

Abstract
Phosphorylation of the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II (Pol II) governs stage-specific interactions with different cellular machines. The CTD consists of Y1S2P3T4S5P6S7 heptad repeats, and sequential phosphorylations of Ser7, Ser5 and Ser2 occur universally across Pol II-transcribed genes. Phosphorylation of Thr4, however, appears to selectively modulate transcription of specific classes of genes. Here, we identify 10 new Thr4 kinases from different kinase structural groups. Irreversible chemical inhibition of the most active Thr4 kinase, Hrr25, reveals a novel role for this kinase in transcription termination of specific class of noncoding snoRNA genes. Genome-wide profiles of Hrr25 reveal a selective enrichment at 3ʹ regions of noncoding genes that display termination defects. Importantly, phospho-Thr4 marks placed by Hrr25 are recognized by Rtt103, a key component of the termination machinery. Our results suggest that these uncommon CTD kinases selectively place phospho-Thr4 marks to regulate expression of targeted genes.
Introduction
In budding yeast, Saccharomyces cerevisiae, the largest subunit of RNA polymerase II (Pol II) bears twenty-six tandemly repeating Y1S2P3T4S5P6S7 heptapeptides1. Sequential post-translational modifications of these residues serve as a scaffold to recruit stage-specific protein complexes to the polymerase2–4. These complexes facilitate multiple processes, including Pol II function through different stages of the transcription cycle, co–transcriptional processing and transport of nascent RNA, remodeling and modification of the underlying chromatin template and even displacement or proteolysis of polymerase that is stalled at sites of DNA damage2–5.
Pol II is globally recruited to gene promoters by general transcription factors (GTF) and the Mediator complex6. Kin28/CDK7, the kinase subunit of TFIIH, phosphorylates Ser5 and Ser7 during transcription initiation7,8. Phospho-Ser5 (pSer5) and phospho-Ser7 (pSer7) marks recruit the capping enzyme complex9,10, which places the m7G cap on the nascent RNA as it emerges from the Pol II exit tunnel. Following promoter clearance, kinase Bur1/CDK9 is recruited by pSer511, and phosphorylates Ser212, priming the CTD for subsequent Ser2 phosphorylation by Ctk1/CDK12/1313. pSer2 marks are critical for productive elongation and recruitment of factors involved in histone modification, co-transcriptional splicing, and transcription termination2–5. This sequential pattern of CTD phosphorylation and stage-specific association of different cellular machines is observed globally at Pol II transcribed genes.
The role of phospho-Thr4 (pThr4), however, remains less well defined. Recent studies have identified a global role for pThr4 in supporting transcription elongation of protein-coding genes14, however, this mark appears to be more closely tied to gene class specific roles, such as 3ʹ end processing of histone mRNA in chicken cells but not in all eukaryotes15, regulating distinct signal-responsive genes in budding versus fission yeast16,17, pre-mRNA splicing18, termination of snoRNA transcripts19, and progression through M phase20. The diverse sets of genes that are tied to pThr4 marks suggests the intriguing possibility that phosphorylation of this position in the CTD heptad provides a means to modulate Pol II activity at targeted genes without perturbing the global patterns of CTD modifications across the genome.
CDK915 and PLK314 were previously identified as Thr4 kinases in chicken cells and human cells, respectively; however, CDK9 can phosphorylate four of the five hydroxyl-bearing residues of the CTD heptad12,15,21,22, and can associate globally with elongating Pol II. While PLK3 assists in transcriptional elongation at protein-coding genes, it is most enriched within nucleoli. Thus, identification of these Thr4 kinases has not shed light on the selective placement and role of pThr4 in regulating transcription of distinct gene-classes. Previous work has shown that CTD kinases recruited to a subset of genes play gene class specific roles23. In this context, we explored the hypothesis that several yet-to-be discovered CTD kinases may be recruited to specific genes to phosphorylate Thr4 and confer gene-selective functions of pThr4.
To identify new Thr4 kinases, we over half of the yeast kinome, examining 70 kinases from all structural groups, and identified 10 that phosphorylate Thr4. Strikingly, many newly identified Thr4 kinases belong to structural groups not known to phosphorylate the CTD. We utilized our newly developed covalent24, as well as well-established non-covalent chemical-genetic25 approaches to conditionally attenuate activity of a select set of Thr4 kinases in vivo. Selectively inhibiting the most active Thr4 kinase, Hrr25, results in termination defects at small nucleolar RNA (snoRNA) transcripts and elongation defects at protein-coding genes. We show that Hrr25 is significantly enriched at the 3ʹ ends of snoRNAs and to a lesser extent across the length of protein-coding genes. Moreover, an important component of the termination machinery binds to Thr4 phosphorylation marks placed by Hrr25. Consistent with a role in transcription termination, irreversible chemical-genetic inhibition of Hrr25 results in: (1) reduced pThr4 levels, (2) delay in engagement of the termination machinery, and (3) defective termination of select snoRNA transcripts. In essence, we report 10 new Thr4 kinases and provide a mechanistic basis for gene-class specific placement and function of the sparingly understood pThr4 mark.
Results
Discovery of new Thr4 kinases
Previous studies reported that Thr4 is phosphorylated by PLK3 in humans14 and by CDK9 in humans and chickens15,26. The ubiquitous association of CDK9 with Pol II at most genes does not readily explain why phosphorylation of Thr4 is essential for regulation of specific sets of signal-responsive genes16,17. We reasoned that additional, as of yet undiscovered, signal/condition-responsive kinases phosphorylate Thr4 leading to differential use of this mark at differentgenes. To test this possibility, 70 of 131 yeast kinases with diverse cellular functions were purified from cells grown in rich medium8 (Fig. 1a, Supplementary Fig. 1a). This set of kinases comprises all nuclear kinases, as well as kinases with diverse cellular functions (e.g., signaling, cell cycle, etc.). Further, while most known CTD kinases are part of the CMGC kinase group, we investigated the possibility that kinases from other structural groups could phosphorylate the CTD. From this diverse set of 70 kinases, we identified 11 that phosphorylate Thr4 at least four-fold above background (Fig. 1b, Supplementary Fig. 1b). Hrr25, Yck2, Yck3, Cmk1, Cmk2, Pho85, Rim11, Ssk2, and Sln1 are novel CTD kinases, whereas Cdc28 is known to promote Ser5 phosphorylation at a specific set of cell cycle genes27. Bur1 is well known to phosphorylate all three Ser residues (Ser2, Ser5 and Ser7) of the CTD, and like CDK9, its human homolog, it phosphorylates Thr412,15,21,28. We also tested the ability of these new Thr4 kinases to phosphorylate other residues on the CTD and discovered the ability of Pho85 to phosphorylate Ser2 and Ser5 residues, and the ability of Cdc28 to phosphorylate the Ser2 residue of the CTD (Supplementary Fig. 1c).
Figure 1 |. Yeast kinases phosphorylate Thr4 in vitro.
