structures of Human Steroidogenic Cytochrome P450 17A1 with Substrates. J. Biol. Chem 2014, 289, 32952–32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Gregory M; Mak PJ; Sligar SG; Kincaid JR Differential Hydrogen Bonding in Human CYP17 Dictates Hydroxylation versus Lyase Chemistry. Angew. Chem. Int. Ed 2013, 52, 5342–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Oloo WN; Meier KK; Wang Y; Shaik S; Munck E; Que L Identification of a Low-spin Acylperoxoiron(III) Intermediate in Bio-inspired Non-Heme Iron-Catalysed Oxidations. Nat. Commun 2014, 5, 3046. [DOI] [PubMed] [Google Scholar]
- (252).Davydov R; Strushkevich N; Smil D; Yantsevich A; Gilep A; Usanov S; Hoffman BM Evidence That Compound I Is the Active Species in Both the Hydroxylase and Lyase Steps by Which P450scc Converts Cholesterol to Pregnenolone: EPR/ENDOR/Cryoreduction/Annealing Studies. Biochemistry 2015, 54, 7089–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Auchus RJ; Miller WL Molecular Modeling of Human P450c17 (17a-hydroxylase/17,20-lyase): Insights into Reaction Mechanisms and Effects of Mutations. Mol Endocrinol 1999, 13, 1169–1182. [DOI] [PubMed] [Google Scholar]
- (254).Ortiz de Montellano PR Oxygen Activation and Reactivity. In Cytochrome P450: Structure, Mechanism, and Biochemistry; 2 ed.; Ortiz de Montellano PR, Ed.; Plenum Press: New York, 1995; pp. 245–303. [Google Scholar]
- (255).Tan L; Falardeau P; Rousseau J Evidence of an Enzymatic 17α leads to 21 Hydroxyl Transfer in the Biosynthesis of Cortexolone from 17α-Hydroperoxyprogesterone by Adrenal Cortex Microsomes. Hormone Res 1975, 6, 213–225. [DOI] [PubMed] [Google Scholar]
- (256).Tan L; Rousseau J Anaerobic Degradation of the Progesterone Side Chain: A Possible New Pathway for the Biosynthesis of Androgen Hormones. Biochem. Biophys. Res. Commun 1975, 65, 1320–1326. [DOI] [PubMed] [Google Scholar]
- (257).Bonomo S; Jørgensen FS; Olsen L Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J. Chem. Info. Model 2017, 57, 1123–1133. [DOI] [PubMed] [Google Scholar]
- (258).Geller DH; Auchus RJ; Mendonça BB; Miller WL The Genetic and Functional Basis of Isolated 17,20-Lyase Deficiency. Nat. Genetics 1997, 17, 201–205. [DOI] [PubMed] [Google Scholar]
- (259).Peng HM; Liu J; Forsberg SE; Tran HT; Anderson SM; Auchus RJ Catalytically Relevant Electrostatic Interactions of Cytochrome P450c17 (CYP17A1) and Cytochrome b5. J. Biol. Chem 2014, 289, 33838–33849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Miller WL; Geller DH; Auchus RJ The Molecular Basis of Isolated 17,20 Lyase Deficiency. Endocr. Res 1998, 24, 817–825. [DOI] [PubMed] [Google Scholar]
- (261).Auchus RJ Steroid 17-Hydroxylase and 17,20-Lyase Deficiencies, Genetic and Pharmacologic. J. Steroid Biochem. Mol. Biol 2017, 165, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Kaku T; Hitaka T; Ojida A; Matsunaga N; Adachi M; Tanaka T; Hara T; Yamaoka M; Kusaka M; Okuda T et al. Discovery of Orteronel (TAK-700), a Naphthylmethylimidazole Derivative, as a Highly Selective 17,20-Lyase Inhibitor with Potential Utility in the Treatment of Prostate Cancer. Bioorg. Med. Chem 2011, 19, 6383–6399. [DOI] [PubMed] [Google Scholar]
- (263).Bonomo S; Hansen CH; Petrunak EM; Scott EE; Styrishave B; Jørgensen FS; Olsen L Promising Tools in Prostate Cancer Res.: Selective Non-Steroidal Cytochrome P450 17A1 Inhibitors. Sci. Rep 2016, 6, 29468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Petrunak EM; Rogers SA; Aube J; Scott EE Structural and Functional Evaluation of Clinically Relevant Inhibitors of Steroidogenic Cytochrome P450 17A1. Drug Metab. Dispos 2017, 45, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Ortiz de Montellano PR Oxygen ACtivation and Transfer. In Cytochrome P-450; Ortiz de Montellano PR, Ed.; Plenum Press: New York, 1986; pp. 217–271. [Google Scholar]
- (266).Ortiz De Montellano PR; De Voss JJ Substrate Oxidation by Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; 3rd ed.; Ortiz de Montellano PR, Ed.; Plenum Publishers: New York, 2005; pp. 183–245. [Google Scholar]
- (267).Yoshimoto FK; Jung IJ; Goyal S; Gonzalez E; Guengerich FP Isotope-Labeling Studies Support the Electrophilic Compound I Iron Active Species, FeO3+, for the Carbon-Carbon Bond Cleavage Reaction of the Cholesterol Side-Chain Cleavage Enzyme, Cytochrome P450 11A1. J. Am. Chem. Soc 2016, 138, 12124–12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Davydov R; Gilep AA; Strushkevich NV; Usanov SA; Hoffman BM Compound I Is the Reactive Intermediate in the First Monooxygenation Step during Conversion of Cholesterol to Pregnenolone by Cytochrome P450scc: EPR/ENDOR/Cryoreduction/Annealing Studies. J. Am. Chem. Soc 2012, 134, 17149–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Greenzaid P; Luz Z; Samuel D A Nuclear Magnetic Resonance Study of the Reversible Hydration of Aliphatic Aldehydes and Ketones. I. Oxygen-17 and Proton Spectra and Equilibrium Constants. J. Am. Chem. Soc 1967, 89, 749–755. [Google Scholar]
- (270).Hitchcock CA; Brown SB; Evans EGV; Adams DJ Cytochrome P-450-dependent 14α-Demethylation of Lanosterol in Candida albicans. Biochem. J 1989, 260, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Rozman D; Waterman MR Lanosterol 14α-demethylase (CYP51) and Spermatogenesis. Drug Metab. Dispos 1998, 26, 1199–1201. [PubMed] [Google Scholar]
- (272).Keber R; Motaln H; Wagner KD; Debeljak N; Rassoulzadegan M; Acimovic J; Rozman D; Horvat S Mouse Knockout of the Cholesterogenic Cytochrome P450 Lanosterol 14α-Demethylase (Cyp51) Resembles Antley-Bixler Syndrome. J. Biol. Chem 2011, 286, 29086–29097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Fischer RT; Trzaskos JM; Magolda RL; Ko SS; Brosz CS; Larsen B Lanosterol 14α-Methyl Demethylase: Isolation and Ccharacterization of the Third Metabolically Generated Oxidative Demethylation Intermediate. J. Biol. Chem 1991, 266, 6124–6132. [PubMed] [Google Scholar]
- (274).Shyadehi AZ; Lamb DC; Kelly SL; Kelly DE; Schunck WH; Wright JN; Corina D; Akhtar M Mechanism of the Acyl-Carbon Bond Cleavage Reaction Catalyzed by Recombinant Sterol 14α-Demethylase of Candida albicans (Other Names are: Lanosterol 14α-Demethylase, P-45014DM, and CYP51). J. Biol. Chem 1996, 271, 12445–12450. [DOI] [PubMed] [Google Scholar]
- (275).Schmid SE; Au WYW; Hill DE; Kadlubar FF; Slikker W Jr. Cytochrome P-450-Dependent Oxidation of the 17α-Ethynyl Group of Synthetic Steroids: D-Homoannulation or Enzyme Inactivation. Drug Metab. Dispos 1983, 11, 531–536. [PubMed] [Google Scholar]
- (276).Guengerich FP Mechanism-based Inactivation of Human Liver Microsomal Cytochrome P-450 IIIA4 by Gestodene. Chem. Res. Toxicol 1990, 3, 363–371. [DOI] [PubMed] [Google Scholar]
- (277).Guengerich FP Inhibition of Oral Contraceptive Steroid-Metabolizing Enzymes by Steroids and Drugs. Am. J. Obstetrics Gynecol 1990, 163, 2159–2163. [DOI] [PubMed] [Google Scholar]
- (278).Williams JG; Williams KI Metabolism of 2–3H- and 4–14C-17α-ethynylestradiol 3-Methyl Ether (Mestranol) by Women. Steroids 1975, 26, 707–720. [DOI] [PubMed] [Google Scholar]
- (279).Abdel-Aziz MT; Williams KI Metabolism of 17-α-Ethynylestradiol and Its 3-Methyl Ether by the Rabbit; An in vivo D-Homoannulation. Steroids 1969, 13, 809–820. [DOI] [PubMed] [Google Scholar]
