

Abstract
Aims
Treatment of prolactinomas with ergoline dopamine agonists can be complicated by intolerance and resistance. This study investigated the pharmacokinetics and pharmacodynamics of the nonergot dopamine agonist ropinirole, to assess its therapeutic potential as a novel therapy for prolactinomas.
Methods
Five female subjects with prolactinomas participated in this dose–response study. Subjects received up to three doses of ropinirole (0.5, 1.0 and 2.0 mg), each on separate occasions. Frequent blood samples for prolactin and ropinirole were collected for 24 h following drug administration. Data were analysed using noncompartmental and compartmental pharmacokinetic–pharmacodynamic (PKPD) techniques.
Results
Seven 24‐h curves revealed increased systemic drug exposure with increasing ropinirole doses. Ropinirole concentrations peaked at 4.4 ± 2.7 h and exhibited a half‐life of 5.8 ± 1.7 h. A dose‐dependent prolactin nadir occurred 4.4 ± 1.2 h after drug intake and prolactin concentrations transiently normalized in two of five subjects. PKPD modelling revealed that single‐dose PK of ropinirole is dose‐independent and can be described with a one‐compartment model with linear absorption and elimination. An indirect response model successfully captures the inhibitory effect of ropinirole on prolactin secretion and incorporates time‐dependent receptor desensitization for three of five subjects whose prolactin concentrations nadired before ropinirole reached Cmax.
Conclusions
This data‐rich study has informed our understanding of the clinical pharmacokinetics and pharmacodynamics of ropinirole, which are successfully captured by the proposed semi‐mechanistic PKPD model. This model can be used to further investigate the PKPD of ropinirole and may facilitate the identification of optimal dose regimens for the treatment of prolactinomas and the establishment of a new therapeutic option for patients impacted by this rare disease.
Keywords: modelling and simulation, pharmacokinetic–pharmacodynamic, pharmacometrics, ropinirole, therapeutics
What is Already Known about this Subject
The ergoline dopamine agonists, bromocriptine and cabergoline, are first line therapies for the treatment of prolactinomas; however, in a subset of patients, their use is limited by medication intolerance and/or resistance.
In comparison to the ergot dopamine agonists, ropinirole hydrochloride is a nonergot dopamine agonist that has greater specificity for the D2 dopamine receptor and has negligible cross reactivity at other receptor subtypes.
In healthy volunteers, ropinirole suppresses prolactin concentrations; however, the pharmacokinetic–pharmacodynamic profile of ropinirole in hyperprolactinaemic individuals and the drug's effect on prolactin concentrations in these patients has not been previously investigated.
What this Study Adds
This study establishes the pharmacokinetic and pharmacodynamic profile of ropinirole in patients with prolactinomas for the first time.
This study shows that ropinirole and prolactin concentrations after single dose administration, can be described by a mechanism‐based pharmacokinetic–pharmacodynamic model.
This study demonstrates that ropinirole acutely suppresses prolactin concentrations in hyperprolactinaemic subjects with prolactinomas and establishes its potential as a therapeutic option for the treatment of these rare tumours.
Introduction
Prolactinomas account for 40% of all pituitary adenomas but, with an incidence of only 60–100 cases per million, they are considered a rare disease 1. Arising from lactotrophic cells of the pituitary gland, they are the most common cause of pathological hyperprolactinaemia and often result in hypogonadism, infertility, low bone density, headaches, galactorrhoea, and, potentially, hypopituitarism and visual loss. While the majority of pituitary tumours are surgically managed, the secretion of prolactin is tonically inhibited by dopamine, and the ergoline dopamine agonists (DAs) bromocriptine and cabergoline are currently first‐line therapies for the treatment of prolactinomas. Both drugs act at D2 dopamine receptors (D2R) to stimulate dopaminergic neurons in the infundibular pathway and inhibit lactotrophic prolactin secretion. While ergot DAs have been shown to lower prolactin concentrations and reduce tumour size, a subset of patients experience medication intolerance or pharmacological resistance, and must pursue alternative therapies including surgical tumour resection, radiotherapy and experimental treatments 2.
In spite of their efficacy, the ergot DAs lack specificity for the D2R subfamily and exhibit receptor cross‐reactivity that impacts tolerability 3. Additionally, cabergoline's affinity for the serotonin 5‐hydroxytryptamine receptor subtype 2B (5HT‐2B) expressed on cardiac valves has generated concerns about its overall safety 4, 5, 6, 7, 8, 9, 10. While long‐term prospective studies are needed to confirm a causative relationship between cabergoline and valvular heart disease in treated hyperprolactinaemics, patients and practitioners remain apprehensive about the implications of its use particularly in cases requiring high doses or prolonged therapy and in patients with pre‐existing valve disease. Given the limitations of currently used ergot DAs, the identification of a pharmacological treatment alternative for the management of prolactinomas may prove beneficial.
Ropinirole hydrochloride, a substituted indole derivative, is a well‐tolerated, low cost, generic, nonergot DA that exhibits greater specificity for the D2R subfamily and has negligible in vitro activity at the 5HT‐2B receptor and other receptor subtypes 3, 4. While it is currently Food and Drug Administration approved for the management of Parkinson's disease, where it acts at nigrostriatal D2Rs to inhibit motor fluctuations and dyskinesias, and restless leg syndrome, where it binds to central nervous system D2Rs to reduce periodic limb movements, ropinirole has also been shown to lower prolactin concentrations in healthy volunteers 11, as effectively as bromocriptine 12. However, its use in patients with hyperprolactinaemia has not been previously investigated. In this study, we prospectively examined the acute effect of ropinirole administration on prolactin concentrations in patients with prolactinomas and evaluated the drug's pharmacokinetic and pharmacodynamic (PKPD) profile in order to assess ropinirole's therapeutic potential as a novel therapy and to help identify optimal dose regimens for the treatment of these rare tumours.
