

Abstract
Bacteria harbor an immense, untapped trove of novel secondary metabolites in the form of ‘silent’ biosynthetic gene clusters (BGCs). These can be identified bioinformatically but are not expressed under normal laboratory growth conditions. Methods to access their products would dramatically expand our pool of bioactive compounds. We report a universal high-throughput method for activating silent BGCs in diverse microorganisms. Our approach relies on elicitor screening to induce the secondary metabolome of a given strain and imaging mass spectrometry to visualize the resulting metabolomes in response to ~500 conditions. Because it does not require challenging genetic, cloning, or culturing procedures, it can be used with both sequenced and unsequenced bacteria. We demonstrate the power of the approach by applying it to diverse bacteria and report the discovery of nine cryptic metabolites with potentially therapeutic bioactivities, including a new glycopeptide chemotype with potent inhibitory activity against a pathogenic virus.
INTRODUCTION
Modern medicine is unimaginable without natural products. Predominantly isolated from microorganisms and plants, these molecules, also referred to as secondary metabolites, form the basis of >70% of antibiotics, >50% of anticancer agents, and overall, more than half of the drugs approved in the United States in the past 35 years1–3. After nearly a century of mining for secondary metabolites, microorganisms appeared to have become an exhausted resource. However, the recent explosion in microbial genome sequences points to a massive, untapped trove of new metabolites4–9. Specifically, members of several bacterial phyla typically harbor 25 or more biosynthetic gene clusters (BGCs) – sets of genes that direct the biosynthesis of a natural product – that are not actively, or only weakly, expressed under standard laboratory conditions. These so-called “silent” or “cryptic” BGCs outnumber the constitutively active ones by a factor of 5–10. As such, finding new methods that access their products could substantially enhance our repertoire of novel natural products and thereby accelerate and aid drug discovery.
The importance of inducing silent BGCs has been recognized by the research community and several approaches have been developed to identify and characterize their small molecule products, including expression of BGCs in a heterologous host, co-culture screening, ribosome engineering, insertion of constitutive or inducible promoters, reporter-guided mutant selection, and endogenous overexpression of regulatory proteins9–17. We added to this canon of approaches by developing high-throughput elicitor screening (HiTES)18–20, a strategy that identifies small molecule elicitors for a given silent BGC. While these methods have collectively begun to illuminate the hidden secondary metabolomes of bacteria, they typically necessitate challenging culturing conditions (i.e. co- or mixed-cultures), molecular biology procedures, and/or genetic manipulations, which slow down the pace and throughput of natural product discovery. A definitive method for accessing cryptic metabolites in varied microorganisms has yet to be developed.
Herein, we work toward that goal and report a genetics-free, endogenous, monoculture strategy for eliciting and detecting the cryptic secondary metabolomes of diverse bacteria. We use HiTES in conjunction with imaging mass spectrometry (IMS), a method we refer to as HiTES-IMS, to induce silent BGCs and to detect the resulting small molecules in a rapid and untargeted fashion. Computational methods are then employed to identify the desired cryptic metabolites as well as their elicitors. Rather than monitor one silent BGC at a time, HiTES-IMS allows us to interrogate the global secondary metabolome of any culturable bacterium in response to 500–1000 conditions. We use this approach in Gram-negative and Gram-positive bacteria and report nine cryptic metabolites, including a new post-translationally modified lasso peptide as well as a new glycopeptide chemotype that in in vitro assays is more potent than the currently used drug against the respiratory syncytial virus (RSV). Our approach is widely applicable to sequenced and unsequenced bacteria or bacterial consortia, and it promises to unearth the vast hidden metabolomes of microorganisms in hopes of expediting the search for new therapeutic agents from microbial sources.
RESULTS
Replacing Genetics with Imaging Mass Spectrometry.
The HiTES approach consists of two components, the activation of silent or lowly expressed BGCs by elicitor screening and a read-out for this process, which so far has relied on genetic reporter assays18,19. The detection step limits the throughput of HiTES, as creating the appropriate genetic constructs is often time-consuming, if not impossible, depending on the strain. In addressing this drawback, we were inspired by recent advances in MS technologies21 and envisioned replacing genetic reporter assays with IMS as a read-out for secondary metabolite production. The workflow would consist of subjecting the wild-type microorganism to elicitor screening followed by imaging the resulting 500–1000 metabolomes, as a function of each molecule in the library, using IMS (Fig. 1). Computational approaches and appropriate visualization would then be used to pinpoint cryptic metabolites.
Fig. 1. I HiTES-IMS workflow.
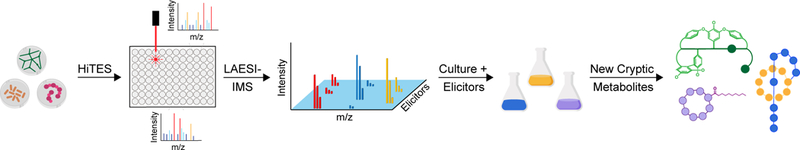
A bacterial culture is arrayed into 96-well plates and subjected to high-throughput elicitor screening. After a suitable incubation period, the cultures are assessed by LAESI-IMS in 96-well format. The observed global metabolome is depicted in a 3D plot that links each elicitor to metabolites, characterized by their m/z and MS intensity values. Large scale cultures with the appropriate elicitor facilitate isolation and characterization of new cryptic metabolites.
To implement this idea, we first selected a test case of the orfamides, sparingly produced metabolites isolated from Pseudomonas protegens Pf-5 (hereafter P. protegens). A genome-isotopic approach was previously used to identify them and ultimately to solve their structures22. As a proof-of-concept for our strategy, wild-type P. protegens was cultured in 96-well plates and subjected to HiTES using a 502-member natural products library. We analyzed the resulting metabolomes using laser ablation-coupled electrospray ionization MS (LAESI-MS), an emerging method in which a mid-IR laser (λ = 2.94 μm) is absorbed by the sample, generating an ablation plume of neutral metabolites that are ionized via electrospray and introduced into the mass spectrometer23–26. LAESI-IMS combines a soft ionization method with broad molecular coverage – including peptides, lipids, and alkaloids among others – with detection sensitivities in the single-digit μM range for many types of metabolites26. Compared to other IMS methods, the advantage of LAESI-IMS is that it can be applied to liquid or solid surfaces and live bacterial cultures with minimal sample preparation at ambient pressure. It shares with other IMS techniques the disadvantage of ion suppression and preferential detection of more ionizable metabolites. Optimization of numerous parameters (see Online Methods) facilitated rapid characterization of the P. protegens metabolome within each of the 502 wells, allowing us to image a 96-well plate liquid culture in less than an hour. The signals observed in each well above a set cut-off value were computationally extracted and amalgamated into a 3D plot depicting the intensity and m/z for each metabolite produced in the presence of a given elicitor (Fig. 2a, Supplementary Fig. 1). By visually inspecting the 502 metabolomes represented in the 3D plot, we could easily detect induction of orfamides – specifically analogs A (2), B (3), and several unknown derivatives – to varying degrees (Fig. 2a,b). Optimal production was triggered by the mild cytotoxin cafestol (1) and the anticancer agent vinorelbine, compounds previously not known to elicit secondary metabolism. Cafestol’s stimulatory activity was further confirmed using HPLC-MS, thus validating the use of HiTES-IMS in inducing silent or lowly expressed BGCs (Fig. 2c, Supplementary Table 1). The simplicity of this approach suggested it could be broadly applied.
Fig. 2. I Proof-of-concept application of HiTES-IMS to P. protegens.
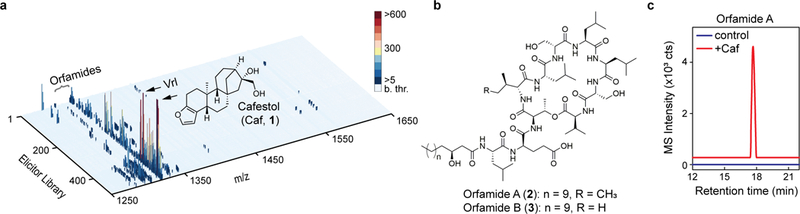
a, 3D plot relating the P. protegens metabolome, in terms of m/z and MS intensity, to each elicitor. MS data were collected in the m/z range of 1200–2000 to focus on orfamide production, in response to a 502-member natural products library. No signals were detected below m/z 1250 or above m/z 1650. MS intensity (in counts) is color-coded according to the color bar shown; ‘b. thr.’ designates signals that were below the 5-count threshold, which were therefore not included in the plot. Orfamides are labeled, as are the best elicitors of orfamide synthesis, cafestol (Caf) and vinorelbine (Vrl). b, Structures of orfamide A and B. c, Validation of Caf as an inducer of orfamide A in flask cultures analyzed by HPLC-MS. Shown are HR-MS extracted ion chromatograms of orfamide A from untreated (blue) and Caf-treated (red) cultures. The HiTES-IMS screen was carried out in a single replicate; production of desired metabolites was validated in three independent biological replicates, with a representative result shown in panel c. All three replicates gave similar levels of induction of orfamides.
