

Abstract
Aims
This programme investigated topical regorafenib, a multikinase inhibitor, in patients with neovascular age‐related macular degeneration (nAMD).
Methods
Topical regorafenib was investigated in an open‐label, phase IIa/b study in which patients with choroidal neovascularization (CNV) secondary to nAMD received regorafenib (25 μl, 30 mg ml–1) three times a day for 12 weeks. The primary endpoint of the phase II/a/b study was mean change in best‐corrected visual acuity (BCVA) from baseline to weeks 4 and 12.
Results
In nAMD patients (N = 51), mean changes in BCVA were +1.2 [90% confidence interval (CI) –0.61, 2.97] and −2.4 (90% CI −4.18, −0.54) letters at weeks 4 and 12, respectively. Ocular treatment‐emergent adverse events (TEAEs) (study eye) were reported in 21 patients by week 12. There was one serious ocular TEAE (visual acuity reduced) that was not drug related. Twenty patients required rescue (intravitreal ranibizumab).
Conclusions
The programme was terminated after phase IIa ended because efficacy was lower than with current nAMD treatments. According to elaborate post hoc analyses, the most likely reason was insufficient exposure in the target compartment (back of the eye).
Keywords: drug development, clinical trials, pharmacometrics, ophthalmology
What is Already Known about this Subject
Neovascular age‐related macular degeneration (nAMD) is currently treated intravitreally by biologics targeting vascular endothelial growth factor (VEGF).
Regorafenib is a multikinase inhibitor that targets nAMD‐relevant kinases VEGF receptor 2/3, with preclinical data suggesting efficacy in disease‐relevant animal models.
A phase I clinical study has confirmed the safety and tolerability of regorafenib eye drops.
What this Study Adds
This study provides first efficacy data of regorafenib eye drops in nAMD patients.
The efficacy of regorafenib eye drops was lower than with current intravitreal nAMD treatments. Therefore, the programme was terminated.
Following extensive post hoc analyses, insufficient exposure in the target compartment (back of the eye) is considered to be the most likely reason for failure.
Introduction
Anti‐VEGF injections are standard of care for neovascular age‐related macular degeneration (nAMD). These large protein biologics target VEGF‐A, a growth factor primarily associated with the development of angiogenesis 1, 2, 3. Fixed dosing in nAMD patients at either monthly (intravitreal ranibizumab and bevacizumab) or bimonthly, after a loading dose (intravitreal aflibercept), intervals yielded the best visual acuity results 4, 5, 6, 7, 8, 9, 10, 11, 12. Given the treatment burden and risks arising from the repeated intravitreal treatment, more convenient, topical therapeutic options that could be self‐administered by the patient would be beneficial.
The small molecule regorafenib inhibits, besides other kinases, disease‐relevant kinases VEGF receptor 2/3 (VEGFR 2/3, belonging to the family of receptor tyrosine kinases, Type IV) 13, 14, 15 and platelet‐derived growth factor receptor β (PDGFRβ, belonging to the family of receptor tyrosine kinases, Type III) 16 at nanomolar concentrations 17, and was therefore developed as a topical eye drop formulation for use in nAMD.
Preclinical studies indicated that regorafenib reached the back of the eye (BoE) when administered as an eye drop. Expression of VEGF receptors has also been shown at the BoE of animals and humans 13, 15. In addition, efficacy was shown in rat and primate laser‐induced choroidal neovascularization (CNV) animal models 18, 19. Following nonclinical local (eye) and systemic toxicity studies, a phase I healthy volunteer study was performed. Regorafenib was well tolerated and there were no safety concerns in humans 20. The objective of the current phase IIa/b study [Developing Regorafenib Eye Drops for wet AMD (DREAM); NCT02222207]was to translate the observed preclinical signs of efficacy to patients and thus to evaluate the efficacy of regorafenib eye drops in improving best‐corrected visual acuity in treatment‐naïve patients with nAMD.
Methods
DREAM was a multicentre, open‐label phase IIa/b study conducted at 34 European sites from 10 October 2014 (first patient first visit) to 17 June 2015 (last patient last visit) that determined the effects of topical regorafenib in patients with nAMD. The study consisted of a phase IIa (proof‐of‐concept) part, designed as a multicentre, single‐arm, open‐label study, and a phase IIb part, constituting a dose‐finding study. Only the phase IIa part was conducted.
Materials
Regorafenib (BAY 73–4506) suspended in 100% light liquid paraffin (Bayer AG, Wuppertal, Germany) at a concentration of 30 mg ml–1 was used. Intravitreal ranibizumab (Lucentis, Genentech, South San Francisco, CA, USA) was used as rescue medication.
Dose rationale
The dose was chosen based on preclinical experiments in rodents and nonhuman primates, in which efficacy was observed on administering 40–50 μl of 20 mg ml–1 regorafenib eye drops twice daily (i.e. 1.6–2 mg day–1) 18, 19. Eye surface scaling from primate to human was applied. In addition, the maximum technically feasible concentration (30 mg ml–1), as well as the highest administration volume without any spill‐over (25 μL) and the maximum dosing frequency considered feasible in the patient population [three times daily (TID)], were considered. The resulting amount of 0.75 mg per administration (or 2.25 mg day–1 when given TID) was also shown to be safe in the phase I study of the programme 20. In order to strengthen this human dose estimate, significant efforts were put into physiology‐based pharmacokinetics (PBPK) modelling in the rabbit as, in addition to pharmacokinetic (PK) data for regorafenib, most of the data in the literature were available for this species 21, 22, 23. However, it turned out that no consistent model could be developed. Based on common physiological parameter values and specific compound‐related parameters, the observations for the different compounds could not be described equally well. Meaningful descriptions for the single compounds could only be achieved after fitting the model individually to observed data. Finally, it could not be excluded that there is still a lack of understanding of the processes involved in drug distribution in the eye after topical application, preventing a sound mechanistic modelling. As a result of these limitations, the aforementioned estimation of the human dose was used.