Methods
Five nonpregnant females, aged 21–77 years, with hyperprolactinaemia (range 39–523 ng ml–1; reference range 1.9–25 ng ml–1) and no evidence of other pituitary hormone hypersecretion, were recruited from the Columbia University Neuroendocrine Unit. Subjects were screened by medical history and chart review and underwent baseline laboratory testing to confirm normal kidney, liver and thyroid function. Macroprolactinaemia, which consists of prolactin complexed to an anti‐prolactin antibody, was ruled out in all subjects using polyethylene glycol precipitation. Subjects had a pituitary adenoma <15 mm on pituitary‐protocoled magnetic resonance imaging and had no evidence of pituitary stalk compression. Additionally, subjects had a median body mass index of 31.9 kg m–2 had no history of major medical conditions, excessive alcohol use, or previous pituitary radiation. One subject was postmenopausal. The other four subjects were premenopausal but had been amenorrhoeic for ≥3 months at the time of participation. Subjects had no DA use for 2 months prior to participation and were not taking other medications known to interfere with prolactin secretion or ropinirole metabolism (Table 1). Two out of five subjects had a history of resistance to ergot DAs; one out of five had a history of ergot DA intolerance, and the other two subjects were DA naïve. This study was approved by the Columbia University Institutional Review Board, and written informed consent was obtained from all subjects prior to participation.
Table 1.
Subject characteristics (n = 5)
| Age (years) Median (IQR: Q1–Q3) | 34 (27–59) |
|---|---|
| Race n (%) | |
| Black/African American | 2 (40%) |
| White/Caucasian | 3 (60%) |
| Female n (%) | 5 (100%) |
| Baseline prolactin (ng ml –1 ) Median (IQR) | 85 (60–394) |
| Body mass index (kg m –2 ) Median (IQR) | 32 (23–35) |
Age, baseline prolactin, and body mass index are reported as medians and interquartile ranges (IQR) are reported as Q1–Q3. Race and sex are reported as percentages of the subject population
Protocol
Subjects participated in an inpatient dose response study to evaluate the PKPD profile of ropinirole. Five subjects completed a total of seven separate 24‐h inpatient study visits in the clinical research centre, during which they received a single dose of immediate release ropinirole (0.5, 1.0 or 2.0 mg) and underwent frequent blood sampling. Blood was initially drawn for ropinirole and prolactin at the time of intravenous catheter placement, 30 min after I intravenous V placement, then immediately prior to administration of ropinirole in order to establish a baseline prolactin level. A single dose of ropinirole was given 30 min after a standardized meal. Blood was then drawn every 30 min for 6 h, every 2 h for 10 h, then every 4 h for the 8 h remaining in the 24‐h study period, and monitoring of vital signs and adverse events occurred concomitantly.
Assays
Serum prolactin was measured by two‐site chemiluminescent enzyme immunometric assay using the Immulite 1000 Analyzer (Siemens Healthcare Diagnostics, Deerfield, IL). The reference range for serum prolactin is 1.9–25 ng ml–1 for adult females. Intra‐ and interassay precision are 6.8 and 9.6%, respectively. Plasma ropinirole was measured by an in‐house ultra‐performance liquid chromatography–tandem mass spectrometry assay using an Agilent 1290/6410 UPLC‐Tandem MS instrument (Agilent Technologies) 13. Ropinirole‐d4‐hydrochloride was used as the internal standard. Intra and interassay CVs are 3.7% and 2.9% respectively over the analytical measurement range (0.25–100 ng ml–1).
Noncompartmental analysis
Ropinirole and prolactin serum concentrations were initially explored using noncompartmental analysis (NCA). Area under the serum concentration time curve between time 0 and time T (AUC0‐T,ropinirole), maximum concentration reached (Cmax), time at which maximum concentration is reached (Tmax) and the terminal half‐life (t½ropinirole) were calculated using Phoenix WinNonlin PKPD software (Certara, Princeton, NJ, USA). The area under the prolactin concentration–time curve between time 0 and time T (AUC0‐T,prolactin), and the nadir were also calculated. Potential relationships between dose and systemic exposure of ropinirole and changes in prolactin concentrations were investigated using exploratory regression and correlation analysis. The small number of patients, which is inherent to investigating drugs in this rare disease population, made PKPD analysis using NCA challenging. However, the density of ropinirole and prolactin measurements within this small cohort allowed for a full PKPD analysis using nonlinear mixed effects modelling.
PKPD modelling
Nonlinear mixed‐effects analysis was conducted using NONMEM (version 7.3, ICON Development Solutions, Ellicott City, Maryland). The ADVAN8 subroutine and the first‐order conditional estimation method with interaction were used. R‐studio (version 1.1) was used for dataset preparation and diagnostic plotting. The model building process was guided by changes in NONMEM objective function value and goodness‐of‐fit plots. Interindividual variances for all PK and PD parameters were modelled assuming a lognormal distribution. The residual error variances were modelled assuming additive and proportional components. The present PKPD model (Figure 2) features a one‐compartment model with linear absorption and elimination to describe ropinirole PK, and an indirect response model (Type I) to capture the inhibition effect of ropinirole on prolactin secretion. Time‐dependent receptor desensitization was incorporated into the subjects whose prolactin concentration reached nadir before the corresponding ropinirole concentration reached Cmax. A hill factor was added to capture the sigmoidicity exhibited in the data. A conventional two‐step approach was used in conducting the fitting. First, a PK model for ropinirole was developed. The parameters to describe ropinirole PK include apparent clearance (CL/F), apparent central volume (Vc/F), first‐order absorption rate (Ka) and lag time (Tlag) on absorption. Second, the empirical Bayes estimates of ropinirole PK parameters obtained in step 1 were fixed, and the predicted individual PK profiles were used as input functions in the PD model to drive the inhibition effect on prolactin secretion. The parameters to depict the ropinirole exposure‐response (Kin, baseline, IC50, α, γ) were fitted simultaneously (more details are described below).
Figure 2.