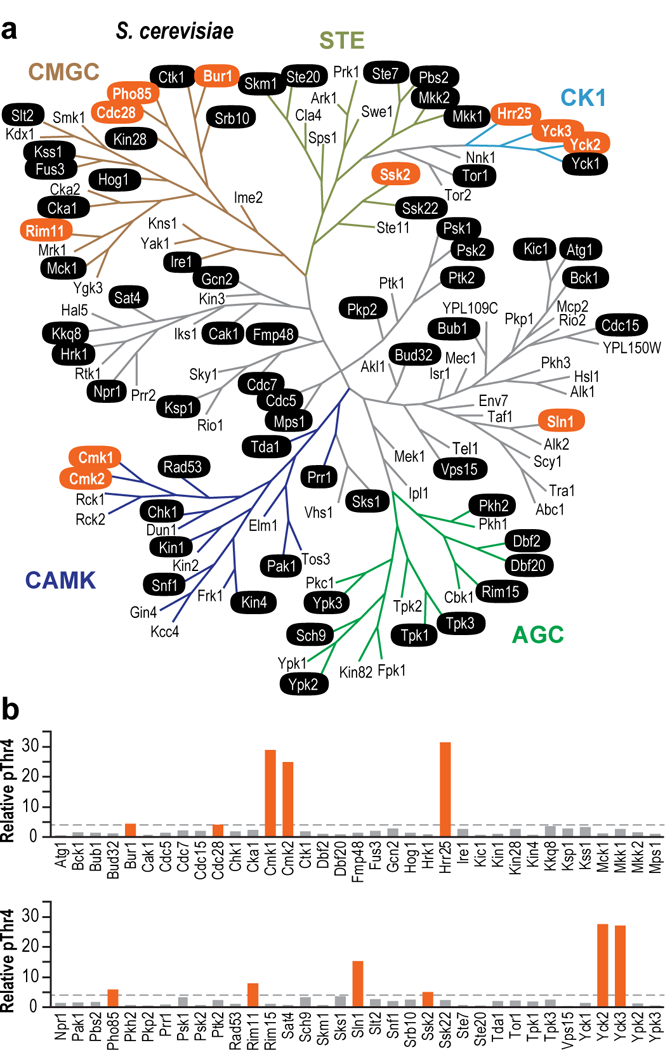
(a) 131 S. cerevisiae kinases organized by maximum likelihood. Colored branches indicate kinase structural groups. In vitro-tested kinases are denoted in black ovals, and significant pThr4 activity is represented in orange ovals. (b) Quantitation of kinase activity is displayed as histograms below the yeast kinome, with orange indicating >4-fold activity over mock (each kinase was tested from at least two independent purifications).
Surprisingly, while PLK3 strongly phosphorylates human Thr414, the yeast ortholog of PLK3, Cdc5, did not phosphorylate Thr4. Further validation of this finding using an alalog sensitive approach suggests that Cdc5 does not phosphorylate Thr4 in vitro or in vivo (Supplementary Fig. 1e). It is important to note that PLKs commonly require substrates with prior phosphorylation marks, and Cdc5 may phosphorylate Thr4 in the presence of a primed substrate.
Three of the most active yeast Thr4 kinases (Hrr25, Yck2, and Yck3) are casein kinases in the CK1 group, whereas the canonical CTD kinases are all cyclin-dependent kinases in the CMGC group. While Hrr25 is known to interact with pSer2/pSer5 CTD29, it was not previously identified as a CTD Thr4 kinase. Instead, this kinase has been implicated in diverse processes including DNA-damage repair30,31, ribosome biogenesis32, meiotic chromosome segregation33, and autophagy34. Another unexpected Thr4 kinase, Pho85, has roles in nutrient and phosphate sensing and cell cycle control35; roles that are consistent with the importance of the pThr4 mark in phosphate metabolism16,17. However, the ability of Pho85 to directly phosphorylate the CTD was previously unknown. Rim 11, another new Thr4 CTD kinase identified in our survey, is required for meiosis36 and ribosome biogenesis37. Finally, four new Thr4 CTD kinases, Ssk2, Cmk1, Cmk2, and Sln1 were implicated in response to multiple environmental stresses38–41. We note that the 70 kinases we tested were purified from cells grown in rich media and it is possible that kinases purified from cells stimulated by different signals and growth conditions may reveal additional signal-responsive Thr4 kinases.
In vivo validation of identified Thr4 kinases
To examine the ability of our newly identified CTD kinases to place the pThr4 mark in vivo, we utilized a chemical-genetic approach that permits in vivo inhibition of a targeted kinase without impacting the activity of the other kinases in the cell. This “bump-hole” approach relies on the substitution of a conserved gatekeeper residue in the ATP binding pocket (e.g. I82 in Hrr25) with a glycine creating a “hole” (Fig. 2a)25. This surgical expansion of the active site does not alter cellular distribution or substrate specificity, but permits ATP analogs bearing a chemical “bump” to selectively dock into and inhibit the engineered “analog sensitive” (-as) kinase.
Figure 2 |. Yeast kinases phosphorylate Thr4 in vivo.
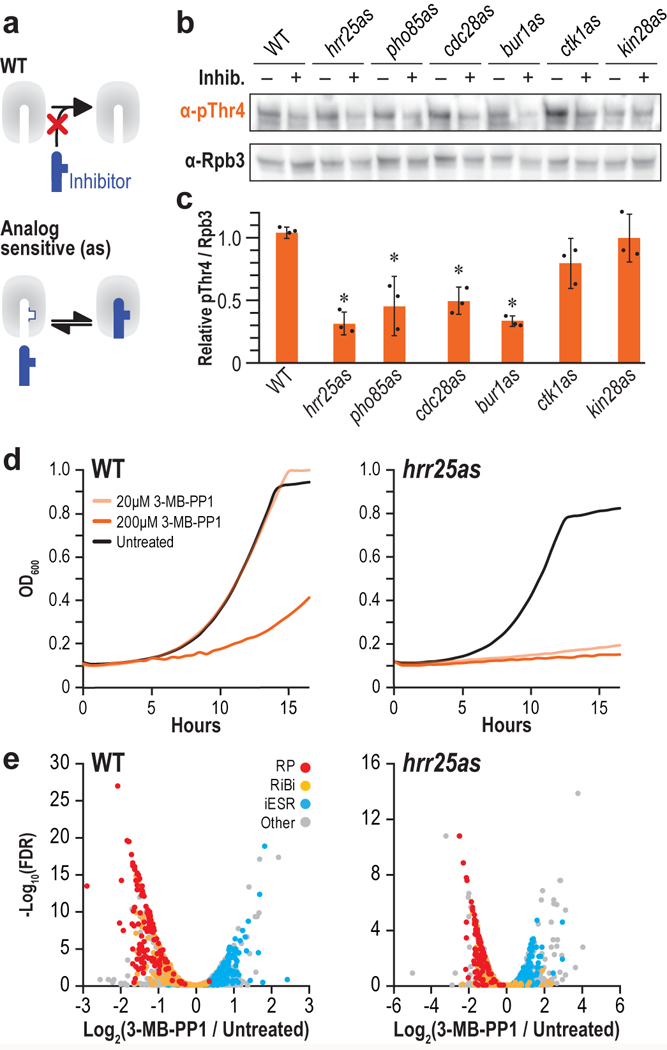
(a) Schematic of the “bump and hole” strategy to inhibit kinases using ATP analogs (blue). (b) Western blots of yeast extract from the indicated strains treated with their respective inhibitors (+) or DMSO (−). Blots were probed with antibodies targeting pThr4 (1G7) or Rpb3 as a loading control. Full length blots are shown in Supplementary Fig. 12. (c) Quantitation of fold change pThr4 when treated with inhibitor, normalized to Rpb3 (n = 3 independent experiments, error bars are S.D., asterisks indicate p < 0.05). (d) Growth curves of WT (left) or hrr25as (right) in the presence of DMSO (black), 20 μM 3-MB-PP1 (light orange), or 200 μM 3-MB-PP1 (dark orange). (n = 3 independent experiments. Representative data from one experiment are shown.) (e) RNA-seq volcano plots showing fold change in expression (x-axis) and significance (y-axis). Ribosome protein genes (red), ribosome biogenesis genes (yellow), and induced ESR genes (blue) are shown for WT (left) or hrr25as (right) cells.
Due to subtle differences in ATP binding sites in different kinases, different cell permeable PP1 analogs show efficacy in docking and inhibiting different analog sensitized kinases. Among the many orthogonal pairs, 3-MB-PP1 non-covalently inhibits hrr25as and bur1as kinases in vivo, 1-NA-PP1 inhibits kin28as, and 1-NM-PP1 inhibits pho85as, cdc28as, and ctk1as21,42–45. Unambiguous reduction in pThr4 levels was observed when the analog-sensitive versions of Hrr25, Pho85, Cdc28, or Bur1 were inhibited with their orthogonally paired small molecule inhibitors (Fig. 2b,c). Parallel treatment of wild type cells bearing unmodified kinases did not result in any reduction of the pThr4 mark. While inhibition of hrr25as and bur1as have similar effects on pThr4 levels, we focused on Hrr25 due to the previously unknown ability of this kinase to phosphorylate the CTD.
Increasing 3-MB-PP1 inhibitor concentration did not eliminate pThr4 marks, but unexpectedly induced toxicity in WT cells (Fig. 2d, left). At 20μM concentrations, 3-MB-PP1 blocked selectively inhibited hrr25as growth but it also induced the global environmental stress response (ESR)46 in WT and hrr25as cells (Fig. 2e).