Clinical evaluation
The key inclusion and exclusion criteria can be found in Table 1. All patients provided written informed consent to participate. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice and was approved by respective ethics committees and institutional review boards of the clinical sites.
Table 1.
Main inclusion and exclusion criteria
| Inclusion criteria | |
| 1 | Men and women aged ≥50 years |
| 2 | Active treatment‐naïve primary subfoveal CNV secondary to nAMD, including juxtafoveal lesions that affected the fovea, as evidenced by fluorescein angiography |
| 3 | Area of CNV occupying at least 50% of total lesion, as determined by fluorescein angiography |
| 4 | Evidence of intraretinal and/or subretinal fluid on optical coherence tomography in the study eye |
| 5 | Best‐corrected visual acuity between 73 and 25 Early Treatment Diabetic Retinopathy Study letters (Snellen activity equivalent of 20/40 to 20/320) in the study eye |
| 6 | Ability to complete all study‐related procedures |
| Exclusion criteria | |
| 1 | Concurrent disease in the study eye, other than AMD, that could compromise visual acuity |
| 2 | Any findings in the study eye that would limit the potential to benefit from study treatment, or could otherwise confound interpretation of the results, such as total lesion size (including structural damage to the centre of the macula, neovascularization, scar, blood) >12 disc areas (30.5 mm2) and subfoveal fibrosis, as assessed by fluorescein angiography |
| 3 | Only one functional eye |
| 4 | Prior ocular or systemic treatment or surgery for nAMD in the study eye, except dietary supplements or vitamins |
| 5 | Prior treatment with any systemic anti‐vascular endothelial growth factor agent, cataract surgery or any other surgery in the study eye within 12 weeks prior to start of study treatment |
| 6 | History or current use of long‐acting steroids, either systemically or injected intraocularly, periocularly or subconjunctivally |
| 7 | Intraocular pressure ≥25 mmHg in the study eye |
| 8 | Uncontrolled glaucoma |
| 9 | Any other condition that would require frequent chronic co‐administration of other topical eye medications that would interfere with study drug administration (e.g. contact lens) |
| 10 | Uncontrolled hypertension |
| 11 | Women of childbearing potential |
AMD, age‐related macular degeneration; CNV, choroidal neovascularization; nAMD, neovascular AMD
All patients received training on the application of eye drops, and self‐administered one 25‐μl drop of regorafenib (30 mg ml–1) to the study eye three times daily for 12 weeks. Patients were seen weekly for 4 weeks, followed by biweekly visits up to week 12. Best‐corrected visual acuity [BCVA; Early Treatment Diabetic Retinopathy Study (ETDRS) letter chart], slit‐lamp examination and optical coherence tomography were performed at each visit, and fundus photography and fluorescein angiography were scheduled at screening, and at weeks 4 and 12. Safety assessments took place at each visit, and patients were observed 30–60 min after administration of eye drops, followed by anterior segment evaluation and measurement of intraocular pressure.
The primary endpoints were the changes in BCVA (ETDRS letters) from baseline to week 4 and to week 12. Secondary endpoints were the proportion of patients with a change in BCVA of ≥0 letters from week 4 to week 12 and the proportion of patients with a loss of ≥10 letters from baseline to week 12. As an exploratory endpoint, the mean change in central retinal thickness (CRT) was recorded at week 4 and week 12. An adverse event (AE) was considered treatment emergent if it was reported after the first day and no later than 30 days after the last administration of study drug. The assessment of the causal relationship between an AE and the administration of study drug was a clinical decision to be reached by the investigator based on all available information at the time. In essence, the investigator needed to answer the question of whether there was a ʻreasonable causal relationshipʼ with the study drug, with the possible answers being ʻyesʼ or ʻnoʼ. Important factors to be considered in assessing this relationship included e.g. temporal proximity to drug administration, recovery on drug discontinuation, recurrence on drug reintroduction, concomitant medication and treatment, as well as underlying, concomitant or intercurrent disease and so forth.
Rescue medication was intravitreal ranibizumab (administered in accordance with the drug label), which was initiated from week 3 through to week 12 if patients had a loss of vision of >5 ETDRS letters (from best previous measurement since baseline, and the actual BCVA was below baseline values) and persistence (no decrease since baseline) or recurrence (from baseline) of subretinal or intraretinal fluid.
The primary efficacy and secondary variables were analysed descriptively. Mean changes for the primary endpoints were presented together with the 90% confidence interval (CI) based on the t‐distribution. Efficacy was analysed in the full analysis set, defined as all patients who received treatment. To allow comparison with data in the literature, missing efficacy observations, as well as observations obtained after the application of rescue treatment, were imputed using a last observation carried forward approach. With the planned sample size of 50, the lower limit of the two‐sided 90% CI for the mean change from baseline to week 4 could be expected to exclude zero letters with a probability of 93%, if a true change of five letters and a standard deviation (SD) of 11 was assumed.
Blood samples for PK analyses were taken before and after administration of regorafenib eye drops during each regular visit up to the end of the study. Regorafenib concentrations in the plasma were determined using a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) method with a lower limit of quantitation (LLOQ) of 0.1 μg l–1.
