

Abstract
We hypothesized that distinct protein expression features of benign and malignant pulmonary nodules may reveal novel candidate biomarkers for the early detection of lung cancer. We performed proteome profiling by liquid chromatography— tandem mass spectrometry to characterize 34 resected benign lung nodules, 24 untreated lung adenocarcinomas (ADCs), and biopsies of bronchial epithelium. Group comparisons identified 65 proteins that differentiate nodules from ADCs and normal bronchial epithelium and 66 proteins that differentiate ADCs from nodules and normal bronchial epithelium. We developed a multiplexed parallel reaction monitoring (PRM) assay to quantify a subset of 43 of these candidate biomarkers in an independent cohort of 20 benign nodules, 21 ADCs, and 20 normal bronchial biopsies. PRM analyses confirmed significant nodule-specific abundance of 10 proteins including ALOX5, ALOX5AP, CCL19, CILP1, COL5A2, ITGB2, ITGAX, PTPRE, S100A12, and SLC2A3 and significant ADC-specific abundance of CEACAM6, CRABP2, LAD1, PLOD2, and TMEM110-MUSTN1. Immunohistochemistry analyses for seven selected proteins performed on an independent set of tissue microarrays confirmed nodule-specific expression of ALOX5, ALOX5AP, ITGAX, and SLC2A3 and cancer-specific expression of CEACAM6. These studies illustrate the value of global and targeted proteomics in a systematic process to identify and qualify candidate biomarkers for noninvasive molecular diagnosis of lung cancer.
Keywords: lung cancer, granuloma, parallel reaction monitoring, adenocarcinoma, indeterminate pulmonary nodule
Graphical Abstract
INTRODUCTION
Lung cancer continues to be the leading cause of cancer-related deaths worldwide.1 One of the major challenges remains a noninvasive method for the early detection of this disease. Most lung cancers are first discovered as lung nodules by chest imaging. In the National Lung Cancer Screening Trial, 39% of low-dose computed tomography (LDCT) study participants had a positive scan during the study, 96.4% of which were false-positive for cancer.2 Lung nodules present a significant public health challenge, both because of their prevalence (an estimated 2 million new nodules a year) and because of the difficulties in making a noninvasive cancer diagnosis.3–9 Positron emission tomography-computed tomography (PET-CT) has been proposed as a diagnostic aid to evaluate indeterminate pulmonary nodules (IPNs), which are those of >5 mm, and which have <65% probability to be confirmed as cancers. However, the specificity of imaging methods for distinguishing lung cancers from benign nodules is decreased dramatically in regions with endemic fungal disease.10
In recent work, nodule characteristics coupled to clinical risk factors have been widely used to predict malignancy.11–16 This approach has primarily relied on nodule size and rate of size increases, as determined by imaging techniques. Several clinical assessment tools have been introduced to predict the probability of malignancy, especially in the higher risk populations.17–19 Therefore, we recognized a need for a novel, noninvasive strategy to distinguish benign nodules from malignant nodules. This motivated us to explore biochemical differences between nodules and cancers that could be combined with molecular imaging to improve diagnostic accuracy.
We hypothesized that benign nodules display distinct patterns of differential protein abundance in comparison with lung cancer and that proteins with characteristically high abundance in nodules may represent new candidate biomarkers for the early detection of lung cancer. We used multidimensional liquid chromatography—tandem mass spectrometry (LC-MS/MS) to characterize proteomes of a collection of benign lung nodules and early-stage (with the exception of one late-stage) lung ADCs. We selected proteins with differential expression between benign nodules, ADCs, and normal lung, and, to enable future imaging-based detection, we further selected proteins that are secreted or have extracellular domains. We validated those differentially expressed proteins by parallel reaction monitoring mass spectrometry (PRM-MS) and immunohistochemistry (IHC) to confirm their ability to distinguish iPNs from cancers.
EXPERIMENTAL PROCEDURES
Study Population and Tissue Collection
Patient characteristics from the discovery and validation sets are presented as a summary in Table 1. Complete clinical characteristics can be found in Tables S-9 and S-10. A retrospective collection of 34 benign lung nodules, 23 early-stage lung ADCs, 1 late-stage lung ADC, 5 normal bronchial, and 5 alveolar epithelium biopsies (hereafter referred to as “normals”) from patients consented at Vanderbilt University Medical Center (VUMC) and the Nashville Veteran Affairs Medical Center (VAMC) was used. For the independent validation set (PRM assays), 20 benign nodules, 21 early-stage ADCs, and 20 normal lung tissues were collected. This study was approved by the Institutional Review Board at both institutions (IRB protocols 000616 and 310233).
Table 1.
Patient Characteristics for Discovery and PRM Validation Cohorts
| discovery cohort |
PRM validation cohort |
|||||||
|---|---|---|---|---|---|---|---|---|
| patients | normal lung (%) | benign nodule (%) | ADC (%) | P value | normal lung (%) | benign nodule (%) | ADC (%) | P value |
| N = 10 | N = 34 | N = 24 | N = 20 | N = 20 | N = 21 | |||
| age ± SD | 68.2 ± 5.8 | 56.4 ± 12.6 | 62.3 ± 7.8 | 0.043a | 56.8 ± 13.8 | 60.5 ± 13.5 | 65.6 ± 8.6 | 0.088a |
| gender | 0.616b | 0.198b | ||||||
| male | 3 (60) | 18 (53) | 10 (42) | 10 (50) | 6 (30) | 12 (57) | ||
| female | 2 (40) | 16 (47) | 14 (58) | 10 (50) | 14 (70) | 9 (43) | ||
| race | 0.792b | 0.380b | ||||||
| Caucasian | 5 (100) | 30 (88) | 20 (83) | 20 (100) | 20 (100) | 20 (95) | ||
| African American | 0 (0) | 2 (6) | 3 (13) | 0 (0) | 0 (0) | 1 (5) | ||
| other | 0 (0) | 2 (6) | 1 (4) | 0 (0) | 0 (0) | 0 (0) | ||
| nodule (mm) ± SD | N/A | 25.3 ± 27.8 | 29.4 ± 18.5 | 0.067c | N/A | 22.9 ± 18.5 | 23.8 ± 12.4 | 0.538c |
| pack years ± SD | 71.0 ± 37.5 | 37.1 ± 30.3 | 42.4 ± 19.1 | 0.080a | 27.8 ± 26.1 | 23.9 ± 33.6 | 39.8 ± 34.4 | 0.216a |
| FEV1% ± SD | 52.0 ± 24.7 | 79.0 ± 22.6 | 71.1 ± 18.8 | 0.063a | 77.3 ± 32.5 | 82.3 ± 25.0 | 69.3 ± 25.4 | 0.286a |
| ADC path stage | ||||||||
| IA-IB | 22 (92) | 17(81) | ||||||
| IIA-IIIA | 1 (4) | 4 (19) | ||||||
| IIIB-IV | 1 (4) | 0 (0) | ||||||
| benign histologies | ||||||||
| acid fast bacilli | 3 (9) | 4 (20) | ||||||
| acute inflam. | 1(3) | 0 (0) | ||||||
| benign tumor | 2 (6) | 0 (0) | ||||||
| fungal | 16 (47) | 11 (55) | ||||||
| mixed etiology | 12 (35) | 5 (25) | ||||||
Kruskal-Wallis test.