- (280).Sisenwine SF; Kimmel HB; Liu AL; Ruelius HW The Metabolic Disposition of Norgestrel in Female Rhesus Monkeys. Drug Metab. Dispos 1979, 7, 1–6. [PubMed] [Google Scholar]
- (281).Mutlib A; Chen H; Schockcor J; Espina R; Chen S; Cao K; Du A; Nemeth G; Prakash S; Gan LS Characterization of Novel Glutathione Adducts of a Non-nucleoside Reverse Transcriptase Inhibitor, (S)-6-Chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-3,4-dih ydro-2(1H)-quinazolinone (DPC 961), in Rats. Possible Formation of an Oxirene Metabolic Intermediate from a Disubstituted Alkyne. Chem. Res. Toxicol 2000, 13, 775–784. [DOI] [PubMed] [Google Scholar]
- (282).Jones G; Prosser DE; Kaufmann M 25-Hydroxyvitamin Δ24-Hydroxylase (CYP24A1): Its Important Role in the Degradation of Vitamin D. Arch. Biochem. Biophys 2012, 523, 9–18. [DOI] [PubMed] [Google Scholar]
- (283).Beckman MJ; Tadikonda P; Werner E; Prahl J; Yamada S; Deluca HF Human 25-Hydroxyvitamin D3-24-Hhydroxylase, a Multicatalytic Enzyme. Biochemistry 1996, Vol 35, 8465–8472. [DOI] [PubMed] [Google Scholar]
- (284).Reddy GS; Tserng KY; Thomas BR; Dayal R; Norman AW Isolation and Identification of 1,23-Dihydroxy-24,25,26,27-tetranorvitamin D3, a New Metabolite of 1,25-Dihydroxyvitamin D3 Produced in Rat Kidney. Biochemistry 1987, 26, 324–331. [DOI] [PubMed] [Google Scholar]
- (285).Reddy GS; Tserng KY Calcitroic Acid, End Product of Renal Metabolism of 1,25-Dihydroxyvitamin D3 Through the C-24 Oxidation Pathway. Biochemistry 1989, 28, 1763–1769. [DOI] [PubMed] [Google Scholar]
- (286).Makin G; Lohnes D; Byford V; Ray R; Jones G Target Cell Metabolism of 1,25-Dihydroxyvitamin D3 to Calcitroic Acid. Evidence for a Pathway in Kidney and Bone Involving 24-oxidation. Biochem. J 1989, 262, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Amunom I; Dieter LJ; Tamasi V; Cai J; Conklin DJ; Srivastava S; Martin MV; Guengerich FP; Prough RA Cytochromes P450s Catalyze the Reduction of α,β-Unsaturated Aldehydes. Chem. Res. Toxicol 2011, 24, 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Nomura T; Kushiro T; Yokota T; Kamiya Y; Bishop GJ; Yamaguchi S The Last Reaction Producing Brassinolide is Catalyzed by Cytochrome P-450s, CYP85A3 in Tomato and CYP85A2 in Arabidopsis. J. Biol. Chem 2005, 280, 17873–17879. [DOI] [PubMed] [Google Scholar]
- (289).Kim TW; Hwang JY; Kim YS; Joo SH; Chang SC; Lee JS; Takatsuto S; Kim SK Arabidopsis CYP85A2, a Cytochrome P450, Mediates the Baeyer-Villiger Oxidation of Castasterone to Brassinolide in Brassinosteroid Biosynthesis. Plant Cell 2005, 17, 2397–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Crombie L; Morgan DO Synthesis of [14,14–2H2]-Linolenic acid and Its Use to Confirm the Pathway to 12-Oxophytodienoic Acid (12-oxoPDA) in Plants: A Conspectus of the Epoxycarbonium Ion Derived Family of Metabolites from Linoleic and Linolenic Acid Hydroperoxides. J. Chem. Soc. Perkin Trans 1 1991, 581–587. [Google Scholar]
- (291).Itoh A; Howe GA Molecular Cloning of a Divinyl Ether Synthase. Identification as a CYP74 Cytochrome P-450. J. Biol. Chem 2001, 276, 3620–3627. [DOI] [PubMed] [Google Scholar]
- (292).Grechkin AN; Bruhlmann F; Mukhtarova LS; Gogolev YV; Hamberg M Hydroperoxide Lyases (CYP74C and CYP74B) Catalyze the Homolytic Isomerization of Fatty Acid Hydroperoxides into Hemiacetals. Biochim. Biophys. Acta 2006, 1761, 1419–1428. [DOI] [PubMed] [Google Scholar]
- (293).Guengerich FP Analysis and Characterization of Enzymes and Nucleic Acids Relevant to Toxicology. In Hayes’ Principles and Methods of Toxicology; 6th ed.; Hayes AW; Kruger CL, Eds.; CRC Press-Taylor & Francis Boca Raton, FL, 2014; pp. 1905–1964. [Google Scholar]
- (294).Guengerich FP Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol 2008, 21, 70–83. [DOI] [PubMed] [Google Scholar]
- (295).Henschler D; Bonse G Metabolic Activation of Chlorinated Ethylenes: Dependence of Mutagenic Effect on Electrophilic Reactivity of the Metabolically Formed Epoxides. Arch. Toxicol 1977, 39, 7–12. [DOI] [PubMed] [Google Scholar]
- (296).Miller RE; Guengerich FP Oxidation of Trichloroethylene by Liver Microsomal Cytochrome P-450: Evidence for Chlorine Migration in a Transition State Not Involving Trichloroethylene Oxide. Biochemistry 1982, 21, 1090–1097. [DOI] [PubMed] [Google Scholar]
- (297).Henschler D; Hoos WR; Fetz H; Dallmeier E; Metzler M Reactions of Trichloroethylene Epoxide In Aqueous Systems. Biochem. Pharmacol 1979, 28, 543–548. [DOI] [PubMed] [Google Scholar]
- (298).Cai H; Guengerich FP Mechanism of Aqueous Decomposition of Trichloroethylene Oxide. J. Am. Chem. Soc 1999, 121, 11656–11663. [Google Scholar]
- (299).Yoshioka T; Krauser JA; Guengerich FP Tetrachloroethylene Oxide: Hydrolytic Products and Reactions with Phosphate and Lysine. Chem. Res. Toxicol 2002, 15, 1096–1105. [DOI] [PubMed] [Google Scholar]
- (300).Yoshioka T; Krauser JA; Guengerich FP Microsomal Oxidation of Tribromoethylene and Reactions of Tribromoethylene Oxide. Chem. Res. Toxicol 2002, 15, 1414–1420. [DOI] [PubMed] [Google Scholar]
- (301).Liebler DC; Guengerich FP Olefin Oxidation by Cytochrome P-450: Evidence for Group Migration in Catalytic Intermediates Formed with Vinylidene Chloride and trans-1-Phenyl-1-butene. Biochemistry 1983, 22, 5482–5489. [DOI] [PubMed] [Google Scholar]
- (302).Ohe T; Mashino T; Hirobe M Novel Metabolic Pathway of Arylethers by Cytochrome P450: Cleavage of the Oxygen-Aromatic Ring Bond Accompanying ipso-Substitution by the Oxygen Atom of the Active Species in Cytochrome P450 Models and Cytochrome P450. Arch. Biochem. Biophys 1994, 310, 402–409. [DOI] [PubMed] [Google Scholar]
- (303).Rietjens IMCM; den Besten C; Hanzlik RP; van Bladeren PJ Cytochrome P450-Catalyzed Oxidation of Halobenzene Derivatives. Chem. Res. Toxicol 1997, 10, 629–635. [DOI] [PubMed] [Google Scholar]
- (304).Kolvenbach B; Schlaich N; Raoui Z; Prell J; Zuhlke S; Schaffer A; Guengerich FP; Corvini PF Degradation Pathway of Bisphenol A: Does ipso Substitution Apply to Phenols Containing a Quaternary α-Carbon Structure in the para Position? Appl. Environ. Microbiol 2007, 73, 4776–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Gabriel FL; Cyris M; Giger W; Kohler HP ipso-Substitution: A General Biochemical and Biodegradation Mechanism to Cleave α-Quaternary Alkylphenols and Bisphenol A. Chem. Biodiversity 2007, 4, 2123–2137. [DOI] [PubMed] [Google Scholar]
- (306).Maeda Y; Ingold KU Kinetic Applications of Electron Paramagnetic Resonance Spectroscopy. 35. The Search for Dialkylaminyl Rearrangement. Ring Opening of N-Cyclobutyl-N-n-propylaminyl. J. Am. Chem. Soc 1980, 102, 328–331. [Google Scholar]
- (307).Griller D; Ingold KU Free-radical Clocks. Acct. Chem. Res 1980, 13, 317–323. [Google Scholar]
- (308).Macdonald TL; Zirvi K; Burka LT; Peyman P; Guengerich FP Mechanism of Cytochrome P-450 Inhibition by Cyclopropylamines. J. Am. Chem. Soc 1982, 104, 2050–2052. [Google Scholar]
- (309).Guengerich FP; Willard RJ; Shea JP; Richards LE; Macdonald TL Mechanism-based Inactivation of Cytochrome P-450 by Heteroatom-Substituted Cyclopropanes and Formation of Ring-opened Products. J. Am. Chem. Soc 1984, 106, 6446–6447. [Google Scholar]
- (310).Augusto O; Beilan HS; Ortiz de Montellano PR The Catalytic Mechanism of Cytochrome P-450: Spin-trapping Evidence for One-electron Substrate Oxidation. J. Biol. Chem 1982, 257, 11288–11295. [PubMed] [Google Scholar]