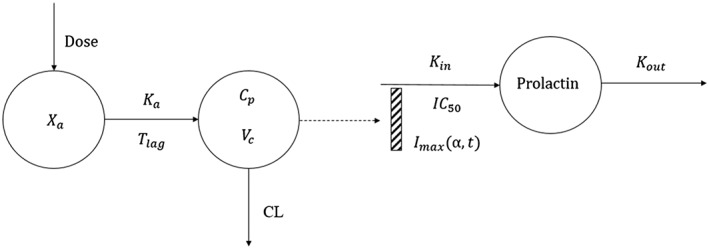
Pharmacokinetic–pharmacodynamic model diagram of ropinirole for the treatment of hyperprolactinaemia. The pharmacokinetics of ropinirole are characterized with a one compartment model with linear absorption and linear elimination. The concentration in the central compartment is used as the driving force for the inhibition of prolactin secretion. The indirect response model is used to capture the exposure response relationship. Xa is the dosing compartment (absorption compartment), Ka is the first order absorption rate constant, Tlag is the lag time for absorption, Cp is the concentration in the central compartment, Vc is the central compartment volume and CL is the clearance ropinirole (l h–1) from the central compartment. Prolactin is the concentration of prolactin in plasma. Imax is the maximum fractional ability of the ropinirole to inhibit prolactin production, which is defined as a function of desensitization slope α and time (t) in patients exhibiting time‐dependent dopamine receptor desensitization (n = 3). IC50 is the plasma concentration of ropinirole that results in 50% of the maximum inhibition. Kin and Kout are the zero‐order production rate and first order elimination rate of prolactin, respectively.
Mathematical relationship of the exposure response
It was hypothesized that the prolactin concentration decrease caused by dopamine D2 receptor activation is due to diminished prolactin production. The indirect response model reflecting this hypothesis is described by the following equation:
Where,
Kin and Kout are the zero‐order production rate and first order elimination rate of prolactin, respectively; Imax is the maximum fractional ability of the ropinirole to inhibit prolactin production; Cprolactin is the prolactin plasma concentration; IC50 is the plasma concentration of ropinirole that results in 50% of Imax; D signifies the on and off switch of receptor desensitization effect; γ is the hill factor to characterize the sigmoidicity in the data; and α is the exponential slope of receptor desensitization.
The error models
All PK parameters (Tlag, Ka, CL/F, V/F) and most PD parameters (Kin, baseline, IC50, α) with between‐subject variability estimated were assumed to be log‐normally distributed. Due to the limited identifiability provided by the data, γ was estimated as the same value across subjects, and Imax was fixed to 1, as this is the maximum the system can possibly be inhibited. For the PK model, the differences between model predicted and observed concentrations were estimated using the additive plus proportional error model. For the PD model, the residual variability was estimated using the proportional error model.
Simulations
Single dose and steady state dosing (non‐desensitizing only) for daily and twice daily dosing of ropinirole were done using the population average parameters returned in the NONMEM analysis and the $simulation function for a single individual.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 14, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 15.
Results
Five patients were included in this study (Table 1). One patient received a ropinirole dose of 0.5 mg, three received a ropinirole dose of 1.0 mg and three received a ropinirole dose of 2.0 mg. Two patients were treated at two dose levels on separate occasions with one receiving a 1.0 mg dose followed by a 2.0 mg dose and the other receiving 0.5 mg followed by a 2.0 mg dose. All patients had pretreatment plasma prolactin concentrations above the normal range (84.8 ng ml–1; median); however, there was a wide range in prolactin pretreatment concentrations between patients (39–523 ng ml–1; Table 1). Ropinirole was well‐tolerated. One subject experienced a mild adverse event characterized by the acute onset of nausea 4.5 h after administration of the highest ropinirole dose studied, 2.0 mg, that lasted approximately 15 min and was followed by a single episode of vomiting. The symptoms self‐resolved and the patient reported no adverse event following the administration of 1.0 mg of ropinirole during a separate visit.
NCA
In general, a single dose of ropinirole led to a transient increase in ropinirole concentrations (Figure 1A) and a transient decrease in prolactin concentrations (Figure 1B). Figure 1A shows the individual plasma ropinirole concentrations and demonstrates that regardless of the dose, ropinirole administration led to an increase in plasma drug concentrations with peak concentrations observed 4.4 h after drug intake. Plasma ropinirole concentrations declined thereafter with an average half‐life of 5.8 h. Among subjects receiving two different doses, higher doses led to an increased Cmax and an increased AUC0‐24h. Figure 1B shows the individual prolactin concentrations for each subject and demonstrates that regardless of the dose, ropinirole administration also led to a decrease in serum prolactin concentrations. The prolactin nadir was observed approximately 4.4 h after drug intake, and prolactin concentrations subsequently increased to pretreatment concentrations by 24 h following drug intake. Higher doses led to a lower AUC0–24h and a lower nadir, in subjects receiving two doses. Table 2 shows the NCA results for ropinirole and prolactin for all subjects at all dose levels.
Figure 1.
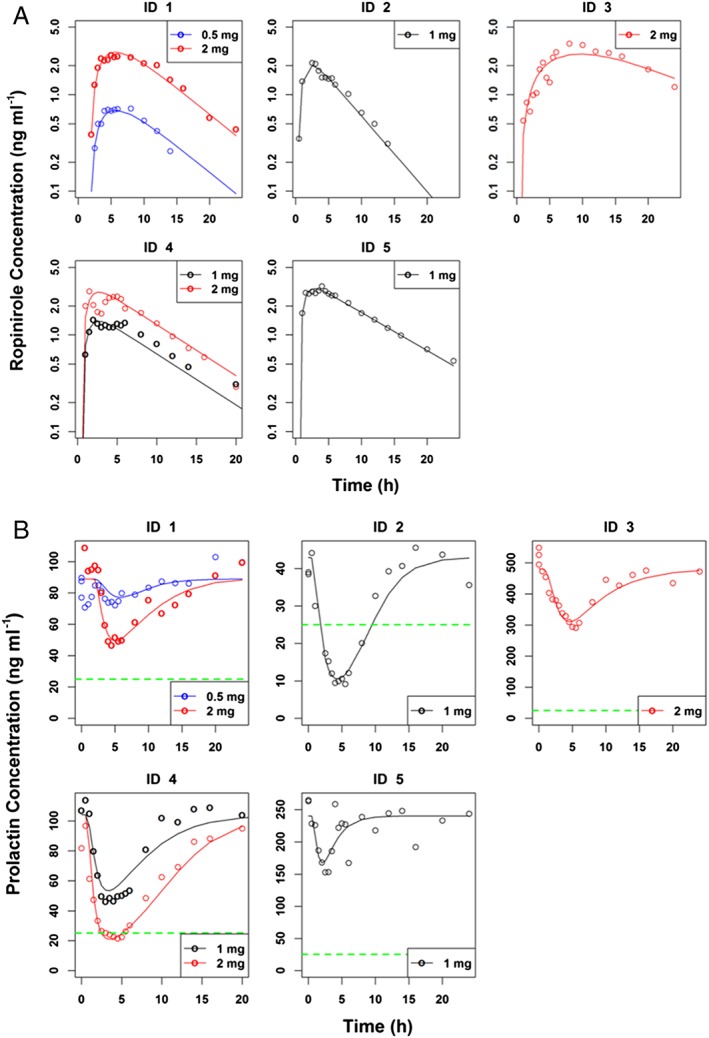
(A) Individual plasma ropinirole concentration versus time profiles after the oral administration of single doses of ropinirole (0.5, 1.0 and 2.0 mg) in five hyperprolactinaemic patients. Symbols represent observations from individual patients and lines are individual predictions. (B) Time profiles of prolactin concentrations after oral administration of ropinirole at the indicated doses to hyperprolactinaemic patients. Solid lines indicate individual model predictions and empty circles are observed prolactin concentrations. Green dashed lines indicate the upper limit of normal for prolactin which is 25 ng ml–1
Table 2.