Pearson chi-square test.
Wilcoxon rank sum test.
Tissue Dissection and Protein Digestion
For both the discovery and validation sets, 10 μm thick tissue sections were macro-dissected under a dissecting microscope (SZ-60 CTV Olympus, Japan) to include either >70% of viable tumor tissue, or the entire benign lung nodule including its peripheral rim. The normal lung tissues were dissected to include bronchial airway epithelium or alveolar tissues specifically. Formalin-fixed, paraffin-embedded (FFPE) samples were deparaffinized, rehydrated, reduced, alkylated, and digested with trypsin as previously described. Digested samples (200 μg protein) were lyophilized,21 and the lyophilized peptide samples were suspended in water prior to solid-phase extraction with a Waters Sep-Pak C18 desalting cartridge (Milford, MA). Prior to use, desalting cartridges were first charged with 1 mL of acetonitrile and then equilibrated with 2 mL of water. Peptide samples were loaded onto the equilibrated column and washed once with 1 mL of water, and the peptides were eluted with 70% acetonitrile containing 0.1% formic acid (Thermo Fisher Scientific, Waltham, MA). These samples were evaporated to dryness in vacuo and stored at −80 °C until further use.
Global LC-MS/MS Analyses
Tryptic digests of lung nodules and ADC were analyzed by multidimensional LC-MS/MS, where the initial separation was by basic reversed-phase LC (bRPLC), as previously described.20 Desalted tryptic peptides, dissolved in 10 mM triethylammonium bicarbonate (TEAB), pH 8.0 were fractionated with an Agilent 1260 Infinity LC system equipped with an XBridge C18 5 μm 4.6 × 250 mm column. Solvent A was 10 mM TEAB in water (Sigma, St. Louis, MO), pH 7.4, and Solvent B was 10 mM TEAB in acetonitrile. Peptides were eluted at room temperature using a flow rate of 0.5 mL/min, and a linear mobile phase gradient was programmed from 100% Solvent A to 5% Solvent B at 10 min, 35% B at 70 min, 70% B at 85 min, and then held at 70% B for an additional 10 min, before returning to 100% A at 100 min, followed by a 5 min equilibration prior to the next analysis. The eluted peptides were collected in 60 fractions, which were concatenated to 15 fractions, as described.20 Concatenated fractions were evaporated to dryness in vacuo, and the dried samples were suspended in 100 μL of 3% acetonitrile with 0.1% formic acid for LC-MS/MS analysis. Tryptic digests from normal tissues were fractionated by isoelectric focusing on immobilized pH gradient strips, as previously described.21
LC-MS/MS analyses of fractionated peptides from normal tissue biopsies were performed on a Thermo LTQ XL mass spectrometer equipped with an Eksigent NanoLC AS1 autosampler and Eksigent NanoLC 1D Plus pump and a Thermo nanospray source. Peptides were separated on a packed capillary tip (Polymicro Technologies, 100 mm × 11 cm) with Jupiter C18 resin (5 mm, 300 A, Phenomenex) using an in-line solid-phase extraction column (100 mm × 6 cm) packed with the same C18 resin using a frit generated with liquid silicate Kasil 1. Mobile phase A consisted of 0.1% formic acid and mobile phase B consisted of 0.1% formic acid in 90% acetonitrile. A 90 min gradient was carried out with a 30 min washing period (100% A) to allow for solid-phase extraction and removal of any residual salts. Following the washing period, the gradient was increased to 25% B by 35 min, followed by an increase to 90% B by 50 min and held for 9 min before returning 95% A. MS/MS spectra were acquired using a data-dependent scanning mode in which one full MS scan (m/z 400–2000) was followed by five MS/MS scans. MS/MS scans were acquired in centroid mode using an isolation width of 3 m/z, an activation time of 30 ms, an activation q of 0.250, and 35% normalized collision energy. MS/MS spectra were collected using a dynamic exclusion of60 s with a repeat of1 s and repeat duration of 1 s.
Analyses of fractionated peptides from ADC were performed on a Thermo LTQ Velos mass spectrometer equipped with an Eksigent NanoLC AS1 autosampler and Eksigent NanoLC 1D Plus pump and a Thermo nanospray source. Peptide LC separations were performed as described above. MS/MS spectra were acquired using a data-dependent scanning mode in which one full MS scan (m/z 400–2000) was followed by five MS/MS scans. MS/MS scans were acquired in centroid mode using an isolation width of 2 m/z, an activation time of 30 ms, an activation q of 0.250, and 30% normalized collision energy. MS/ MS spectra were collected using a dynamic exclusion of 60 s with a repeat of 1 s and repeat duration of 1 s.
Analyses of fractionated peptides from nodules were performed on a Thermo Orbitrap Elite mass spectrometer equipped with a Thermo Easy n-LC1000 LC system and autosampler. Peptide LC separations were performed as described above. Full MS scans were acquired on the Orbitrap from m/z 300–2000 at a resolution of 60 000 using an automatic gain control (AGC) value of 5 × 105. The minimum threshold was set to 500 ion counts. Precursor ions were fragmented with the LTQ using an isolation width of 2 m/z units, a maximum injection time of 50 ms, and an AGC value of 1 × 103. Normalized collision energy was set to 35 m/z.
Protein Identification from MS/MS Data
Tandem spectra were searched against the human RefSeq protein database (version 54; September 2012, with 34589 protein entries) using the Myrimatch search algorithm (v 2.1.132)22 and MS-GF+ Beta (v9979) search algorithm,23 and Pepitome search algorithm (v 1.0.42) for the spectral library searches.24 The database incorporated both the forward as well as reversed sequences to allow for determination of false discovery rates. The searches were performed, allowing for static modifications of +57 Da on cysteines (for carboxyamidomethy-lation from iodoacetamide) and dynamic modifications of +16 Da on methionines and formation of N-terminal pyroglutamine (−17 Da). Matches to semitryptic peptides were allowed. Peptide and fragment ion tolerances were set to ±1.25 m/z and 0.5 Da, respectively. The data were filtered and assembled with the IDPicker algorithm (v. 3.1.642.0), requiring at least two unique peptides (minimum peptide length of seven amino acids) and six spectra per protein in the entire data set.25 These settings resulted in an overall protein FDR of 5.6% and a peptide FDR of 1.9%. The combined shotgun proteomics data from all nodule, tumor, and normal samples identified 5575 protein groups, a minimal number of proteins that explained all spectral matches that fulfilled the filtering parameters.26
Targeted Analysis by PRM-MS
We developed PRM assays to quantify the 43 candidate biomarker proteins via 170 tryptic peptides in a single LC- PRM-MS run. We obtained synthetic peptides of moderate (~85%) purity from New England Peptide (Gardner, MA) to use as standards to establish chromatographic properties and to verify sequence-specific transitions in PRM. A PRM assay was developed using the experimental design and data analysis tool Skyline 1.1 software.27 To develop a scheduled PRM method, a “master mix” of unlabeled synthetic standards representing all target peptides was spiked into a matrix background made from four nodule samples. This master mix sample was analyzed in an unscheduled PRM run to determine retention times and representative fragment ions. A total of 170 peptides were monitored in each scheduled PRM run across the entire set of validation samples.