Rational design of irreversibly sensitized Hrr25
Next, we utilized our recently developed chemical-genetic inhibition strategy that permits irreversible inhibition of the targeted kinase in vivo without the induction of ESR24. In this approach, the “irreversibly sensitized” (-is) kinase is altered at two positions, one of which forms a covalent bond with chloromethylketone (CMK) moiety of a small molecule inhibitor (Fig. 3a). In hrr25is, the I82 gatekeeper residue was substituted with a glycine to allow CMK to selectively bind in the expanded ATP binding pocket of hrr25is, while preventing CMK from entering the active site of any WT kinase (Fig. 3b). Sequence alignment suggested that a cysteine at position 23 (I23C) in the ATP binding pocket would permit the nucleophilic sidechain to covalently conjugate to the chloromethylketone moiety of CMK, thus, irreversibly inhibiting the rationally designed hrr25is kinase (Fig. 3a,b).
Figure 3 |. Irreversible inhibition of Hrr25.
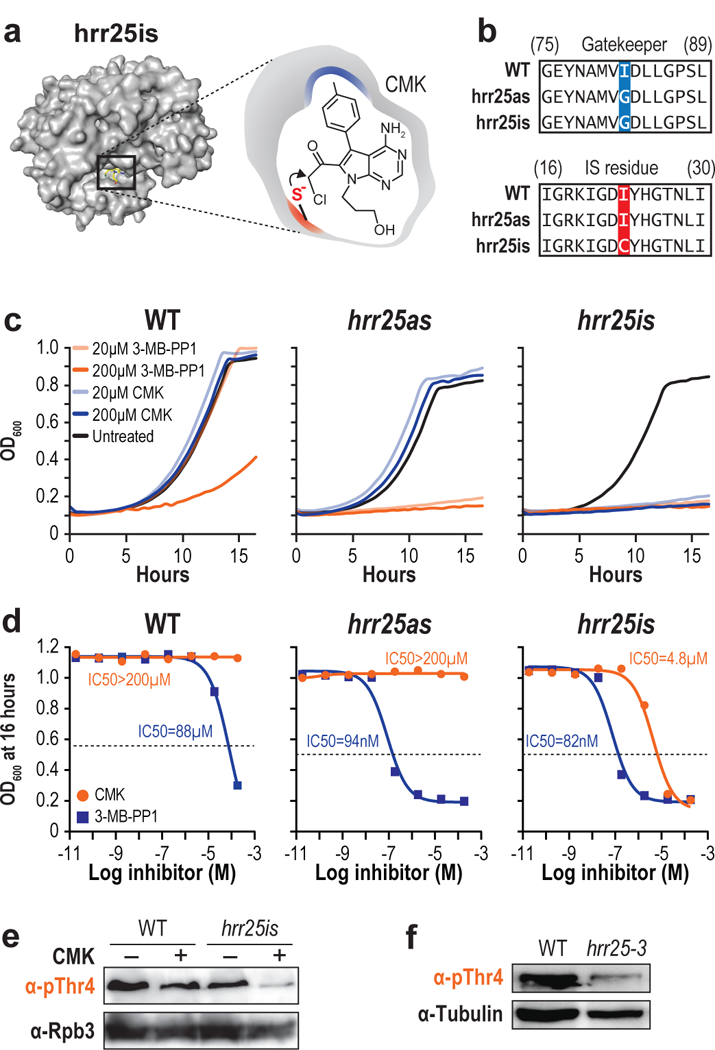
(a) Structure of hrr25is (left) and a schematic of CMK docking (right). (b) Sequence alignment of WT, hrr25as, and hrr25is with the gatekeeper substitution (blue) and “IS residue” substitution (red) shown. (c) Growth curves of WT (top), hrr25as (middle), or hrr25is (bottom) in the presence of DMSO (black), 20 μM 3-MB-PP1 (light orange), 200 μM 3-MB-PP1 (dark orange), 20 μM CMK (light blue), or 200 μM CMK (dark blue). (n = 3 independent experiments. Representative data from one experiment are shown.) (d) WT, Hrr2as, and Hrr25is cells were treated with either CMK or 3-MB-PP1 at concentrations ranging from 20 pM to 200 μM. For each concentration, the OD600 after 16 hours of inhibition is plotted. IC50 values were calculated with GraphPad Prism. (n = 2 independent experiments. Representative data from one experiment are shown.) (e) Western blots of yeast extract from WT or hrr25is cells treated with either DMSO (−) or CMK (+). Blots were probed with antibodies targeting pThr4 (1G7) or Rpb3 as a loading control. (n = 3 independent experiments). (f) Western blots of yeast extract from WT or hrr25–3 cells. Blots were probed with antibodies targeting pThr4 or Tubulin as a loading control. (n = 3 independent experiments). Full length blots are shown in Supplementary Fig. 12.
Modeling CMK in the active site of Hrr25
To determine if CMK would dock in the modified ATP binding pockets or at other unintended sites on the kinase, we modeled CMK with unaltered Hrr25, hrr25as, and hrr25is using AutoDock47. In the cluster of lowest binding energy, the methylphenyl group of CMK fits into the pocket created by I82G substitution. Further, the CMK group is in close proximity to I23C substitution, allowing for efficient covalent crosslinking. (Fig. 3c). Next, we docked 3-MB-PP1 into hrr25as (Supplementary Fig. 3). We observed similar binding of 3-MB-PP1 to the active site of hrr25is, consistent with the ability of 3-MB-PP1 to inhibit growth of hrr25is cells (Fig. 3c).
Next we experimentally validated our docking by calculating the apparent IC50 of CMK or 3-MB-PP1 for WT, hrr25as, and hrr25is cells (Fig. 3d, Supplementary Fig. 4a). As expected, CMK did not inhibit WT or hrr25as cells, but did inhibit hrr25is with an IC50 of 4.8 μM. We observed comparable IC50 values for 3-MB-PP1 of 94 nM and 82 nM for hrr25as and hrr25is cells, respectively. While 3-MB-PP1 appeared more potent than CMK in hrr25is, 3-MB-PP1 inhibits WT cells with an IC50 of 88 μM, suggesting that some of the reduction in growth for hrr25as and hrr25is cells might occur due to general toxicity of the small molecule. In contrast, no general toxicity to WT cells was observed with CMK even at 200μM concentrations (Fig. 3c,d).
Irreversible inhibition of the sensitized Hrr25 allele
No reduction in pThr4 levels were observed when CMK was added to WT cells, but a clear reduction in pThr4 upon inhibition of hrr25is was evident (Fig. 3e). While greatly diminished, inhibition does not eliminate the pThr4 mark. Reassuringly, the reduction in bulk pThr4 levels in chemically inhibited hrr25is strain was comparable to the catalytically inactive hrr25–3 mutant allele (Fig. 3f). To confirm that this reduction is due to kinase inhibition, and not due to an inherent reduction in activity due to the double mutation, we performed in vitro kinase assays with purified WT Hrr25, Hrr25as, and Hrr25is. We observed comparable levels of Thr4 phosphorylation, confirming that the “as” and “is” mutations do not significantly perturb kinase activity in the absence of inhibitor (Supplementary Fig. 1f).
Next, we examined the timeframes within which Bur1 and Hrr25 phosphorylate Thr4. We determined the levels of pThr4 at different time intervals after the chemical-genetic inhibition of bur1as with 3-MB-PP1 and hrr25is with CMK (Supplementary Fig. 4c,d). Consistent with our results from Fig. 2b,c, we observed similar perturbations in the levels of pThr4 at each timepoint, suggesting that the pThr4 activity of Bur1 and Hrr25 are comparable.
As with the covalently inhibited hrr25is strain, we observed slower growth, and diminished pThr4 levels at both protein coding and noncoding genes in the hrr25–3 strain (Supplementary Fig. 5). It remains to be determined whether the residual pThr4 is placed by a general CTD kinase such as Bur1 or if it represents the activity of other unidentified kinases. Interestingly, while CMK treatment of hrr25is results in a complete cessation of growth, hrr25–3 only results in slow growth. We hypothesize that in cells with a chronically defective Hrr25, other kinases can compensate for the lack of Hrr25. With our rapid and targeted chemical-genetic inhibition strategy, this adaptive compensation does not occur quickly enough resulting in lethality.