- (311).Ortiz de Montellano PR; Beilan HS; Kunze KL N-Methylprotoporphyrin IX: Chemical Synthesis and Identification as the Green Pigment Produced by 3,5-Diethoxycarbonyl-1,4-dihydrocollidine Treatment. Proc. Natl. Acad. Sci. U. S. A 1981, 78, 1490–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Böcker RH; Guengerich FP Oxidation of 4-Aryl- and 4-Alkyl-substituted 2,6-Dimethyl-3,5-bis(alkoxycarbonyl)-1,4-dihydropyridines by Human Liver Microsomes and Immunochemical Evidence for the Involvement of a Form of Cytochrome P-450. J. Med. Chem 1986, 29, 1596–1603. [DOI] [PubMed] [Google Scholar]
- (313).Guengerich FP; Bocker RH Cytochrome P-450-catalyzed Dehydrogenation of 1,4-Dihydropyridines. J. Biol. Chem 1988, 263, 8168–8175. [PubMed] [Google Scholar]
- (314).Bondon A; Macdonald TL; Harris TM; Guengerich FP Oxidation of Cycloalkylamines by Cytochrome P-450. Mechanism-based Inactivation, Adduct Formation, Ring Expansion, and Nitrone Formation. J. Biol. Chem 1989, 264, 1988–1997. [PubMed] [Google Scholar]
- (315).Stearns RA; Ortiz de Montellano PR Cytochrome P-450 Catalyzed Oxidation of Quadricyclane. Evidence for a Radical Cation Intermediate. J. Am. Chem. Soc 1985, 107, 4081–4082. [Google Scholar]
- (316).Jiang Y; He X; Ortiz de Montellano PR Radical Intermediates in the Catalytic Oxidation of Hydrocarbons by Bacterial and Human Cytochrome P450 Enzymes. Biochemistry 2006, 45, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Cryle MJ; Ortiz de Montellano PR; De Voss JJ Cyclopropyl Containing Fatty Acids as Mechanistic Probes for Cytochromes P450. J. Org. Chem 2005, 70, 2455–2469. [DOI] [PubMed] [Google Scholar]
- (318).Stok JE; De Voss J Expression, Purification, and Characterization of BioI: A Carbon-carbon Bond Cleaving Cytochrome P450 Involved in Biotin Biosynthesis in Bacillus subtilis. Arch. Biochem. Biophys 2000, 384, 351–360. [DOI] [PubMed] [Google Scholar]
- (319).Cryle MJ; Schlichting I Structural Insights from a P450 Carrier Protein Complex Reveal how Specificity is Achieved in the P450(BioI) ACP complex. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 15696–15701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Cryle MJ; De Voss JJ Carbon-carbon Bond Cleavage by Cytochrome P450 (BioI) (CYP107H1). J. Chem. Soc., Chem. Commun 2004, 86–87. [DOI] [PubMed] [Google Scholar]
- (321).Belcher J; McLean KJ; Matthews S; Woodward LS; Fisher K; Rigby SE; Nelson DR; Potts D; Baynham MT; Parker DA et al. Structure and Biochemical Properties of the Alkene Producing Cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J. Biol. Chem 2014, 289, 6535–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Hsieh CH; Huang XY; Amaya JA; Rutland CD; Keys CL; Groves JT; Austin RN; Makris TM The Enigmatic P450 Decarboxylase OleT Is Capable of, but Evolved To Frustrate, Oxygen Rebound Chemistry. Biochemistry 2017, 56, 3347–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Amaya JA; Rutland CD; Leschinsky N; Makris TM A Distal Loop Controls Product Release and Chemo- and Regioselectivity in Cytochrome P450 Decarboxylases. Biochemistry 2018, 57, 344–353. [DOI] [PubMed] [Google Scholar]
- (324).Grant JL; Mitchell ME; Makris TM Catalytic Strategy for Carbon-carbon Bond Scission by the Cytochrome P450 OleT. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 10049–10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Vasilou V; Petersen DR Aldehyde Dehydrogenases. In Biotransformation; 2 ed.; Guengerich FP, Ed.; Elsevier: Oxford, U.K., 2010; Vol. 4; pp. 131–147. [Google Scholar]
- (326).Roberts ES; Vaz ADN; Coon MJ Catalysis by Cytochrome P-450 of an Oxidative Reaction in Xenobiotic Aldehyde Metabolism: Deformylation with Olefin Formation. Proc. Natl. Acad. Sci. U. S. A 1991, 88, 8963–8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Reed JR; Quilici DR; Blomquist GJ; Reitz RC Proposed Mechanism for the Cytochrome P450-catalyzed Conversion of Aldehydes to Hydrocarbons in the House Fly, Musca domestica. Biochemistry 1995, 34, 16221–16227. [DOI] [PubMed] [Google Scholar]
- (328).Qiu Y; Tittiger C; Wicker-Thomas C; Le Goff G; Young S; Wajnberg E; Fricaux T; Taquet N; Blomquist GJ; Feyereisen R An Insect-specific P450 Oxidative Decarbonylase for Cuticular Hydrocarbon Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 14858–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Ortiz de Montellano PR; De Voss JJ Oxidizing Species in the Mechanism of Cytochromes P450. Nat. Prod. Rep 2002, 19, 477–493. [DOI] [PubMed] [Google Scholar]
- (330).Chowdhury G; Calcutt MW; Guengerich FP Oxidation of N-Nitrosoalkylamines by Human Cytochrome P450 2A6: Sequential Oxidation to Aldehydes and Carboxylic Acids and Analysis of Reaction Steps. J. Biol. Chem 2010, 285, 8031–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Chowdhury G; Calcutt MW; Nagy LD; Guengerich FP Oxidation of Methyl and Ethyl Nitrosamines by Cytochrome P450 2E1 and 2B1. Biochemistry 2012, 51, 9995–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Varfaj F; Zulkifli SNA; Park H-G; Challinor VL; De Voss JJ; de Montellano PRO Carbon-Carbon Bond Cleavage in Activation of the Prodrug Nabumetone. Drug Metab. Dispos 2014, 42, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Turpeinen M; Hofmann U; Klein K; Murdter T; Schwab M; Zanger UM A Predominate Role of CYP1A2 for the Metabolism of Nabumetone to the Active Metabolite, 6-Methoxy-2-naphthylacetic acid, in Human Liver Microsomes. Drug Metab. Dispos 2009, 37, 1017–1024. [DOI] [PubMed] [Google Scholar]
- (334).Brandon EF; van Ooijen RD; Sparidans RW; Lazaro LL; Heck AJ; Beijnen JH; Schellens JH Structure Elucidation of Aplidine Metabolites Formed in vitro by Human Liver Microsomes using Triple Quadrupole Mass Spectrometry. J. Mass Spectrom 2005, 40, 821–831. [DOI] [PubMed] [Google Scholar]
- (335).Brandon EF; Sparidans RW; van Ooijen RD; Meijerman I; Lazaro LL; Manzanares I; Beijnen JH; Schellens JH In vitro Characterization of the Human Biotransformation Pathways of Aplidine, a Novel Marine Anti-Cancer Drug. Investig. New Drugs 2007, 25, 9–19. [DOI] [PubMed] [Google Scholar]
- (336).Guengerich FP; Isin EM Unusual Metabolic Reactions and Pathways. In Handbook of Metabolic Pathways of Xenobiotics; Lee P; Aizawa H; Gau L; Prakash C; Zhong D, Eds.; John Wiley & Sons: Chichester, UK, 2014; Vol. 1; pp. 147–197. [Google Scholar]
- (337).Yoo HH; Chung HJ; Lee J; Lee CS; Kang MJ; Kim DH Enzymatic C-Demethylation of 1-[2-(5-tert-Butyl-[1,3,4] oxadiazole-2-carbonyl)-4-fluoro-pyrrolidin-1-yl]-2-(2-hydroxy-1,1-dimethyl-ethylamino)-ethanone (LC15–0133) in Rat Liver Microsomes. Drug Metab. Dispos 2008, 36, 485–489. [DOI] [PubMed] [Google Scholar]
- (338).Prakash C; Wang W; O’Connell T; Johnson KA CYP2C8- and CYP3A-mediated C-Demethylation of (3-{[(4-tert-Butylbenzyl)-(pyridine-3-sulfonyl)-amino]-methyl}-phenoxy)-ac etic acid (CP-533,536), an EP2 Receptor-selective Prostaglandin E2 Agonist: Characterization of Metabolites by High-resolution Liquid Chromatography-Tandem Mass Spectrometry and Liquid Chromatography/Mass Spectrometry-Nuclear Magnetic Resonance. Drug Metab. Dispos 2008, 36, 2093–2103. [DOI] [PubMed] [Google Scholar]
- (339).Dalvie DK; Kalgutkar AS; Khojasteh-Bakht SC; Obach RS; O’Donnell JP Biotransformation Rreactions of Five-membered Aromatic Heterocyclic Rings. Chem. Res. Toxicol 2002, 15, 269–299. [DOI] [PubMed] [Google Scholar]