Samples for PRM analyses were prepared in the same manner as for global proteomics, except that no bRPLC fractionation was performed. Desalted peptide samples were dissolved to 0.5 μg/ μL in 3% aqueous acetonitrile containing 0.1% formic acid and a mixture of three labeled reference peptide (LRP) standards (β- actin peptide U-13C,15N-ArgGYSFTTTAER, alkaline phosphatase (AP) peptide U-13C,15N-Arg-AAQGITAPGGAR, and β-galactosidase (BG) peptide U-13C,15N-Arg-APLDNDIGVSEATR) were spiked into the samples at a final concentration of 12.5 fmol/μL.
PRM analyses were performed on a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with an Easy-nLC 1000 autosampler. Peptides were resolved on an PicoFrit Emitter column (11 cm × 75 μm ID, New Objective, Wortham, MA) with a 10 μm ID opening, packed with ReproSil C18-AQ resin of 3 μm particle size (Dr. Maisch, Ammerbuch-Entringen, Germany). Liquid chromatography was performed at room temperature with a mobile-phase gradient program using 0.1% formic acid in water (Solvent A) and 0.1% formic acid in acetonitrile (Solvent B). The column was programmed with a linear gradient from 2 to 35% Solvent B at a flow rate of 300 nL/min over 40 min, followed by an increase to 90% B over 4 min and then held at 90% B for 6 min before returning to initial conditions of 2% B. For electrospray ionization, 1800 V was applied and a 250 °C capillary temperature was used. Each sample was analyzed using an acquisition method that combined a full-scan SIM event, followed by 14 PRM scans, as triggered by a scheduled inclusion list, with a 4 min retention time window. Retention times were established with analyses of unlabeled standards for all peptides. The SIM scan event was collected using an m/z 380–1500 mass selection, an Orbitrap resolution of 17 500 (at m/z 200), target AGC value of 3 × 106, and a maximum injection time of 30 ms. The PRM scan events used an Orbitrap resolution of 17 500, an AGC value of 5 × 105. and maximum fill time of 80 ms with an isolation width of 0.5 m/z. Fragmentation was performed with a normalized collision energy of 27, and MS/MS scans were acquired starting at m/z 150.
Tissue Microarray Preparation
Tissue microarrays (TMAs) were prepared from paraffin- embedded formalin-fixed (FFPE) blocks. Archived tissue blocks from consecutive anatomic resections acquired from 1989 to 2012 were retrieved from the files of the pathology departments at Vanderbilt University Medical Center and at the Tennessee Valley Health Care System, Nashville, TN. The TMAs consisted of 41 ADCs, 45 other nonsmall cell lung cancers, and 63 benign lung nodules and were constructed according to protocols previously described.28,29 Demographic and clinical characteristics of the 149 patients represented in three TMAs are summarized in Table 2. Complete clinical characteristics for all patients can be found in Table S-11.
Table 2.
Patient Characteristics for IHC Validation Cohort
| patients | no cancer(%) | cancer(%) | P value |
|---|---|---|---|
| N = 63 | N = 86 | ||
| age ± SD | 66.3 ± 20.9 | 62.9 ± 10.4 | 0.616a |
| gender | 0.381b | ||
| male | 31 (49) | 49 (57) | |
| female | 32 (51) | 37 (43) | |
| race | 0.179b | ||
| Caucasian | 58 (92) | 84 (98) | |
| African American | 3 (5) | 2 (2) | |
| other | 2 (3) | 0 (0) | |
| nodule size (mm) ± SD | 22.0 ± 16.0 | 30.0 ± 20.0 | 0.014a |
| smoking status | <0.001b | ||
| never smoker | 20 (32) | 6 (7) | |
| ex-smoker | 33 (53) | 55 (63) | |
| current smoker | 10 (16) | 25 (29) | |
| pack years ± SD | 27.0 ± 30.1 | 45.5 ± 31.7 | <0.001a |
| FEV1% ± SD | 67.0 ± 22.5 | 65.3 ± 24.1 | 0.660a |
| lung cancer histologies | |||
| ADC | 41 (48) | ||
| SCC | 31 (36) | ||
| NSCLC | 7 (8) | ||
| LCC | 5 (6) | ||
| carcinoid | 2 (2) | ||
| path stages | |||
| IA-IB | 55 (64) | ||
| IIA-IIIA | 19 (22) | ||
| IIIB-IV | 12 (14) | ||
| benign histologies | |||
| acid fast bacilli | 5 (8) | ||
| acute inflam. | 3 (5) | ||
| benign tumor | 2 (2) | ||
| fungal | 25 (40) | ||
| multiple etiologies | 25 (40) | ||
| normal lung | 3 (5) | ||
Wilcoxon Rank Sum Test.
Pearson Chi-Square Test.
Immunohistochemistry
Five micron tissue sections were cut from FFPE lung TMAs and were placed on the Leica Bond Max IHC stainer (Leica Biosystems, Buffalo Grove, IL). All steps excluding dehydration, clearing, and coverslipping are performed on the Bond Max. Heat-induced antigen retrieval was performed on the Bond Max using Epitope Retrieval 1 or 2 solution for 10–20 min depending on the analyte. Primary antibodies for ALOX5 (Abcam ab115764, Cambridge, MA, 1:100), ITGAX (Leica PA0554, Newcastle Upon Tyne, NE, U.K.), BST1 (Novus NBP2–14363, Littleton, CO, 1:100), ALOX5AP (Abcam ab151515, Cambridge, MA, 1:100), SLC2A3 (Abcam ab15312, Cambridge, MA), CRABP2 (Origene TA800069, Rockville, MD, 1:100), COL4A5 (Novus NBP1–55880, Littleton, CO, 1:150), CEA- CAM6 (Thermo Scientific PA5–29551, Rockford, IL, 1:1,000), and LAD1 (Thermo Scientific PA5–22344, Rockford, IL, 1:100) were used at the specified concentrations. For visualization, the Bond Refine Polymer detection system was used. IHC analysis was performed as previously reported by our group.29 Results of the IHC staining were analyzed by a pathologist (RE).
TMA slides were scanned using the Leica SCN400 Slide Scanner (Leica Biosystems, Buffalo Grove, IL) and analyzed with the Leica Biosystems Digital Image Hub. TMAs were evaluated based on number of cells staining positive for each marker in the entire tissue area. Values were normalized to the total number of cells calculated by nuclear counterstain yielding in the percent positive as reported. On the basis of an automated 0–255 light intensity scale, cells were considered to be positive if the intensity of the stain was below 220, with 0 being the greatest intensity, and negative if intensity values were above 220.
Experimental Design and Statistical Rationale
For global LC-MS/MS proteome analyses, protein spectral counts were compared using a Quasi-likelihood model and filtered for low spectral counts (one spectrum/protein across the entire data set). A group analysis of protein spectral counts was carried out to identify proteins that were differentially expressed in one of the groups at the quasi.FDR < 0.05 and a 4-fold increase in protein expression or higher. Group-wise comparisons between shotgun data sets of benign nodules with lung ADCs and normal lung tissue identified 65 “nodule-specific” proteins that differentiated nodules from ADCs and control tissues and a second group of 66 “cancer-specific” proteins that differentiate ADCs from nodules and control tissues. We selected 25 nodule- specific proteins and 18 cancer-specific proteins that met additional criteria of (1) uniform expression across the discovery sample set and (2) annotation as membrane-bound or secreted proteins.