Application of HiTES-IMS to Streptomycetes.
With the success in P. protegens, we next applied HiTES-IMS to Streptomyces spp., the most prolific genera of bacterial secondary metabolite producers known. We chose Streptomyces canus NRRL B3980, which is related to the amphomycin producer and contains over 20 BGCs that have not been linked to a natural product27,28. The results of HiTES-IMS with S. canus using again a 502-member natural products library are shown in 3D representation (Fig. 3a, Supplementary Fig. 2). Several metabolites were induced in the m/z range of 250–650 (Supplementary Fig. 2). To optimize our chances of finding new metabolites, we focused on compounds with higher molecular weights (Fig. 3a). In this range, three clusters of peaks, representing three compound families appeared to be elicited: we detected compounds with m/z 1260–1295, which further analysis identified as the amphomycins. The best elicitors for the amphomycins were the flavonol morin and the antimalarial quinine. A second set of induced compounds was also detected with m/z 1220–1245. Finally, we observed induction of a third compound family, distinct from the other two, with m/z of 1563 and 1579 elicited by the cyclin-dependent kinase inhibitor kenpaullone (4) (Fig. 3a, b). These findings highlight the ability of our approach to induce multiple cryptic BGCs in a parallel fashion. Whereas in our previous renditions of HiTES we monitored the expression of only a single BGC at a time, the current approach allows us to monitor all BGCs that can be captured by our detection method in one experiment.
Fig. 3. I Discovery of a novel, cryptic lasso peptide by HiTES-IMS.
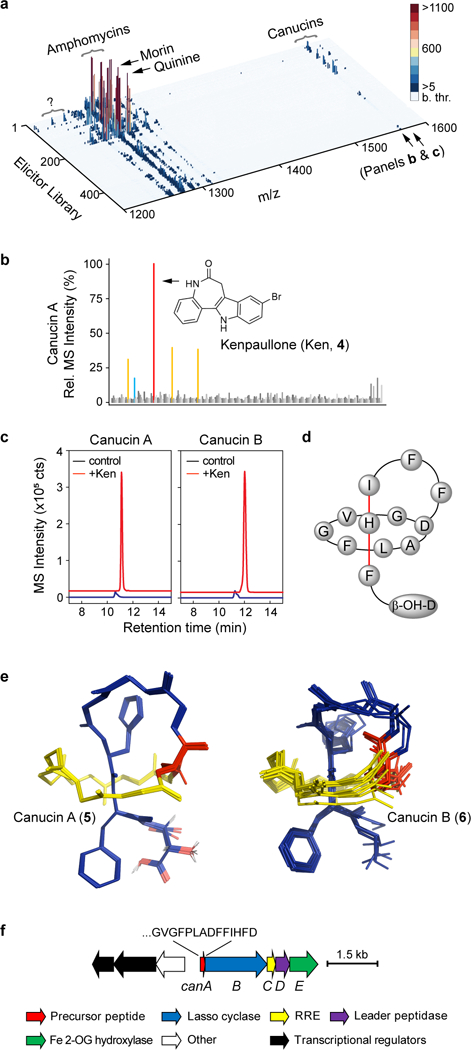
a, Secondary metabolome of S. canus in response to 502 elicitors. MS data were collected in the m/z range of 250–1600 (see Supplementary Fig. 2). The high m/z range is shown to focus on canucins; ‘b. thr.’ on the color bar (in counts) designates below threshold of detection. Morin and quinine, which induce amphomycin synthesis, are marked. Canucins are pointed out as is an uncharacterized set of induced metabolites (question mark). b, 2D component of the 3D plot focusing on canucin A (m/z 1579). Kenpaullone (Ken) was the most effective elicitor. c, Induction of canucin A and B by Ken in flask cultures analyzed by HPLC-MS. The HR-MS extracted ion chromatogram traces are offset in the X- and Y-axes for clarity. d, Illustration of the topology of canucin A, with His12 and Phe13 providing steric locks. e, Overlay of the top-10 computed structures for canucin A and B using NMR NOESY constraints and the CYANA algorithm. Both exhibit a lasso topology. f, BGC for canucins (can) as identified by bioinformatic studies. The C-terminal sequence of CanA is shown along with predicted functions of the tailoring enzymes. The HiTES-IMS screen was carried out in a single replicate; production of desired metabolites was validated in three independent biological replicates, with representative results shown in panel c. All three replicates gave similar levels of induction of canucins.
From the cryptic metabolites elicited, we focused further efforts on the compounds with m/z 1579 and 1563, to which we have assigned the trivial names canucin A and B, respectively (Supplementary Table 1). We validated the results observed in 96-well plates and found strong induction of both compounds by kenpaullone (approx. 12-fold in 96-well plates and flask cultures), consistent with the screening results (Fig. 3c). Large-scale production cultures with kenpaullone as inducer allowed us to isolate sufficient material to solve the structures of canucins by 1D/2D NMR. Analysis of 1H, gCOSY and TOCSY NMR data showed that canucin A (5) was a peptide with 14 recognizable α−1Hs. HSQC and HMBC analysis revealed 13 of these as canonical amino acids, while one was the modified β-hydroxy-Asp (Supplementary Table 2 and Supplementary Fig. 3). Further analysis by NMR and HR-MS suggested that canucin A harbored an isopeptide bond between residues Gly1 and Asp8, a feature that is typical for lasso peptides29,30. Indeed, NOESY correlations between the C-terminal tail residues and those surrounding the isopeptide bond suggested that canucin A contained a lasso topology (Supplementary Fig. 3). To verify, we collected high-resolution NOESY spectra with various mixing times and solved the 3D structure of canucin A using the CYANA algorithm, which utilizes molecular dynamics simulations in a peptide’s torsion angle space to compute structures that agree best with the NOESY data31,32. The ten best structures converged on a lasso topology, in which His12 and Phe13 provide steric locks above and below the ring, respectively (Fig. 3d,e). At 14 amino acids, canucin A is one of the smallest lasso peptides discovered to date.
Repeated efforts to determine the stereochemistry at the β-carbon of the C-terminal Asp residue by Mosher analysis failed, possibly due to steric hindrance. We subsequently used CYANA to calculate which stereoisomer best fits the observed NOESY correlations. The S-stereoisomer gave a lower f-function, indicative of a better match between the calculated structure and the set of constraints, as well as a lower backbone-rmsd (Supplementary Table 3). We suggest the S-configuration at the β-carbon of Asp, a prediction to be tested by future experiments. We also identified a second analog, canucin B (6). Structural elucidation by HR-MS and NMR identified it as the des-hydroxy variant of canucin A (Supplementary Tables 1 and 4). CYANA calculations confirmed that canucin B harbors a lasso topology, again revealing His12 and Phe13 as steric locks in a topology akin to that of canucin A (Fig. 3e). These results imply that hydroxylation at the C-terminal Asp in canucin A occurs after the threaded lasso motif has been installed, though the order remains to be determined experimentally.
Post-translationally modified lasso peptides are rare, and a β-hydroxylated amino acid has not previously been observed within this compound family29,30,33,34. To gain insights into the biosynthesis of canucin A, we examined the genome sequence of S. canus. We identified a BGC, which we annotate as can, with a typical lasso peptide synteny and a precursor peptide, whose C-terminal sequence perfectly matched that of the canucins (Fig. 3f). Aside from the precursor peptide, the can BGC contains a typical protease and an Asn synthetase, which removes the precursor peptide and forms the threaded lasso motif, respectively29,30. In addition, we annotate canE, encoding a putative α-KG-dependent mononuclear Fe enzyme, members of which have been shown to hydroxylate unactivated carbon positions35,36 (Supplementary Table 5). CanE is likely involved in the synthesis of β-OH-Asp. The discovery of canucins shows that HiTES-IMS can be applied to streptomycetes to unveil new cryptic metabolites.