- (340).Marchetti E; Mattalia G; Bergesi G Ditazol Metabolism in Rat, Rabbit and Man. Arzneimittel-Forschung 1973, 23, 1291–1295. [PubMed] [Google Scholar]
- (341).Maurer H; Kleff I On the Metabolism of Ditazole in Mman. Arzneimittel-Forschung 1988, 38, 1843–1845. [PubMed] [Google Scholar]
- (342).Isin EM; Guengerich FP Complex Reactions Catalyzed by Cytochrome P450 Enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [DOI] [PubMed] [Google Scholar]
- (343).Helliwell CA; Chandler PM; Poole A; Dennis ES; Peacock WJ The CYP88A Cytochrome P450, ent-Kaurenoic Acid Oxidase, Catalyzes Three Steps of the Gibberellin Biosynthesis Pathway. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 2065–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Rojas MC; Hedden P; Gaskin P; Tudzynki B The P450–1 gene of Gibberella fujikuroi Encodes a Multifunctional Enzyme in Gibberellin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 5838–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Lee S; Badieyan S; Bevan DR; Herde M; Gatz C; Tholl D Herbivore-induced and Floral Homoterpene Volatiles are Biosynthesized by a Single P450 Enzyme (CYP82G1) in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 21205–21210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Stanjek V; Miksch M; Lueer P; Matern U; Boland W Biosynthesis of Psoralen: Mechanism of a Cytochrome P450 Catalyzed Oxidative Bond Cleavage. Angew. Chem. Int. Ed 1999, 38, 400–402. [DOI] [PubMed] [Google Scholar]
- (347).Larbat R; Kellner S; Specker S; Hehn A; Gontier E; Hans J; Bourgaud F; Matern U Molecular Cloning and Functional Characterization of Psoralen Synthase, the First Committed Monooxygenase of Furanocoumarin Biosynthesis. J. Biol. Chem 2007, 282, 542–554. [DOI] [PubMed] [Google Scholar]
- (348).Larbat R; Hehn A; Hans J; Schneider S; Jugdé H; Schneider B; Matern U; Bourgaud F Isolation and Functional Characterization of CYP71AJ4 Encoding for the First P450 Monooxygenase of Angular Furanocoumarin Biosynthesis. J. Biol. Chem 2009, 284, 4776–4785. [DOI] [PubMed] [Google Scholar]
- (349).Lin H-C; Tsunematsu Y; Dhingra S; Xu W; Fukutomi M; Chooi Y-H; Cane DE; Calvo AM; Watanabe K; Tang Y Generation of Complexity in Fungal Terpene Biosynthesis: Discovery of a Multifunctional Cytochrome P450 in the Fumagillin Pathway. J. Am. Chem. Soc 2014, 136, 4426–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Zhu D; Seo M-J; Ikeda H; Cane DE Genome Mining in Streptomyces. Discovery of an Unprecedented P450-Catalyzed Oxidative Rearrangement That Is the Final Step in the Biosynthesis of Pentalenolactone. J. Am. Chem. Soc 2011, 133, 2128–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Rontein D; Onillon S; Herbette G; Lesot A; Werck-Reichhart D; Sallaud C; Tissier A CYP725A4 from Yew Catalyzes Complex Structural Rearrangement of Taxa-4(5),11(12)-diene into the Cyclic Ether 5(12)-Oxa-3(11)-cyclotaxane. J. Biol. Chem 2008, 283, 6067–6075. [DOI] [PubMed] [Google Scholar]
- (352).Yamamoto H; Katano N; Ooi A; Inoue K Secologanin Synthase Which Catalyzes the Oxidative Cleavage of Loganin into Secologanin is a Cytochrome P450. Phytochemistry 2000, 53, 7–12. [DOI] [PubMed] [Google Scholar]
- (353).Arunachalam U; Massey V Studies on the Oxidative Half-reaction of p-Hydroxyphenylacetate 3-Hydroxylase. J. Biol. Chem 1994, 269, 11795–11801. [PubMed] [Google Scholar]
- (354).Ziegler DM Recent Studies on the Structure and Function of Multisubstrate Flavin-containing Monooxygenases. Annu. Rev. Pharmacol. Toxicol 1993, 33, 179–199. [DOI] [PubMed] [Google Scholar]
- (355).Yu H; Hausinger RP; Tang HZ; Xu P Mechanism of the 6-Hydroxy-3-succinoyl-pyridine 3-Monooxygenase Flavoprotein from Pseudomonas putida S16. J. Biol. Chem 2014, 289, 29158–29170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Walsh CT; Chen YCJ Enzymic Baeyer-Villiger Oxidations by Flavin-dependent Monooxygenases. Angew. Chem. Int. Ed 1988, 27, 333–343. [Google Scholar]
- (357).Fiorentini F; Geier M; Binda C; Winkler M; Faber K; Hall M; Mattevi A Biocatalytic Characterization of Human FMO5: Unearthing Baeyer-Villiger Reactions in Humans. ACS Chem. Biol 2016, 11, 1039–1048. [DOI] [PubMed] [Google Scholar]
- (358).Hu Y; Dietrich D; Xu W; Patel A; Thuss JAJ; Wang J; Yin W-B; Qiao K; Houk KN; Vederas JC et al. A Carbonate-forming Baeyer-Villiger Monooxygenase. Nat. Chem. Biol 2014, 10, 552–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (359).Ziegler DM; Mitchell CH Microsomal Oxidase IV: Properties of a Mixed-function Amine Oxidase Isolated from Pig Liver Microsomes. Arch. Biochem. Biophys 1972, 150, 116–125. [DOI] [PubMed] [Google Scholar]
- (360).Fiorentini F; Romero E; Fraaije MW; Faber K; Hall M; Mattevi A Baeyer-Villiger Monooxygenase FMO5 as Entry Point in Drug Metabolism. ACS Chem. Biol 2017, 12, 2379–2387. [DOI] [PubMed] [Google Scholar]
- (361).Lai WG; Farah N; Moniz GA; Wong YN A Baeyer-Villiger Oxidation Specifically Catalyzed by Human Flavin-containing Monooxygenase 5. Drug Metab. Dispos 2011, 39, 61–70. [DOI] [PubMed] [Google Scholar]
- (362).Meng J; Zhong D; Li L; Yuan Z; Yuan H; Xie C; Zhou J; Li C; Gordeev MF; Liu J et al. Metabolism of MRX-I, a Novel Antibacterial Oxazolidinone, in Humans: The Oxidative Ring Opening of 2,3-Dihydropyridin-4-one catalyzed by non-P450 enzymes. Drug Metab. Dispos 2015, 43, 646–659. [DOI] [PubMed] [Google Scholar]
- (363).Sparrow LG; Ho PPK; Sundaram TK; Zach D; Nyns EJ; Snell EE The Bacterial Oxidation of Vitamin B6 : VII. Purification, Properties, and Mechanism of Action of an Oxygenase which Cleaves the 3-Hydroxypyridine Ring. J. Biol. Chem 1969, 244, 2590–2600. [PubMed] [Google Scholar]
- (364).Nelson MJK; Snell EE Enzymes of Vitamin B6 Degradation: Purification and Properties of 5-Pyridoxic Acid Oxygenase from Arthrobacter sp. J. Biol. Chem 1986, 261, 15115–15120. [PubMed] [Google Scholar]
- (365).Chaiyen P; Brissette P; Ballou DP; Massey V Unusual Mechanism of Oxygen Atom Transfer and Product Rearrangement in the Catalytic Reaction of 2-Methyl-3-hydroxypyridine-5-carboxylic Acid Oxygenase. Biochemistry 1997, 36, 8060–8070. [DOI] [PubMed] [Google Scholar]
- (366).Powlowski J; Ballou DP; Massey V Studies of the Oxidative Half-reaction of Anthranilate Hydroxylase (Deaminating) with Native and Modified Substrates. J. Biol. Chem 1990, 265, 4969–4975. [PubMed] [Google Scholar]
- (367).Entsch B; van Berkel WJH Structure and Mechanism of para-Hydroxybenzoate Hydroxylase. FASEB J 1995, 9, 476–483. [DOI] [PubMed] [Google Scholar]
- (368).Wessiak A; Bruice TC On the Nature of the Intermediate between 4a-Hydroperoxyflavin and 4a-Hydroxyflavin in the Hydroxylation Reaction of p-Hydroxybenzoate Hydroxylase. Synthesis of 6-Aminopyrimidine-2,4,5(3H)-triones and the Mechanism of Aromatic Hydroxylation by Flavin Monooxygenases. J. Am. Chem. Soc 1981, 103, 6996–6998. [Google Scholar]
- (369).Marshall SA; Payne KAP; Leys D The UbiX-UbiD system: The Biosynthesis and Use of Prenylated Flavin (prFMN). Arch. Biochem. Biophys 2017, 632, 209–221. [DOI] [PubMed] [Google Scholar]
