

Abstract
Aim
Interleukin (IL)‐7 signalling modulates T cell activity and is implicated in numerous autoimmune diseases. The present study investigated the safety, pharmacokinetics, target engagement, pharmacodynamics and immunogenicity of GSK2618960, an IL‐7 receptor‐α subunit (CD127) monoclonal antibody.
Methods
A double‐blind (sponsor‐unblind) study of a single intravenous infusion of either GSK2618960 (0.6 mg kg–1 or 2.0 mg kg–1) or placebo was carried out in 18 healthy subjects over 24 weeks.
Results
GSK2618960 was well tolerated; there were no serious or significant adverse events. The observed half‐life was 5 (±1) days (2.0 mg kg–1), with nonlinear pharmacokinetics. Full receptor occupancy (>95%) was observed until day 8 (0.6 mg kg–1) and day 22 (2.0 mg kg–1). Maximal inhibition of IL‐7‐mediated signal transducer and activator of transcription 5 (STAT5) phosphorylation was observed in 5/6 subjects until day 22 (2.0 mg kg–1). Mean circulating IL‐7 and soluble receptor (CD127) levels were increased above baseline during days 2 and 15 (0.6 mg kg–1) and days 2 and 22 (2.0 mg kg–1). No meaningful changes were observed in absolute numbers or proportions of immune cell populations or inflammatory cytokine profiles (IL‐6, tumour necrosis factor‐α, interferon‐γ, IL‐2). Persistent antidrug antibodies (ADAs) were detected in 5/6 subjects administered a dose of 0.6 mg kg–1 (neutralizing in 2/6) and in 6/6 subjects administered 2.0 mg kg–1 (neutralizing in 5/6).
Conclusion
GSK2618960 was well tolerated and blocked IL‐7 receptor signalling upon full target engagement. Although there was no discernible impact on peripheral T cell subsets in healthy subjects, GSK2618960 may effectively modulate the autoinflammatory activity of pathogenic T cells in diseased tissue. A relatively short half‐life is likely the result of target‐mediated rather than ADA‐mediated clearance.
Keywords: drug safety, immunology, monoclonal antibodies, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject
Interleukin‐7 (IL‐7) and thymic stromal lymphopoietin share a common receptor component, CD127, and have been implicated in several autoimmune and allergic inflammatory diseases, respectively.
IL‐7 signalling may promote the survival, activation and differentiation of lymphocytes.
What this Study Adds
Single doses of GSK2618960 (0.6 mg kg–1 or 2.0 mg kg–1) were well tolerated, increased circulating IL‐7 and soluble CD127, and elicited antidrug antibodies with neutralizing capability, but did not have an impact on lymphocyte populations, inflammatory cytokines or differentiation markers.
Drug concentrations required for full in vivo receptor occupancy agreed with previous in vitro studies.
Introduction
An increasing body of both preclinical and clinical evidence suggests that raised expression levels of interleukin (IL)‐7 and/or the IL‐7 receptor (IL‐7R) are associated with various disease states, including rheumatoid arthritis 1, 2, 3, juvenile idiopathic arthritis 4, spondyloarthritis 5, psoriasis 6, primary Sjögren's syndrome 7, 8, 9, 10, colitis 11, type 1 diabetes 12, 13 and multiple sclerosis 14, 15. Genome‐wide association studies have also identified genetic variations in IL‐7R as a risk factor for some autoimmune diseases 16, 17.
IL‐7 is a pleiotropic cytokine that is constitutively produced by stromal cells in nonhaematological tissues (intestinal epithelium, lungs, skin, liver) as well as by lymphoid tissue (thymus and bone marrow), and by lymphatic endothelial cells 18, 19. IL‐7 plays a key role in T cell ontogeny 20 and in the homeostatic maintenance of the peripheral T cell pool 21. Activation of the IL‐7/IL‐7R pathway in lymphocytes leads to survival (antiapoptotic activity), differentiation of mature naïve and memory T cells, and proliferation 19. In addition, IL‐7 plays a role in survival and function of innate lymphoid cells, including natural killer cells 22, lymphoid tissue inducer cells 23 and other diverse cell types 19. IL‐7 promotes TH1 and TH17 effector cell function 8, 15, 24, 25, and stimulates cytotoxic activity of CD8+ T cells 26 and the secretion of proinflammatory cytokines by monocytes 27. IL‐7 also stimulates peripheral blood mononuclear cells to secrete T cell‐attracting cytokines 9, and acts via CD4+ T cells and monocytes in co‐culture to augment the activation of B cells 28.