Application of HiTES-IMS to Rare Actinomycetes.
With the success of HiTES-IMS with common, prolific Gram-negative and Gram-positive bacteria, we next sought to apply this approach to rare actinomycetes, a group of bacteria that account for some structurally fascinating and functionally potent metabolites, including the antibiotic of-last-resort vancomycin as well as the anticancer agent calicheamicin37–40. Rare actinomycetes are an ideal test case for HiTES-IMS because in addition to housing an abundance of silent BGCs, they are difficult to manipulate genetically, thus all but precluding transcriptional reporter assays. To apply HiTES-IMS, we chose Amycolatopsis keratiniphila NRRL B24117 as a test case. Its genome has not been sequenced, but efforts to find further BGCs that encode glycopeptide antibiotics (GPAs) using PCR had shown that it contains a vancomycin-like BGC, though a product has not yet been reported41. We subjected A. keratiniphila to HiTES-IMS and observed numerous induced metabolites in both low and high m/z ranges (Fig. 4a, Supplementary Fig. 4), suggesting that this strain boasts a substantial hidden metabolome. Again, induction of silent BGCs occurred in a high-throughput fashion. In the m/z range typical for GPAs, a compound with m/z 1286 was induced by galangin and the indole-containing alkaloids evodiamine and ajmaline. Additionally, a compound with m/z 1425 was induced by similar elicitors, including galangin and evodiamine (Supplementary Fig. 4). Lastly, compounds with m/z 1654 and 1811 were observed in the induced metabolome, primarily by dihydroergocristine (Dhe, 7) and vincamine (Vin, 8; Fig. 4a,b). A. keratiniphila appeared to respond to indole-bearing alkaloids of diverse origins by activating numerous, otherwise silent, BGCs leading to the production of cryptic metabolites.
Fig. 4. I Induction of novel glycopeptides using HiTES-IMS.
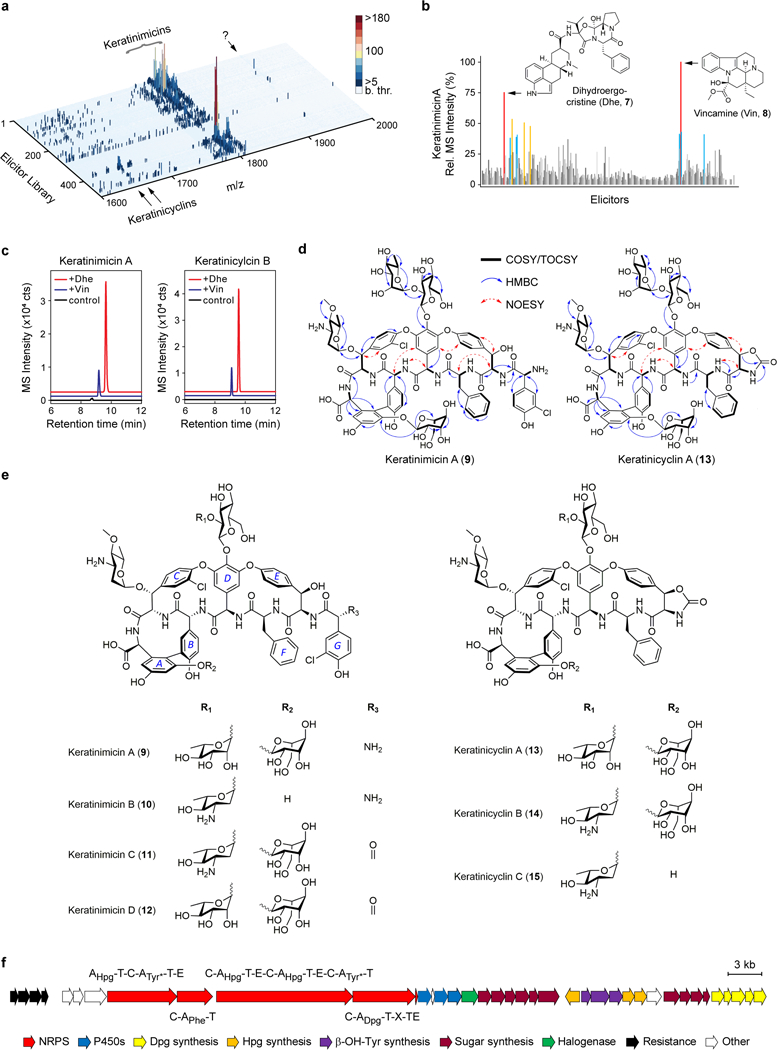
a, Secondary metabolome of A. keratiniphila in response to 502 elicitors. MS data were collected in the m/z range of 250–2000 (Supplementary Fig. 4). A magnified view is shown to focus on glycopeptides; ‘b. thr.’ designates below MS detection threshold. Keratinimicins, keratinicyclins, and uncharacterized induced metabolites (question mark) are indicated. b, 2D component of the 3D plot focusing on keratinimicin A (m/z 1811). Dihydroergocristine (Dhe) and vincamine (Vin) were the most effective elicitors. c, Validation of Dhe and Vin as elicitors of keratinimicin and keratinicylin in 96-well cultures analyzed by HPLC-MS. Shown are HR-MS extracted ion chromatograms from untreated (black), Vin-treated (blue), and Dhe-treated (red) cultures. The traces are offset in the X- and Y-axes for clarity. d, Relevant NMR correlations used to solve the structures of keratinimicin A and keratinicyclin A. e, Structures of four keratinimicin and three keratinicyclin derivatives with varying substitution patterns. The nomenclature to identify different rings in glycopeptides is shown in keratinimicin A. f, The ker BGC as identified by bioinformatic analysis after sequencing the genome of A. keratiniphila. The predicted domain composition for each NRPS is shown as are the predicted functions of the remaining enzymes in the BGC. Tyr* denotes modified Tyr. The HiTES-IMS screen was carried out in a single replicate; production of desired metabolites was validated in three independent biological replicates, with representative results shown in panel c. All three replicates gave similar levels of induction of keratinimicins and keratinicyclins.
Of the compounds detected, one with m/z 1811 appeared to contain a chlorine atom as judged by the HR-MS isotope distribution pattern, a common modification in GPAs (Supplementary Table 1). Follow-up experiments validated the strong stimulatory activity by Dhe and Vin, demonstrating a marked elicitation of this metabolite (15-fold in 96-well plates and 4-fold in flask cultures with Dhe, Fig. 4c), consistent with the HiTES-IMS screening results. Suspecting that it might be the putative glycopeptide, we isolated seven analogs from large-scale production cultures in the presence of Dhe. HR-MS analysis suggested that these compounds fall into two families, one that we refer to as keratinimicins, and a second, the keratinicyclins. 1D/2D NMR analysis showed that keratinimicin A (9) contains seven highly modified amino acids, with recognizable α−1Hs at 4.41, 4.45, 4.52, 5.62, 4.23, 4.48, and 4.42 ppm (Fig. 4d, Supplementary Table 6). TOCSY, HSQC, and HMBC analysis identified these as the 3-Cl-derivative of 4-hydroxyphenylglycine (Hpg), β-OH-Tyr, Phe, two crosslinked Hpg residues, a glycosylated β-OH-3-Cl-Tyr, and a crosslinked 3,5-dihydroxyphenylglycine (Dpg) (Supplementary Fig. 5). Characteristically, the crosslinks occurred between rings A–B, via a carbon-carbon bond, and between rings C–D and D–E via aryl ether bonds as elucidated by HMBC and ROESY spectra (Fig. 4e). Four glycosyl groups were identified by NMR analysis, mannose, actinosamine, and a glucose-rhamnose disaccharide on rings A, C, and D, respectively, thus completing the two-dimensional structure of keratinimicin A (Supplementary Fig. 5 and Supplementary Table 6). To assign the chiral centers, we chose a combined spectroscopic and bioinformatic approach. Shot-gun sequencing of the entire genome of A. keratiniphila allowed us to pinpoint a GPA cluster, which we annotate as ker, using the canonical synteny previously described (Fig. 4f)39. To the best of our knowledge, ker is the first BGC reported for a class II GPA. Bioinformatic analysis revealed an identical domain organization as the archetypal class I GPAs (Supplementary Table 7). We therefore propose a pattern of d- and l-amino acids as shown (from C- to N-terminus: l-l-d-d-l-d-d, Fig. 4e)38. With this pattern in mind, the R-configuration was tentatively assigned for the β-OH groups using NMR ROESY correlations, thus completing the proposed three-dimensional structure. Keratinimicin A is similar to the actinoidins, notably actinoidin B, except that it carries a different disaccharide at residue D42,43.