- (370).Bailey SS; Payne KAP; Fisher K; Marshall SA; Cliff MJ; Spiess R; Parker DA; Rigby SEJ; Leys D The Role of Conserved Residues in Fdc Decarboxylase in Prenylated Flavin Mononucleotide Oxidative Maturation, Cofactor Isomerization, and Catalysis. J. Biol. Chem 2018, 293, 2272–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Arunrattanamook N; Marsh ENG Kinetic Characterization of Prenyl-Flavin Synthase from Saccharomyces cerevisiae. Biochemistry 2018, 57, 696–700. [DOI] [PubMed] [Google Scholar]
- (372).Ewing TA; Fraaije MW; Mattevi A; van Berkel WJH The VAO/PCMH Flavoprotein Family. Arch. Biochem. Biophys 2017, 632, 104–117. [DOI] [PubMed] [Google Scholar]
- (373).Brady SF; Chao CJ; Clardy J New Natural Product Families from an Environmental DNA (eDNA) Gene Cluster. J. Am. Chem. Soc 2002, 124, 9968–9969. [DOI] [PubMed] [Google Scholar]
- (374).Rachid S; Revermann O; Dauth C; Kazmaier U; Müller R Characterization of a Novel Type of Oxidative Decarboxylase Involved in the Biosynthesis of the Styryl Moiety of Chondrochloren from an Acylated Tyrosine. J. Biol. Chem 2010, 285, 12482–12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Mathews CK; van Holde KE Fatty ACcid Oxidation: The β-Oxidation Pathway. In Biochemistry; Benjamin/Cummings: Redwood City, CA, 1990, pp. 580–583. [Google Scholar]
- (376).Bai Y; McCoy JG; Levin EJ; Sobrado P; Rajashankar KR; Fox BG; Zhou M X-ray Structure of a Mammalian Stearoyl-CoA Desaturase. Nature 2015, 524, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Rettie AE; Rettenmeier AW; Howald WN; Baillie TA Cytochrome P-450 Catalyzed Formation of δ4-VPA, a Toxic Metabolite of Valproic Acid. Science 1987, 235, 890–893. [DOI] [PubMed] [Google Scholar]
- (378).Kramlinger VM; Nagy LD; Fujiwara R; Johnson KM; Phan TT; Xiao Y; Enright JM; Toomey MB; Corbo JC; Guengerich FP Human Cytochrome P450 27C1 Catalyzes 3,4-Desaturation of Retinoids. FEBS Lett 2016, 590, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Cooper HL; Mishra G; Huang X; Pender-Cudlip M; Austin RN; Shanklin J; Groves JT Parallel and Competitive Pathways for Substrate Desaturation, Hydroxylation, and Radical Rearrangement by the Non-heme Diiron Hydroxylase AlkB. J. Am. Chem. Soc 2012, 134, 20365–20375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Shanklin J; Guy JE; Mishra G; Lindqvist Y Desaturases: Emerging Models for Understanding Functional Diversification of Diiron-containing Enzymes. J. Biol. Chem 2009, 284, 18559–18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Guy JE; Abreu IA; Moche M; Lindqvist Y; Whittle E; Shanklin J A Single Mutation in the Castor Δ9-18:0-Desaturase Changes Reaction Partitioning from Desaturation to Oxidase Chemistry. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17220–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Broadwater JA; Whittle E; Shanklin J Desaturation and Hydroxylation. Residues 148 and 324 of Arabidopsis FAD2, in Addition to Substrate Chain Length, Exert a Major Influence in Partitioning of Catalytic Specificity. J. Biol. Chem 2002, 277, 15613–15620. [DOI] [PubMed] [Google Scholar]
- (383).Coffey R; Ganz T Iron Homeostasis: An Anthropocentric Perspective. J. Biol. Chem 2017, 292, 12727–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Schultz IJ; Chen C; Paw BH; Hamza I Iron and Porphyrin Trafficking in Heme Biogenesis. J. Biol. Chem 2010, 285, 26753–26759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Zhang AS; Enns CA Iron Homeostasis: Recently Identified Proteins Provide Insight into Novel Control Mechanisms. J. Biol. Chem 2009, 284, 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Masters BSS; Schacter BA The Catalysis of Heme Degradation by Purified NADPH-Cytochrome c Reductase in the Absence of Other Microsomal Proteins. Ann. Clin. Res 1976, 8, 18–27. [PubMed] [Google Scholar]
- (387).Guengerich FP; Strickland TW Metabolism of Vinyl Chloride: Destruction of the Heme of Highly Purified Liver Microsomal Cytochrome P-450 by a Metabolite. Mol. Pharmacol 1977, 13, 993–1004. [PubMed] [Google Scholar]
- (388).Levin W; Lu AYH; Jacobson M; Kuntzman R Lipid Peroxidation and the Degradation of Cytochrome P-450 Heme. Arch. Biochem. Biophys 1973, 158, 842–852. [DOI] [PubMed] [Google Scholar]
- (389).Guengerich FP Destruction of Heme and Hemoproteins Mediated by Liver Microsomal Reduced Nicotinamide Adenine Dinucleotide Phosphate-Cytochrome P-450 Reductase. Biochemistry 1978, 17, 3633–3639. [DOI] [PubMed] [Google Scholar]
- (390).Thurman RG; Chance B Inhibition of Catalase in Perfused Rat Liver by Sodium Azide. Ann. New York Acad. Sci 1969, 168, 348–353. [DOI] [PubMed] [Google Scholar]
- (391).Beers RF Jr.; Sizer IW Progressive Inhibition of the Catalase-Hydrogen Peroxide System by Acetate, Chloride and Azide. Arch. Biochem. Biophys 1956, 60, 115–125. [DOI] [PubMed] [Google Scholar]
- (392).Schaefer WH; Harris TM; Guengerich FP Characterization of the Enzymatic and Nonenzymatic Peroxidative Degradation of Iron Porphyrins and Cytochrome P-450 Heme. Biochemistry 1985, 24, 3254–3263. [DOI] [PubMed] [Google Scholar]
- (393).Fischer H; Müller A Über die Pentdyopent-reaktion. I. Mitteilung. Z. Physiol. Chem 1937, 246, 43–58. [Google Scholar]
- (394).He K; Bornheim LM; Falick AM; Maltby D; Yin H; Correia MA Identification of the Heme-modified Peptides from Cumene Hydroperoxide-Inactivated Cytochrome P450 3A4. Biochemistry 1998, 37, 17448–17457. [DOI] [PubMed] [Google Scholar]
- (395).Bissell DM; Guzelian PS Degradation of Endogenous Hepatic Heme by Pathways Not Yielding Carbon Monoxide: Studies in Normal Rat Liver and in Primary Hepatocyte Culture. J. Clin. Invest 1980, 65, 1135–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Meunier B; Robert A Heme as Trigger and Target for Trioxane-containing Antimalarial Drugs. Acct. Chem. Res 2010, 43, 1444–1451. [DOI] [PubMed] [Google Scholar]
- (397).Matsui T; Unno M; Ikeda-Saito M Heme Oxygenase Reveals its Strategy for Catalyzing Three Successive Oxygenation Reactions. Acct. Chem. Res 2010, 43, 240–247. [DOI] [PubMed] [Google Scholar]
- (398).Trakshel GM; Maines MD Multiplicity of Heme Oxygenase Isozymes: HO-1 and HO-2 are Different Molecular Species in Rat and Rabbit. J. Biol. Chem 1989, 264, 1323–1328. [PubMed] [Google Scholar]
- (399).Noguchi M; Yoshida T; Kikuchi G Purification and Properties of Biliverdin Reductases from Pig Spleen and Rat Liver. J. Biochem. (Tokyo) 1979, 86, 833–848. [DOI] [PubMed] [Google Scholar]
- (400).Gordon ER; Goresky CA; Chang TH; Perlin AS The Isolation and Characterization of Bilirubin Diglucuronide, the Major Bilirubin Conjugate in Dog and Human Bile. Biochem. J 1976, 155, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Bock KW; Kohle C Contributions of the Ah Receptor to Bilirubin Homeostasis and its Antioxidative and Atheroprotective Functions. Biol. Chem 2010, 391, 645–653. [DOI] [PubMed] [Google Scholar]
- (402).Zimmerman HJ The Differential Diagnosis of Jaundice. Med. Clin. North Am 1968, 52, 1417–1444. [PubMed] [Google Scholar]
- (403).Verma A; Hirsch DJ; Glatt CE; Ronnett GV; Snyder SH Carbon Monoxide: A Putative Neural Messenger. Science 1993, 259, 381–384. [DOI] [PubMed] [Google Scholar]
- (404).Morita T; Perrella MA; Lee ME; Kourembanas S Smooth Muscle Cell-Derived Carbon Monoxide is a Regulator of Vascular cGMP. Proc. Natl. Acad. Sci. U. S. A 1995, 92, 1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Marks GS Heme Oxygenase: the Physiological Role of One of Its Metabolites, Carbon Monoxide and Interactions with Zinc Protoporphyrin, Cobalt Protoporphyrin and Other Metalloporphyrins. Cell. Mol. Biol 1994, 40, 863–870. [PubMed] [Google Scholar]