Assessment of BoE exposure
In order to understand our results better, they were compared with other preclinical and clinical data obtained with regorafenib which have previously been published 18, 19, 24 or for which there are data on file (some of the primate data). All published and thus far unpublished data were obtained according to respective state authority regulations and were approved by the local ethics committees. The BoE exposure in patients was estimated, as no direct measurement can be performed. This was done under the following assumptions. The area under the concentration–time curve from time 0 to 24 h [AUC(0–24 h)] was considered to be the most robust measure for comparing exposures to regorafenib in studies with different animal species and in humans. Although it was not possible formally to check whether the AUC was the PK/pharmacodynamic driver, it was chosen as it includes information on both the level and duration of exposure. All AUCs were calculated from concentration data in steady state after 7–28 days (depending on the study) of daily dosing; these are summarized in Table 2.
Table 2.
Exposure of regorafenib in plasma, eye, and BoE, based on regimens used in animal and human studies
| Unbound fraction | Dose a (mg day –1 ) | AUC(0–24) (μg h l –1 ) | AUC u(0–24) b (μg h l –1 ) | ||
|---|---|---|---|---|---|
| Rat topical 14 | Plasma | 0.0072 | 0.4 | 222 | 1.6 |
| Eye | 0.4 | 15 100 | 109 | ||
| Monkey topical c [data on file] | Plasma | 0.022 | 1d | 108 | 2.38 |
| 13 | BoE | 2 | 1362 | 30.0 | |
| Human topical (from DREAM study) | Plasma | 0.005 | 2.25 | 76 | 0.38 |
| BoE (estimated) | 2.25 | 963 | 4.82 | ||
| Human oral 17 | Plasma | 0.005 | 160 | 58 300 | 292 |
| Plasma | 10 | 5670 | 28.5 |
All data except the human topical data are from sources other than the presented phase IIa (DREAM) study. Data sources are indicated by reference numbers. AUC(0–24), area under the curve from time 0 to 24 h; AUCu, area under unbound concentration curve; BoE, back of the eye; DREAM, Developing Regorafenib Eye drops for neovascular Age‐related Macular degeneration
Dose per eye in case of topical applications
Unbound AUC
AUC values were calculated from dose‐normalized data and then multiplied with dose per eye
Both eyes treated, 1 mg per eye. A total dose of 2 mg is therefore comparable to results for exposure in eye
In order to allow an assessment of the in vivo regorafenib exposure compared with the in vitro half‐maximal inhibitory concentration (IC50; cellular IC50 = 3 nM or 1.5 μg l–1) for VEGF receptor inhibition of regorafenib, all in vivo AUC values were converted into unbound AUCs (AUCu) using the unbound fractions determined for different species.
As BoE concentrations cannot be measured in humans, we tried to estimate the unbound concentrations that were likely to be reached in patients by transferring relationships found in monkeys. For this purpose, the ratio between the AUCu for BoE and plasma in monkeys (see Table 2) was calculated, and found to be 12.6. In order to strengthen this assumption, a second approach to derive an AUC ratio (BoE vs. plasma) was applied. Here, the steady‐state partition coefficient for BoE tissue (KBoE/plasma = CBoE/Cplasma) is 10, estimated on the basis of logD = 3.8, log (phospholipid membrane/water partition coefficient) = 4.4 and unbound fraction in plasma fu = 0.022 (for details on the model, see Schmitt 25). Therefore, measured values and the physicochemical model revealed similar ratios, indicating that regorafenib concentrations in eye tissue and plasma are in steady‐state equilibrium in monkeys. For the estimation below, a value of 12.6 was applied.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 27, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 28.
Results
A total of 52 nAMD patients received treatment in DREAM, and 51 were included in the full analysis set (also see Figure 1). Baseline characteristics for the 51 patients are shown in Table 3. Following treatment with regorafenib 30 mg ml–1, the mean (SD) change in BCVA from baseline was +1.2 (7.5) letters (90% CI –0.61, 2.97) at week 4 and −2.4 (7.7) letters (90% CI −4.18, −0.54) at week 12 (Figure 2A). The median (quartiles) of the changes at week 4 and week 12, respectively, were 1 (Q1: −2; Q3: +5) and − 3 (Q1: −8; Q3: +2). The proportion of patients with a gain of ≥0 letters from week 4 to week 12 was 42.0% (n = 21). Sixteen per cent (n = 8) of the patients lost ≥10 letters from baseline to week 12. Patients with occult CNV (n = 42) lost fewer letters than did those with classic CNV (n = 8; 0.9 vs. 10.1 letters at week 12). The mean (SD) change in CRT was +26 (74) and +47 (137) μm at weeks 4 and 12 (Figure 2B). A total of 20 patients required rescue medication, which led to an improvement in BCVA and CRT (Figure 3).
Figure 1.
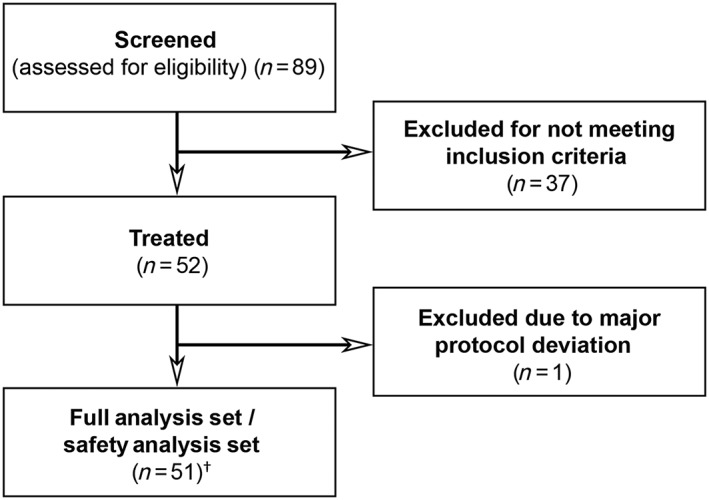
Study flow chart. † Includes one patient who died after the first dose of study drug. No efficacy measures were obtained from this patient
Table 3.