To explore the therapeutic utility of blocking IL‐7 signalling, a humanized Fc‐disabled immunoglobulin G1 (IgG1) monoclonal antibody (mAb) directed against the alpha component (IL‐7Rα; CD127) of the heterodimeric IL‐7R (GSK2618960) was developed. CD127 is also a component of the thymic stromal lymphopoietin receptor (TSLPR) and is required for competent TSLP signalling 29, 30. The TSLPR complex has been associated with a number of allergic inflammatory diseases 30, 31, 32. Therefore, GSK2618960 may have the potential to inhibit both pharmacologies and may have utility in autoimmune and allergic inflammatory diseases. Although IL‐7Rα signalling is crucial to most T lymphocytes, regulatory T cells (Treg) exhibit relatively low‐to‐undetectable expression of the receptor 33. Thus, it was hypothesized that blocking IL‐7R signalling may selectively spare Treg function while exerting inhibitory effects on other potentially pathogenic T cell types, further supporting the rationale as a potential therapeutic target.
A first‐in‐human study of single intravenous (IV) infusions of GSK2618960 (ranging from 0.001 mg kg–1 to 0.15 mg kg–1) demonstrated favourable safety and tolerability profiles within which no antidrug antibodies (ADAs) were detected (Study I7R116702; NCT01808482) 34, 35. The present study (Study 200 902) assessed the safety, tolerability, pharmacokinetics (PK), target engagement, pharmacodynamics (PD) and immunogenicity of two single ascending doses of GSK2618960 in healthy subjects.
Methods
Study design
This was a phase I randomized, double‐blind (sponsor‐unblind), placebo‐controlled study performed in a single centre in Cambridge, UK, between November 2014 and September 2015. The study was conducted in accordance with Good Clinical Practice and the Declaration of Helsinki 2013, and local regulations. The protocol was approved by the local ethics committee (14/LO/1670, National Research Ethics Service Committee, London, UK) and all study subjects provided written informed consent. The study was registered on Clinicaltrials.gov (identifier: NCT02293161).
Two single ascending dose cohorts (A and B), each consisting of nine healthy subjects, were sequentially randomized (2:1) to receive either GSK2618960 or placebo as a single IV infusion over 1 h. The randomization schedule was generated by the sponsor's clinical statistics department using validated proprietary software. The IV infusion was prepared by pharmacy staff and dispensed in a blinded fashion. Both the active IV infusion and placebo were colourless.
Both sentinel and staged dosing were implemented. A dose escalation committee was employed (sponsor unblind: only the central medical monitors were to review unblinded safety data when deemed necessary). The starting dose level of 0.6 mg kg–1 (Cohort A) was a fourfold increase over the highest dose level tested in the first‐in‐human study (I7R116702; see Supporting Information). The dose level of GSK2618960 that was predicted to provide no more than 30 days’ maximal receptor occupancy (RO) was 2.0 mg kg–1 (planned for Cohort B). The no observed adverse effect dose level in the 4‐weekly repeat IV bolus Good Laboratory Practice cynomolgus monkey toxicity study was 300 mg kg–1 (GlaxoSmithKline data on file).
Subjects were screened up to 28 days (42 days for those subjects consenting to vaccination) before admission to the clinical unit on day −1 (predose). All subjects remained in the unit for at least 24 h following dosing, and were monitored at least weekly during the period of full RO and then every 4 weeks thereafter until week 24 (in case of latent lymphopenia).
Stopping criteria were: a serious adverse event (SAE); a lymphocyte count <0.5 × 109 l–1 (sustained for 2 weeks); infusion reactions (National Cancer Institute Common Terminology Criteria for Adverse Events grade 3 or greater) or evidence of immune activation leading to moderate to severe clinical manifestations in one subject administered active treatment; and AEs of moderate or severe intensity (consistent across subjects and reasonably attributable to study drug). The liver stopping criterion was alanine aminotransferase levels ≥3 × the upper limit of normal.