Three additional keratinimicin analogs were identified and structurally elucidated as well. Relative to variant A, keratinimicin B (10) only contains three glycosyl moieties: actinosamine and a glucose-acosamine disaccharide on rings C and D, respectively, while mannose on ring A is missing (Supplementary Table 8). Keratinimicin C (11) is N-terminally capped by an unusual m-chloro-p-hydroxyphenylglyoxylic acid moiety, with again a different bouquet of sugar substituents (Supplementary Table 9). Keratinimicin D (12) also contains the m-chloro-p-hydroxyphenylglyoxylic acid residue, with yet a different combination of sugars on ring D, relative to keratinimicin C (Supplementary Table 10).
The structure of keratinicyclin A (13) was characterized by extensive analysis of spectroscopic data leading to the structure shown (Fig. 4d,e). It consists of a 6-mer peptidic backbone, containing the same sequence as the keratinimicins sans the N-terminal 3-Cl-Hpg residue. It bears the same aromatic crosslinks as the keratinimicins, with an A–B biaryl bond as well as the C-O-D and D-O-E aryl ether crosslinks. The glycosyl groups were also identified by NMR analysis as described above (Supplementary Table 11 and Supplementary Fig. 6). Most notably, the keratinicyclins contain an N-terminal 2-oxazolidinone, a functional group present in the clinically used antibiotics linezolid and tedizolid44. Thus, the keratinicyclins combine the characteristic features of the GPAs with those of the oxazolidinone antibiotics, the first such combination reported thus far. Within the GPAs, the keratinicyclins represent a new chemotype.
We solved the structures of two additional keratinicyclins. Relative to derivative A, keratinicyclin B (14) contains a glucose-acosamine disaccharide on ring D, rather than glucose-rhamnose (Supplementary Table 12). Keratinicyclin C (15) also contains a glucose-acosamine disaccharide on ring D but lacks mannose on ring A (Supplementary Table 13).
Initial in-house assays revealed strong antimicrobial activity for the keratinimicins, but not for the keratinicyclins. Keratinimicins A and C were submitted for broad bioactivity tests against bacterial pathogens. Because some GPAs have been documented to harbor antiviral properties38, keratinicyclin B was assessed against a panel of pathogenic human viruses. Keratinimicins showed potent antibacterial activity against numerous Gram-positive pathogens, with MICs (minimal inhibitory concentrations) akin to those of vancomycin against Streptococci, Clostridium difficile, and Enterococcus faecalis (Table 1, Supplementary Table 14). They were ineffective against vancomycin-resistant Enterococci, suggesting a similar mode of action as vancomycin. Keratinicyclin B did not exhibit notable antibacterial activity but was a potent inhibitor against the respiratory syncytial virus (RSV). Indeed, the MIC determined against RSV was ~20-fold more potent than that of the currently-used drug, Ribavirin (Table 1)45. The discoveries of these cryptic antibacterial and antiviral agents highlight the utility of HiTES-IMS in unearthing novel metabolites with potentially therapeutic bioactivities.
Table 1.
I MIC values (in μM) for keratinimicins and keratinicyclin B against select pathogenic Gram-positive bacteria and viruses.a
| Strain | Keratini- micin A |
Keratini- micin C |
Keratini- cyclin B |
Control drugb |
|---|---|---|---|---|
| S. aureus | 4.4 | 4.5 | >39 | 2.1 (V), 1.5 (C) |
| S. aureus MRSA | 2.2 | 4.5 | >39 | 0.7 (V), 12.1 (C) |
| S. pneumoniae PSPP | 0.1 | 0.6 | 19.5 | 0.4 (V), 1.5 (C) |
| S. pyogenes | 0.3 | 1.1 | 19.5 | 0.2 (V), 0.8 (C) |
| S. agalactiae | 0.6 | 2.2 | 39 | 0.2 (V), 0.8 (C) |
| E. faecalis VSE | 2.2 | 4.5 | >39 | 2.8 (V), 6.0 (C) |
| E. faecalis VRE | >35 | >35 | >39 | >44 (V), >12.1 (C) |
| B. subtilis | 0.3 | 1.1 | 19.5 | 0.1 (V), 0.2 (C) |
| C. difficile | 0.3 | 0.3 | 9.8 | 1.5 (M) |
| Influenza A | –c | – | 92 | 0.7 (O) |
| Respiratory Syncytial Virus (RSV) | – | – | 0.4 | 7.8 (R) |
See Online Methods and Supplementary Table 14 for additional details.
Control drugs are abbreviated as follows: V, vancomycin; C, ciprofloxacin, M, metronidazole; O, oseltamivir; R, ribavirin.
Not determined.
DISCUSSION
Silent BGCs are a treasure trove of potential new secondary metabolites. Advances in DNA technology and extensive genome sequencing have compiled a massive genome database, which needs to be mined to harvest the fruits of decades of innovation. The ideal method to do so is one that does not necessitate challenging genetic or cloning procedures of the typically large BGCs. Moreover, the method should activate silent BGCs in a high-throughput fashion, preferably in an endogenous host, to avoid difficulties associated with heterologous expression. Lastly, a mono-culture approach is preferred to eliminate the possibility of irreproducible interactions that sometimes plague mixed- or co-culture screens. Herein, we have implemented HiTES-IMS, a method that satisfies all these criteria. We highlight its utility by applying it to sequenced and unsequenced bacteria, notably a rare actinomycete, members of which class are difficult to manipulate genetically. The typical output is a picture of the secondary metabolome of a given bacterium in response to ~500 conditions, which demonstrates activation of silent BGCs in a high-throughput fashion. Leveraging these advantages, we report a lasso peptide with an unprecedented post-translational modification, new glycopeptide antibiotics with bioactivities similar to or better than those of vancomycin, and a novel glycopeptide chemotype, which combines features of the GPAs with those of the oxazolidinone antibiotics and exhibits better antiviral activity in vitro than the currently used drug Ribavarin against RSV.
HiTES-IMS adds to the cadre of available approaches for imaging bacterial metabolomes. MALDI-TOF- and DESI-based IMS have been pioneered for assessing bacterial cultures and interspecies interactions21,46–48. LAESI-IMS has been employed for spatially resolving secondary metabolite production and for strain selection.23,24,49 High-throughput elicitation has previously been reported in a compound- and cluster-specific manner along with ion-mobility UPLC-MS to assess a small number of eliciting conditions.18,50,51 By combining the two overarching methods – high-throughput elicitation and IMS – HiTES-IMS facilitates interrogation of cryptic bacterial metabolomes in response to hundreds of conditions.
In contrast to primary metabolism, the complete secondary metabolome of any given prolific bacterium is not yet known7,52. Thus, in addition to unearthing novel cryptic metabolites, we propose that HiTES-IMS may be used to uncover global secondary metabolomes, that is, the full biosynthetic capacity of selected bacterial species, as detected by MS. While this was in theory possible with previous renditions of HiTES and other methods, the advantage of the current approach, and of IMS in general, is that the final product of a BGC, the secondary metabolite, provides the read-out, rather than transcriptional or translational assays. Additionally, by linking new cryptic metabolites to an elicitor, the mechanism of elicitation may be investigated, which in turn can identify regulatory circuits that kick-start secondary metabolism in response to sub-inhibitory concentrations of toxins53–55. HiTES-IMS is the most general approach so far for activation of silent BGCs, and it is poised to simultaneously shed light onto the products and regulation of cryptic metabolism in diverse bacteria.
ONLINE METHODS
Bacterial strains and culture media.
Pseudomonas protegens Pf-5 was obtained from the ATCC. Amycolatopsis keratiniphila subsp. keratiniphila NRRL B-24117 and Streptomyces canus NRRL B-3980 were acquired from the ARS (NRRL) culture collection. All culture media were obtained from Becton-Dickinson. Other media components were obtained from Sigma-Aldrich.