Baseline characteristics of patients included in the phase IIa study
| Characteristic [mean (SD) or n (%)] | Regorafenib (n = 51) |
|---|---|
| Age, years, mean (SD) | 75 (8.4) |
| Female, n (%) | 29 (56.9) |
| Race, n (%) | |
| White | 44 (86.3) |
| Not reported | 7 (13.7) |
| BCVA, letters, mean (SD) | 59 (11.9) |
| Median (Q1, Q3) | 62 (52, 67) |
| CNV lesion size, mm2, mean (SD) | 7.1 (5.6) |
| Total lesion size, mm2, mean (SD) | 7.3 (5.9) |
| Type of CNV, n (%) | |
| Predominantly classic | 8 (15.7) |
| Occult with fibrovascular PED | 42 (82.4) |
| Occult with serous PED | 1 (2.0) |
| CRT, μm, mean (SD) | 463.7 (113.0) |
BCVA, best‐corrected visual acuity; CNV, choroidal neovascularization; CRT, central retinal thickness; PED, pigment epithelial detachment; Q, quartile; SD, standard deviation
Figure 2.
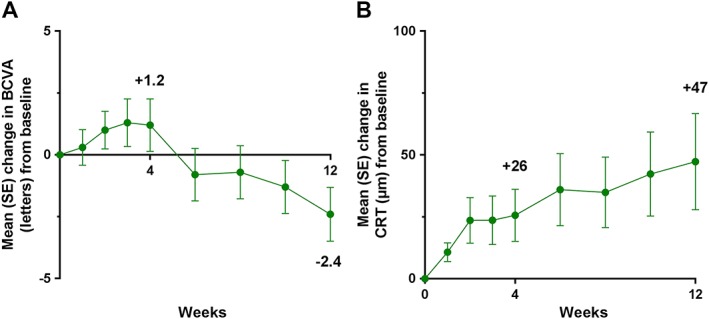
Mean changes in BCVA (ETDRS letters) (A) and CRT (B) from baseline in 51 patients with neovascular age‐related macular degeneration following treatment with regorafenib 30 mg ml–1 three times daily for 12 weeks. BCVA, best‐corrected visual acuity; CRT, central retinal thickness; ETDRS, Early Treatment Diabetic Retinopathy Study; SE, standard error
Figure 3.
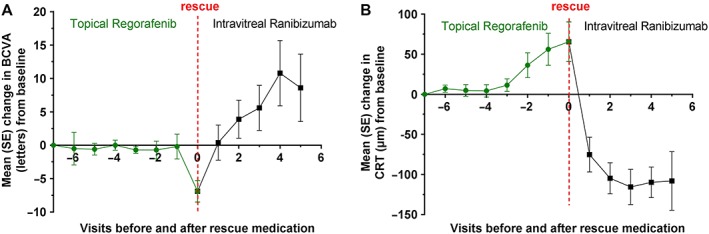
Mean changes in BCVA (ETDRS letters) (A) and CRT (B) in patients with neovascular age‐related macular degeneration (n = 20) who received rescue medication (intravitreal ranibizumab) after prior regorafenib treatment. BCVA, best‐corrected visual acuity; CRT, central retinal thickness; ETDRS, Early Treatment Diabetic Retinopathy Study; SE, standard error
The drug was well tolerated; ocular treatment‐emergent AEs (TEAEs) were reported in the study eye of 21 patients (41.2%) by week 12 (Table 4); the maximum severity was mild to moderate in the majority of patients (95.2%). The only AEs that occurred in 5% or more of patients were ʻvisual acuity reducedʼ (7.8%) and ʻvisual acuity tests abnormalʼ (11.8%); both affected the study eye.
Table 4.
Patients with TEAEs (safety analysis set) at week 12 who were included in the phase IIa study
| Event [n (%)] | Regorafenib (n = 51) |
|---|---|
| Any TEAE | 33 (64.7) |
| Any ocular TEAE in the study eye | 21 (41.2) |
| Mild | 12 (23.5) |
| Moderate | 8 (15.7) |
| Severe | 1 (2.0) |
| Treatment related | 8 (15.7) |
| Any non‐ocular TEAE | 18 (35.3) |
| Treatment related | 1 (2.0) |
| Any serious TEAE | 3 (5.9) |
| Any serious ocular TEAE in the study eye | 1 (2.0) |
| Treatment related | 0 (0.0) |
| Any non‐ocular serious TEAE | 2 (3.9) |
| Treatment related | 0 (0.0) |
| Death | 1 (2.0) |
TEAE, treatment‐emergent adverse event
There was one serious ocular TEAE (visual acuity reduced) and two serious non‐ocular TEAEs (ankle fracture and acute myocardial infarction, resulting in one death), but none were drug related according to the assessment of the investigator. Regorafenib plasma concentrations were similar to those observed after administration of corresponding regorafenib eye drop doses in the phase I study 20 (Figure 4).