Hrr25 inhibition leads to snoRNA readthrough
To identify the role of Hrr25-mediated Thr4 phosphorylation, we investigated its impact on the transcriptome by RNA-seq. Upon chemical inhibition of hrr25is, 3ʹ-extensions were observed on many snoRNAs, but not globally at other non-coding or protein-coding RNAs. To calculate the 3ʹ-extension index (EI), we took the fold change in normalized reads in hrr25is between CMK and DMSO treated samples in the 200 base pair window downstream of the processed 3ʹ-ends of snoRNAs, and normalized to WT (Fig. 4a, inset). As an independent validation of our approach, the 3ʹ-extensions in hrr25is were significantly correlated with 3ʹ-extensions in strains with an alanine substitution at Thr4 (T4A) (Supplementary Fig. 6, Supplementary Table 1)19.
Figure 4 |. Hrr25-dependent Thr4 phosphorylation is required for snoRNA termination.
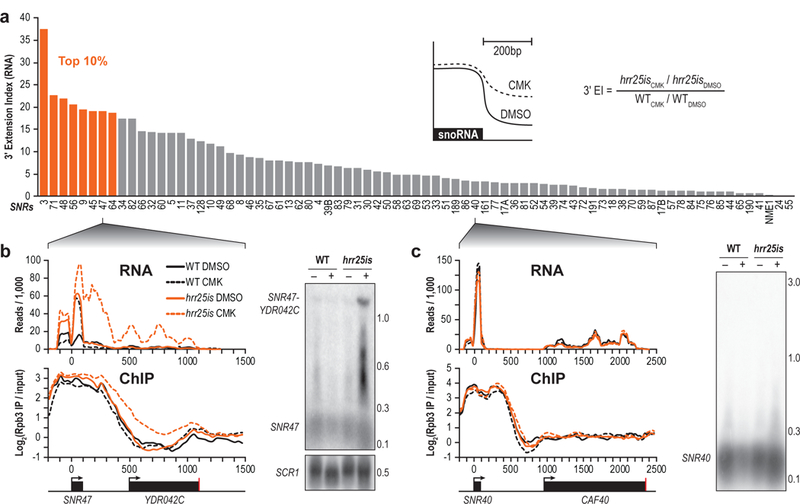
(a) Method used to calculate the 3ʹ-extension index (inset), and Quantification of the 3ʹ extension indices for all snoRNAs. Extension indices in the top 10 percentile are shown in orange. The equation used to calculate the 3ʹ extension index is depicted (right). (b) RNA-seq traces (top) or Rpb3 ChIP (bottom) from WT (black) or hrr25is (orange) cells treated with either DMSO (solid lines) or CMK (dotted lines) at the SNR47 locus. A Northern blot probing for SNR47 is shown (right) with the number of kilobases indicated. Loading control, SCR1, is shown. (n = 2 independent experiments). (c) Similar data are shown at the SNR40 locus. (n = 2 independent experiments). Full length blots are shown in Supplementary Fig. 12.
We previously reported that in cells with Pol II bearing T4A substitutions, many non-coding snoRNAs used the cleavage and polyadenylation signal (CPS) of a downstream protein coding gene to terminate transcription19. In hrr5is cells treated with CMK, we observed similar readthrough at several snoRNA genes. SNR47 displayed a strong readthrough defect and appeared to terminate at the CPS of YDR042C (Fig. 4b, top). Northern blots confirm this 3ʹ extension (Fig. 4b, right), and interestingly, intermediate bands on the Northern blot at around 0.4 and 0.6 kb appear to correlate with strong dips in the RNA-seq traces, suggesting potential additional Pol II termination or pause sites. Using Rpb3 ChIP, we also observed retention of Pol II downstream of SNR47 and through YDR042C, further confirming readthrough upon inhibition of hrr25is cells (Fig. 4b, bottom).
While we previously observed strong readthrough defects of SNR13 in T4A19, SNR13 in hrr25is was not read through all the way to the downstream termination site. Instead, we observed a 3ʹ extension that appeared to terminate at the transcription start site (TSS) of the next gene (Supplementary Fig. 7, top). Consistent with this observation, Pol II was retained until the TSS (Supplementary Fig. 7, bottom), and Northern blots confirm this 0.3 kb transcript when hrr25is was inhibited with CMK (Supplementary Fig. 7, right). Efficient termination was observed at some snoRNAs. SNR40 was properly terminated shown by RNA-seq, Northern blot, and Pol II ChIP (Fig. 4c), consistent with the proper termination observed in T4A19.
To examine whether pThr4 marks placed by other kinases are also involved in snoRNA termination, we inhibited bur1is cells with 3-MB-PP1 and performed RNA-seq. Surprisingly, we did not observe similar readthrough defects at snoRNAs (Supplementary Figure 6g, Supplementary Table 1), suggesting a gene class specific role for Hrr25 in terminating snoRNA transcripts.
Genome-wide Hrr25 profiles mirror RNA Pol II profiles
Since Hrr25 is known to associate with Pol II bearing pSer2 and pSer5 marks29, we compared the average ChIP occupancy of Hrr25 and Rpb3 across each gene. Indeed, we observed a strong correlation (R = 0.74, p < 0.00001) (Supplementary Fig. 8a), suggesting that Hrr25 associates with the CTD of actively transcribing Pol II across most genes. Therefore, we normalized Hrr25 to Rpb3 and clustered profiles using k-means clustering (Fig. 5a). We examined the gene ontology associated with each of these clusters and found that Cluster 2, in which Hrr25 was enriched near the TSS, was particularly rich in genes known to respond to DNA damage stimulus (Fig 5a, Supplementary Fig. 8b). This is consistent with a known role for Hrr25 in responding to DNA damage30,31. Further, Clusters 4 and 5 are enriched with genes involved in ribosome biogenesis, consistent with a role for Hrr25 in ribosome assembly32.
Figure 5 |. Hrr25 profiles correlate with Pol II at protein coding genes.
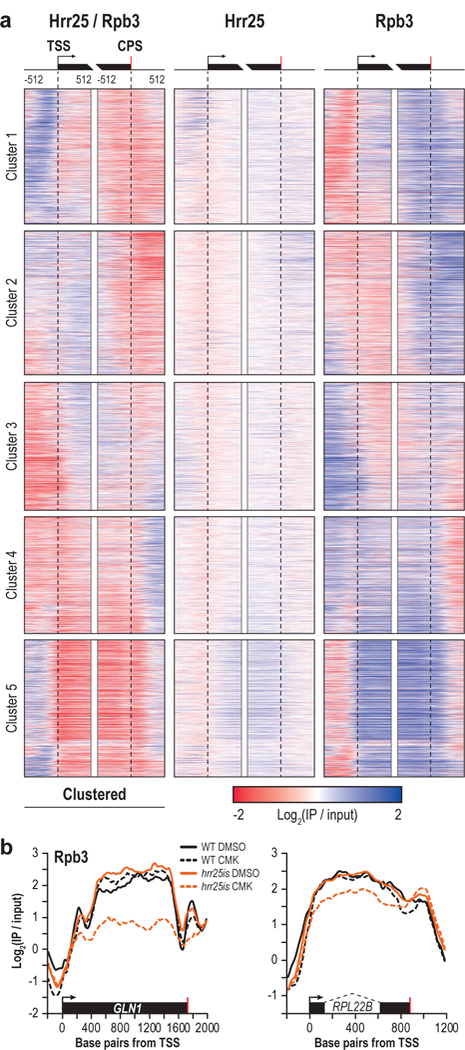
(a) Heatmap of Hrr25 ChIP normalized to Rpb3 flanking the TSS (arrow) and CPS (red bar) of protein coding genes (left). Genes were sorted into five clusters via k-means clustering. Heatmap of Hrr25 (middle) or Rpb3 (right) at the same genes are shown. (b) Representative Rpb3 ChIP traces in WT (black) or hrr25is (orange) cells treated with either DMSO (solid lines) or CMK (dotted lines) at GLN1 (left) and RPL22B (right). Dotted lines indicate an intron.