Noncompartmental pharmacokinetic–pharmacodynamic analysis of single dose 24‐h plasma concentration curves
| Subject ID | Dose (mg) | Tlag (h) | Tmax (h) | Cmax (ng ml–1) | AUC (h ng ml–1) | T1/2 (h) | Pre‐treatment PRL (ng ml–1) | Nadir (%PRL) | Time of Nadir (h) | Desensitizationa | %Inhibition/AUCb |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A | 0.5 | 2.0 | 8 | 0.7 | 6.5 | 3.8 | 85 | 85 | 5.0 | 1 | 2.3 |
| 1B | 2.0 | 1.5 | 5 | 2.6 | 34 | 5.6 | 109 | 43 | 4.5 | 1 | 1.7 |
| 2 | 1.0 | 0 | 2.5 | 2.1 | 15 | 3.9 | 39 | 61 | 5.5 | 0 | 2.6 |
| 3 | 2.0 | 0.5 | 8 | 3.4 | 52 | 7.6 | 523 | 56 | 5.5 | 1 | 0.9 |
| 4A | 1.0 | 0.5 | 2 | 1.4 | 14 | 6.7 | 107 | 43 | 3.0 | 0 | 4.0 |
| 4B | 2.0 | 0.5 | 1.5 | 2.8 | 25 | 4.8 | 82 | 26 | 4.5 | 0 | 2.9 |
| 5 | 1.0 | 0.5 | 4 | 3.2 | 36 | 7.9 | 264 | 58 | 2.5 | 1 | 1.2 |
| Mean ± SD | N/a | 0.8 ± 0.7 | 4.4 ± 2.7 | N/a | N/a | 5.8 ± 1.7 | 173 ± 170 | N/a | 4.4 ± 1.2 | N/a | 2.2 ± 1.0 |
The subject will be marked as desensitization = 1, if its time of nadir < Tmax. Otherwise, desensitization = 0.
%Inhibition/AUC is calculated as (100 – nadir)/AUC
PKPD data are reported for each study visit (n = 7) and mean ± standard deviation is reported when applicable. N/a indicates not applicable
AUC, area under the serum concentration time curve; Cmax, maximum drug concentration; Tlag, lag time for absorption; Tmax, time at maximum drug concentration; T1/2, terminal half‐life
Acute prolactin normalization following a single dose of ropinirole was observed in two of five subjects (Figure 1B). Subject 2, a 34‐year‐old woman with GI intolerance to ergot DAs and persistent tumour, hyperprolactinaemia, galactorrhoea and menstrual irregularity 6 months following transsphenoidal microprolactinoma resection, exhibited prolactin normalization in response to 1.0 mg of ropinirole. Subject 4, a 41‐year‐old female with galactorrhoea, hyperprolactinaemia secondary to a small microadenoma and a partially empty sella, demonstrated prolactin normalization following a 2.0 mg ropinirole dose. Notably, in the two subjects (Subjects 3 and 5) with known resistance to ergot DAs, acute prolactin suppression was observed. Prolactin concentrations suppressed by 42.2% following 1.0 mg ropinirole in Subject 5 and by 44% in Subject 3 following 2.0 mg ropinirole. Overall, a single dose of ropinirole ranging from 1.0–2.0 mg suppressed prolactin concentrations by 39–74% (Table 2 and Figure 1B).
PKPD modelling
The PK of ropinirole were best described using a one‐compartmental PK model with first order absorption and elimination and an absorption lag time (Figure 2). Compartmental PK parameters are listed in Table 3. All population means of the PK parameters had relative standard errors of the estimates (%RSE) <40%. For a given dose, PK profiles among individual subjects were highly variable, as indicated by moderate to high (38%–63%) between‐subject variability in major PK parameters (CL/F, V/F, Tlag, Ka). Based on the obtained population PK parameters, the derived time at maximum drug concentration (Tmax) was approximately 3.4 h post dose, the terminal half‐life (t½) was approximately 6 h, and apparent volume of distribution (V/F) was approximately 444 l. These model‐derived PK parameters are similar to those using non‐compartmental analysis (Table 2). The predicted and individual plasma ropinirole concentrations in Figure 1A (and additional diagnostic plots in Supporting information Figure S1) demonstrate that the described linear PK model is able to adequately describe plasma ropinirole concentrations across three dose levels.
Table 3.