Subjects
Healthy males or females of nonchildbearing potential, 18–65 years of age with normal laboratory values, body weight ≥50 kg, body mass index 19.0–32.0 kg m–2 and a confirmed positive vaccination status for childhood vaccines (or consent to vaccination) were enrolled. Subjects were excluded if they had a history of a significant disease, had received a live vaccine within 1 month of screening (or were planning to receive a live vaccine within 6 months of completing dosing), or had a history of, or were at high risk for, tuberculosis.
Study assessments and procedures
Safety and tolerability assessments (co‐primary endpoints)
Vital signs, routine haematology, clinical chemistry (including proinflammatory cytokines) and urinalysis (predose, and on days 1, 2, 3, 4, 8, 15, 22, 29, 43, 57, 85, 113, 141 and 169), full physical examination (baseline, and on days 2 and 169), 12‐lead electrocardiogram (ECG) (days 1, 2, 15 and 169), and 24‐h Holter ECG (day 1) were performed; AEs were recorded throughout the study. Virology screening and serology (for vaccination status) were also undertaken and a baseline blood sample for comparative retrospective viral testing was archived in case of symptoms of infection.
PK assessments
PK profiling of GSK2618960 was conducted on plasma samples using a validated electrochemiluminescence immunoassay (ECLIA), with a lower limit of quantification of 50 ng ml–1.
PD/biomarker assessments
The extent and duration of GSK2618960 target engagement was assessed by evaluating RO on CD3+ T lymphocytes in a whole‐blood flow cytometry assay. Full RO was defined as the occupation of >95% IL‐7Rα molecules on peripheral blood T cells. To assess whether GSK2618960 target engagement effectively blocked IL‐7R signalling, phosphorylation levels of signal transducer and activator of transcription 5 (STAT5) expression in CD3+ CD4+ T cells were assessed by flow cytometry after ex vivo incubation of whole blood with exogenous IL‐7 stimulant.
Total plasma IL‐7 levels (free and bound to soluble IL‐7Rα) were measured using a validated assay kit (Meso‐Scale Discovery, Rockville, MD, USA; see Supporting Information). Total soluble IL‐7Rα (CD127) levels in the plasma were measured using a validated ECLIA method (Meso Scale Discovery; see Supporting Information). Plasma TSLP levels were measured by a validated Singulex Erenna homogeneous assay format (see Supporting Information).
The effect of GSK2618960 on circulating lymphocyte populations was assessed using two approaches: (i) flow cytometry in whole blood: T, CD4 (and CD8), Th1/ Th2/ Th17, Tc1/Tc2/Tc17, cytotoxic T, Treg, naïve T, central and effector memory T, recent thymic emigrants, natural killer and B cells; and (ii) epigenetic quantification of CD3/TH17, CD3/FoxP3, CD3/TfH and B cells was performed at Epiontis (Berlin, Germany).
The effect of GSK2618960 on circulating inflammatory markers was assessed in the plasma: for IL‐17A and IL‐17F, a bead‐based Singulex Erenna immunoassay kit (EMD Millipore, St Charles, MO, USA) was used; for interferon‐ γ (IFN‐γ), IL‐1β, IL‐2, IL‐4, IL‐6, IL‐8, IL‐10, IL‐12p70, IL‐13 and tumour necrosis factor‐α, a validated 10‐plex assay kit (Meso Scale Discovery) was used.
Immunogenicity (incidence and titre of ADAs) was assessed on days −1, 15 and 22 (Cohort B), 29, 85 and 169 (week 24) and analysed using a validated bridging ECLIA method (Meso Scale Discovery; see Supporting Information). ADAs were characterized for their neutralizing capability and titre. The titre was defined as the reciprocal of the maximum dilution still yielding a positive response (> the screening cut point).
Statistical analysis
No formal statistical hypothesis was tested. The sample size of 18 subjects was chosen based on feasibility considerations. The safety population comprised all subjects who received at least one dose of study drug (or partial administration) and was used for assessment of safety, PD/biomarkers and immunogenicity. The PK population comprised all subjects in the safety population for whom ≥80% of the study drug volume was administered and a PK sample was acquired. Plasma GSK2618960 concentration–time data were analysed by noncompartmental methods (WinNonlin 6.3; Certara, Princeton, NJ, USA). A locally weighted regression (locally estimated scatterplot smoothing; LOESS) was used to explore the PK/PD relationship between concentration of GSK2618960 and RO as a post hoc analysis. For data analysis, placebo‐treated subjects from each dose cohort were combined as the placebo treatment group (n = 6).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 36, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 37.