To investigate the genome-wide consequences of hrr25is inhibition on transcription, we performed Pol II ChIP-seq in strains treated with CMK. Pol II levels across some protein coding genes were dramatically reduced (Fig. 5b) suggesting a potential role for Hrr25 in transcription elongation. A role for pThr4 in elongation of protein coding genes is consistent with previous results suggesting a similar role (mediated by PLK3) in human cells14. Ribosome protein genes were significantly enriched (p < 1e-14) among genes with elongation defects upon hrr25is inhibition, further linking Hrr25 to ribosome biogenesis.
Gene-class specific differences in Hrr25 occupancy
While Hrr25 associated with elongating Pol II, our results indicated a dramatic difference in Hrr25 levels at protein-coding versus non-coding genes, the two major classes of Pol II transcribed genes. We averaged the Hrr25 ChIP signals flanking the 3ʹends of all protein-coding genes, and while some genes displayed increased Hrr25 levels (Fig. 5a), overall Hrr25 levels were quite low in comparison to snoRNA genes (Fig. 6a).
Figure 6 |. Hrr25-dependent Thr4 phosphorylation recruits RTT103.
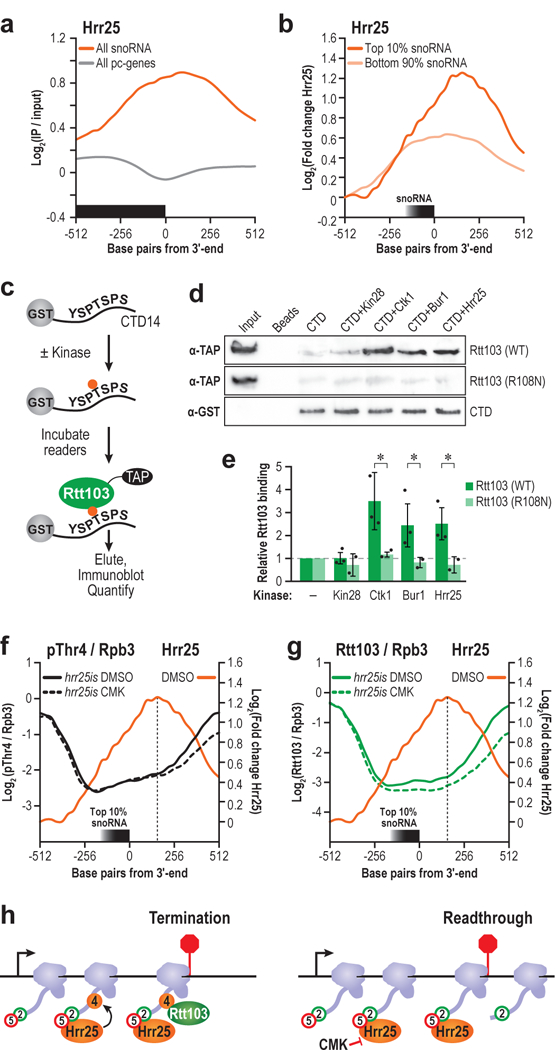
(a) Hrr25 ChIP centered at the 3ʹ end of snoRNA genes (orange) or protein coding genes (grey). (b) Hrr25 ChIP centered at the 3ʹ end of the snoRNAs with the top 10% 3ʹ extension indices (dark orange) or bottom 90% (light orange) (c) Schematic of assay used to confirm direct binding. (d) Rtt103 (WT) or Rtt103 (R108N) was incubated with kinase-treated or mock-treated beads, eluted, and analyzed via western blot. α-GST CTD loading control is shown. Input refers to unbound Rtt103, loaded at 10% the abundance of Rtt103 incubated with CTD. Full length blots are shown in Supplementary Fig. 12. (e) Quantification of the fold change in Rtt103 vs. the beads only control for unphosphorylated CTD, or kinase-treated CTD is shown. (n=3 independent experiments, error bars are S.D., * p<0.05). (f) Profiles of pThr4 normalized to Rpb3 in hrr25is treated with either DMSO (solid black) or CMK (dotted black) at the snoRNAs with the highest 3ʹ extension indices. Hrr25 profile is overlaid (orange) (g) Similar profiles of Rtt103 in hrr25is treated with either DMSO (solid green) or CMK (dotted green), with the Hrr25 profile overlaid (orange). (h) Model of role of Hrr25 in terminating snoRNA genes. Arrow is TSS, and the red bar is the 3ʹ end. Hrr25 is recruited by pSer5 and pSer2 marks to phosphorylate Thr4. Rtt103 binds pThr4 and terminates transcription.
Closer inspection of Hrr25 profiles across snoRNAs with high readthrough (top 10% 3ʹextension indices) vs low readthrough (bottom 90% 3ʹextension indices) (Fig. 4a), revealed higher levels of Hrr25, and a slight 3ʹoffset at the snoRNAs that show maximal termination defects (Fig. 6b). These results indicate that even within this class of non-coding genes, a subset is more sensitive to the activity of Hrr25-dependent levels of pThr4.
Hrr25-dependent pThr4 marks are read by Rtt103
Next, we asked if the pattern of pThr4 marks placed by Hrr25 on the CTD is recognized by a bona fide “reader” of these marks. We previously showed that the termination factor Rtt103 interacts with the CTD phosphorylated at Thr419. We examined the extent to which Hrr25 kinase activity contributes to Rtt103 association by binding a GST-CTD fusion protein phosphorylated by four CTD kinases (Fig. 6c). Significant binding was observed between Rtt103 and the CTD phosphorylated by Hrr25, confirming that the pattern of pThr4 marks placed by Hrr25 on the CTD is specifically recognized by Rtt103 (Fig. 6d,e). Consistent with our prior observations, Ctk1-treated CTD (phosphorylated at Ser2) (Supplementary Fig. 9) bound Rtt103 slightly better than Hrr25-treated CTD19. Bur1-treated CTD bound Rtt103 to a comparable level as Hrr25, and as expected, Rtt103 did not bind to pSer5/pSer7-CTD, phosphorylated by Kin28. Arg108 in Rtt103 enables binding to pThr4 and pSer2, and substituting this residue dramatically compromises binding to CTD phosphorylated by any of the four CTD kinases tested (Fig. 6e).
Next, we analyzed the pThr4 levels (normalized to Pol II) in hrr25is cells treated with CMK or the solvent control (DMSO). When treated with CMK, pThr4 levels decreased directly underneath the Hrr25 peak (~200 bp) downstream of the 3ʹend of SNR genes with high extension indices (Fig. 6f). We inspected Rtt103 levels (normalized to Pol II) at the same set of genes and observed a similar reduction upon hrr25is inhibition (Fig. 6g). Further, at representative protein coding gene GLN1, we observed reduced pThr4 levels 600bp upstream of the CPS, and a concomitant reduction in Rtt103 levels ~100bp upstream of the CPS (Supplementary Fig. 10). These data show a clear correlation between hrr25is inhibition, the deposition of pThr4, and the levels of Rtt103 across a gene. We propose that this reduction in Rtt103 contributes to the observed termination defects (Fig. 6h).
Discussion
Using an unbiased activity-based kinase assay, we surveyed 70 yeast kinases encompassing all kinase structural group and over half the kinome for their ability to phosphorylate Thr4 of the Pol II CTD. Of the 11 hits, we identified 10 new pThr kinases from diverse structural groups and we demonstrated that Bur1, the CDK9 homolog, phosphorylates Thr4. We focused on Hrr25 of the Casein Kinase 1 (CK1) group, a kinase not known to phosphorylate the CTD (Fig. 1a). Akin to the Bur1-mediated “priming” of the CTD for subsequent phosphorylation by Ctk113, kinases in the CK1 group act on pre-phosphorylated substrates48. The substrate recognition motif for CK1 kinases is pS/pT-Xn-S/T, where n = 1–3. Consistent with this mechanism, Hrr25 was previously identified from mass spectrometric studies as a protein that bound to CTD bearing high levels of pSer2 and pSer5 marks29. Borrowing from the parlance of the histone code hypothesis, here we demonstrate that Hrr25 is a “reader-writer” kinase. It “reads” previously placed phospho-CTD marks, and subsequently “writes” additional marks, enabling sequential and therefore temporal encoding of information on the CTD of Pol II at a later stage of productive elongation.