For PRM analyses, the top three most intense fragment ions were used for peptide peak area quantitation, and the peptide signals for candidate biomarker proteins were normalized by the LRP method.30 Peptide peak areas were calculated as the sum of the peak areas for the three most intense fragment ions, and this summed peak area was normalized to the summed peak area for the BG LRP peptide, which had the lowest coefficient of variation (CV) across all of the PRM analyses. Although available samples were sufficient to permit only a single processed replicate per sample, each LC—MS-ready sample was injected twice (technical replicates). The summed peak areas for each individual peptide were averaged, and an intraclass correlation coefficient (ICC) was calculated from the averaged values; the ICC indicates the fraction of overall measurement variation associated with differences between the three experimental groups. Pearson correlation was used to compare normalized peak areas for the replicate analyses across the data set. Only peptides with an ICC above 0.6 and a Pearson correlation above 0.7 were considered for further statistical comparisons, while the remaining peptides were not significantly different between biological classes. ANOVA was performed across the three groups to significance of differences.
For IHC analyses, data comparing two experimental conditions were analyzed by two-tailed Wilcoxon ranked sum test. Only results with P < 0.05 or P < 0.01 were considered to be statistically significant. All experimental data are presented as a representative of three independent experiments. For the TMAs, the average scores of duplicate biopsies (cores) were used for IHC analysis. Maximal immunostain scores from replicates were used for IHC, and data obtained from the tumor registry allowed survival analysis. Clinical data elements were obtained from the Bioinformatics Core of the Vanderbilt Ingram Cancer Center (VICC). Data analysis included Spearman correlation coefficients and Kaplan—Meier survival estimates with Cox proportional hazards regression models. Survival analysis was calculated from date of diagnosis to date of death or last date of contact for those alive at the time of the analysis. Curves were compared with the log-rank test.
RESULTS
Identification of Differentially Abundant Proteins among IPNs, Normal Lung, and Lung ADCs
To identify protein abundance features characteristic of benign nodules, we first performed global proteomic analyses of normal bronchial and alveolar epithelium (N = 10), IPNs (N = 34), and ADCs (N = 24) (Table 1). These discovery data sets were generated with three different MS platforms over a 4 year period as part of three previously unpublished studies. The three data sets were searched together and combined into a single protein assembly for further analysis. The combined shotgun proteomics data set from all normal, benign nodules, and tumor samples identified 8420 proteins corresponding to 5575 protein groups (2% peptide FDR, 5.6% protein FDR) (Supplemental Data Set 1). Protein abundance differences between tissue types were estimated from spectral counts. The logic flow for identification of tissue type-specific proteins is summarized in Figure 1.
Figure 1.
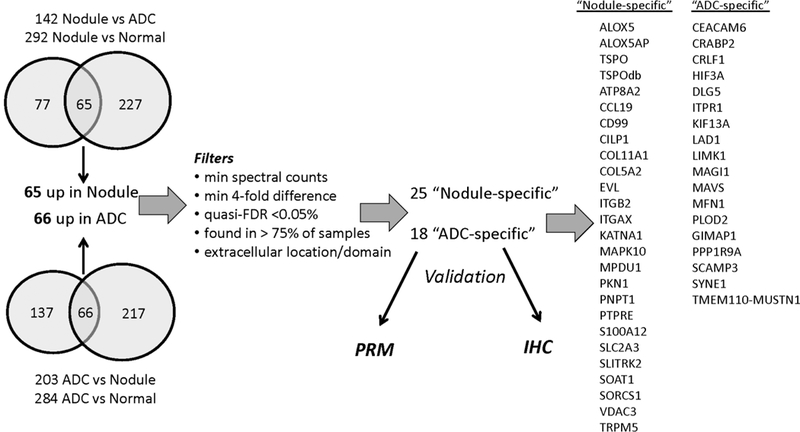
Logic flow for identification of putative benign nodule-specific and ADC-specific proteins.
First, proteins with low spectral counts (<1 spectrum/protein across the entire data set) were removed. A Quasi-likelihood model then was used to estimate spectral count-based differences between tissues, and significant proteins were required to have a quasi.FDR < 0.05 and a ≥4-fold difference in protein expression. According to these criteria, 142 proteins were higher in lung nodules than in cancers, and 292 proteins were higher in nodules than in normal tissues. Proteins in both of these groups (65 proteins) were designated as putative nodule-specific proteins for subsequent analyses (see below). Similarly, we identified proteins that discriminated between lung ADC and lung nodules (203 proteins) and between lung ADC versus normal (284 proteins). Proteins in both of these groups (66 proteins) were considered ADC-specific. Of these 131 proteins identified as nodule-specific or ADC-specific, we selected 25 nodule-specific proteins and 18 ADC-specific proteins that met additional filtering criteria: (1) spectral counts ≥ 4-fold different between nodule/ADC and normal tissue, (2) quasi.FDR < 0.05% for spectral count-based abundance differences, (3) putative nodule-or ADC-specific proteins were required to be expressed in at least 75% of the discovery samples in the respective group, and (4) had Uniprot annotation as secreted proteins or proteins with extracellular domains.
Verification of Putative Nodule- and ADC-specific Proteins by PRM Analysis in an Independent Sample Set
To verify the specificity of putative nodule-specific and ADC- specific proteins, we developed PRM assays to quantify 43 candidate biomarker proteins by measuring 165 proteotypic, tryptic peptides in a single LC-PRM run. These PRM analyses were applied to an independent cohort of FFPE samples consisting of 20 benign nodules, 21 early-stage ADCs, and 20 normal bronchial biopsies (Table 1). All proteins, peptides, and measured transitions are shown in Table S-1, and measured peptide peak areas are listed in Table S-2. Of the 43 protein candidates, 27 (17 for benign nodules, 10 for ADCs) met criteria for quantification by PRM measurement of at least one peptide. These criteria were: (1) targeted peptides were detectable by at least three MS/MS transitions, (2) all detected PRM transitions coeluted chromatographically with a retention consistent with that of an external synthetic peptide standard in the same LC system, and (3) the intensity order of the detected transitions corresponded to those for the standard. Data for the PRM measurements for these 62 peptides from 27 proteins are shown in Tables S-3 and S-4.
Criteria for significantly different abundance of peptides from these 27 proteins was based on (1) calculated ICC values >0.6, (2) a Pearson correlation >0.7 for technical replicates across the data set, and (3) three-way ANOVA indicating significant difference between the groups (p < 0.05) (Table S-6). A total of 31 peptides corresponding to 16 proteins met these criteria for significance (Table S-6). Among these 16 proteins, 10 were represented by two or more peptides.