Results
Study population results
All 18 subjects were treated per protocol, completed the study and were included in the safety and PK populations (Figure 1). Demographics and baseline characteristics are summarized in Table 1. Concomitant medications were taken by approximately 78% of subjects and were in accordance with the protocol.
Figure 1.
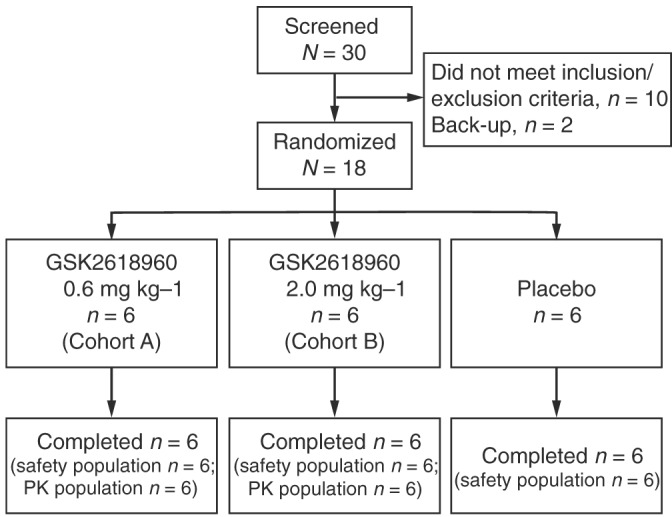
Subject disposition. PK, pharmacokinetic
Table 1.
Summary of subject demographics and baseline characteristics (safety population)
| Demographics | Placebo (N = 6) | GSK2618960 0.6 mg kg–1 (N = 6) | GSK2618960 2.0 mg kg–1 (N = 6) |
|---|---|---|---|
| Age, years | 30.3 (9.2) | 30.3 (5.2) | 36.7 (7.8) |
| Sex, n (%) | |||
| Female | 0 (0) | 0 (0) | 0 (0) |
| Male | 6 (100) | 6 (100) | 6 (100) |
| Body mass index , kg m –2 | 28.9 (2.5) | 25.1 (2.8) | 26.6 (2.9) |
| Height, cm | 173.7 (8.4) | 184.2 (5.5) | 178.83 (8.4) |
| Weight, kg | 87.9 (14.6) | 85.6 (14.1) | 85.2 (10.5) |
| Ethnicity , n (%) | |||
| Hispanic or Latino | 0 (0) | 0 (0) | 0 (0) |
| Not Hispanic or Latino | 6 (100) | 6 (100) | 6 (100) |
| Race , n (%) | |||
| White – Arabic/North African Heritage | 1 (17) | 0(0) | 0 (0) |
| White – White/Caucasian/European Heritage | 5 (83) | 6 (100) | 6 (100) |
Values given are mean (standard deviation) unless specified otherwise
Safety
There were no clinically significant safety or tolerability concerns following a single IV dose of either GSK2618960 0.6 mg kg–1 or 2.0 mg kg–1. There were no deaths or SAEs, and none of the subjects were withdrawn owing to AEs. A total of 54 AEs were reported as either of mild (n = 50) or moderate (n = 4) intensity, and all resolved prior to study completion without the need for long‐term follow‐up. The most commonly reported AEs were nasopharyngitis (nine subjects) and headache (five subjects), which resolved fully; eight subjects across the three treatment groups used paracetamol as needed. Although nasopharyngitis, headache and conjunctivitis were deemed to be causally related to the study drug (blinded assessment), the frequencies of nasopharyngitis and headache were spread across the three treatment groups (Table 2). No trends or clinically significant signals were observed for any of the laboratory parameters, vital signs or ECGs.
Table 2.
Summary of all adverse events deemed drug related (safety population)
| Adverse event | Placebo (N = 6) | GSK2618960 0.6 mg kg–1 (N = 6) | GSK2618960 2.0 mg kg–1 (N = 6) |
|---|---|---|---|
| Nasopharyngitis | 3 (50%) | 4 (67%) | 2 (33%) |
| Headache | 2 (33%) | 3 (50%) | 0 (0%) |
| Conjunctivitis | 1 (17%) | 0 (0%) | 0 (0%) |
PK
The main PK parameters estimated by noncompartmental analysis are shown in Table 3. After administration of GSK2618960 0.6 mg kg–1, the mean clearance was 0.6 ml h–1 kg–1 and the mean volume of distribution was 52.3 ml kg–1; after administration of GSK2618960 2.0 mg kg–1, the mean clearance was 0.3 ml h–1 kg–1 and the mean volume of distribution was 55.9 ml kg–1. The decreased clearance at increased dose was likely to have been a result of saturating target‐mediated drug disposition.