The hitherto recorded physiological roles of Hrr25 are quite diverse. One role of Hrr25 is in mediating cellular response to DNA damage30,31. While it is unclear how the DNA damage signal activates Hrr25, it is thought that Hrr25 phosphorylates the transcription factor Swi6, which induces DNA repair gene synthesis31. Consistent with this role, we observed Hrr25 enrichment patterns upstream of the transcription start sites of DNA repair genes in Cluster 2 (Fig. 5a, Supplementary Fig. 8a). It is possible that Hrr25 is poised at these genes waiting for a DNA damage signal in order to facilitate productive transcription of genes encoding DNA repair proteins. Another well documented biological role of Hrr25 is in ribosome biogenesis32. In support of this role, we observed higher Hrr25 levels across the gene body of Cluster 5 (enriched with genes involved in ribosome biogenesis), closely mirroring Rpb3 occupancy (Fig. 5a). Upon hrr25is inhibition, we observed a reduction in Pol II occupancy across ribosome biogenesis genes (p < 1e-14) (Fig. 5b). This finding when viewed in the context of previous literature on Hrr25 directly phosphorylating a component of the 40S ribosome32 suggests a two-pronged mode of regulation. Hrr25-mediated placement of pThr4 marks on Pol II may modulate the transcription and abundance of ribosome proteins whereas Hrr25-mediated phosphorylation of ribosome subunits regulates the assembly of the translational machinery.
We applied our recently developed covalent inhibition strategy to irreversibly inhibit a sensitized hrr25is allele24. Upon covalent and irreversible inhibition of Hrr25, we observed termination defects at a subset of snoRNA genes. This result shows remarkable agreement with our previous work characterizing snoRNA termination defects in the T4A strain where substitution of Thr4 with alanine eliminates the ability of kinases to place a phospho-Thr4 mark on the Pol II CTD19. More specifically, we compared the 3ʹ extension indices in hrr25is to T4A19 and observed a significant correlation (p = 0.005) (Supplementary Fig. 6a). We further compared the 3ʹ extension indices in hrr25is to T4V18. The extent of readthrough in T4V was over an order of magnitude lower than hrr25is (Supplementary Fig. 6e), suggesting that alanine is a better choice when seeking a non-phosphorylatable threonine mimic. Taken together, our chemical and genetic approaches to attenuate pThr4 marks result in similar outcomes, thereby confirming a gene-class specific role for Hrr25 in snoRNA termination (Fig. 6h). We further provide evidence that the termination factor Rtt103 is specifically recruited to pThr4 marks placed by Hrr25.
In essence, we identify 10 novel Thr4 kinases and reveal that two CTD kinases (Cdc28 and Bur1) also phosphorylate Thr4. In particular, we focused on Hrr25, a kinase long known to interact with the CTD, but never known to phosphorylate Thr429. It remains to be seen if the other Thr4 kinases identified in our screen are responsible for other gene-class specific roles of pThr4 including phosphate metabolism16,17, splicing18, or M-phase progression20. Finally, by “irreversibly sensitizing” (-is) a second kinase, we demonstrate that our structurally guided chemical-genetic approach could be rationally applied to target other kinases to explore their functions in vivo.
Online Methods
Phylogenetic tree
The primary structure of 131 S. cerevisiae kinases were aligned with ClustalW. An unrooted tree was developed using a maximum likelihood model with MEGA6, and the tree was plotted with Hypertree.
Yeast Kinase purification.
C-terminally TAP-tagged kinases (GE Healthcare) were purified as previously described8. Cells were grown in 200 mL selective media to an OD600 of 1.0, and cells were pelleted and transferred to a 1.5 mL tube. Pellets were resuspended in 500μL TAP buffer A (20 mM HEPES pH 7.9, 300 mM potassium acetate, 0.5 mM EDTA pH 8.0, 10% glycerol, 0.05% NP40), with 1 mM DTT, 1 mM protease inhibitors (PMSF, benzamidine, pepstatin), and 1 mM phosphatase inhibitors (NaF, NaN3, and Na3VO4) freshly added. Approximately 200 μL silica beads (OPS Diagnostics) were added, and cells were lysed via bead beating for 15 minutes. A 22 gauge needle was heated with a Bunsen burner and used to puncture a hole in the tube, which was then placed in a 2mL tube and spun at 7,000 RPM for 30 seconds. 50 μL of a 50/50 slurry of IgG-Sepharose 6 Fast Flow beads (GE Healthcare) in TAP buffer A was added to the supernatant. After 3 hours nutating at 4°C, beads were pelleted and washed five times with TAP buffer A (500 μL each). Kinases were eluted overnight at 4°C in 25μL TAP buffer A with 1mM DTT and 10 U AcTEV (Invitrogen).
GST-CTD purification.
The CTD substrate, GST-CTD14 (fourteen repeats of YSPTSPS fused to GST) was expressed in BL21(DE3). Cells were grown to midlog phase at 37°C and induced with 1 mM IPTG overnight at 16°C. Cells were pelleted (5,000 g for 20 minutes) and resuspended in 5 mL Buffer B (1X PBS, 10% glycerol, 0.01% NP40) with freshly added protease inhibitors (1mM PMSF, 1mM benzamidine, 1.45mM pepstatin). Cells were lysed with 3 rounds of sonication at power level 5 using a Heat Systems-Ultrasonics Inc. W-220 Sonicator. Cell debris was pelleted at 13,000 rpm for 20 minutes at 4°C and a 200 μL 50/50 slurry of Gutathione-Sepharose 4B beads (GE Healthcare) in Buffer B was added to the supernatant. After 3 hours nutating at 4°C, beads were pelleted and washed once with Buffer B. Beads were resuspended in 1 mL FastAP buffer (10 mM Tris-HCl pH 8.0, 5 mM MgCl2, 100 mM KCl, 0.02% TritonX-100, 100 ug/mL BSA) and incubated with 100 U FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific) for 1 hour at 37°C. Beads were washed four times with Buffer B, then once with Buffer C (5 mM HEPES pH 7.4, 10 mM potassium glutamate, 10mM magnesium acetate, 0.5 mM EDTA pH 8.0, 10% glycerol, 0.1% NP-40). Reduced glutathione (Fisher Scientific) was added to Buffer C to 50 mM, and the pH was adjusted to 7.0. GST-CTD14 was eluted overnight in 200 μL Buffer C with 50 mM reduced glutathione. Any remaining alkaline phosphatase was heat inactivated at 75°C for 5 minutes.
In vitro kinase assay.
Each assay was performed with 5 μL of tandem affinity purified (TAP) kinase and 1 μM GST-CTD14 in a 30μL volume of Buffer D49 (20mM HEPES pH 7.5, 10% glycerol, 2.5 mM EGTA, 15 mM magnesium acetate, 100 mM potassium acetate) with 1 mM DTT, 1 mM ATP, freshly added protease inhibitors (1mM PMSF, 1mM benzamidine, 1.45mM pepstatin), and 1 mM phosphatase inhibitors (NaF, NaN3, and Na3VO4). Reactions were performed at 30°C for two hours and resolved via SDS-PAGE. Western blot analysis was performed using standard procedures. Blots were probed with antibodies targeting pThr4 (1G7), or the TAP-tag (Thermo Scientific). Quantitation was performed using ImageJ.
In vivo inhibition.
100 mL cultures were grown to an OD600 of 0.3 at 30°C, split to 2×50mL cultures, and inhibitor was added to a final concentration of 20μM (or an equivalent volume of DMSO). 3-MB-PP1 (hrr25as and bur1as) was purchased from Toronto Research Chemicals, 1-NA-PP1 (kin28as) and 1-NM-PP1 (cdc28as, pho85as, and ctk1as) were from MedChem Express. Cells were grown in the presence of inhibitor for one hour prior to pelleting (4,000 RPM, 5 minutes) and flash freezing. Pellets were resuspended in 200 μL 1X-NET-seq buffer50 (20mM HEPES pH 7.4, 110mM KOAc, 0.5% Triton X-100, 0.1% Tween-20, 10mM MnCl2) with 1mM PMSF, 1mM benzamidine, 1.45mM pepstatin, 1mM NaN3, and 1mM NaF. Silica beads (OPS Diagnostics) were added, and cells were lysed via bead beating for 15 minutes. A 22 gauge needle was heated with a Bunsen burner and used to puncture a hole in the tube, which was then placed in a 2mL tube and spun at 7,000 RPM for 10 seconds. Debris was resuspended, and 50U DNase I (Sigma) was added. Samples were mixed on a rotating, rocking mixer for 30 minutes at 4°C, and debris was pelleted. Supernatant was processed as normal for western blot analysis.