Figure 4.
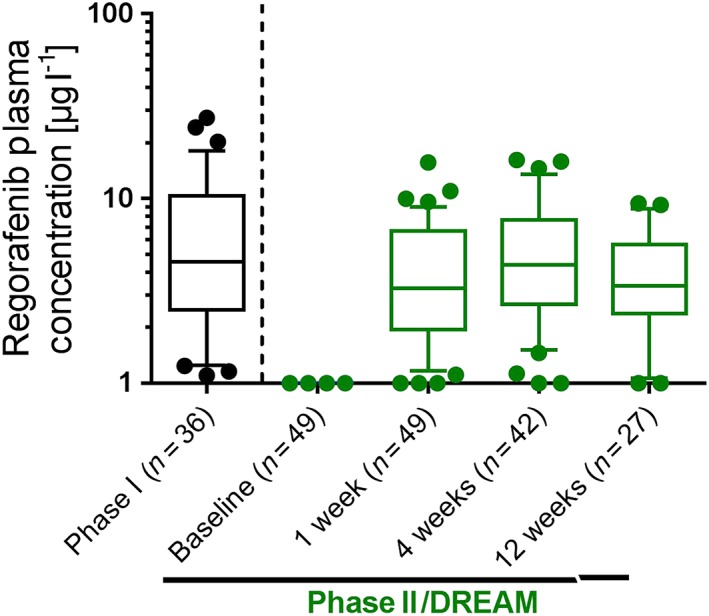
Plasma concentrations of regorafenib after topical administration of 30 mg ml–1, 25 μl three times daily in steady state. There was no significant difference between phase I, in which regorafenib was administered by a study nurse to healthy volunteers, and phase II, when patients self‐administered the eye drops. Data presented as box‐plots (median, boxes 25th and 75th percentile; whiskers 10th and 90th percentile; single values outside 10th and 90th percentile). DREAM, Developing Regorafenib Eye drops for neovascular Age‐related Macular degeneration
Derived from the aforementioned assumptions and the AUC of regorafenib in plasma, the estimated human AUC(0–24 h)BoE was 963 μg h l–1, and the respective calculated AUCu, BoE was 4.82 μg h l–1 (see Table 2 and Figure 5).
Figure 5.
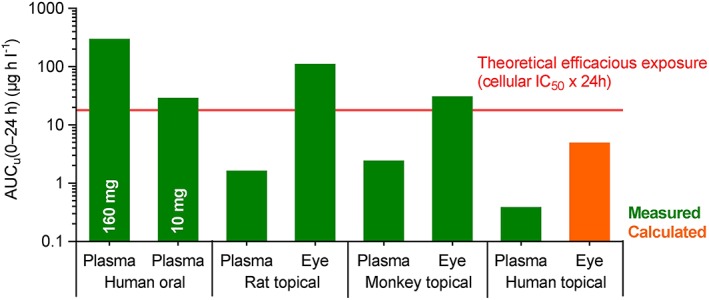
Model showing plasma and eye exposure of regorafenib based on routes used in animal and human studies. AUCu (0–24 h), area under unbound concentration curve from time 0 to 24 h. Data from rat studies published in 19, data from primate studies partly published in 18. Remaining data on file. Data on systemic exposure taken from the study report of the study referenced in [25]. IC50 , half‐maximal inhibitory concentration
Discussion
Regorafenib is a multikinase inhibitor targeting nAMD‐relevant kinases VEGF‐ and PDGFRs at nanomolar concentrations 17. Preclinical results from rat and monkey laser‐CNV models showed that the efficacy of topical regorafenib was similar to that observed with intravitreal anti‐VEGF agents. In addition, sufficient concentrations of regorafenib were reached in the BoE of these species to explain the in vivo efficacy 18, 19. Unfortunately, these preclinical efficacy results did not translate into the clinical studies. None of the predefined success criteria based on the primary readout (mean change in BCVA at weeks 4 and 12) were met in the phase IIa/b study in patients with nAMD. Instead, patients treated with regorafenib eye drops showed BCVA and CRT time courses that were similar to a natural disease course (e.g. compared with the sham treatment arm in the MARINA study, which showed a decline in BCVA by approximately four letters within the first 3 months 7). The study did not continue after completion of phase IIa because of insufficient efficacy. Regorafenib was well tolerated, and the safety profile was similar to that observed in phase I studies in healthy human volunteers 20.
There are a number of potential reasons for the failed translation of efficacy from preclinical to clinical studies. First, insufficient patient compliance to self‐administer the eye drops could have affected outcomes; however, this is unlikely as plasma concentrations in the phase IIa/b study were in the same range as those observed in phase I, in which eye drops were administered by a study nurse (see Figure 4). Although the skipping of single administrations could not be fully excluded from this analysis, patients must have taken the majority of doses, or otherwise the systemic exposure would have been expected to be lower.
Second, although targeting the same pathway, the molecular targets that are inhibited by protein therapeutics (i.e. VEGF) and kinase inhibitors (i.e. VEGF receptor) are different. However, expression of the relevant VEGF receptors has been described both in animal species used for preclinical experiments and in humans 13, 15. Therefore, a similar effect can be assumed for neutralizing the ligand and for inhibiting the receptor tyrosine kinase further downstream. This is supported by anecdotal clinical evidence which also supports the potential efficacy of small‐molecule oral multireceptor tyrosine kinase inhibitors in nAMD 26, 29. One additional difference between ligand‐ and receptor targeting may be the tissue compartment that needs to be reached. Whereas ligand‐neutralizing agents such as ranibizumab or aflibercept are most likely to act in the vitreous, a kinase inhibitor will need to penetrate deeper into the tissues to reach the affected blood vessels. The physicochemical properties of regorafenib are considered to be suitable for such tissue penetration, further supported by its antitumour activity in gastrointestinal stromal tumours 30.