Screens of P. protegens Pf-5.
P. protegens was streaked out onto an LB-agar plate from frozen culture stocks and grown at 30°C overnight. Colonies were used to inoculate 5 mL of LB in a 14 mL sterile culture tube, which was cultured for 12–13 h at 30°C/250 rpm. The overnight culture was then used to inoculate 550 mL LB in a sterile Erlenmeyer flask to an initial optical density at 600 nm (OD600 nm) of 0.01. The culture was distributed into six sterile, deep-well 96-well plates (0.9 mL per well) using a MultiFlo Microplate Dispenser (BioTek). Subsequently, elicitors were added from a commercially-available 502-member natural products library (Enzo Scientific, cat# BML-2865) using a CyBi-Well automated liquid transfer robot (CyBio). Each well received 2.5 μL of an elicitor (from a stock concentration of 10 mM). The plates were sealed with air-permeable membranes and grown at 25°C/200 rpm. After 44 h, the plates were spun down, supernatants loaded onto a 96-well Strata C8-resin (Phenomenex), and the material eluted with 600 μL of 50% and 600 μL 100% MeCN into fresh 96 well plates. The eluates were dried in a speedvac, resuspended in 30 μL of 40% MeCN (in water), and imaged by LAESI-MS (see below).
Screens of S. canus and A. keratiniphila.
Freshly-collected spores of S. canus or A. keratiniphila (~107) were transferred to 50 mL YEME medium (3% (w/v) yeast extract, 5% peptone, 3% malt extract, 1% glucose, 34% sucrose, and 5 mM MgCl2•6H2O) in a 250 mL Erlenmeyer flask fitted with a stainless-steel spring and cultured at 30°C/250 rpm for 3 days. Mycelia were then collected by centrifugation (10 min, 3000g, RT) and diluted into 300 mL of R4 medium to give a final concentration of 0.05% (w/v). R4 medium consisted of 0.5% (w/v) glucose, 0.1% yeast extract, 0.5% MgCl2•6H2O, 0.2% CaCl2•2H2O, 0.15% proline, 0.118% valine, 0.28% TES, 50 mg/L casamino acid, 100 mg/L K2SO4, and 1x trace element solution, which contains 40 mg/L ZnCl2, 200 mg/L FeCl3•6H2O, 10 mg/L CuCl2•2H2O, 10 mg/L MnCl2•4H2O, 10 mg/L Na2B4O7•10H2O, and 10 mg/L (NH4)6Mo7O24•4H2O)56. Subsequently 300 μL were dispensed into six deep-well 96-well plates using a MultiFlo Microplate Dispenser. The wells were supplemented with candidate elicitors from the same 502-member natural products library used above with the aid of a CyBi-Well automated liquid transfer robot. Each well received 0.85 μL of an elicitor (from a stock concentration of 10 mM). The plates were sealed with air-permeable membranes and grown at 30°C/250 rpm. After 5 days, the samples were desalted as described above for P. protegens and imaged by LAESI-IMS (see below).
Analysis by LAESI-IMS.
A laser ablation electrospray ionization (LAESI) DP1000 system (Protea Bioscience) coupled to an LTQ XL mass spectrometer (Thermo) was used for IMS analysis. The extension tube connecting the two instruments was kept at 130°C with an external heater and the sample stage was kept at 10°C during analysis. Sheath gas flow was set to 2.0 L/h. Eighty laser pulses were applied to each well to ablate samples using an 80% laser energy setting (~850 μJ) and a 10 Hz frequency. A solution of 2:1 MeCN/water with 0.1% acetic acid (v/v) was supplied as the electrospray solution by a syringe pump running at a flow rate of 1 μL/min. We found this solution to be effective in detection of a variety of structurally distinct metabolites, including alkaloids, quinolones, small aromatic compounds, GPAs, and other peptides. The emitter was connected to high voltage power operating at +4000 V or +4500 V in positive ion detection mode. All data were visualized in ProteaPlot software.
After data collection, the signals observed in each well were extracted using GMSU-LAESI software (Gubbs, Inc), which gave all m/z values and the corresponding intensities per well (i.e. per elicitor). The data were binned in 1 m/z units for 3D-plotting. Signals with an intensity value lower than a set threshold were not included in the 3D plots. The data were plotted in MatLab using the bar3 function. Simple visual inspection of 3D plots generated in this fashion allowed us to identify metabolites whose production was induced or enhanced by a given elicitor. Because low-MW metabolites generally ionized better than high-MW compounds, a higher threshold was used for visualizing the former (see intensity scale bars in 3D plot figures).
The 3D plot provides a visual read-out of the effect of all small molecules in the library on all MS-detected secondary metabolites. The effect of the small molecules in the library on the production of a single metabolite was best assessed in the component 2D plots (see Figs. 3b & 4b). The component 2D plots were extracted from the corresponding 3D data in MatLab and plotted in Excel. The 2D plots were normalized to the highest intensity peak. Hits identified in this manner were validated using flask cultures and HPLC-MS analysis, as described below. Elicitation of orfamides, canucins, keratinimicins, and keratinicyclins was also observed in large-scale cultures. While it was not the case with the compounds that we pursued, it is possible that the production of other compounds will not translate well from 96-well plates to flask cultures.
Validation of HiTES elicitors.
To validate induction of the cryptic metabolite identified in the high-throughput screen, flask cultures of each condition were grown and analyzed by HPLC-MS. To validate orfamide production, 5 mL overnight cultures of P. protegens were prepared as described above. The culture was then diluted into 2 × 125 mL Erlenmeyer flasks, each containing 20 mL of LB to give an initial OD600 nm of 0.01. One flask was supplemented with cafestol (22 μM final concentration); the other was supplemented with the same volume of DMSO (control). Both cultures were incubated at 25°C/150 rpm. After 44 h, cells were removed by centrifugation, and 10 mL of each supernatant was desalted on a C8 PrepSep solid-phase extraction column (Fisher). After loading, the columns were washed with H2O and bound material eluted with a 2-step gradient of 50% MeCN (in H2O) and 100% MeCN. The fractions were dried in vacuo, redissolved in 100 μL MeOH, and analyzed by HPLC-Qtof-MS (see below).
To validate canucin production, seed cultures of S. canus were prepared as described above. The culture was diluted into 2 × 250 mL Erlenmeyer flasks, each containing 50 mL of R4 medium to a final mycelial concentration of 0.05% (w/v). Kenpaullone was added to one flask (final concentration of 17 μM), while the other served as control and received the same volume of DMSO. The cultures were grown at 30°C/200 rpm for 5 days. The supernatants were extracted twice with 30 mL of ethyl acetate. The organic phases were combined, dried in vacuo, re-dissolved in 200 μL MeOH, and the two samples analyzed by HPLC-Qtof-MS (see below).
To validate keratinimicin and keratinicyclin production, seed cultures of A. keratiniphila were prepared as above. The culture was diluted into 3 × 1 mL wells in a deep 96-well plate containing 0.9 mL of R4 medium at a final mycelial concentration of 0.05% (w/v). The 96-well plate was supplemented with a final concentration of 7.8 μM vincamine (well 1), 2 μM Dhe (well 2), and the same volume of DMSO (well 3, control). The plate was then grown at 30°C/200 rpm for 5 days. Cells and mycelia were removed by centrifugation, and the wells worked up as noted above for the 96-well screens and analyzed by HPLC-Qtof-MS. Alternatively, the seed culture was diluted into 3 × 250 mL Erlenmeyer flasks carrying 50 mL of R4 medium at a mycelial concentration of 0.05% (w/v). The flasks were supplemented with 7.8 μM vincamine (flask 1), 2 μM Dhe (flask 2), and the same volume of DMSO (flask 3, control), and then grown at 30°C/200 rpm for 5 days. Cells and mycelia were removed, and 10 mL of each supernatant was loaded onto a C18 SPE column (Phenomenex, 100 mg), washed with H2O, and eluted with 50% and 100% MeCN. The fractions were dried in vacuo, re-dissolved in 100 μL MeOH, and analyzed by HPLC-Qtof-MS. The deep-well plate method resulted in better induction of keratinimicin and keratinicyclin. All validations were carried out in three independent biological replicates.
HPLC-MS and HPLC analysis.