Table 3.
Summary statistics of derived plasma GSK2618960 PK parameters
| Parameter | GSK2618960 treatment | Meana | SD | Median | Min | Max |
|---|---|---|---|---|---|---|
| AUC (0–inf) (h*μg ml –1 ) | 0.6 mg kg–12.0 mg kg–1 | 1005.56858.2 | 127.7988.0 | 994.96619.1 | 8275467 | 11658235 |
| AUC (0–t) (h*μg ml –1 ) | 0.6 mg kg–12.0 mg kg–1 | 924.06615.0 | 148.8989.5 | 939.16315.1 | 6935414 | 11177900 |
| CL (ml h –1 kg –1 ) | 0.6 mg kg–12.0 mg kg–1 | 0.60.3 | 0.080.04 | 0.60.3 | 0.50.2 | 0.70.4 |
| Cmax (μg ml –1 ) | 0.6 mg kg–12.0 mg kg–1 | 11.243.2 | 1.57.6 | 11.141.0 | 9.0036.1 | 13.357.8 |
| Vss (ml kg –1 ) | 0.6 mg kg–12.0 mg kg–1 | 52.355.9 | 7.64.8 | 50.555.0 | 46.350.5 | 67.263.8 |
| t½ (h) | 0.6 mg kg–12.0 mg kg–1 | 57.4123.0 | 7.626.8 | 56.6126.9 | 48.086.8 | 68.1155.0 |
| tmax (h) | 0.6 mg kg–12.0 mg kg–1 | 2.53.0 | 1.22.7 | 2.02.0 | 1.11.0 | 4.08.0 |
AUC, area under the concentration–time curve; CL, systemic clearance of parent drug; Cmax, maximum observed concentration; inf, infinity; PK, pharmacokinetic; SD, standard deviation; t, time of last observed quantifiable concentration; t½, terminal phase half‐life; tmax, time of occurrence of Cmax; Vss, volume of distribution at steady state
Arithmetic mean
Figure 2 shows the GSK2618960 individual plasma concentrations. In the 0.6 mg kg–1 group, the mean concentration (n = 6) at 7 days post‐infusion was 2.05 μg ml–1 (range: 1.46–2.70 μg ml–1); at 14 days post‐infusion, the mean value (n = 3) was 0.055 μg ml–1 (range: 0–0.14 μg ml–1) (three samples were below the limit of quantitation and were set to zero for the calculation). In the 2.0 mg kg–1 group, the mean concentration (n = 6) at 14 days and 21 days post‐infusion was 8.14 μg ml–1 (range: 6.08–9.66 μg ml–1) and 3.05 μg ml–1 (range: 0.42–5.18 μg ml–1), respectively; and 5/6 subjects had a plasma concentration >1 μg ml–1.
Figure 2.
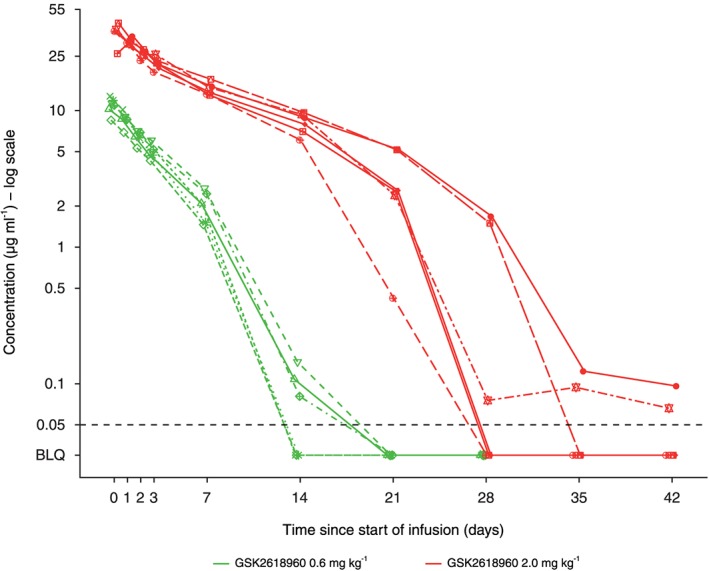
GSK2618960 plasma concentrations in individual subjects following a single intravenous infusion. Symbols represent individual subjects. The dashed horizontal line represents the lower limit of quantification (LLQ = 50 ng ml–1). BLQ, below the limit of quantification
Target engagement
Following dosing of GSK2618960 0.6 mg kg–1, RO >95% was observed for all subjects at 1 h and day 8 (Figure 3). After this time, individual subjects' RO values diminished at different rates, and by day 15 the mean value was 39.3% (range: 0–75.9%). Following dosing of GSK2618960 2 mg kg–1, RO >95% was observed at 1 h and last observed in 5/6 subjects on day 22. After this time, all individual subjects' RO values diminished at different rates, and by day 29 the mean value was 60.3% (range: 24.2–97.6%).