Third, the included population of nAMD patients may have been less responsive to anti‐VEGF therapy. However, the inclusion and exclusion criteria were very similar to those of previous studies on intravitreal protein therapeutics 4, 5, 6, 7, 8. In addition, when patients were administered rescue treatment according to predefined criteria, the changes in BCVA and CRT were rapid and in line with the known response to anti‐VEGF treatment (also see Figure 3).
Fourth, exposure of regorafenib in the target compartment (i.e. BoE) might have been insufficient in humans. As this hypothesis could not be tested according to actual BoE exposure, because this cannot be measured in humans, we used available preclinical data for estimation. Under several assumptions, mainly that eye/tissue concentration ratios in humans are equal to those observed in monkeys (see above), human BoE exposure calculated from the respective plasma exposure results in an AUC(0–24 h)BoE of 963 μg h l–1, or an AUCu, BoE of 4.82 μg h l–1. In order to put this into perspective with local exposure levels at the target needed to achieve efficacy, a threshold for efficacy based on the in vitro potency of regorafenib was defined as a theoretical AUC, calculated as the product of the cellular IC50 of regorafenib at the VEGFR2 and an exposure time of 24 h (cellular IC50 = 3 nM or 1.5 μg l–1; according to assay conditions, a fraction unbound (fu, i.e. the fraction of drug not bound to plasma proteins) of 50% can be assumed—i.e. 0.75 μg l–1; 0.75 μg l–1 × 24 h = 18 μg h l–1). To validate this theoretical threshold, plasma exposure data from early clinical studies of the oncology programme, with oral administration of regorafenib and therefore systemic delivery to the cancer target, were included in the comparison. Exposure was taken from one very low dose, which was the highest dose tested at which no biological activity was observed clinically (10 mg) and from the approved dose in oncology indications (160 mg; regorafenib as an oral medication is currently approved for the treatment of patients with colorectal cancer, gastrointestinal stromal tumour or hepatocellular carcinoma) 24. Putting this, as well as the exposures obtained in different species, into perspective (see Figure 5, which is based on data from Table 4), the 10 mg oral dose just reached the theoretical threshold, and the clinically used oral dose (160 mg) was well above it. The efficacy observed in rats and monkeys can likewise be explained by this approach, as measured exposures in the eye tissues exceed the threshold value. At the same time, the absence of systemic side effects in rats, monkeys and humans can be explained, as plasma exposure in all species is clearly below the theoretical threshold. Thus, there is some validation for this theoretical threshold. However, under these assumptions, the unbound concentration reached at the BoE using regorafenib eye drops would have been fourfold below the theoretical efficacious threshold (see Figure 5).
Overall, we could not provide clear proof confirming or denying any of the aforementioned potential reasons for the failed translation of efficacy from preclinical to clinical studies. However, considering all of the aspects discussed above, with the dosing regimen used, we consider a limited exposure at the BoE to be the most likely explanation. This does not exclude the possibility that topically administered regorafenib can be efficacious if given at higher dosing frequencies, yet dosing more frequently than three times per day, probably close to 10 times per day, was not considered feasible for long‐term treatment and was therefore not tested.
A previous clinical programme examined pazopanib, another small‐molecule kinase inhibitor with a similar kinase profile. The pazopanib eye drops were not given alone but in addition to ranibizumab therapy. They failed to achieve either superior gains, as compared with injections alone, or a reduction in the number of injections given during the 52‐week period 31. Regorafenib has different physicochemical properties to pazopanib. In animal experiments, the formulation for regorafenib has been optimized, resulting in an oily suspension in 100% light liquid paraffin. This was assumed to increase residence time at the corneal surface. Overall, however, neither of these topical approaches have shown benefit over injectable anti‐VEGF therapy.
Considering the failure to translate the findings from the preclinical to clinical studies, one could question whether the monkey laser‐CNV model can be considered predictive for the efficacy of topical application of regorafenib eye drops and, in general, whether the screening cascade as applied can be used to develop any treatment for nAMD other than intravitreally administered therapeutics targeting VEGF. Although the laser‐CNV model shows similarities to nAMD (i.e. Bruch's membrane is ruptured, which is a known feature of the pathophysiology of nAMD), the acute nature of the model and its self‐limitation represent substantial differences to the disease 32, 33. Moreover, although it can be regarded as an efficacy prediction model for intravitreally administered protein therapeutics binding VEGF (intravitreal aflibercept and ranibizumab) 4, 5, 6, 8, 32, 34, no precedent has been established in relation to topical treatments.
In summary, regorafenib eye drops did not show sufficient efficacy compared with historic levels observed with other nAMD treatments. Consequently, the clinical programme (i.e. the DREAM study) was terminated after completion of phase IIa. There was no indication that this was due to noncompliance, as plasma concentrations in the phase II study were similar to those observed in the phase I study. Insufficient exposure in the target compartment (i.e. BoE) was considered to be the most likely explanation for this lack of efficacy. In this case, the translation of convincing animal data to clinical efficacy was not successful. Future approaches to develop non‐invasive therapies for nAMD should consider the lack of positive predictive power of the monkey laser‐CNV model for the human situation in the case of topical therapy. In preparation for follow‐on projects, more research is required to understand quantitatively how PK and pharmacodynamics in humans correlate with the corresponding data from animal models. Furthermore, PBPK models of the eye may need to be further developed and be used early on (e.g. in healthy volunteer studies), to predict tissue‐specific exposure in patients.