Autodock.
The structure of Hrr25 was from PDB ID 4XHL. To create a structure of Hrr25as, I82 of the WT structure was substituted with a glycine in PyMol51. To create a structure of Hrr25is, I23C of the Hrr25as structure was substituted with a cysteine. The casein kinase inhibitor (N-(2-aminoethyl)-5-chloroisoquinoline-8-sulfonamide) was removed from the active site in PyMol. The structure of 3-MB-PP1 was taken from PDB ID 2WEI. For each structure, 100 models of docked 3-MB-PP1 or CMK were created. Briefly, an initial population of 150 individuals were used for each docking operation (100 runs) with a maximum number of 27 000 generations. The maximum number of energy evaluations was set to 2 500 000. We allowed crossover and mutation to take place at a rate of 0.8 and 0.02, respectively, and structures with RMSD ≤ 2 Å were clustered.
RNA-seq.
hrr25is was grown to OD600 ~0.3 at 30°C in YPD. Cells were treated with either DMSO or 20μM CMK (MedChem Express) for 1hr, were harvested, and total RNA was extracted with hot phenol52. 50 μg total RNA was treated with DNase (Invitrogen) and rRNA was depleted using Ribo-Zero beads (Epicentre). RNA was fragmented, reverse transcribed, and barcoded using the ScriptSeq kit (Epicentre). RNA-seq libraries were sequenced on an Illumina HiSeq 2500 System. 100 bp single-end reads were obtained, adapter sequences were clipped, and reads were mapped to the S288c genome using STAR53. HTSeq was used to count reads54, and DESeq55 was used to analyze differential expression among two replicates. Traces from RNA-seq were from a single replicate. Read coverage was normalized in deepTools56 using –normalizeUsingRPKM and plotted. 3ʹ extension indices were calculated using the RPKM-normalized reads using an average of both replicates.
Northern Blot analysis.
25mL cultures were grown to OD 0.5, pelleted, resuspended in TE lysis buffer (10mM Tris-HCl pH 7.5, 10mM EDTA, 0.5% SDS) and flash frozen. The following day, cells were thawed, and RNA was extracted with 700μL hot phenol. 2μL GlycoBlue was added to the aqueous layer, and RNA was extracted again. 700μL chloroform was added to the aqueous layer and was added to a Heavy Phase Lock Gel Tube and spun for 5 munutes at room temp (16,000g). RNA was precipitated from the aqueous layer with 10% 3M NaOAc and 2.5 volumes cold 100% EtOH and frozen at −80°C overnight. The next day, RNA was pelleted, washed with 70% EtOH and resuspended in 50μL RNase-free water. 20ug RNA was run on a 1.5% agarose gel in 1X MOPS, 6% formaldehyde. RNA was transferred to Hybond-N membrane overnight using 20x SSC buffer and was crosslinked to the membrane via UV irradiation. Primers were designed to SNR4757, SNR1358, SNR40, and SCR1, containing a T7 promoter for in vitro transcription of a radioactive probe (Supplementary Table 2). 32P-UTP was incorporated into probes via in vitro transcription (AmpliScribe T7-Flash transcription kit – Epicentre). G25 columns (GE Healthcare) were used to remove unincorporated UTP. Blots were incubated in ULTRAhyb solution (Ambion) at 68°C for 30 minutes prior to addition of the radioactive probe overnight. Blots were washed twice with 2x SSC, 0.1% SDS and twice with 0.1x SSC, 0.1% SDS. Phosphorimage screens were exposed overnight and scanned the next morning on a Typhoon FLA9000 (GE Healthcare).
Validation of direct binding to pThr4.
GST-CTD was expressed in bacteria, bound to glutathione beads (Amersham Biosciences), and subjected to dephosphorylation using alkaline phosphatase (FastAP, Thermo Fisher). CTD was phosphorylated at position 4 by Hrr25 as described above, and Rtt103-TAP was incubated for 1 hour at 23°C. Beads were washed, and proteins were eluted with Laemmli sample buffer prior to western blot analysis probing for the TAP-tag. GST-CTD was also eluted from the beads and subjected to dot blot analysis probing for CTD modifications and the GST tag.
Creation of hrr25-HA.
pRS315-hrr25-I82G (gift from Ed Hurt) was used. Primers Hrr25-NcoI-F and Hrr25-NcoI-R were used to amplify Hrr25 from an internal NcoI site up to the stop codon. Primers Hrr25-pFA6a-F and Hrr25-pFA6a-R were used to amplify the HA3-kanMX cassette from pFA6a-3HA-kanMX6 plasmid, and contain an AatII restriction site. The two products were fused via PCR, cut with NcoI and AatII, and ligated into a similarly cut pRS315-hrr25-I82G vector. Hrr25as was converted back to WT or to –is using SDM primers listed in Supplementary Table 2. Plasmids were transformed into the Hrr25 shuffle strain, and pRS316-Hrr25-WT was shuffled out using 5-FOA.
Chromatin immunoprecipitation.
ChIP was done as previously described with minor modifications21. Cells were grown in rich media to OD600 ~0.5, crosslinked, pelleted, and flash frozen. Cells were lysed in lysis buffer (50mM HEPES-KOH, 140mM NaCl 1mM EDTA, 1% Triton X-100, 0.1% Na-Deoxycholate), and 1mM freshly added protease inhibitors (1mM PMSF, 1mM benzamidine, 1.45mM pepstatin). Chromatin was sonicated on a Misonix 4000 sonicator, input was reserved, chromatin was immunoprecipitated overnight (3μL α-Rpb3 (W0012 from Neoclone, currently BioLegend), 10μL α-pThr4 (1G7), 3μL α-HA (Abcam – ab9110), or 3μL α-FLAG (Sigma – M2). DNA was reverse crosslinked, amplified, and labeled using ligation-mediated PCR, and was hybridized to full yeast genome tiled microarrays (NimbleGen). Data were normalized as previously described21. qPCR of pThr4 ChIP in hrr25–3 was performed at multiple loci through PMA1, PYK1, SNR19, and SNR20. Primer sequences are listed in Supplementary Table 2.
Supplementary Material
Acknowledgements
We thank Kristy Moeller for preliminary data, and Professors Jeffry Corden, Ed Hurt, Brenda Andrews, Arno Greenleaf, Steve Hahn, Michael Stark, and Martin Webb for plasmids and strains. We thank members of the lab, especially Juan Rodríguez-Molina, for thoughtful comments, suggestions, and advice. This work was funded by NSF MCB1413547 and NIH GM117362 to AZA, NIH R01 GM083989 to APG, Deutsche Forschungsgemeinschaft, SFB1064, Chromatin Dynamics to DE, The Department of Biotechnology, Government of India (GAP0103) to MSA. AA and MFA are supported by an Indo-US postdoctoral fellowship from SERB, Government of India.
Footnotes
Competing Financial Interests
AZA is the sole member of VistaMotif, LLC and founder of the educational US nonprofit WINStep Forward.
Data Availability:
All ChIP and RNA-seq data are available at the Gene Expression Omnibus under the accession number: GSE107166.