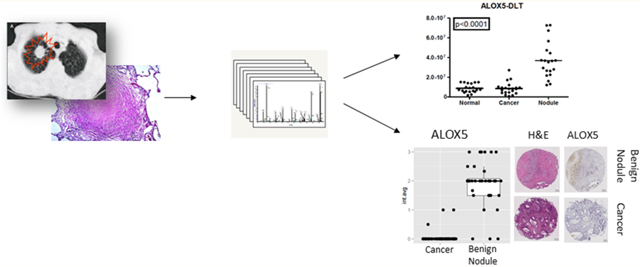
Of the 17 PRM-quantifiable, putative markers for benign nodules, 10 were confirmed by PRM to be significantly elevated in nodules compared with normal and ADC tissues. These proteins were ALOX5, ALOX5AP, CCL19, CILP1, COL5A2, ITGB2, ITGAX, PTPRE, S100A12, and SLC2A3. Representative plots of the abundance distributions for peptides from these proteins are shown in Figure 2.
Figure 2.
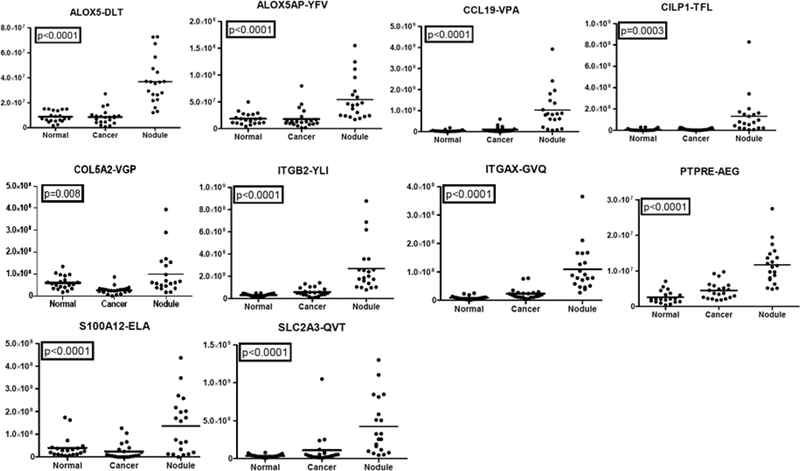
PRM measurement of representative peptides from putative benign nodule-specific proteins. Replicate PRM analyses were performed for peptides from 17 candidate proteins in 20 benign nodules, 22 ADC, and 20 normal bronchial biopsies. Representative plots for peptides that passed thresholds for ICC, Spearman correlation, and ANOVA are shown.
Of the 10 PRM-quantifiable, putative markers for ADCs, four were confirmed by PRM to be significantly elevated in ADCs compared with nodules and normal tissue. These proteins were CEACAM6, CRABP2, LAD1, PLOD2, and TMEM110- MUSTN1. Representative plots of the abundance distributions for peptides from these proteins are shown in Figure 3. Plots for all peptides that met significance criteria are shown in Figures S-1 and S-2. Receiver operating characteristic (ROC) curves were generated, and area under the curve (AUC) values were generated (Figure S-4A).
Figure 3.
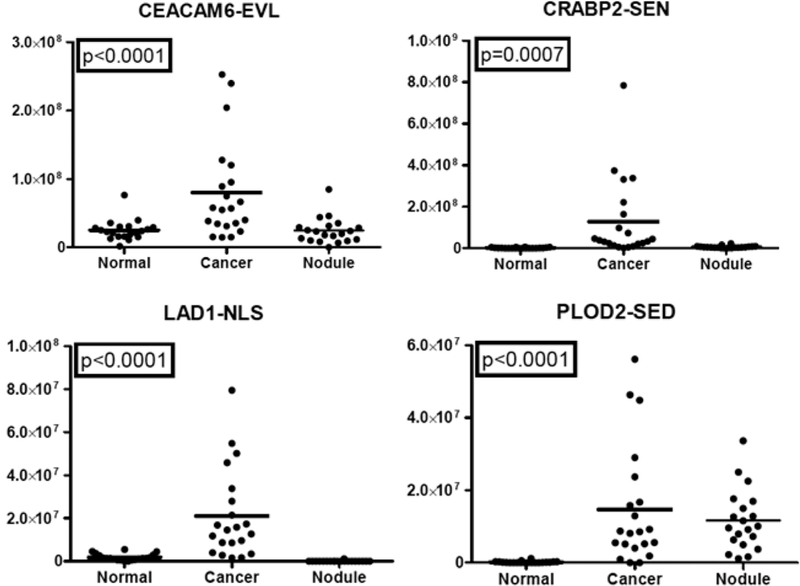
PRM measurement of representative peptides from putative ADC-specific proteins. Replicate PRM analyses were performed for peptides from 10 candidate proteins in 20 benign nodules, 22 ADC, and 20 normal bronchial biopsies. Representative plots for peptides that passed thresholds for ICC, Spearman correlation, and ANOVA are shown.
Validation by Immunohistochemistry
IHC analysis was performed on another independent set of benign nodules, ADCs, and normal lung tissues assembled in TMAs (Table 2). These IHC analyses targeted a seven-protein subset of the candidate biomarkers analyzed by PRM, based on the quality of the antibodies available, and included ALOX5, ALOX5AP, ITGAX, and SLC2A3 as well as the ADC-specific proteins CRABP2, CEACAM6, and LAD1 (Table S-7). The total number of cells tested for staining intensity was 7873 ± 2210 and 7069 ± 1917 on average (±STDEV) among lung cancers and benign lung nodules, respectively. ALOX5, ALOX5AP, ITGAX, and SLC2A3 were significantly overexpressed (p < 0.001 for all four proteins) in benign nodules as compared with NSCLCs (Table 3 and Figure 4). ALOX5 showed a strong intracellular pattern in 92% of the benign nodules and in 5% of the cancers. ALOX5AP showed strong membranous staining in 74% of the benign nodules and in no cancers, ITGAX in 92% of benign nodules and no cancers, and SLC2A3 in 96% of benign nodules and 30% of the cancers. IHC staining for LAD1 and CRABP2 did not distinguish benign nodules from cancers, whereas CEACAM6 was overexpressed in 81% of all cancers and 21% of benign nodules (p < 0.001) (Figure S-3). We found no significant association for all seven candidate biomarkers staining intensity with gender, stage, smoking status, or age among cancer patients (Table S-8). ROC curve analysis showed AUC values of 96% for ALOX5 and ITGAX, 89% for ALOX5AP, 86% fir SLC2A3, 85% for CEACAM6, 68% for CRABP2, and 61% for LAD1 (Figure S-4B).
Table 3.
Summary of IHC Analyses
| candidate biomarker | relative intensity ± STDEV in NSCLC (N = 86) | relative intensity ± STDEV in benign nodule (N = 63) | P value |
|---|---|---|---|
| ALOX5 | 0.04 ± 0.19 | 1.90 ± 0.79 | <0.001 |
| ALOX5AP | 0.00 ± 0.00 | 0.93 ± 0.71 | <0.001 |
| ITGAX | 0.00 ± 0.00 | 2.15 ± 0.81 | <0.001 |
| SLC2A3 | 0.46 ± 0.81 | 1.59 ± 0.69 | <0.001 |
| CEACAM6 | 1.47 ± 0.95 | 0.20 ± 0.61 | <0.001 |
| CRABP2 | 1.76 ± 0.89 | 1.24 ± 0.72 | 0.004 |
| LAD1 | 0.58 ± 0.56 | 0.34 ± 0.43 | 0.054 |
| CD4 memb | 6.8 ± 5.2 | 18.3 ± 14.3 | <0.001 |
| CD8 memb | 15.0 ± 11.0 | 24.4 ± 10.7 | <0.001 |
| FOXP3 nuc | 21.0 ± 11.0 | 30.0 ± 16.0 | <0.001 |
Figure 4.