Competing Interests
A.M.J. is a consultant for Bayer AG, Division Pharma, Novartis, and OD‐DS. S.W. declares financial support from Alcon, Allergan, Bayer AG, Division Pharma and Novartis, and nonfinancial support from Heidelberg Engineering, Optos and Zeiss. P.K.K. is a consultant for Alcon, Allergan, Bayer AG, Division Pharma, Genentech, Kanghong, Neurotech, Novartis, Ohr, Ophthotech and Regeneron. D.B. is a consultant for Alcon, Allergan, Bayer AG, Division Pharma, Genentech, Neurotech, Novartis, Ohr and Roche. T.S., R.S., O.Z., G.D., J.H., U.B., A.R., T.Z., W.S., B.S. and M.K.B. were employees of Bayer AG, Division Pharma at the time the study was performed and hold shares in Bayer. Editorial assistance for a previous version of the manuscript was provided by PAREXEL, and funded by Bayer AG, Pharmaceuticals. All studies were funded by Bayer AG, Division Pharma, Leverkusen, Germany. The sponsor participated in the study design and conduct, data collection, data management, data analysis, data interpretation, and preparation, review and approval of the manuscript.
The data included in this manuscript are not in the current scope of clinical trial data sharing according to Bayer's transparency policy. This scope was carefully defined based on the European Federation of Pharmaceutical Industries and Associations (EFPIA) and Pharmaceutical Research and Manufacturers of America (PhRMA) principles on responsible clinical trial data sharing.
Contributors
A.R. contributed to formulation development, including data collection; J.H. and T.Z. contributed to the phase I study, including data collection, analysis and interpretation; R.S., O.Z., B.S., M.K.B., J.H., U.B. and W.S. contributed to the phase IIa/b study, including data collection, analysis and interpretation; T.S. contributed to the clinical statistics; G.D. was the project manager. The steering committee comprised: A.M.J. (head), S.W., P.K.K., D.B., T.S., R.S., O.Z., B.S. and M.K.B. A.M.J was the principal investigator. All authors were involved in the writing and review of this manuscript.
Joussen, A. M. , Wolf, S. , Kaiser, P. K. , Boyer, D. , Schmelter, T. , Sandbrink, R. , Zeitz, O. , Deeg, G. , Richter, A. , Zimmermann, T. , Hoechel, J. , Buetehorn, U. , Schmitt, W. , Stemper, B. , and Boettger, M. K. (2019) The Developing Regorafenib Eye drops for neovascular Age‐related Macular degeneration (DREAM) study: an open‐label phase II trial. Br J Clin Pharmacol, 85: 347–355. 10.1111/bcp.13794.
The clinical data were presented at the Association for Research in Vision and Ophthalmology (ARVO) 2016 Meeting, 1–5 May 2016, Seattle, WA, USA.
References
- 1. Ho QT, Kuo CJ. Vascular endothelial growth factor: biology and therapeutic applications. Int J Biochem Cell Biol 2007; 39: 1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baffi J, Byrnes G, Chan CC, Csaky KG. Choroidal neovascularization in the rat induced by adenovirus mediated expression of vascular endothelial growth factor. Invest Ophthalmol Vis Sci 2000; 41: 3582–3589. [PubMed] [Google Scholar]
- 3. Miller JW. Vascular endothelial growth factor and ocular neovascularization. Am J Pathol 1997; 151: 13–23. [PMC free article] [PubMed] [Google Scholar]
- 4. Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, et al Ranibizumab versus verteporfin for neovascular age‐related macular degeneration. N Engl J Med 2006; 355: 1432–1444. [DOI] [PubMed] [Google Scholar]
- 5. Brown DM, Michels M, Kaiser PK, Heier JS, Sy JP, Ianchulev T, et al Ranibizumab versus verteporfin photodynamic therapy for neovascular age‐related macular degeneration: two‐year results of the ANCHOR study. Ophthalmology 2009; 116: 57–65.e5. [DOI] [PubMed] [Google Scholar]
- 6. Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, et al Intravitreal aflibercept (VEGF trap‐eye) in wet age‐related macular degeneration. Ophthalmology 2012; 119: 2537–2548. [DOI] [PubMed] [Google Scholar]
- 7. Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, et al Ranibizumab for neovascular age‐related macular degeneration. N Engl J Med 2006; 355: 1419–1431. [DOI] [PubMed] [Google Scholar]
- 8. Schmidt‐Erfurth U, Kaiser PK, Korobelnik JF, Brown DM, Chong V, Nguyen QD, et al Intravitreal aflibercept injection for neovascular age‐related macular degeneration: ninety‐six‐week results of the VIEW studies. Ophthalmology 2014; 121: 193–201. [DOI] [PubMed] [Google Scholar]
- 9. Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE, et al Comparison of Age‐related Macular Degeneration Treatments Trials Research Group: ranibizumab and bevacizumab for treatment of neovascular age‐related macular degeneration: two‐year results. Ophthalmology 2012; 119: 1388–1398.22555112 [Google Scholar]
- 10. Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Catt Research Group: ranibizumab and bevacizumab for neovascular age‐related macular degeneration. N Engl J Med 2011; 364: 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al Alternative treatments to inhibit VEGF in age‐related choroidal neovascularisation: 2‐year findings of the IVAN randomised controlled trial. Lancet 2013; 382: 1258–1267. [DOI] [PubMed] [Google Scholar]