HPLC-MS analysis was performed on an Agilent 1260 Infinity Series HPLC system equipped with an automated liquid sampler, a diode array detector, and a 6120 Series ESI mass spectrometer using a reversed phase Luna C18 column (Phenomenex, 5 μm, 150 × 4.6 mm). The mobile phases consisted of H2O and MeCN (+ 0.1% formic acid). Elution was carried out isocratically with 5% MeCN in water for 3 min followed by gradients of 5%–70% MeCN over 20 min, and then 70%–100% over 5 min, at a flow rate of 0.6 mL/min. High-resolution (HR) HPLC-MS and HR-tandem HPLC-MS were carried out on an Agilent 6540 Accurate Mass Qtof LC-MS, consisting a 1260 Infinity Series HPLC system, an automated liquid sampler, a diode array detector, a JetStream ESI source, and the 6540 Series Qtof. Samples were resolved on a Luna C18 column (Phenomenex, 5 μm, 100 × 4.6 mm). The mobile phase consisted of water and MeCN (+0.1% formic acid). Elution for orfamides was carried out isocratically with 10% MeCN in water (3 min) followed by a gradient of 10%–95% MeCN over 8 min, and by an isocratic elution at 95% MeCN over 25 min at a flow rate of 0.4 mL/min. Elution of canucins, keratinimicins and kertinicyclins was carried out isocratically with 5% MeCN in water (3 min) followed by a gradient of 5%–95% MeCN in water over 15 min, at a flow rate of 0.4 mL/min.
HPLC purifications were carried out on an Agilent preparative HPLC system equipped with a 1260 Infinity series binary pump, a diode array detector, and an automated fraction collector. Semi-preparative or analytical-scale purifications were performed on an Agilent HPLC system containing a 1260 Infinity Series binary pump or a 1290 Infinity quaternary pump. Each system was equipped with an automatic liquid sampler, a temperature-controlled column compartment, a diode array detector, and an automated fraction collector.
Large-scale growth of S. canus and A. keratiniphila.
Large-scale fermentation was carried out following a similar procedure as for small-scale fermentations described above. 3-day seed cultures were prepared as above, mycelia isolated, and used to inoculate several 2 L Erlenmeyer flasks containing 200 mL of R4 medium to a final mycelial concentration of 0.01% (w/v). The culture was then supplemented with the optimal concentration of the elicitor (see above). Typically, 8–12 L of total culture was used for compound isolation. The flasks were incubated at 30°C/250 rpm for 7 days, at which point the desired products were purified (see below).
Purification of canucin A and B.
Canucins were purified from 12 L fermentation of S. canus in the presence of kenpaullone (at a final concentration of 17 μM). After a 7-day fermentation, the supernatant was extracted twice with an equal volume of ethyl acetate. The combined organic phase was dried over Na2SO4, dried in vacuo, resuspended in 45 mL MeOH, and purified on an Agilent Preparative HPLC. The sample was resolved with repeated injections onto a preparative Luna C18 column (Phenomenex, 5 μm, 21.2 × 250 mm) operating at a flow rate of 12 mL/min with mobile phases consisting of water and MeCN (+0.1% formic acid). Upon injection, elution was carried out isocratically with 20% MeCN for 2 min, followed by a gradient of 20%–100% MeCN over 25 min. Peaks containing canucin A and B, as judged by HPLC-MS, were pooled, dried in vacuo, resuspended in MeOH, and further purified on a semi-preparative/analytical HPLC system. The peptides were purified on a semi-preparative XDB-C8 column (Agilent, 5 μm, 10 × 250 mm) operating at a flow rate of 2.5 mL/min with a gradient of 30%–50% MeCN (in water) over 30 min followed by a gradient of 50%–100% MeCN over 5 min. Peaks containing pure canucin B were combined and lyophilized to dryness. Peaks containing canucin A were pooled, dried in vacuo, resuspended in MeOH, and further purified on a semi-preparative Luna C18 column (Phenomenex, 5 μm, 10 × 250 mm) with a gradient of 33%–55% MeCN over 30 min followed by a gradient of 55%–100% MeCN over 5 min. Peaks containing pure canucin A were combined, and lyophilized to dryness. This procedure gave 3.6 mg of canucin A and 1.7 mg canucin B.
Purification of keratinimicins and keratinicyclins.
Keratinimicins and keratinicyclins were purified from 8 L fermentations of A. keratiniphila in the presence of 2 μM Dhe. After a 7-day fermentation, the resulting supernatant was loaded on a pre-packed C18 column (Phenomenex, 50 μm, 65 Å, 10 g) and eluted with 20%, 50% and 100% MeCN in water step-wise. The 20% fraction containing keratinimicins and keratinicyclins was dried in vacuo, resuspended in 50 mL MeOH, and further purified by preparative HPLC using a Luna C18 column (Phenomenex, 5 μm, 21.2 × 250 mm) operating at a flow rate of 12 mL/min with mobile phases consisting of water and MeCN (+0.1% formic acid). Upon injection, elution was carried out isocratically with 5% MeCN in water for 2 min, followed by a gradient of 5%–40% MeCN in water over 20 min, and a gradient of 40%–100% MeCN over 5 min. Fractions were collected in 1 min intervals over the time range of 5–25 min. Peaks containing keratinimicin A-D, as judged by HPLC-MS analysis, were pooled, dried in vacuo, resuspended in MeOH and further purified on a semi-preparative/analytical HPLC system. The sample was applied to a an RP Amide-C16 column (Supelco, 5 μm, 10 × 250 mm) operating at a flow rate of 2.5 mL/min with the same mobile phases as above and a gradient of 8%–16% MeCN in water over 30 min followed by a gradient of 16%–100% MeCN over 5 min. Peaks containing pure keratinimicin C and D were combined and lyophilized to dryness. Peaks containing keratinimicin A and B were pooled, dried in vacuo, resuspended in MeOH and further purified on a semi-preparative XDB-C8 column (Agilent, 5 μm, 10 × 250 mm) with a gradient of 5%–15% MeCN in water over 30 min followed by a gradient of 15%–100% MeCN over 5 min. Peaks containing pure keratinimicin A and B were combined and lyophilized to dryness. This procedure gave 8.5 mg of keratinimicin A, 1.6 mg keratinimicin B, 5.1 mg keratinimicin C, and 0.8 mg keratinimicin D.
Peaks containing keratinicyclin A–C from the preparative Luna C18 column were pooled, dried in vacuo, resuspended in MeOH and further purified on a semi-preparative/analytical HPLC system. The sample was applied to a RP Amide-C16 column (Supelco, 5 μm, 10 × 250 mm) operating at a flow rate of 2.5 mL/min with the same mobile phase as above and a gradient of 10%–20% MeCN in water over 30 min followed by a gradient of 20%–100% MeCN over 5 min. Peaks containing pure keratinicyclin A and C were combined, and lyophilized to dryness. Peaks containing keratinicyclin B were pooled, dried in vacuo, resuspended in MeOH and further purified on a semi-preparative XDB-C8 column (Agilent, 5 μm, 10 × 250 mm) with a gradient of 5%–15% MeCN in water over 30 min followed by a gradient of 15%–100% MeCN over 5 min. This procedure gave 2.7 mg of keratinicyclin A, 5.3 mg keratinicyclin B, and 1.2 mg keratinicyclin C.
Structural elucidation of canucins, keratinimicins, and keratinicyclins.
HR-MS data for all compounds and their inferred molecular formula are listed in Supplementary Table 1. For structural elucidation, 1D/2D NMR spectra were acquired at the Princeton University Department of Chemistry NMR Facilities on an A8 Avance III HD 800 MHz NMR spectrometer (Bruker) with a triple resonance cryoprobe. The NMR samples of keratinimicin A-D and keratinicyclin A-C were prepared in DMSO-d6, and those of canucin A and B were prepared in CD3OH. 1D/2D NMR spectra of canucin A, keratinimicin A, and keratinicyclin A are shown (Supplementary Figs. 3, 5, 6). NMR tables listing chemical shift assignments for all compounds can be found in Supplementary Tables 2, 4, 6, and 8–13.
3D Structure calculations of canucins.