- (406).Matsui T; Iwasaki M; Sugiyama R; Unno M; Ikeda-Saito M Dioxygen Activation for the Self-Degradation of Heme: Reaction Mechanism and Regulation of Heme Oxygenase. Inorg. Chem 2010, 49, 3602–3609. [DOI] [PubMed] [Google Scholar]
- (407).Masters BSS; Nelson EB; Schacter BA; Baron J; Isaacson EL NADPH-Cytochrome c Reductase and Its Role in Microsomal Cytochrome P-450-Dependent Reactions. Drug Metab. Dispos 1973, 1, 121–128. [PubMed] [Google Scholar]
- (408).Davydov R; Fleischhacker AS; Bagai I; Hoffman BM; Ragsdale SW Comparison of the Mechanisms of Heme Hydroxylation by Heme Oxygenases-1 and −2: Kinetic and Cryoreduction Studies. Biochemistry 2016, 55, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Ortiz de Montellano PR The Mechanism of Heme Oxygenase. Curr. Opin. Chem. Biol 2000, 4, 221–227. [DOI] [PubMed] [Google Scholar]
- (410).Liu Y; Ortiz de Montellano PR Reaction Intermediates and Single Turnover Rate Constants for the Oxidation of Heme by Human Heme Oxygenase-1. J. Biol. Chem 2000, 275, 5297–5307. [DOI] [PubMed] [Google Scholar]
- (411).Davydov RM; Yoshida T; Ikeda-Saito M; Hoffman BM Hydroperoxy-Heme Oxygenase Generated by Cryoreduction Catalyzes the Formation of α-meso-Hydroxyheme as Detected by EPR and ENDOR. J. Am. Chem. Soc 1999, 121, 10656–10657. [Google Scholar]
- (412).Lad L; Wang J; Li H; Friedman J; Bhaskar B; Ortiz de Montellano PR; Poulos TL Crystal Structures of the Ferric, Ferrous, and Ferrous-NO Forms of the Asp140Ala Mutant of Human Heme Oxygenase-1: Catalytic Implications. J. Mol. Biol 2003, 330, 527–538. [DOI] [PubMed] [Google Scholar]
- (413).Wilks A; Heinzl G Heme Oxygenation and the Widening Paradigm of Heme Degradation. Arch. Biochem. Biophys 2014, 544, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Docherty JC; Schacter BA; Firneisz GD; Brown SB Mechanism of Action of Heme Oxygenase: A Study of Heme Degradation to Bile Pigment by 18O labeling. J. Biol. Chem 1984, 259, 13066–13069. [PubMed] [Google Scholar]
- (415).Matsui T; Nakajima A; Fujii H; Matera KM; Migita CT; Yoshida T; Ikeda-Saito M O2- and H2O2-Dependent Verdoheme Degradation by Heme Oxygenase: Reaction Mechanisms and Potential Physiological Roles of the Dual Pathway Degradation. J. Biol. Chem 2005, 280, 36833–36840. [DOI] [PubMed] [Google Scholar]
- (416).Uchida T; Sekine Y; Matsui T; Ikeda-Saito M; Ishimori K A Heme Degradation Enzyme, HutZ, from Vibrio cholerae. J. Chem. Soc., Chem. Commun 2012, 48, 6741–6743. [DOI] [PubMed] [Google Scholar]
- (417).Celis AI; Streit BR; Moraski GC; Kant R; Lash TD; Lukat-Rodgers GS; Rodgers KR; DuBois JL Unusual Peroxide-Dependent, Heme-Transforming Reaction Catalyzed by HemQ. Biochemistry 2015, 54, 4022–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Streit BR; Celis AI; Shisler K; Rodgers KR; Lukat-Rodgers GS; DuBois JL Reactions of Ferrous Coproheme Decarboxylase (HemQ) with O2 and H2O2 Yield Ferric Heme b. Biochemistry 2017, 56, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Milazzo L; Hofbauer S; Howes BD; Gabler T; Furtmuller PG; Obinger C; Smulevich G Insights into the Active Site of Coproheme Decarboxylase from Listeria monocytogenes. Biochemistry 2018, 57, 2044–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Breckau D; Mahlitz E; Sauerwald A; Layer G; Jahn D Oxygen-dependent Coproporphyrinogen III Oxidase (HemF) from Escherichia coli Is Stimulated by Manganese. J. Biol. Chem 2003, 278, 46625–46631. [DOI] [PubMed] [Google Scholar]
- (421).Nambu S; Matsui T; Goulding CW; Takahashi S; Ikeda-Saito M A New Way to Degrade Heme: The Mycobacterium tuberculosis enzyme MhuD Catalyzes Heme Degradation Without Generating CO. J. Biol. Chem 2013, 288, 10101–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Matsui T; Nambu S; Goulding CW; Takahashi S; Fujii H; Ikeda-Saito M Unique Coupling of Mono- and Dioxygenase Chemistries in a Single Active Site Promotes Heme Degradation. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 3779–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Matsui T; Nambu S; Ono Y; Goulding CW; Tsumoto K; Ikeda-Saito M Heme Degradation by Staphylococcus aureus IsdG and IsdI Liberates Formaldehyde Rather than Carbon Monoxide. Biochemistry 2013, 52, 3025–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Buis JM; Cheek J; Kalliri E; Broderick JB Characterization of an Active Spore Photoproduct Lyase, a DNA Repair Enzyme in the Radical S-Adenosylmethionine Superfamily. J. Biol. Chem 2006, 281, 25994–26003. [DOI] [PubMed] [Google Scholar]
- (425).Boss L; Oehme R; Billig S; Birkemeyer C; Layer G The Radical SAM Enzyme NirJ Catalyzes the Removal of Two Propionate Side Chains During Heme d1 Biosynthesis. FEBS J 2017, 284, 4314–4327. [DOI] [PubMed] [Google Scholar]
- (426).Khaliullin B; Ayikpoe R; Tuttle M; Latham JA Mechanistic Elucidation of the Mycofactocin-Biosynthetic Radical S-Adenosylmethionine Protein, MftC. J. Biol. Chem 2017, 292, 13022–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Bruender NA; Bandarian V The Radical S-Adenosyl-L-methionine Enzyme MftC Catalyzes an Oxidative Decarboxylation of the C-Terminus of the MftA Peptide. Biochemistry 2016, 55, 2813–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Wolf G The Enzymatic Cleavage of β-Carotene: End of a Controversy. Nutr. Rev 2001, 59, 116–118. [DOI] [PubMed] [Google Scholar]
- (429).dela Sena C; Riedl KM; Narayanasamy S; Curley RW Jr.; Schwartz SJ; Harrison EH The Human Enzyme That Converts Dietary Provitamin A Carotenoids to Vitamin A Is a Dioxygenase. J. Biol. Chem 2014, 289, 13661–13666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Kiefer C; Hessel S; Lampert JM; Vogt K; Lederer MO; Breithaupt DE; von Lintig J Identification and Characterization of a Mammalian Enzyme Catalyzing the Asymmetric Oxidative Cleavage of Provitamin A. J. Biol. Chem 2001, 276, 14110–14116. [DOI] [PubMed] [Google Scholar]
- (431).Poliakov E; Gentleman S; Cunningham FX; Miller-Ihli NJ; Redmond TM Key Role of Conserved Histidines in Recombinant Mouse β-Carotene 15,15′–Monooxygenase-1 Activity. J. Biol. Chem 2005, 280, 29217–29223. [DOI] [PubMed] [Google Scholar]
- (432).Hu K-Q; Liu C; Ernst H; Krinsky NI; Russell RM; Wang X-D The Biochemical Characterization of Ferret Carotene-9′, 10′-Monooxygenase Catalyzing Cleavage of Carotenoids in Vitro and in Vivo. J. Biol. Chem 2006, 281, 19327–19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Sui X; Golczak M; Zhang J; Kleinberg KA; von Lintig J; Palczewski K; Kiser PD Utilization of Dioxygen by Carotenoid Cleavage Oxygenases. J. Biol. Chem 2015, 290, 30212–30223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Amengual J; Widjaja-Adhi MAK; Rodriguez-Santiago S; Hessel S; Golczak M; Palczewski K; von Lintig J Two Carotenoid Oxygenases Contribute to Mammalian Provitamin A Metabolism. J. Biol. Chem 2013, 288, 34081–34096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Harrison PJ; Bugg TDH Enzymology of the Carotenoid Cleavage Dioxygenases: Reaction Mechanisms, Inhibition and Biochemical Roles. Arch. Biochem. Biophys 2014, 544, 105–111. [DOI] [PubMed] [Google Scholar]