- 12. Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Wordsworth S, et al Ranibizumab versus bevacizumab to treat neovascular age‐related macular degeneration: one‐year findings from the IVAN randomized trial. Ophthalmology 2012; 119: 1399–1411. [DOI] [PubMed] [Google Scholar]
- 13. Kim I, Ryan AM, Rohan R, Amano S, Agular S, Miller JW, et al Constitutive expression of VEGF, VEGFR‐1, and VEGFR‐2 in normal eyes. Invest Ophthalmol Vis Sci 1999; 40: 2115–2121. [PubMed] [Google Scholar]
- 14. Roskoski R Jr. VEGF receptor protein‐tyrosine kinases: structure and regulation. Biochem Biophys Res Commun 2008; 375: 287–291. [DOI] [PubMed] [Google Scholar]
- 15. Witmer AN, Blaauwgeers HG, Weich HA, Alitalo K, Vrensen GF, Schlingemann RO. Altered expression patterns of VEGF receptors in human diabetic retina and in experimental VEGF‐induced retinopathy in monkey. Invest Ophthalmol Vis Sci 2002; 43: 849–857. [PubMed] [Google Scholar]
- 16. Jo N, Mailhos C, Ju M, Cheung E, Bradley J, Nishijima K, et al Inhibition of platelet‐derived growth factor B signaling enhances the efficacy of anti‐vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol 2006; 168: 2036–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al Regorafenib (BAY 73‐4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011; 129: 245–255. [DOI] [PubMed] [Google Scholar]
- 18. Boettger MK, Klar J, Richter A, von Degenfeld G. Topically administered regorafenib eye drops inhibit grade IV lesions in the non‐human primate laser CNV model. Presented at: ARVO 2015 Annual Meeting, 3–7 May 2015; Denver, CO, USA; Poster B0199.
- 19. Klar J, Boettger MK, Freundlieb J, Keldenich J, Elena PP, von Degenfeld G. Effects of the multi‐kinase inhibitor regorafenib on ocular neovascularization. Presented at: ARVO 2015 Annual Meeting, 3–7 May 2015; Denver, CO, USA; Poster C0118.
- 20. Zimmermann T, Höchel J, Becka M, Boettger MK, Rohde B, Schug B, et al Topical administration of regorafenib eye drops: phase I dose‐escalation study in healthy volunteers. Br J Clin Pharmacol 2018; 84: 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Araie M, Takase M, Sakai Y, Ishii Y, Yokoyama Y, Kitagawa M. Beta‐adrenergic blockers: ocular penetration and binding to the uveal pigment. Jpn J Ophthalmol 1982; 26: 248–263. [PubMed] [Google Scholar]
- 22. Chang SC, Bundgaard H, Buur A, Lee VH. Improved corneal penetration of timolol by prodrugs as a means to reduce systemic drug load. Invest Ophthalmol Vis Sci 1987; 28: 487–491. [PubMed] [Google Scholar]
- 23. Sakanaka K, Kawazu K, Tomonari M, Kitahara T, Nakashima M, Nishida K, et al Ocular pharmacokinetic/pharmacodynamic modeling for timolol in rabbits using a telemetry system. Biol Pharm Bull 2008; 31: 970–975. [DOI] [PubMed] [Google Scholar]
- 24. Mross K, Frost A, Steinbild S, Hedbom S, Buchert M, Fasol U, et al A phase I dose‐escalation study of regorafenib (BAY 73‐4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 2012; 18: 2658–2667. [DOI] [PubMed] [Google Scholar]
- 25. Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro 2008; 22: 457–467. [DOI] [PubMed] [Google Scholar]
- 26. Giddabasappa A, Lalwani K, Norberg R, Gukasyan HJ, Paterson D, Schachar RA, et al Axitinib inhibits retinal and choroidal neovascularization in in vitro and in vivo models. Exp Eye Res 2016; 145: 373–379. [DOI] [PubMed] [Google Scholar]
- 27. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 2017; 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thiele S, Liegl RG, Konig S, Siedlecki J, Langer J, Eibl K, et al Multikinase inhibitors as a new approach in neovascular age‐related macular degeneration (AMD) treatment: in vitro safety evaluations of axitinib, pazopanib and sorafenib for intraocular use. Klin Monbl Augenheilkd 2013; 230: 247–254. [DOI] [PubMed] [Google Scholar]
- 30. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013; 381: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Csaky KG, Dugel PU, Pierce AJ, Fries MA, Kelly DS, Danis RP, et al Clinical evaluation of pazopanib eye drops versus ranibizumab intravitreal injections in subjects with neovascular age‐related macular degeneration. Ophthalmology 2015; 122: 579–588. [DOI] [PubMed] [Google Scholar]
- 32. Dobi ET, Puliafito CA, Destro M. A new model of experimental choroidal neovascularization in the rat. Arch Ophthalmol 1989; 107: 264–269. [DOI] [PubMed] [Google Scholar]
- 33. Nork TM, Dubielzig RR, Christian BJ, Miller PE, Miller JM, Cao J, et al Prevention of experimental choroidal neovascularization and resolution of active lesions by VEGF trap in nonhuman primates. Arch Ophthalmol 2011; 129: 1042–1052. [DOI] [PubMed] [Google Scholar]
- 34. Meyer CH, Holz FG. Preclinical aspects of anti‐VEGF agents for the treatment of wet AMD: ranibizumab and bevacizumab. Eye 2011; 25: 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]