A NOESY spectrum of canucin A acquired in CD3OH at 295 K with a mixing time of 500 ms exhibited the greatest number of correlations, while avoiding spin diffusion, and was therefore used for structure calculations. Cross-peak positions and volumes in this spectrum were measured in MestReNova and assigned manually. These were given as initial input data for the calculations, which were performed in CYANA 2.1 on a Linux cluster. The isopeptide bond was incorporated via explicit distance constraints for the N–C bond between the N of Gly1 and the Cγ of Asp8. Specifically, both upper and lower limits for the N–Cγ bond length were set to 1.4 Å, with weighting factors of 1.00. These distances were based on the average bond length of an amide bond. The unnatural amino acid β-OH-Asp in canucin A was encoded into the CYANA residue library using CYLIB software57. Seven cycles of combined NOESY assignment and structure calculation were performed, followed by a final structure calculation. Calibration parameters for extraction of distance constraints from cross-peak volumes were determined automatically. For each cycle and for the final calculation, 100 initial conformers were generated, and a simulated annealing schedule, composed of 10000 torsion angle dynamic steps, was applied to each conformer. Statistics were generated for the 10 conformers with the lowest final target functions (see Supplementary Table 3). The calculated conformers were visualized in PyMoL.
Assignment of the location of sugars on keratinimicin A.
The location of mono- and di-saccharide substituents was assigned based on 1D/2D NMR spectra: Sugar Cg (see Supplementary Table 6) was easily assigned by observation of HMBC correlations between Cg1 and Cβ of the amino acid residue (residue C). Specifically, two key HMBC correlations were observed: Cg1-1H at 4.67 ppm correlated with Cβ at 74.8 ppm; and Cβ1-1H at 5.11 ppm correlated with Cg1 at 94.2 ppm).
For sugars Dg and Dgꞌ, we first confirmed the assignments of the amino acids in the peptide scaffold and subsequently assigned the sugar signals. Two signals with shifts at 5.17 and 5.65 ppm were assigned as side-chain 1Hs on residue D. These showed HMBC correlations to the α-carbon of residue D at 54.6 ppm as well as with an aromatic carbon at 132.4 ppm (C4, residue D), in addition to 3-bond correlations to the opposing C2 (104.9 ppm) and C6 (108.4 ppm). They also weakly interacted with each other, characteristic of a W-coupling interaction. ROSEY interactions from these protons (i.e. to the C3-1H and C5-1H on residue E at 7.22 and 7.13 ppm, respectively) were all consistent with the assignment of these signal as 1Hs on the residue D side-chain. A signal (δH/δC: 5.67/99.8) was found to correlate with C4 on residue D (132.4 ppm). Additional COSY and HMBC correlations from this signal were consistent with its assignment of the anomeric-position on sugar Dg. This C1-1H on sugar Dg was found to correlate to another spin system via HMBC, specifically a signal with δH/δC of 5.18/100.6 ppm. This signal was assigned to the anomeric position on sugar Dg’, an assignment consistent with COSY and HMBC data on this Dg spin system.
After assigning two of the three overlapping signals (δH/δC: 5.17/104.9 & 5.18/100.6), the remaining one (5.18/98.1 ppm) was assigned to the anomeric position on sugar Ag, consistent among others with an HMBC correlation from C1-1H on Ag to C5 (155.2 ppm) on the residue A side-chain. A similar process was used to assign the location of sugars in keratinimicin B–D.
Assignment of β-OH stereochemistry in keratinimicin A.
To assign the β-OH stereocenter in keratinimicin A, we noted NOE correlations from the β−1H of residue C (5.11 ppm) to the α−1H (4.23 ppm) and aromatic C2-1H (7.89 ppm) on residue C, but not to the aromatic C6-1H (7.36 ppm), indicating that the Cα−1H, Cβ−1H, and C2-1H were close in space. The α-carbon is in the S-configuration, as deduced from NRPS domain analysis of the ker gene cluster. This information together led us to propose that the R-configuration for the β−1H on residue C. A similar analysis was conducted with residue E: The β−1H (5.20 ppm) showed a strong NOE with the α−1H (4.52 ppm) and the aromatic C6-1H (7.05 ppm), but not the C2-1H (7.78 ppm), suggesting the α−1H, Cβ−1H, and C6-1H were close in space. Given the R-configuration of the α-carbon on residue E, we predict the R-configuration for its β-carbon.
Genomic DNA isolation and sequencing.
A. keratiniphila was cultured in 25 mL YEME medium for 2 days and the mycelium was subsequently harvested to isolate the genomic DNA using the Promega Wizard Genomic DNA Purification Kit as per manufacturer’s instructions. Genomic DNA of high quality was obtained at a concentration of 990 ng/μL and a UV260/280 value of 1.8.
To sequence the ker gene cluster, genomic DNA was submitted to the Lewis Sigler Institute Sequencing Core Facility, where short DNA fragment libraries were prepared via an Illumina MiSeq Reagent Kit, and the fragments then sequenced on an Illumina MiSeq System. The raw sequence data were assembled with Unicycler and SPAdes software. A total of 229 contigs covering 9.1 Mbp were obtained. Genome annotation was carried out via the RAST server 2.0. The data were then searched using the OxyB sequence from A. orientalis as a query, which allowed us to identify the ker cluster. By examining the sequence 75 kb upstream and downstream of the OxyB homolog, we were able to assign the cluster boundaries. Predicted protein functions were assigned using the IMG and antiSMASH databases. The sequence for the whole ker gene cluster was uploaded to NCBI (accession number MH428036).
Antibacterial and antiviral assays.
Antibacterial assays were carried out by Micromyx, LLC in accordance with methods from the Clinical and Laboratory Standards Institute. Minimal inhibitory concentrations were determined with the following strains (listed in Table 1 and Supplementary Table 14): S. aureus ATCC 29213, S. aureus MMX 2011, S. pneumoniae ATCC 49619, S. pyogenes MMX 6253, S. agalactiae MMX 6189, E. faecalis ATCC 29212, E. faecalis MMX 486, B. subtilis ATCC 6633, E. coli ATCC 25922, K. pneumoniae MMX 214, P. aeruginosa ATCC 27853, A. baumannii ATCC 19606, V. cholerae BAA-2163, C. difficile ATCC 700057, and B. fragilis ATCC 25285.
Antiviral assays were performed by Virapur in accordance with methods from the Clinical and Laboratory Standards Institute. Minimal inhibitory concentrations were determined with the following viruses and host cells (listed in Table 1 and Supplementary Table 14): Influenza A/Perth/16/2009 in MDCK cells, Influenza B/Wisconsin/1/2010 in MDCK cells, Herpes Simplex 1 Strain MacIntyre in Vero cells, Herpex Simplex 2 Strain G in Vero cells, Vaccinia virus WR in Vero cells, Rhinovirus 8 in HeLa cells and Respiratory Syncytial Virus in Hep2 cells.
All assays (antibacterial and antiviral) were carried out in triplicates and yielded identical MIC values for all replicates (Table 1). As such a range or an error was not available to report.
Statistics and Reproducibility.
All HiTES-IMS screens (Figs. 2a, 3a, 4a, Supplementary Figs. 2, 4) were carried out in a single replicate; production of the desired cryptic metabolites was validated in three independent biological replicates (Figs. 2c, 3b, 4b,). All replicates gave similar levels of induction of orfamides, canucins, keratinimicins, and keratinicyclins with the respective elicitors. Note that Figs. 3b and 4b are 2D slices from the 3D plots in Figs. 3a and 4a, respectively. Full 1D/2D NMR datasets for canucins, keratinimicins, and keratinicyclins (Supplementary Figs. 3, 5, 6 and Supplementary Tables 2, 4, 6, 8–13) were collected once. Antibacterial and antiviral MIC measurements were carried out in three independent replicates and identical MIC values were obtained in all cases; these are listed in Table 1 and Supplementary Table 14.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the National Institutes of Health (DP2-AI-124786 to M.R.S.), the Burroughs Wellcome Fund, and the Princeton IP Accelerator Fund for support of this work. K.M.D. was supported by an Arnold O. Beckman Postdoctoral Fellowship. L.B.B. was supported by a National Science Foundation Graduate Research Fellowship.
Footnotes
DATA AVAILABILITY
The data supporting the findings of this study are available within the paper and the supplementary material. NMR data used to characterize the cryptic metabolites are available from the corresponding author upon reasonable request. The DNA sequence of the ker gene cluster from A. keratiniphila has been submitted to GenBank (accession number MH428036). The LAESI-IMS data for S. canus and A. keratiniphila, including the raw data for Figs. 3a and 4a as well as the source code used to generate the 3D plots, have been submitted to GNPS (accession number MassIVE MSV000082658).