References
- 1.Corden JL, Cadena DL, Ahearn JM & Dahmus ME A Unique Structure at the Carboxyl Terminus of the Largest Subunit of Eukaryotic RNA Polymerase-II. Proc Natl Acad Sci U S A 82, 7934–7938, (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang DW, Rodriguez-Molina JB, Tietjen JR, Nemec CM & Ansari AZ Emerging Views on the CTD Code. Genetics research international 2012, 347214, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eick D & Geyer M The RNA polymerase II carboxy-terminal domain (CTD) code. Chemical reviews 113, 8456–8490, (2013). [DOI] [PubMed] [Google Scholar]
- 4.Corden JL RNA polymerase II C-terminal domain: Tethering transcription to transcript and template. Chemical reviews 113, 8423–8455, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phatnani HP & Greenleaf AL Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936, (2006). [DOI] [PubMed] [Google Scholar]
- 6.Kornberg RD Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 30, 235–239, (2005). [DOI] [PubMed] [Google Scholar]
- 7.Feaver WJ, Svejstrup JQ, Henry NL & Kornberg RD Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell 79, 1103–1109, (1994). [DOI] [PubMed] [Google Scholar]
- 8.Akhtar MS et al. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 34, 387–393, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho EJ, Takagi T, Moore CR & Buratowski S mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 11, 3319–3326, (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bharati AP et al. The mRNA capping enzyme of Saccharomyces cerevisiae has dual specificity to interact with CTD of RNA Polymerase II. Sci Rep 6, 31294, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu H, Hu C & Hinnebusch AG Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol. Cell 33, 752–762, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray S, Udupa R, Yao S, Hartzog G & Prelich G Phosphorylation of the RNA polymerase II carboxy-terminal domain by the Bur1 cyclin-dependent kinase. Mol. Cell. Biol. 21, 4089–4096, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones JC et al. C-terminal repeat domain kinase I phosphorylates Ser2 and Ser5 of RNA polymerase II C-terminal domain repeats. J. Biol. Chem. 279, 24957–24964, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hintermair C et al. Threonine-4 of mammalian RNA polymerase II CTD is targeted by Polo-like kinase 3 and required for transcriptional elongation. EMBO J. 31, 2784–2797, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsin JP, Sheth A & Manley JL RNAP II CTD phosphorylated on threonine-4 is required for histone mRNA 3’ end processing. Science 334, 683–686, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosonina E et al. Threonine-4 of the budding yeast RNAP II CTD couples transcription with Htz1-mediated chromatin remodeling. Proc Natl Acad Sci U S A 111, 11924–11931, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwer B, Bitton DA, Sanchez AM, Bahler J & Shuman S Individual letters of the RNA polymerase II CTD code govern distinct gene expression programs in fission yeast. Proc Natl Acad Sci U S A 111, 4185–4190, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harlen KM et al. Comprehensive RNA Polymerase II Interactomes Reveal Distinct and Varied Roles for Each Phospho-CTD Residue. Cell Rep 15, 2147–2158, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemec CM et al. Different phosphoisoforms of RNA polymerase II engage the Rtt103 termination factor in a structurally analogous manner. Proc Natl Acad Sci U S A 114, E3944–E3953, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hintermair C et al. Specific threonine-4 phosphorylation and function of RNA polymerase II CTD during M phase progression. Sci Rep 6, 27401, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tietjen JR et al. Chemical-genomic dissection of the CTD code. Nat. Struct. Mol. Biol. 17, 1154–1161, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y et al. Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol. Cell. Biol. 29, 4852–4863, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coudreuse D et al. A gene-specific requirement of RNA polymerase II CTD phosphorylation for sexual differentiation in S. pombe. Curr. Biol. 20, 1053–1064, (2010). [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Molina JB, Tseng SC, Simonett SP, Taunton J & Ansari AZ Engineered Covalent Inactivation of TFIIH-Kinase Reveals an Elongation Checkpoint and Results in Widespread mRNA Stabilization. Mol. Cell 63, 433–444, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bishop AC et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401, (2000). [DOI] [PubMed] [Google Scholar]
- 26.Hsin JP, Xiang K & Manley JL Function and control of RNA polymerase II C-terminal domain phosphorylation in vertebrate transcription and RNA processing. Mol. Cell. Biol. 34, 2488–2498, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chymkowitch P et al. Cdc28 kinase activity regulates the basal transcription machinery at a subset of genes. Proc Natl Acad Sci U S A 109, 10450–10455, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keogh MC, Podolny V & Buratowski S Bur1 kinase is required for efficient transcription elongation by RNA polymerase II. Mol. Cell. Biol. 23, 7005–7018, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phatnani HP, Jones JC & Greenleaf AL Expanding the functional repertoire of CTD kinase I and RNA polymerase II: novel phosphoCTD-associating proteins in the yeast proteome. Biochemistry 43, 15702–15719, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoekstra MF et al. HRR25, a putative protein kinase from budding yeast: association with repair of damaged DNA. Science 253, 1031–1034, (1991). [DOI] [PubMed] [Google Scholar]
- 31.Ho Y, Mason S, Kobayashi R, Hoekstra M & Andrews B Role of the casein kinase I isoform, Hrr25, and the cell cycle-regulatory transcription factor, SBF, in the transcriptional response to DNA damage in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 94, 581–586, (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schafer T et al. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature 441, 651–655, (2006). [DOI] [PubMed] [Google Scholar]
- 33.Petronczki M et al. Monopolar attachment of sister kinetochores at meiosis I requires casein kinase 1. Cell 126, 1049–1064, (2006). [DOI] [PubMed] [Google Scholar]
- 34.Pfaffenwimmer T et al. Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg19. EMBO Rep 15, 862–870, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Measday V et al. A family of cyclin-like proteins that interact with the Pho85 cyclin-dependent kinase. Mol. Cell. Biol. 17, 1212–1223, (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell AP & Bowdish KS Selection for early meiotic mutants in yeast. Genetics 131, 65–72, (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yabuki Y et al. Glycogen synthase kinase-3 is involved in regulation of ribosome biogenesis in yeast. Biosci Biotechnol Biochem 78, 800–805, (2014). [DOI] [PubMed] [Google Scholar]
- 38.Posas F & Saito H Activation of the yeast SSK2 MAP kinase kinase kinase by the SSK1 two-component response regulator. EMBO J. 17, 1385–1394, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanchez-Piris M et al. The serine/threonine kinase Cmk2 is required for oxidative stress response in fission yeast. J. Biol. Chem. 277, 17722–17727, (2002). [DOI] [PubMed] [Google Scholar]
- 40.Posas F et al. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 ““two-component”” osmosensor. Cell 86, 865–875, (1996). [DOI] [PubMed] [Google Scholar]
- 41.Chasman D et al. Pathway connectivity and signaling coordination in the yeast stress-activated signaling network. Mol. Syst. Biol. 10, 759, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferreira-Cerca S et al. ATPase-dependent role of the atypical kinase Rio2 on the evolving pre-40S ribosomal subunit. Nat. Struct. Mol. Biol. 19, 1316–1323, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y et al. Two cyclin-dependent kinases promote RNA polymerase II transcription and formation of the scaffold complex. Mol. Cell. Biol. 24, 1721–1735, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia B, Stollar EJ & Davidson AR The importance of conserved features of yeast actin-binding protein 1 (Abp1p): the conditional nature of essentiality. Genetics 191, 1199–1211, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanin EI et al. Chemical inhibition of the TFIIH-associated kinase Cdk7/Kin28 does not impair global mRNA synthesis. Proc Natl Acad Sci U S A 104, 5812–5817, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gasch AP et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11, 4241–4257, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris GM et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30, 2785–2791, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flotow H et al. Phosphate Groups as Substrate Determinants for Casein Kinase-I Action. J. Biol. Chem. 265, 14264–14269, (1990). [PubMed] [Google Scholar]
- 49.Ansari AZ, Ogirala A & Ptashne M Transcriptional activating regions target attached substrates to a cyclin-dependent kinase. Proc Natl Acad Sci U S A 102, 2346–2349, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Churchman LS & Weissman JS Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrödinger L The PyMOL Molecular Graphics System, Version 1.3. (2010). [Google Scholar]
- 52.Gasch AP in Methods Enzymol. Vol. Volume 350 (eds Christine Guthrie & Gerald R. Fink) 393–414 (Academic Press, 2002). [DOI] [PubMed] [Google Scholar]
- 53.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anders S & Huber W Differential expression analysis for sequence count data. Genome Biol 11, R106, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–191, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chanfreau G, Rotondo G, Legrain P & Jacquier A Processing of a dicistronic small nucleolar RNA precursor by the RNA endonuclease Rnt1. EMBO J. 17, 3726–3737, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim M et al. Distinct pathways for snoRNA and mRNA termination. Mol. Cell 24, 723–734, (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.