Summary of pharmacokinetic–pharmacodynamic model parameters and interindividual variability estimates for ropinirole's effect on hyperprolactinaemia
| Parameter | Population Mean | IIV | ||
|---|---|---|---|---|
| Value | %RSE | Value | %RSE | |
| CL (l h –1 ) | 50.7 | 13.7 | 0.0986 | 62.4 |
| V (l) | 444 | 11.8 | 0.0464 | 38.4 |
| Ka (h −1 ) | 0.609 | 40.2 | 0.668 | 48.4 |
| Tlag (h) | 0.595 | 25.2 | 0.32 | 63.1 |
| Kin (ng ml –1 h –1 ) | 154 | 40 | 0.929 | 33.1 |
| L max | 1 | Fixed | NA | NA |
| IC50 (ng ml –1 ) | 1.12 | 8.6 | 0.0326 | 65 |
| Baseline (ng ml –1 ) | 109 | 32.5 | 0.859 | 42 |
| α | 0.226 | 31.7 | 0.355 | 73 |
| γ | 2.1 | 11.4 | NA | NA |
| Residual errors | ||||
|---|---|---|---|---|
| Additive | Proportional | |||
| Observations | value | %RSE | value | %RSE |
| PK | 0.0092 | 25.1 | 0.0209 | 38.5 |
| PD | NA | NA | 0.0125 | 12.2 |
Baseline, initial prolactin concentration in plasma; CL, clearance; IIV, Interindividual variability; Imax, maximal fraction of inhibition; IC50, plasma concentration of ropinirole that results in 50% of Imax; Ka, first order absorption rate constant; Kin, zero order production rate of prolactin; NA, not applicable; Tlag, lag time for absorption; V, volume of central compartment; α, desensitization slope; γ, Hill coefficient
Plotting prolactin concentrations against ropinirole concentrations revealed a hysteresis relationship. Therefore, an indirect response model (Type I) was used to describe the exposure–response relationship between ropinirole and prolactin 16. However, in several subjects (ID = 1,3,5), the prolactin concentration reaches its nadir before the ropinirole concentration reaches Cmax. Therefore, a time‐dependent desensitization term was multiplied by the Imax term in the model to account for this backward shift of peak drug effect from peak drug concentration. The sigmoidal Imax model for the inhibition of prolactin input rate (Kin) was superior to the basic Imax (>100 reduction in the objective function value, P < 0.0001) PD parameters are listed in Table 3, which also lists the additive and proportional components of the residual error variance for PK and PD observations. Individual predicted and observed serum prolactin concentrations in Figure 1B (and additional diagnostic plots in Supplementary Figure S2) demonstrate that prolactin concentrations are also adequately described by the PKPD model at all three dose levels – 0.5, 1.0 and 2.0 mg. This includes the application of the desensitization term in subjects 1, 3 and 5.
Prolactin response simulations
Figures 3a and 3b show the results of simulations of prolactin response after single dose daily, bid and steady state administration of ropinirole. The prolactin response is shown for doses from 0.5 to 6 mg. For steady state, only the non‐desensitizing patients are included.
Figure 3.
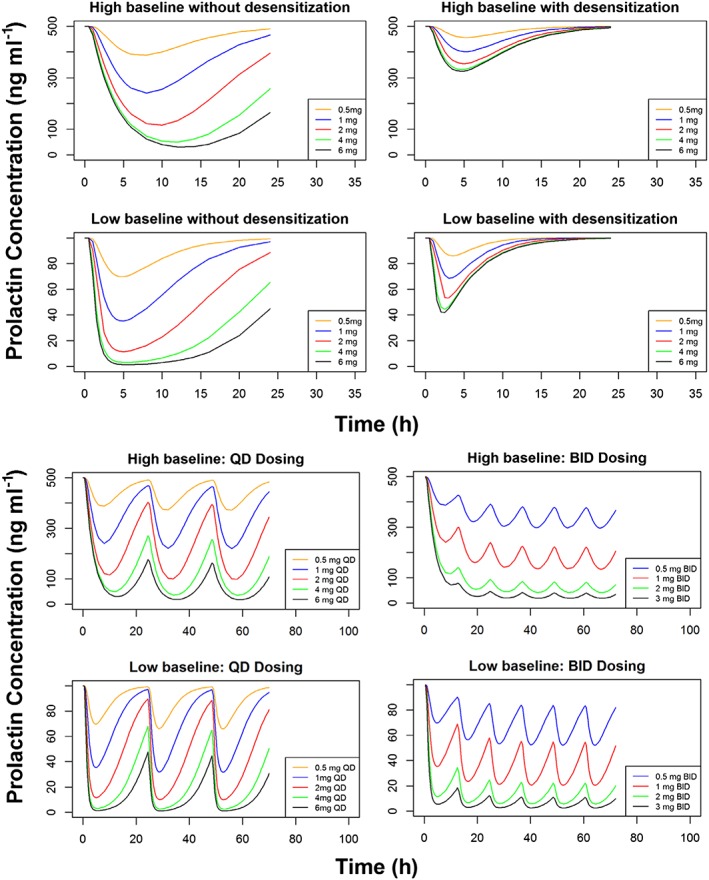
Simulations of ropinirole's drug effect under various conditions including after a single dose (top) and at steady state in nondesensitizing subjects after once and twice daily dosing (bottom)
Discussion
Ropinirole is a nonergoline DA with a unique structure that probably influences its receptor specificity 17. In comparison to its ergot counterparts, it is highly specific for the D2 receptor subfamily, which includes D2 and D3 receptor subtypes, and exhibits selectivity comparable to that of dopamine 18. Given that all DAs exert their effects by binding to postsynaptic D2 receptors on normal or tumoural lactotrophs and inhibiting prolactin synthesis and secretion through G protein‐mediated pathways, ropinirole's strong affinity for D2 receptors may potentiate its efficacy for the treatment of prolactinomas 2. Our results show, for the first time, the effects of a single dose of the nonergot DA ropinirole on prolactin concentrations in hyperprolactinaemic subjects and establish that ropinirole effectively lowers serum prolactin concentrations in patients with prolactinomas in the acute setting. The effect of a single dose of ropinirole on prolactin concentrations was previously evaluated in 14 healthy subjects, at doses ranging from 0.8 to 2.5 mg. In these subjects, ropinirole lowered serum prolactin concentrations relative to placebo and a single 2.5 mg ropinirole dose decreased mean serum prolactin concentrations by 42% 11. Due to the rarity of prolactinomas, which have a prevalence of only 100 cases per million in the general population, large clinical studies in this population can be difficult to conduct. Nonetheless, the high‐density data set obtained from this small cohort of patients resulted in a number of important observations. In comparison to healthy volunteers, among hyperprolactinaemic subjects we observed a moderate degree of prolactin suppression even at the lowest dose studied (0.5 mg; Table 2). Higher single doses of 1.0 and 2.0 mg of ropinirole suppressed prolactin concentrations by 39–74% and prolactin normalization was observed in 2/5 subjects at these doses. Among patients who received two ropinirole doses, higher doses led to greater a degree of prolactin suppression.