Figure 3.
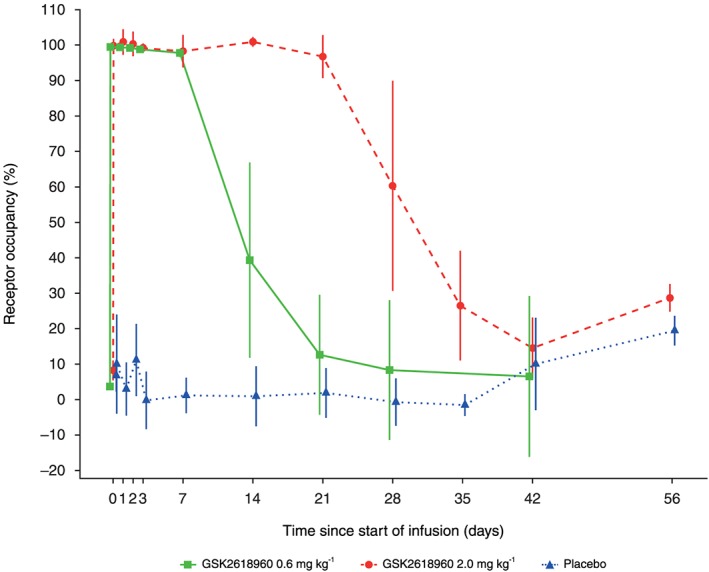
Receptor occupancy of interleukin‐7 receptor subunit‐α (IL‐7Rα) following a single administration of GSK2618960. Mean (± standard deviation) values
PD
Inhibition of IL‐7R signalling (phosphorylated STAT5)
IL‐7R signal inhibition (derived from residual ex vivo IL‐7‐driven STAT5 phosphorylation) was maximally observed at 1 h postdose and last observed on day 8 in the 0.6 mg kg–1 group (see caveat in Supporting Information) and last observed in 5/6 subjects in the 2.0 mg kg–1 group on day 22 (Figure 4).
Figure 4.
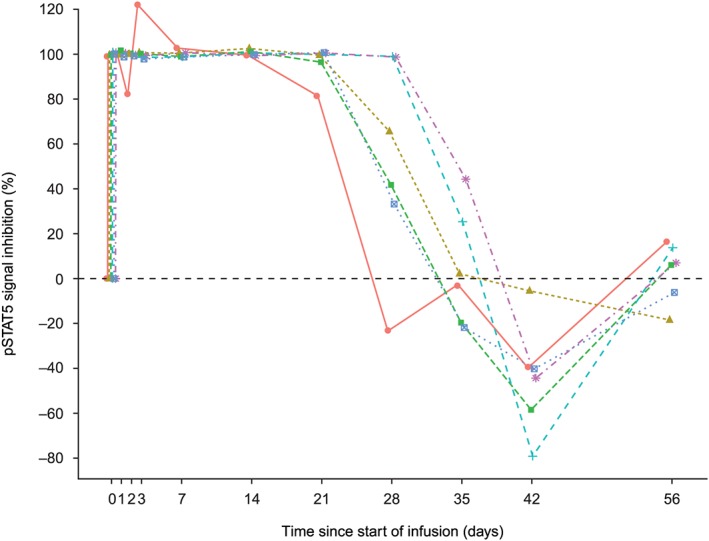
Extent and duration of phosphorylated signal transducer and activator of transcription 5 (pSTAT5) signal inhibition in individual subjects following a single 2.0 mg kg–1 intravenous infusion of GSK2618960. Percentage inhibition of pSTAT5 signal upon ex vivo interleukin‐7 exposure. Coloured symbols represent individual subjects
Effect of GSK2618960 on the phosphorylation of STAT5
Addition of GSK2618960 to CD3+ CD4+ T cells (prior to dosing, and in the absence of IL‐7) induced the phosphorylation of STAT5 (receptor agonism). The mean of replicate determinations was 6.94% (standard deviation 2.79%) of the maximum achieved with IL‐7 stimulation alone (positive control, 100%) and values were observed in 17/18 subjects (data not shown). There was no change in receptor agonism (as defined by STAT5 phosphorylation) over time (assessed ex vivo using exogenous GSK2618960) when comparing baseline samples with those obtained throughout the Cohort B treatment period (samples from Cohort A were not evaluable owing to a procedural technicality – see Supporting Information).