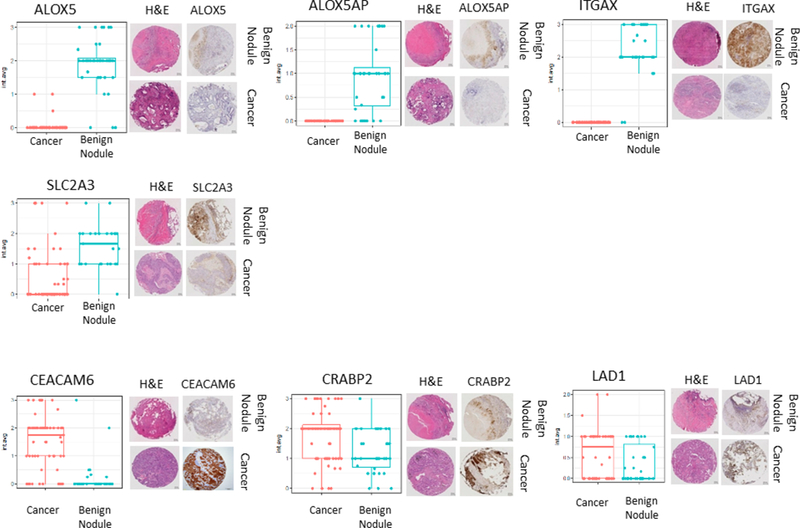
Validation of differential expression of seven candidate biomarkers by immunohistochemistry. Presented are box plots and distributions of IHC scores between 63 benign nodules and 86 lung cancers based on average staining intensity. For each antibody staining, representative images of hematoxylin and eosin stains of the nodule of interest and matched immunostains on an adjacent tissue section of a tissue microarray are provided.
Immunophenotyping of Lung Nodules
We next hypothesized that the nature of infiltrating lymphocytes in the tissue microenvironment of IPNs may be specific to nature of the nodules (benign or malignant) and that the infiltrating lymphocytes may be associated with the candidate biomarkers identified above. We therefore quantified the abundance of CD4, CD8, or FOXP3 infiltrating lymphocytes in our TMAs and tested its association with the expression of ALOX5, ALOX5AP, ITGAX, SLC2A3, CEACAM6, CRABP2, and LAD1. We obtained two replicate measurements and determined cytoplasmic, membranous, and nuclear staining as well as total cell number (average or maximum of repeated measurement). Spearman correlation was used to test the association between seven markers and percent positive of each of the cytoplasmic, membranous, and nuclear stains for the three cell types as well as total cell number. Representative images of CD4, CD8, and FOXP3 stains are shown in Figure 5A Benign nodules were associated with a stronger infiltration of CD4, CD8 T cells, and FOXP3 cells with a p value of <0.001 for all three markers (Figure 5B). The average staining intensity for the T cell markers was stronger in benign nodules for CD4 and CD8 but weaker for FOXP3 cells (Table 3). The associations between candidate biomarker immunostains and CD4, CD8, and FOXP3 stains were nonsignificant (Table 4), with the exception of a strong association between SLC2A3 staining and CD4 and FOXP3 staining intensity as well as an association between ITGAX and CD8 T cells.
Figure 5.
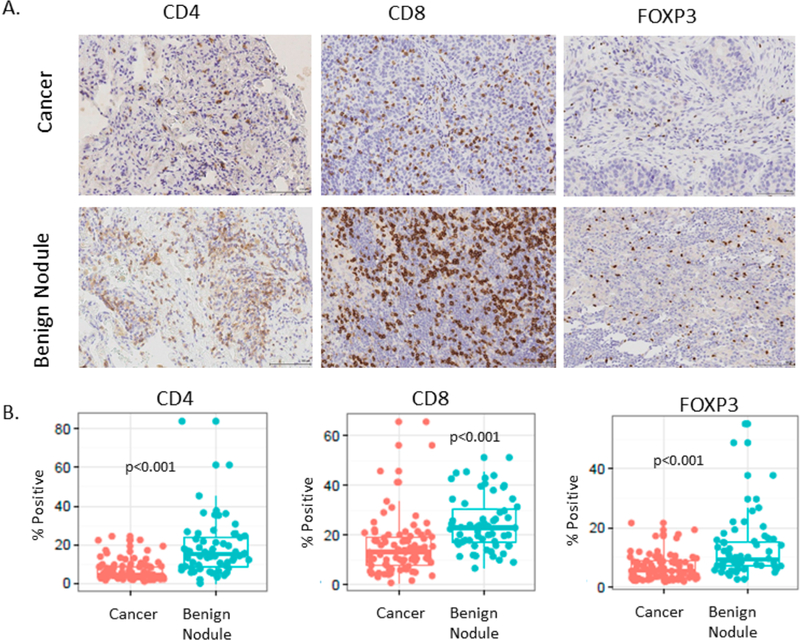
Immunophenotyping of lung nodules both benign and malignant. (A) Representative images of immunostains for CD4, CD8, or FOXP3. (B) Box plot and distribution of the percentage of positive infiltrating lymphocytes in 86 NSCLCs and 63 benign nodules.
Table 4.
Association between Staining Intensity for Seven Candidate Biomarkers and Staining Intensity for Tumor-Infiltrating T cells in Nodules, ADC, and SCC of the Lunga
| CD4 memb max P values |
CD8 memb max P values |
FOXP3 nuc max P values |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| biomarker candidates | nodule | ADC | SCC | nodule | ADC | SCC | nodule | ADC | SCC |
| ALOX5 | 0.127 | 0.582 | n/a | 0.176 | 0.304 | n/a | 0.474 | 0.638 | n/a |
| ALOX5AP | 0.071 | n/a | n/a | 0.735 | n/a | n/a | 0.500 | n/a | n/a |
| ITGAX | 0.231 | n/a | n/a | 0.006 | n/a | n/a | 0.854 | n/a | n/a |
| SLC2A3 | 0.544 | 0.037 | 0.088 | 0.436 | 0.042 | 0.858 | 0.781 | 0.841 | 0.004 |
| CEACAM6 | 0.326 | 0.968 | 0.350 | 0.683 | 0.212 | 0.096 | 0.547 | 0.147 | 0.786 |
| CRABP2 | 0.670 | 0.061 | 0.805 | 0.846 | 0.568 | 0.799 | 0.886 | 0.955 | 0.286 |
| LAD1 | 0.377 | 0.565 | 0.807 | 0.439 | 0.782 | 0.155 | 0.404 | 0.931 | 0.921 |
n/a not available because not expressed in cancer.
DISCUSSION
We report that proteomic analysis of benign and malignant nodules may uncover novel candidate biomarkers that could then be evaluated through other molecular diagnostic strategies. While our long-term goal is to identify noninvasive strategies for the early detection of lung cancer, we report here a first step toward this goal by completing a tissue-based discovery analysis with early validation efforts. We characterized the proteomes of a collection of resected benign lung nodules, lung ADCs, and airway epithelium, derived candidates based on the assumption that future molecular imaging probes would most likely detect plasma-membrane-associated proteins. We validated our candidate by PRM assays and by IHC in TMAs assembled for validation. We identified seven proteins that clearly discriminate between the groups and suggest that these candidates represent potential candidates for further validation.