The single‐dose pharmacokinetics of ropinirole in this hyperprolactinaemic cohort were similar to those described earlier in healthy volunteers 11. Systemic exposure to ropinirole seems to increase with dose as does the transient decrease in prolactin concentrations. At 5.8 h, the half‐life of ropinirole is markedly shorter than that of cabergoline but is comparable to the half‐life of bromocriptine. As with bromocriptine, twice daily ropinirole dosing may be most beneficial for the treatment of prolactinomas. However, given that bromocriptine has been shown to be therapeutically useful when given once daily, the efficacy of daily ropinirole administration in patients with hyperprolactinaemia should also be evaluated 19. Notably, we observed significant interpatient variability in both the PK and PD profiles of ropinirole in this small cohort, which warranted a more sophisticated analysis of the PK and PD of ropinirole in these patients.
Accordingly, using nonlinear mixed effect modelling (NONMEM), a PKPD model was derived that adequately describes concentrations of plasma ropinirole and serum prolactin after a single ropinirole doses ranging from 0.5 to 2.0 mg. While it is difficult to generalize the interindividual variability estimates on the parameters due to the small sample size, this distinct analysis did reveal a number of important observations. First, the analysis demonstrated that the PK parameters of ropinirole were dose‐independent and the observed concentrations were dose proportional for the 0.5–2.0 mg dosage range evaluated in this study. Second, the ability to describe prolactin concentrations using an indirect effect model coupled with a nonlinear inhibitory model impacting the production of prolactin, is consistent with our knowledge about the mechanism of action of ropinirole with respect to changing prolactin concentrations. Third, our estimated prolactin turnover half‐time of 0.49 h is consistent with those reported by Petersson et al. ranging from 0.34 to 1.28 h 20, providing further physiological support for this semi‐mechanistic PKPD model. In addition, the PKPD model effectively describes the PK and PD of ropinirole in patients with a wide range of pretreatment prolactin concentrations, suggesting that it may be applicable to patients with different degrees of disease severity. The PKPD analysis revealed that a subset of patients reached prolactin nadirs before maximum ropinirole concentrations were achieved. The pharmacological behaviour of these tumours can be described by introducing time‐dependent desensitization into the PKPD model, a phenomenon that has also been observed in in vitro studies evaluating the effects of DAs on D2 receptors 21. In the case of prolactinomas, this observed pharmacological desensitization reflects the clinical phenomenon of DA resistance, which is known to affect 24% of prolactinoma patients treated with bromocriptine and 11% of those treated with cabergoline 22. Although there are varying definitions of DA resistance in the literature, it is generally regarded as either the failure to normalize prolactin concentrations or the inability to achieve a 50% reduction in hyperprolactinaemia 22, 23, 24, 25, 26. The molecular mechanisms underlying the behaviour of resistant prolactinomas are not fully understood. At least some resistant prolactinomas exhibit a reduced D2 receptor density 23, 27, while a subset of resistant tumours has been shown to exhibit alterations in G protein signalling pathways and disruptions in nerve growth factor‐mediated autocrine signalling pathways 22, 28. There are clinical data demonstrating that DA resistance is not a class effect and that patients who are resistant to one DA may respond to another. In this cohort, two out of three of the patients exhibiting desensitization, are also known to be resistant to ergot DAs (the third patient was DA naïve). However, in spite of their history of DA resistance, prolactin concentrations declined in both patients following single doses of ropinirole (1.0 and 2.0 mg) – a noteworthy finding – that highlights the treatment potential of nonergot DAs in this subset of patients. This observed response to single‐dose ropinirole may reflect ropinirole's strong affinity for the D2 receptor, which is greater than that of bromocriptine and at least comparable to that of cabergoline and may facilitate the suppression of prolactin concentrations even in the setting of receptor downregulation. Additionally, the effect could also result from improved receptor specificity. Regardless of the underlying mechanism, the ability of this PKPD model to simulate the effects of various doses of ropinirole in patients across the spectrum of disease severity and in the absence or presence of time‐dependent desensitization (Figures 3a and 3b) can facilitate the identification of optimal dose regimens for ropinirole in prolactinoma patients, on an individual and a population level. Furthermore, it is possible that screening for desensitization during initial treatment may be beneficial, as it could provide information about the likelihood of resistance to ropinirole up front and could guide subsequent treatment planning.