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Newman DJ & Cragg GM Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Cragg GM & Newman DJ Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta. 1830, 3670–3695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cragg GM, Grothaus PG & Newman DJ Impact of natural products on developing new anti-cancer agents. Chem. Rev. 109, 3012–3043 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Bentley SD et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Ikeda H et al. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 21, 526–531 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Oliynyk M et al. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol. 25, 447–453 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Nett M, Ikeda H & Moore BS Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26, 1362–1384 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X & Cheng YQ Genome-guided discovery of diverse natural products from Burkholderia sp. J. Ind. Microbiol. Biotechnol. 41, 275–284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baltz RH Gifted microbes for genome mining and natural product discovery. J. Ind. Microbiol. Biotechnol. 44, 573–588 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Okada BK & Seyedsayamdost MR Antibiotic dialogues: induction of silent biosynthetic gene clusters by exogenous small molecules. FEMS Microbiol. Rev. 41, 19–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochi K & Hosaka T New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl. Microbiol. Biotechnol. 97, 87–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutledge PJ & Challis GL Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 13, 509–523 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Zhu H, Sandiford SK & van Wezel GP Triggers and cues that activate antibiotic production by actinomycetes. J. Ind. Microbiol. Biotechnol. 41, 371–386 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Nah HJ, Pyeon HR, Kang SH, Choi SS & Kim. E.S. Cloning and heterologous expression of a large-sized natural product biosynthetic gene cluster in streptomyces species. Front. Microbiol. 8, 394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren H, Wang B & Zhao H Breaking the silence: new strategies for discovering novel natural products. Curr. Opin. Biotechnol. 48, 21–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon V & Nodwell JR Activating secondary metabolism with stress and chemicals. J. Ind. Microbiol. Biotechnol. 41, 415–424 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Guo F et al. Targeted activation of silent natural product biosynthesis pathways by reporter-guided mutant selection. Metab. Eng. 28, 134–142 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Seyedsayamdost MR High-throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc. Natl. Acad. Sci. USA 111, 7266–7271 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu F, Nazari B, Moon K, Bushin LB & Seyedsayamdost MR Discovery of a cryptic antifungal compound from Streptomyces albus J1074 using high-throughput elicitor screens. J. Am. Chem. Soc. 139, 9203–9212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen PC & Seyedsayamdost MR Though much is taken, much abides: finding new antibiotics using old ones. Biochemistry 56, 4925–4926 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watrous AD & Dorrestein PC Imaging mass spectrometry in microbiology. Nat. Rev. Microbiol. 9, 683–694 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross H et al. The genomisotopic approach: a systematic method to isolate products of orphan biosynthetic gene clusters. Chem. Biol. 14, 53–63 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Nemes P & Vertes A Laser ablation electrospray ionization for atmospheric pressure, in vivo, and imaging mass spectrometry. Anal. Chem. 79, 8098–8106 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Li H, Balan P & Vertes A Molecular imaging of growth, metabolism, and antibiotic inhibition in bacterial colonies by laser ablation electrospray ionization mass spectrometry. Angew. Chem. Int. Ed. Engl. 55, 15035–15039 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Fincher JA et al. Enhanced sensitivity and metabolite coverage with remote laser ablation electrospray ionization-mass spectrometry aided by coaxial plume and gas dynamics. Analyst 142, 3157–3164 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Li H & Vertes A Solvent gradient electrospray for laser ablation electrospray ionization mass spectrometry. Analyst 142, 2921–2927 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Heinemann B, Kaplan MA, Muir RD & Hooper IR Amphomycin, a new antibiotic. Antibiot. Chemother. 3, 1239–1242 (1953). [PubMed] [Google Scholar]
- 28.Bodanszky M, Sigler GF & Bodanszky A Structure of the peptide antibiotic amphomycin. J. Am. Chem. Soc. 95, 2352–2357 (1973). [DOI] [PubMed] [Google Scholar]
- 29.Maksimov MO, Pan SJ & Link AJ Lasso peptides: structure, function, biosynthesis, and engineering. Nat. Prod. Rep. 29, 996–1006 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Hegemann JD, Zimmermann M, Xie X & Marahiel MA Lasso peptides: an intriguing class of bacterial natural products. Acc. Chem. Res. 48, 1909–1919 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Güntert P, Mumenthaler C & Wüthrich K Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Herrmann T, Güntert P & Wüthrich K Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR. 24, 171–189 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Zhu S et al. Insights into the unique phosphorylation of the lasso peptide paeninodin. J. Biol. Chem. 291, 13662–13678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tietz JI et al. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol. 13, 470–478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price JC, Barr EW, Tirupati B, Bollinger JM Jr. & Krebs C The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Krebs C, Galonić Fujimori D, Walsh CT & Bollinger JM Jr. Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res. 40, 484–492 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tiwari K & Gupta RK Rare actinomycetes: a potential storehouse for novel antibiotics. Crit. Rev. Biotechnol. 32, 108–132 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Nicolaou KC, Boddy CN, Bräse S & Winssinger N Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. Engl. 38, 2096–2152 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Hubbard BK & Walsh CT Vancomycin assembly: nature’s way. Angew. Chem. Int. Ed. Engl. 42, 730–765 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Shen B, Liu W & Nonaka K Enediyne natural products: biosynthesis and prospect towards engineering novel antitumor agents. Curr. Med. Chem. 10, 2317–2325 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Everest GJ & Meyers PR Evaluation of the antibiotic biosynthetic potential of the genus Amycolatopsis and description of Amycolatopsis circi sp. nov., Amycolatopsis equina sp. nov. and Amycolatopsis hippodromi sp. nov. J. Appl. Microbiol. 111, 300–311 (2011).21615633 [Google Scholar]
- 42.Heald SL, Mueller L & Jeffs PW Actinoidins A and A2: structure determination using 2D NMR methods. J. Antibiot. 40, 630–645 (1987). [DOI] [PubMed] [Google Scholar]
- 43.Berdnikova TF, Lomakina NN & Potapova NP Structure of actinoidins A and B. Antibiotiki 27, 252–258. [PubMed] [Google Scholar]
- 44.Diekema DJ & Jones RN Oxazolidinone antibiotics. Lancet 358, 1975–1982 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Jorquera PA & Tripp RA Respiratory syncytial virus: prospects for new and emerging therapeutics. Expert Rev. Respir. Med. 11, 609–615 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Yang YL, Xu Y, Straight P & Dorrestein PC Translating metabolic exchange with imaging mass spectrometry. Nat. Chem. Biol. 5, 885–887 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kersten RD et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat. Chem. Biol. 7, 794–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Traxler MF, Watrous JD, Alexandrov T, Dorrestein PC & Kolter R Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. MBio 4, e00459–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du L et al. Unique amalgamation of primary and secondary structural elements transform peptaibols into potent bioactive cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 114, E8957–E8966 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Craney A, Ozimok C, Pimentel-Elardo SM, Capretta A & Nodwell JR Chemical perturbation of secondary metabolism demonstrates important links to primary metabolism. Chem. Biol. 19, 1020–1027 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Goodwin CR et al. Structuring microbial metabolic responses to multiplexed stimuli via self-organizing metabolomics maps. Chem. Biol. 22, 661–670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okada BK, Wu Y, Mao D, Bushin LB & Seyedsayamdost MR Mapping the trimethoprim-induced secondary metabolome of Burkholderia thailandensis. ACS Chem. Biol. 11, 2124–2130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davies J, Spiegelman GB & Yim G The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol. 9, 445–453 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Yim G, Wang HH & Davies J Antibiotics as signalling molecules. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 362, 1195–1200 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romero D, Traxler MF, López D & Kolter R Antibiotics as signal molecules. Chem. Rev. 111, 5492–5505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
ONLINE METHODS REFERENCES
- 56.Hu H & Ochi K Novel Approach for Improving the Productivity of Antibiotic-Producing Strains by Inducing Combined Resistant Mutations. Appl. Environ. Microbiol. 67, 1885–1892 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yilmaz EM & Güntert P NMR structure calculation for all small molecule ligands and non-standard residues from the PDB Chemical Component Dictionary. J. Biomol. NMR. 63, 21–37 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.