The novelty of this research effort is three-fold. First, this is the first study to approach the diagnosis of IPNs by looking at the proteomic composition of these nodules and comparing to lung ADCs. Second, this report demonstrates the value of proteomic approaches to discover new candidate biomarkers. This in-depth search for proteomic candidate biomarker of IPNs was validated by two orthogonal methods in two independent data sets. We used PRM analysis and IHC to our candidates and demonstrated promising results. Third, our discovery effort specifically targeted membrane-associated proteins, which should be most readily detected by molecular imaging. While targeting the identification of organ-specific biomarkers of cancers has led to disappointing results, it remains a key strategy to advance diagnostics for lung cancer. Molecular imaging will play a significant role in basic, translational, and clinical research as related to functional genomics. Protein targets located at the cell surface of either the cancer cells or inflammatory cells in granulomas represent good candidates for molecular imaging. Clearly, the abundance and specificity of the target for the diagnosis of IPNs are critical to make molecular imaging approaches successful. The proteins identified have no overlap with other biomarkers previously reported.31,32
Because both granulomas and tumors are characterized by a rich immune microenvironment and because T lymphocytes are key effectors of the adaptive immunity,33,34 we characterized their prevalence in TMAs. Immunohistochemical analyses were performed on paraffin-embedded lung cancer tissue and the relation between Tregs (FOXP3+), CD4, and CD8 T cells and the diagnosis of granulomatous disease versus lung ADCs. We found that Tregs, CD4, and CD8 T cells were in greater abundance in granulomas as compared with lung ADCs without apparent specificity. Interestingly, some of the inflammatory cells were associated with expression of proteins identified in this study as candidate biomarkers, such as SLC2A3 and ITGAX expression (Table 4). The significance of these findings is yet to be determined. The immune response related to such different pathological processes as infection and malignancy is regulated by complex mechanisms that may demonstrate cellular specificity. The integration of such phenotypic findings with genotype, for example, should translate to more accurate diagnostic approaches. This hypothesis warrants further investigation.
The limitations of this study include the heterogeneous nature of the nodule population, the primary focus on comparing lung nodules to lung ADCs (and not all lung cancers), as well as the intended focus on membrane-associated proteins. This study also does not address any sort of clinical risk prediction tool that can be investigated in future studies. We are evaluating candidates for future studies, including the development of new molecular imaging probes to test this concept. Ongoing work includes the design of molecular imaging probes and evaluation of animal models to test these candidate biomarkers. These next studies can evaluate novel candidate biomarkers for noninvasive molecular diagnosis of lung cancer.
Supplementary Material
Data Set S-1: Summary and analysis settings, spectrum, peptide, protein, and modification information.
Table S-1: PRM protein validation panel for biomarker candidates.
Table S-2: PRM protein validation – total peptide peak area exported from Skyline file for all 43 protein biomarker candidates.
Table S-3: PRM protein validation – total peptide peak area; quantifiable peptides for 27 protein biomarker candidates.
Table S-4: PRM protein validation – total peptide peak area normalized to BG peptide standard for 27 protein biomarker candidates.
Table S-5: PRM protein validation – total peptide peak area normalized to BG peptide standard for 27 protein biomarker candidates – technical replicates averaged.
Table S-6: PRM protein validation – ICC, Pearson correlation for replicates and ANOVA for 27 protein biomarker candidates.
Table S-7: Summary of IHC observations.
Table S-8: Summary of IHC associations in patients with cancer.
Table S-9: Complete patient characteristics: discovery.
Table S-10: Complete patient characteristics: PRM validation.
Table S-11: Complete patient characteristics: IHC validation.
Figure S-1: PRM results for all peptides up in nodules.
Figure S-2: PRM results for all peptides up in cancer.
Figure S-3: IHC images (40×) showing staining pattern.
Figure S-4: ROC analysis.
ACKNOWLEDGMENTS
We acknowledge the Translational Pathology Shared Resource supported by NCI/NIH Cancer Center Support Grant 2P30 CA068485–14 and the Vanderbilt Mouse Metabolic Phenotyp- ing Center Grant 5U24DK059637–13. This study was supported by the National Institutes of Health (R01CA102353, U01CA152662, and the Lung SPORE P50CA90949 to P.P.M.; U01CA152647 to D.C.L.), Department of Defense (W81XWH- 11–2-0161 and CDMRP LC090615P3 to P.P.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- ADC
adenocarcinoma
- AP
E. coli alkaline phosphatase
- BG
E. coli beta-galactosidase
- CV
coefficient of variation
- FDR
false discovery rate
- IHC
immunohistochemistry
- IPN
indeterminate pulmonary nodule
- LC-MS
liquid chromatography—mass spectrometry
- LDCT
low-dose computed tomography
- LRP
labeled reference peptide
- MRM
multiple reaction monitoring
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- NSCLC
non-small cell lung cancer
- PET-CT
positron emission tomography-computed tomography
- PRM
parallel reaction monitoring
- SCC
squamous cell carcinoma
- SCX
strong cation exchange chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.7b00245.
Notes
The authors declare no competing financial interest.