We found acute ropinirole administration to be well‐tolerated. Overall, the ropinirole doses used in this study were relatively low, considering maximum ropinirole doses range from 4 mg day–1 for the treatment of restless leg syndrome to 24 mg day–1 for the treatment of Parkinson's. Unlike its ergot counterparts, ropinirole does not have an affinity for the D1 receptor, and its affinity for 5HT‐1, 5HT‐2, GABA, benzodiazepine, muscarinic, and α‐ and β‐adrenoreceptors is negligible, as confirmed by radio‐ligand binding studies 3, 29. These pharmacological characteristics make ropinirole an appealing therapeutic alternative that could prove more tolerable. Bromocriptine is not tolerated in 12–36% patients, due to side effects including nausea and vomiting, headaches, dizziness and fatigue 5, 19, 30, 31. In spite of a higher incidence of orthostatic hypotension, cabergoline is associated with fewer side effects overall, but its high affinity for the serotonin 5HT‐2B expressed in heart valves, may stimulate mitogenesis and fibroblastic proliferation, and promote the development of cardiac valve disease as has been observed in both Parkinson's and prolactinoma patients treated with the drug 4, 6, 7, 10, 26. While a single prospective study did not identify an increased risk of significant cardiac valve disease in the first 5 years of cabergoline treatment, the median cumulative dose exposure was relatively low (149 mg) and the 60 month follow‐up period is well below the treatment duration required for a significant percentage of prolactinoma patients, limiting its generalizability 32. Valvular endpoints were not evaluated in this acute study; however, haematological and biochemical safety labs, including renal and hepatic function, were examined prior to and following drug administration and were unchanged. Side effects were closely monitored as well and overall one subject experienced a mild adverse event, characterized by the acute onset of nausea and vomiting at the highest single dose of ropinirole tested (2.0 mg). Side effects were not observed following administration of a lower 1.0 mg dose to the same subject. In spite of its nonergoline properties, dopaminergic gastrointestinal side effects at higher ropinirole doses are possible, given that dopamine and all DAs stimulate the chemoreceptor trigger zone in the postrema of the medulla. In clinical practice, when ropinirole is prescribed for the Food and Drug Administration‐approved indications of Parkinson's disease and restless leg syndrome, gastrointestinal complications are attenuated by the initiation of a low starting dose (0.25 mg) followed by gradual up‐titration. While this study required the acute administration of single ropinirole doses exceeding the recommended starting dose, were ropinirole used for the treatment of prolactinomas, a low starting dose may mitigate dopaminergic side effects in most patients. Overall, this study provides novel data about the ropinirole/prolactin concentration effect relationship that may inform its future clinical use. However, the results are limited by the small sample size and will ultimately require confirmation in a larger cohort of hyperprolactinaemic patients. The absence of placebo data is also a study limitation, although the diurnal rhythm in prolactin concentrations that is classically observed in healthy individuals is ablated in the majority of patients with prolactinomas and secretion patterns are considerably less variable 33, 34, 35, 36.
Conclusions
While comprehensive evaluation of the long‐term use of ropinirole for the treatment of prolactinomas and hyperprolactinaemia is clearly indicated, this novel report suggests that ropinirole may represent a new pharmacological alternative that could prove particularly useful in patients with ergot DA intolerance and resistance and in those with pre‐existing cardiac valve disease. This data‐rich PKPD study in a small cohort of patients has informed our understanding of the clinical pharmacology of ropinirole, which has been successfully captured by this newly developed semi‐mechanistic PKPD model of ropinirole in hyperprolactinaemic individuals. This model can now be used to further investigate the PK and PD of ropinirole in a larger patient cohort, potentially using a limited sampling strategy during multiple dose regimens. Such studies will further assist in the identification of optimal dose regimens for ropinirole for the treatment of prolactinomas and may facilitate the establishment of a new therapeutic option for patients impacted by this rare disease.
Competing Interests
There are no competing interests to declare.
We would like to acknowledge the nurses of the Irving Institute of Clinical Research for their technical expertise and to gratefully thank the patients in this protocol for their participation. This work was supported by the National Center for Advancing Translational Sciences (National Institutes of Health Grants UL1‐TR‐000040 and UL1‐RR‐024156), Irving Institute for Clinical and Translational Research/Clinical Trials Office Pilot Award (G.P‐W), and the Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program Grant #71951 (G.P‐W) and the National Institutes of Health Grant DK112093 (G.P‐W).
Supporting information
Figure S1 Goodness of fit for ropinirole pharmacokinetics model
Figure S2 Goodness of fit for ropinirole pharmacodynamics model
Liu, S. , Hu, C. , Peters, J. , Tsang, A. , Cremers, S. , Bies, R. , and Page‐Wilson, G. (2019) Pharmacokinetics and pharmacodynamics of ropinirole in patients with prolactinomas. Br J Clin Pharmacol, 85: 366–376. 10.1111/bcp.13802.
PI Statement: The authors confirm that the PI for this paper is Dr. Gabrielle Page‐Wilson and that she had direct clinical responsibility for patients.
References
- 1. Ciccarelli A, Daly AF, Beckers A. The epidemiology of prolactinomas. Pituitary 2005; 8: 3–6. [DOI] [PubMed] [Google Scholar]
- 2. Molitch ME. Pharmacologic resistance in prolactinoma patients. Pituitary 2005; 8: 43–52. [DOI] [PubMed] [Google Scholar]
- 3. Tulloch IF. Pharmacologic profile of ropinirole: a nonergoline dopamine agonist. Neurology 1997; 49 (1 Suppl. 1): S58–S62. [DOI] [PubMed] [Google Scholar]
- 4. Roth BL. Drugs and valvular heart disease. N Engl J Med 2007; 356: 6–9. [DOI] [PubMed] [Google Scholar]
- 5. Webster J, Piscitelli G, Polli A, Ferrari CI, Ismail I, Scanlon MF. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline comparative study group. N Engl J Med 1994; 331: 904–909. [DOI] [PubMed] [Google Scholar]
- 6. Schade R, Andersohn F, Suissa S, Haverkamp W, Garbe E. Dopamine agonists and the risk of cardiac‐valve regurgitation. N Engl J Med 2007; 356: 29–38. [DOI] [PubMed] [Google Scholar]
- 7. Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopamine agonists for Parkinson's disease. N Engl J Med 2007; 356: 39–46. [DOI] [PubMed] [Google Scholar]
- 8. Colao A, Galderisi M, Di Sarno A, Pardo M, Gaccione M, D'Andrea M, et al Increased prevalence of tricuspid regurgitation in patients with prolactinomas chronically treated with cabergoline. J Clin Endocrinol Metab 2008; 93: 3777–3784. [DOI] [PubMed] [Google Scholar]
- 9. Kars M, Delgado V, Holman ER, Feelders RA, Smit JW, Romijn JA, et al Aortic valve calcification and mild tricuspid regurgitation but no clinical heart disease after 8 years of dopamine agonist therapy for prolactinoma. J Clin Endocrinol Metab 2008; 93: 3348–3356. [DOI] [PubMed] [Google Scholar]
- 10. Caputo C, Prior D, Inder WJ. The third case of cabergoline‐associated valvulopathy: the value of routine cardiovascular examination for screening. J Endocr Soc 2018; 2: 965–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Acton G, Broom C. A dose rising study of the safety and effects on serum prolactin of SK&F 101468, a novel dopamine D2‐receptor agonist. Br J Clin Pharmacol 1989; 28: 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fukushima T. Pituitary hormones in Parkinson's disease before and after inititaion of dopamine agonists. Mov Disord 2000; 15: 118. [Google Scholar]
- 13. Bharathi DV, Jagadeesh B, Kumar SS, Lakshmi RN, Hotha KK, Naidu A, et al Highly sensitive method for the determination of ropinirole with a lower limit of quantitation of 3.45 pg/mL in human plasma by LC‐ESI‐MS/MS: application to a clinical pharmacokinetic study. Biomed Chromatogr 2009; 23: 557–562. [DOI] [PubMed] [Google Scholar]
- 14. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174 (Suppl. 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm 1993; 21: 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deleu D, Northway MG, Hanssens Y. Clinical pharmacokinetic and pharmacodynamic properties of drugs used in the treatment of Parkinson's disease. Clin Pharmacokinet 2002; 41: 261–309. [DOI] [PubMed] [Google Scholar]
- 18. Eden RJ, Costall B, Domeney AM, Gerrard PA, Harvey CA, Kelly ME, et al Preclinical pharmacology of ropinirole (SK&F 101468‐a) a novel dopamine D2 agonist. Pharmacol Biochem Behav 1991; 38: 147–154. [DOI] [PubMed] [Google Scholar]
- 19. Verhelst JA, Froud AL, Touzel R, Wass JA, Besser GM, Grossman AB. Acute and long‐term effects of once‐daily oral bromocriptine and a new long‐acting non‐ergot dopamine agonist, quinagolide, in the treatment of hyperprolactinemia: a double‐blind study. Acta Endocrinol 1991; 125: 385–391. [DOI] [PubMed] [Google Scholar]
- 20. Peterson K. Population Pharmacodynamic modeling and methods for D2‐receptor antagonists. Uppsala University. 2012.