- (436).Kim Y-S; Kim N-H; Yeom S-J; Kim S-W; Oh D-K In Vitro Characterization of a Recombinant Blh Protein from an Uncultured Marine Bacterium as a β-Carotene 15,15′-Dioxygenase. J. Biol. Chem 2009, 284, 15781–15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).McAndrew RP; Sathitsuksanoh N; Mbughuni MM; Heins RA; Pereira JH; George A; Sale KL; Fox BG; Simmons BA; Adams PD Structure and Mechanism of NOV1, a Resveratrol-cleaving Dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 14324–14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Brefort T; Scherzinger D; Limon MC; Estrada AF; Trautmann D; Mengel C; Avalos J; Al-Babili S Cleavage of Resveratrol in Fungi: Characterization of the Enzyme Rco1 from Ustilago maydis. Fungal Genetics Biol 2011, 48, 132–143. [DOI] [PubMed] [Google Scholar]
- (439).Vogel JT; Tan BC; McCarty DR; Klee HJ The Carotenoid Cleavage Dioxygenase 1 Enzyme has Broad Substrate Specificity, Cleaving Multiple Carotenoids at Two Different Bond Positions. J. Biol. Chem 2008, 283, 11364–11373. [DOI] [PubMed] [Google Scholar]
- (440).dela Seña C; Narayanasamy S; Riedl KM; Curley RW; Schwartz SJ; Harrison EH Substrate Specificity of Purified Recombinant Human β-Carotene 15,15′-Oxygenase (BCO1). J. Biol. Chem 2013, 288, 37094–37103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).dela Sena C; Sun J; Narayanasamy S; Riedl KM; Yuan Y; Curley RW; Schwartz SJ; Harrison EH Substrate Specificity of Purified Recombinant Chicken Carotene 9,10-Oxygenase (BCO2). J. Biol. Chem 2016, 291, 14609–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Millett ES; Efimov I; Basran J; Handa S; Mowat CG; Raven EL Heme-containing Dioxygenases Involved in Tryptophan Oxidation. Curr. Opin. Chem. Biol 2012, 16, 60–66. [DOI] [PubMed] [Google Scholar]
- (443).Basran J; Booth ES; Lee M; Handa S; Raven EL Analysis of Reaction Intermediates in Tryptophan 2,3-Dioxygenase: A Comparison with Indoleamine 2,3-Dioxygenase. Biochemistry 2016, 55, 6743–6750. [DOI] [PubMed] [Google Scholar]
- (444).Efimov I; Basran J; Thackray SJ; Handa S; Mowat CG; Raven EL Structure and Reaction Mechanism in the Heme Dioxygenases. Biochemistry 2011, 50, 2717–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Booth ES; Basran J; Lee M; Handa S; Raven EL Substrate Oxidation by Indoleamine 2,3-Dioxygenase: Evidence for a Common Reaction Mechanism. J. Biol. Chem 2015, 290, 30924–30930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Jerina DM; Daly JW; Witkop B; Zaltzman-Nirenberg P; Udenfriend S 1,2-Naphthalene Oxide as an Intermediate in the Microsomal Hydroxylation of Naphthalene. Biochemistry 1970, 9, 147–156. [DOI] [PubMed] [Google Scholar]
- (447).Stanier RY; Ingraham JL Protocatechuic Acid Oxidase. J. Biol. Chem 1954, 210, 799–808. [PubMed] [Google Scholar]
- (448).Hayaishi O Crystalline Oxygenases of Pseudomonads. Bacteriol. Rev 1966, 30, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Acheson JF; Bailey LJ; Brunold TC; Fox BG In-crystal Reaction Cycle of a Toluene-bound Diiron Hydroxylase. Nature 2017, 544, 191–195. [DOI] [PubMed] [Google Scholar]
- (450).Kovaleva EG; Lipscomb JD Crystal structures of Fe2+ Dioxygenase Superoxo, Alkylperoxo, and Bound Product Intermediates. Science 2007, 316, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Kovaleva EG; Lipscomb JD Intermediate in the O-O Bond Cleavage Reaction of an Extradiol Dioxygenase. Biochemistry 2008, 47, 11168–11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Groce SL; Lipscomb JD Aromatic Ring Cleavage by Homoprotocatechuate 2,3-Dioxygenase: Role of His200 in the Kinetics of Interconversion of Reaction Cycle Intermediates. Biochemistry 2005, 44, 7175–7188. [DOI] [PubMed] [Google Scholar]
- (453).Mbughuni MM; Chakrabarti M; Hayden JA; Meier KK; Dalluge JJ; Hendrich MP; Munck E; Lipscomb JD Oxy Intermediates of Homoprotocatechuate 2,3-Dioxygenase: Facile Electron Transfer between Substrates. Biochemistry 2011, 50, 10262–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Mbughuni MM; Meier KK; Munck E; Lipscomb JD Substrate-Mediated Oxygen Activation by Homoprotocatechuate 2,3-Dioxygenase: Intermediates Formed by a Tyrosine 257 Variant. Biochemistry 2012, 51, 8743–8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Olson JA Jr.; Lindberg M; Bloch K On the Demethylation of Lanosterol to Cholesterol. J. Biol. Chem 1957, 226, 941–956. [PubMed] [Google Scholar]
- (456).Gautschi F; Bloch K On the Structure of an Intermediate in the Biological Demethylation of Lanosterol. J. Am. Chem. Soc 1957, 79, 684–689. [Google Scholar]
- (457).Fukushima H; Grinstead GF; Gaylor JL Total Enzymic Synthesis of Cholesterol from Lanosterol. Cytochrome b5-dependence of 4-Methyl Sterol Oxidase. J. Biol. Chem 1981, 256, 4822–4826. [PubMed] [Google Scholar]
- (458).Gachotte D; Sen SE; Eckstein J; Barbuch R; Krieger M; Ray BD; Bard M Characterization of the Saccharomyces cerevisiae ERG27 Gene Encoding the 3-Keto Reductase Involved in C-4 Sterol Demethylation. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 12655–12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Li L; Kaplan J Characterization of Yeast Methyl Sterol Oxidase (ERG25) and Identification of a Human Homologue. J. Biol. Chem 1996, 271, 16927–16933. [DOI] [PubMed] [Google Scholar]
- (460).Rahier A Dissecting the Sterol C-4 Demethylation Process in Higher Plants. From Structures and Genes to Catalytic Mechanism. Steroids 2011, 76, 340–352. [DOI] [PubMed] [Google Scholar]
- (461).Porter TD Electron Transfer Pathways in Cholesterol Synthesis. Lipids 2015, 50, 927–936. [DOI] [PubMed] [Google Scholar]
- (462).He M; Smith LD; Chang R; Li X; Vockley J The role of Sterol-C4-Methyl Oxidase in Epidermal Biology. Biochim. Biophys. Acta 2014, 1841, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Lindberg M; Gautschi F; Bloch K Ketonic Intermediates in the Demethylation of Lanosterol. J. Biol. Chem 1963, 238, 1661–1664. [PubMed] [Google Scholar]
- (464).Lv JM; Hu D; Gao H; Kushiro T; Awakawa T; Chen GD; Wang CX; Abe I; Yao XS Biosynthesis of Helvolic Acid and Identification of an Unusual C-4-Demethylation Process Distinct from Sterol Biosynthesis. Nat. Commun 2017, 8, 1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Hagel JM; Facchini PJ Dioxygenases Catalyze the O-Demethylation Steps of Morphine Biosynthesis in Opium Poppy. Nat. Chem. Biol 2010, 6, 273–275. [DOI] [PubMed] [Google Scholar]
- (466).Martinez S; Hausinger RP Catalytic Mechanisms of FeII-and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Martinez S; Fellner M; Herr CQ; Ritchie A; Hu J; Hausinger RP Structures and Mechanisms of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme: Substrate Binding Creates a Twist. J. Am. Chem. Soc 2017, 139, 11980–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Matsuda Y; Awakawa T; Wakimoto T; Abe I Spiro-Ring Formation is Catalyzed by a Multifunctional Dioxygenase in Austinol Biosynthesis. J. Am. Chem. Soc 2013, 135, 10962–10965. [DOI] [PubMed] [Google Scholar]
- (469).Cox RJ; Al-Fahad A Chemical Mechanisms Involved during the Biosynthesis of Tropolones. Curr. Opin. Chem. Biol 2013, 17, 532–536. [DOI] [PubMed] [Google Scholar]