Data are available through ProteomeXchange Accession PXD005116.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer statistics, 2017. Ca- Cancer J. Clin. 2017, 67, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Aberle DR; Adams AM; Berg CD; Black WC; Clapp JD; Fagerstrom RM; Gareen IF; Gatsonis C; Marcus PM; et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365 (5), 395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Massion PP Biomarkers to the rescue in a lung nodule epidemic. J. Clin. Oncol. 2014, 32 (8), 725–6. [DOI] [PubMed] [Google Scholar]
- (4).Massion PP; Walker RC Indeterminate pulmonary nodules: risk for having or for developing lung cancer? Cancer Prev. Res. 2014, 7 (12), 1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gould MK; Tang T; Liu IL; Lee J; Zheng C; Danforth KN; Kosco AE; Di Fiore JL; Suh DE Recent Trends in the Identification of Incidental Pulmonary Nodules. Am. J. Respir. Crit. Care Med. 2015, 192 (10), 1208–14. [DOI] [PubMed] [Google Scholar]
- (6).Patz EF Jr.; Pinsky P; Gatsonis C; Sicks JD; Kramer BS; Tammemagi MC; Chiles C; Black WC; Aberle D R Overdiagnosis in low-dose computed tomography screening for lung cancer. JAMA Intern Med. 2014, 174 (2), 269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Swensen SJ; Jett JR; Hartman TE; Midthun DE; Mandrekar SJ; Hillman SL; Sykes AM; Aughenbaugh GL; Bungum AO; Allen KL CT screening for lung cancer: five-year prospective experience. Radiology 2005, 235 (1), 259–65. [DOI] [PubMed] [Google Scholar]
- (8).Croswell JM; Baker SG; Marcus PM; Clapp JD; Kramer BS Cumulative incidence of false-positive test results in lung cancer screening: a randomized trial. Ann. Intern. Med. 2010, 152 (8), 505–12 W176—80.. [DOI] [PubMed] [Google Scholar]
- (9).Henschke CI; Yankelevitz DF; Mirtcheva R; McGuinness G; McCauley D; Miettinen OS CT screening for lung cancer: frequency and significance of part-solid and nonsolid nodules. AJR, Am. J. Roentgenol. 2002, 178 (5), 1053–7. [DOI] [PubMed] [Google Scholar]
- (10).Deppen SA; Blume JD; Kensinger CD; Morgan AM; Aldrich MC; Massion PP; Walker RC; McPheeters ML; Putnam JB Jr.; Grogan EL Accuracy of FDG-PET to diagnose lung cancer in areas with infectious lung disease: a meta-analysis. JAMA 2014, 312 (12), 1227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Brandman S; Ko JP Pulmonary nodule detection, characterization, and management with multidetector computed tomography. J. Thorac Imaging. 2011, 26 (2), 90–105. [DOI] [PubMed] [Google Scholar]
- (12).Sayyouh M; Vummidi DR; Kazerooni EA Evaluation and management of pulmonary nodules: state-of-the-art and future perspectives. Expert Opin. Med. Diagn. 2013, 7 (6), 629–44. [DOI] [PubMed] [Google Scholar]
- (13).Furman AM; Dit Yafawi JZ; Soubani AO An update on the evaluation and management of small pulmonary nodules. Future Oncol. 2013, 9 (6), 855–65. [DOI] [PubMed] [Google Scholar]
- (14).Gould MK; Donington J; Lynch WR; Mazzone PJ; Midthun DE; Naidich DP; Wiener RS Evaluation of individuals with pulmonary nodules: when is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013, 143 (5), e93S–e120S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Matsuguma H; Mori K; Nakahara R; Suzuki H; Kasai T; Kamiyama Y; Igarashi S; Kodama T; Yokoi K Characteristics of subsolid pulmonary nodules showing growth during follow-up with CT scanning. Chest 2013, 143 (2), 436–43. [DOI] [PubMed] [Google Scholar]
- (16).Pinsky PF; Nath PH; Gierada DS; Sonavane S; Szabo E Short- and long-term lung cancer risk associated with noncalcified nodules observed on low-dose CT. Cancer Prev. Res. 2014, 7 (12), 1179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gould MK; Ananth L; Barnett PG A clinical model to estimate the pretest probability of lung cancer in patients with solitary pulmonary nodules. Chest 2007, 131 (2), 383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Patel VK; Naik SK; Naidich DP; Travis WD; Weingarten JA; Lazzaro R; Gutterman DD; Wentowski C; Grosu HB; Raoof S A practical algorithmic approach to the diagnosis and management of solitary pulmonary nodules: part 2: pretest probability and algorithm. Chest 2013, 143 (3), 840–6. [DOI] [PubMed] [Google Scholar]
- (19).Deppen SA; Blume JD; Aldrich MC; Fletcher SA; Massion PP; Walker RC; Chen HC; Speroff T; Degesys CA; Pinkerman R; et al. Predicting lung cancer prior to surgical resection in patients with lung nodules. J. Thorac. Oncol. 2014, 9 (10), 1477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang Y; Yang F; Gritsenko MA; Wang Y; Clauss T; Liu T; Shen Y; Monroe ME; Lopez-Ferrer D; Reno T; et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11 (10), 2019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sprung RW; Martinez MA; Carpenter KL; Ham AJ; Washington MK; Arteaga CL; Sanders ME; Liebler DC Precision of multiple reaction monitoring mass spectrometry analysis of formalin-fixed, paraffin-embedded tissue. J. Proteome Res. 2012, 11 (6), 3498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tabb DL; Fernando CG; Chambers MC MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 6 (2), 654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kim S; Pevzner PA MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dasari S; Chambers MC; Martinez MA; Carpenter KL; Ham AJ; Vega-Montoto LJ; Tabb DL Pepitome: evaluating improved spectral library search for identification complementarity and quality assessment. J. Proteome Res. 2012, 11 (3), 1686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ma ZQ; Dasari S; Chambers MC; Litton MD; Sobecki SM; Zimmerman LJ; Halvey PJ; Schilling B; Drake PM; Gibson BW; et al. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 2009, 8 (8), 3872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang B; Chambers MC; Tabb DL Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007, 6 (9), 3549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kononen J; Bubendorf L; Kallionimeni A; Barlund M; Schraml P; Leighton S; Torhorst J; Mihatsch MJ; Sauter G; Kallionimeni OP Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med. 1998, 4 (7), 844–7. [DOI] [PubMed] [Google Scholar]
- (29).Massion PP; Taflan PM; Jamshedur Rahman SM; Yildiz P; Shyr Y; Edgerton ME; Westfall MD; Roberts JR; Pietenpol JA; Carbone DP; et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Chest 2003, 63 (21), 7113–21. [PubMed] [Google Scholar]
- (30).Zhang H; Liu Q; Zimmerman LJ; Ham AJ; Slebos RJ; Rahman J; Kikuchi T; Massion PP; Carbone DP; Billheimer D; et al. Methods for peptide and protein quantitation by liquid chromatography-multiple reaction monitoring mass spectrometry. Mol. Cell. Proteomics 2011, 10 (6), M110.006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Vachani A; Pass HI; Rom WN; Midthun DE; Edell ES; Laviolette M; Li XJ; Fong PY; Hunsucker SW; Hayward C; et al. Validation of a multiprotein plasma classifier to identify benign lung nodules. J. Thorac. Oncol. 2015, 10 (4), 629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li XJ; Hayward C; Fong PY; Dominguez M; Hunsucker SW; Lee LW; McLean M; Law S; Butler H; Schirm M; et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Sci. Transl Med. 2013, 5 (207), 207ra142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Schalper KA; Brown J; Carvajal-Hausdorf D; McLaughlin J; Velcheti V; Syrigos KN; Herbst RS; Rimm DL Objective measurement and clinical significance of TILs in non-small cell lung cancer. J. Natl. Cancer Inst. 2015, 107 (3), dju435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ganesan AP; Johansson M; Ruffell B; Yagui-Beltran A; Lau J; Jablons DM; Coussens LM Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J. Immunol. 2013, 191 (4), 2009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Set S-1: Summary and analysis settings, spectrum, peptide, protein, and modification information.
Table S-1: PRM protein validation panel for biomarker candidates.
Table S-2: PRM protein validation – total peptide peak area exported from Skyline file for all 43 protein biomarker candidates.
Table S-3: PRM protein validation – total peptide peak area; quantifiable peptides for 27 protein biomarker candidates.
Table S-4: PRM protein validation – total peptide peak area normalized to BG peptide standard for 27 protein biomarker candidates.
Table S-5: PRM protein validation – total peptide peak area normalized to BG peptide standard for 27 protein biomarker candidates – technical replicates averaged.
Table S-6: PRM protein validation – ICC, Pearson correlation for replicates and ANOVA for 27 protein biomarker candidates.
Table S-7: Summary of IHC observations.
Table S-8: Summary of IHC associations in patients with cancer.
Table S-9: Complete patient characteristics: discovery.
Table S-10: Complete patient characteristics: PRM validation.
Table S-11: Complete patient characteristics: IHC validation.
Figure S-1: PRM results for all peptides up in nodules.
Figure S-2: PRM results for all peptides up in cancer.
Figure S-3: IHC images (40×) showing staining pattern.
Figure S-4: ROC analysis.