- 21. Barton AC, Black LE, Sibley DR. Agonist‐induced desensitization of D2 dopamine receptors in human Y‐79 retinoblastoma cells. Mol Pharmacol 1991; 39: 650–658. [PubMed] [Google Scholar]
- 22. Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev 2006; 27: 485–534. [DOI] [PubMed] [Google Scholar]
- 23. Pellegrini I, Rasolonjanahary R, Gunz G, Bertrand P, Delivet S, Jedynak CP, et al Resistance to bromocriptine in prolactinomas. J Clin Endocrinol Metab 1989; 69: 500–509. [DOI] [PubMed] [Google Scholar]
- 24. Brue T, Pellegrini I, Gunz G, Morange I, Dewailly D, Brownell J, et al Effects of the dopamine agonist CV 205‐502 in human prolactinomas resistant to bromocriptine. J Clin Endocrinol Metab 1992; 74: 577–584. [DOI] [PubMed] [Google Scholar]
- 25. Di Sarno A, Landi ML, Cappabianca P, Di Salle F, Rossi FW, Pivonello R, et al Resistance to cabergoline as compared with bromocriptine in hyperprolactinemia: prevalence, clinical definition, and therapeutic strategy. J Clin Endocrinol Metab 2001; 86: 5256–5261. [DOI] [PubMed] [Google Scholar]
- 26. Delgrange E, Maiter D, Donckier J. Effects of the dopamine agonist cabergoline in patients with prolactinoma intolerant or resistant to bromocriptine. Eur J Endocrinol 1996; 134: 454–456. [DOI] [PubMed] [Google Scholar]
- 27. Kukstas LA, Domec C, Bascles L, Bonnet J, Verrier D, Israel JM, et al Different expression of the two dopaminergic D2 receptors, D2415 and D2444, in two types of lactotroph each characterised by their response to dopamine, and modification of expression by sex steroids. Endocrinology 1991; 129: 1101–1103. [DOI] [PubMed] [Google Scholar]
- 28. Missale C, Losa M, Boroni F, Giovanelli M, Balsari A, Spano PF. Nerve growth factor and bromocriptine: a sequential therapy for human bromocriptine‐resistant prolactinomas. Br J Cancer 1995; 72: 1397–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lam YW. Clinical pharmacology of dopamine agonists. Pharmacotherapy 2000; 20 (1 Pt 2): 17S–25S. [DOI] [PubMed] [Google Scholar]
- 30. Vance ML, Evans WS, Thorner MO. Drugs five years later. Bromocriptine. Ann Intern Med 1984; 100: 78–91. [DOI] [PubMed] [Google Scholar]
- 31. Homburg R, West C, Brownell J, Jacobs HS. A double‐blind study comparing a new non‐ergot, long‐acting dopamine agonist, CV 205‐502, with bromocriptine in women with hyperprolactinaemia. Clin Endocrinol (Oxf) 1990; 32: 565–571. [DOI] [PubMed] [Google Scholar]
- 32. Auriemma RS, Pivonello R, Perone Y, Grasso LF, Ferreri L, Simeoli C, et al Safety of long‐term treatment with cabergoline on cardiac valve disease in patients with prolactinomas. Eur J Endocrinol 2013; 169: 359–366. [DOI] [PubMed] [Google Scholar]
- 33. Seki K, Uesato T, Kato K, Shima K. Twenty‐four hour secretory pattern of prolactin in hyperprolactinaemic patients with pituitary micro‐ and macroadenomas. Acta Endocrinol 1984; 106: 433–436. [DOI] [PubMed] [Google Scholar]
- 34. Wardlaw SL, Kim J, Matera C. Effect of chronic prolactin infusion on pituitary prolactin and hypothalamic proopiomelanocortin. J Neuroendocrinol 1997; 9: 81–85. [DOI] [PubMed] [Google Scholar]
- 35. Cowden EA, Ratcliffe JG, Thomson JA, Macpherson P, Doyle D, Teasdale GM. Tests of prolactin secretion in diagnosis of prolactinomas. Lancet 1979; 1: 1155–1158. [DOI] [PubMed] [Google Scholar]
- 36. Boyar RM, Kapen S, Finkelstein JW, Perlow M, Sassin JF, Fukushima DK, et al Hypothalamic‐pituitary function in diverse hyperprolactinemic states. J Clin Invest 1974; 53: 1588–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Goodness of fit for ropinirole pharmacokinetics model
Figure S2 Goodness of fit for ropinirole pharmacodynamics model