- (470).Jakubczyk D; Caputi L; Hatsch A; Nielsen CA; Diefenbacher M; Klein J; Molt A; Schroder H; Cheng JZ; Naesby M et al. Discovery and Reconstitution of the Cycloclavine Biosynthetic Pathway-Enzymatic Formation of a Cyclopropyl Group. Angew. Chem. Int. Ed 2015, 127, 5206–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Ward JL; Gaskin P; Brown RGS; Jackson GS; Hedden P; Phillips AL; Willis CL; Beale MH Probing the Mechanism of Loss of Carbon-20 in Gibberellin Biosynthesis. Synthesis of Gibberellin 3α,20-Hemiacetal and 19,20-Lactol Analogues and Their Metabolism by a Recombinant GA 20-Oxidase. J. Chem. Soc., Perk Trans 1 2002, 232–241. [Google Scholar]
- (472).Matsuda Y; Wakimoto T; Mori T; Awakawa T; Abe I Complete Biosynthetic Pathway of Anditomin: Nature’s Sophisticated Synthetic Route to a Complex Fungal Meroterpenoid. J. Am. Chem. Soc 2014, 136, 15326–15336. [DOI] [PubMed] [Google Scholar]
- (473).Bräuer A; Beck P; Hintermann L; Groll M Structure of the Dioxygenase AsqJ: Mechanistic Insights into a One-Pot Multistep Quinolone Antibiotic Biosynthesis. Angew. Chem. Int. Ed 2016, 55, 422–426. [DOI] [PubMed] [Google Scholar]
- (474).Mader SL; Brauer A; Groll M; Kaila VR I. Catalytic Mechanism and Molecular Engineering of Quinolone Biosynthesis in Dioxygenase AsqJ. Nat. Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Huang JL; Tang Y; Yu CP; Sanyal D; Jia X; Liu X; Guo Y; Chang WC Mechanistic Investigation of Oxidative Decarboxylation Catalyzed by Two IronII- and 2-Oxoglutarate-Dependent Enzymes. Biochemistry 2018, 57, 1838–1841. [DOI] [PubMed] [Google Scholar]
- (476).Zhang Z; Barlow JN; Baldwin JE; Schofield CJ Metal-catalyzed Oxidation and Mutagenesis Studies on the IronII binding site of 1-Aminocyclopropane-1-carboxylate Oxidase. Biochemistry 1997, 36, 15999–16007. [DOI] [PubMed] [Google Scholar]
- (477).Thrower J; Mirica LM; McCusker KP; Klinman JP Mechanistic Investigations of 1-Aminocyclopropane 1-Carboxylic Acid Oxidase with Alternate Cyclic and Acyclic Substrates. Biochemistry 2006, 45, 13108–13117. [DOI] [PubMed] [Google Scholar]
- (478).Merkens H; Kappl R; Jakob RP; Schmid FX; Fetzner S Quercetinase QueD of Streptomyces sp. FLA, a Monocupin Dioxygenase with a Preference for Nickel and Cobalt. Biochemistry 2008, 47, 12185–12196. [DOI] [PubMed] [Google Scholar]
- (479).Jeoung JH; Nianios D; Fetzner S; Dobbek H Quercetin 2,4-Dioxygenase Activates Dioxygen in a Side-On O2-Ni Complex. Angew. Chem. Int. Ed 2016, 55, 3281–3284. [DOI] [PubMed] [Google Scholar]
- (480).Oka T; Simpson FJ Quercetinase, a Dioxygenase Containing Copper. Biochem. Biophys. Res. Commun 1971, 43, 1–5. [DOI] [PubMed] [Google Scholar]
- (481).Brown SB; Rajananda V; Holroyd JA; Evans EG A Study of the Mechanism of Quercetin Oxygenation by 18O labelling. A Comparison of the Mechanism with That of Haem Degradation. Biochem. J 1982, 205, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Dai Y; Pochapsky TC; Abeles RH Mechanistic Studies of Two Dioxygenases in the Methionine Salvage Pathway of Klebsiella pneumoniae. Biochemistry 2001, 40, 6379–6387. [DOI] [PubMed] [Google Scholar]
- (483).Pollegioni L; Tonin F; Rosini E Lignin-degrading Enzymes. FEBS J 2015, 282, 1190–1213. [DOI] [PubMed] [Google Scholar]
- (484).Abdel-Hamid AM; Solbiati JO; Cann IK Insights into Lignin Degradation and Its Potential Industrial Applications. Adv. Appl. Microbiol 2013, 82, 1–28. [DOI] [PubMed] [Google Scholar]
- (485).Goodwin DC; Aust SD; Grover TA Evidence for Veratryl Alcohol as a Redox Mediator in Lignin Peroxidase-Catalyzed Oxidation. Biochemistry 1995, 34, 5060–5065. [DOI] [PubMed] [Google Scholar]
- (486).Khindaria A; Yamazaki I; Aust SD Veratryl Alcohol Oxidation by Lignin Peroxidase. Biochemistry 1995, 34, 16860–16869. [DOI] [PubMed] [Google Scholar]
- (487).Hammel KE; Tien M; Kalyanaraman B; Kirk TK Mechanism of Oxidative Cα-Cβ Cleavage of a Lignin Model Dimer by Phanerochaete chrysosporium Ligninase. Stoichiometry and Involvement of Free Radicals. J. Biol. Chem 1985, 260, 8348–8353. [PubMed] [Google Scholar]
- (488).Chung N; Aust SD Veratryl Alcohol-Mediated Indirect Oxidation of Pentachlorophenol by Lignin Peroxidase. Arch. Biochem. Biophys 1995, 322, 143–148. [DOI] [PubMed] [Google Scholar]
- (489).Khindaria A; Grover TA; Aust SD Evidence for Formation of the Veratryl Alcohol Cation Radical by Lignin Peroxidase. Biochemistry 1995, 34, 6020–6025. [DOI] [PubMed] [Google Scholar]
- (490).Hammel KE; Kalyanaraman B; Kirk TK Substrate Free Radicals are Intermediates in Ligninase Catalysis. Proc. Natl. Acad. Sci. U. S. A 1986, 83, 3708–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Candeias LP; Harvey PJ Lifetime and Reactivity of the Veratryl Alcohol Radical Cation: Implications for Lignin Peroxidase Catalysis. J. Biol. Chem 1995, 270, 16745–16748. [DOI] [PubMed] [Google Scholar]
- (492).Houtman CJ; Maligaspe E; Hunt CG; Fernandez-Fueyo E; Martinez AT; Hammel KE Fungal Lignin Peroxidase Does Not Produce the Veratryl Alcohol Cation Radical as a Diffusible Ligninolytic Oxidant. J. Biol. Chem 2018, 293, 4702–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Haemmerli SD; Leisola MSA; Fiechter A Polymerisation of Lignins by Ligninases from Phanerochaete chrysosporium. FEMS Microbiol. Lett 1986, 35, 33–36. [Google Scholar]
- (494).Bumpus JA; Tien M; Wright D; Aust SD Oxidation of Persistent Environmental Pollutants by a White Rot Fungus. Science 1985, 228, 1434–1436. [DOI] [PubMed] [Google Scholar]
- (495).Hammel KE; Kalyanaraman B; Kirk TK Oxidation of Polycyclic Aromatic Hydrocarbons and Dibenzo[p]dioxins by Phanerochaete chrysosporium Ligninase. J. Biol. Chem 1986, 261, 16948–16952. [PubMed] [Google Scholar]
- (496).Marnett LJ; Wlodawer P; Samuelsson B Co-oxygenation of Organic Substrates by the Prostaglandin Synthetase of Sheep Vesicular Gland. J. Biol. Chem 1975, 250, 8510–8517. [PubMed] [Google Scholar]
- (497).Cavalieri EL; Devanesan PD; Rogan EG Radical Cations in the Horseradish Peroxidase and Prostaglandin H Synthase Mediated Metabolism and Binding of Benzo[a]pyrene to Deoxyribonucleic Acid. Biochem. Pharmacol 1988, 37, 2183–2187. [DOI] [PubMed] [Google Scholar]
- (498).Anzenbacher P; Niwa T; Tolbert LM; Sirimanne SR; Guengerich FP Oxidation of 9-Alkylanthracenes by Cytochrome P450 2B1, Horseradish Peroxidase, and Iron Tetraphenylporphine/Iodosylbenzene Systems: Anaerobic and Aerobic Mechanisms. Biochemistry 1996, 35, 2512–2520. [DOI] [PubMed] [Google Scholar]
- (499).Cameron MD; Timofeevski S; Aust SD Enzymology of Phanerochaete chrysosporium with Respect to the Degradation of Recalcitrant Compounds and Xenobiotics. Appl. Microbiol. Biotechnol 2000, 54, 751. [DOI] [PubMed] [Google Scholar]
- (500).Sugano Y DyP-type Peroxidases Comprise a Novel Heme Peroxidase Family. Cell. Mol. Life Sci 2009, 66, 1387–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Yoshida T; Sugano Y A Structural and Functional Perspective of DyP-type Peroxidase Family. Arch. Biochem. Biophys 2015, 574, 49–55. [DOI] [PubMed] [Google Scholar]
- (502).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of Dioxygen by Copper Metalloproteins and Insights from Model Complexes. J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Bauerle MR; Schwalm EL; Booker SJ Mechanistic Diversity of Radical S-Adenosylmethionine (SAM)-dependent Methylation. J. Biol. Chem 2015, 290, 3995–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Lanz ND; Booker SJ Auxiliary Iron-sulfur Cofactors in Radical SAM Enzymes. Biochim. Biophys. Acta-Mol. Cell Res 2015, 1853, 1316–1334. [DOI] [PubMed] [Google Scholar]