Exploratory markers
Circulating biomarkers: IL‐7, TSLP and soluble IL‐7Rα (sCD127)
Baseline IL‐7 levels ranged from 2.23 pg ml–1 to 26.45 pg ml–1. In subjects receiving GSK2618960 0.6 mg kg–1, the mean increase in IL‐7 levels was first detected at 4 h (Figure 5A) (and by which time full target engagement had already been achieved). IL‐7 concentration reached peak levels during days 2–15, and returned to baseline by day 22. In subjects receiving GSK2618960 2.0 mg kg–1, IL‐7 concentration reached peak levels during days 2–22 and returned to baseline by day 29 (Figure 5A). Only four subjects had measurable TSLP levels pre‐ and postdose (range: 9.49–80.55 pg ml–1), with no significant change from baseline following dosing (data not shown).
Figure 5.
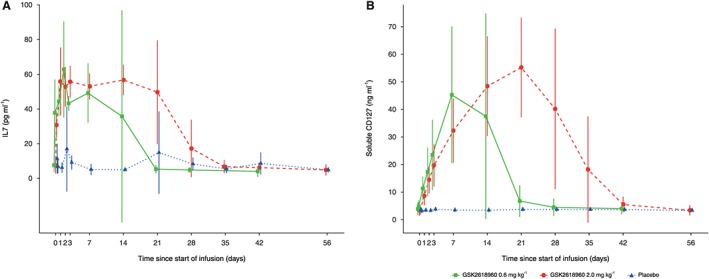
Circulating (A) interleukin‐7 (IL‐7) and (B) soluble IL‐7 receptor‐α subunit (CD127) in plasma following a single intravenous infusion of GSK2618960. Mean (± standard deviation) values
Figure 5B illustrates circulating sCD127 levels (free and drug bound) before and after treatment (baseline range: 1.02–8.49 ng ml–1). Levels were increased from baseline during days 8–15 and returned to baseline by day 22 in the 0.6 mg kg–1 group, and during days 22–29, with a return to baseline levels by day 43 in the 2.0 mg kg–1 group.
Circulating lymphocyte populations
Except for B cells (CD45+, CD3−, CD19+), there were no consistent changes in the flow cytometric assessment of either the absolute numbers or percentages in any of the cell lineages in either of the two GSK2618960 or placebo groups from baseline. There was an unexpected, small, transient decrease in circulating B cells across all three treatment groups from baseline to day 8. The mean group absolute values (CD19+ as a percentage of total lymphocytes) at these time points and the change from baseline (± standard deviation) were: placebo, from 13.1% to 11.8% (−1.3 ± 2.1%); 0.6 mg kg–1, from 10.8% to 8.9% (−1.9 ± 1.2%); and 2.0 mg kg–1, from 9.5% to 7.1% (−2.5 ± 1.2%). Notably, treatment with GSK2618960 did not appear to change the absolute number of Tregs (CD45+ CD3+ CD4+ CD25high CD127−). CD25 and programmed death‐1 activation markers on selected cell lineages did not change consistently.
Epigenetic quantification of cell subtypes corroborated that there were no consistent changes in the specific cell subtypes selected, but that there were small transient unexpected decreases in circulating B cells.
Inflammatory markers
Proinflammatory cytokine plasma levels measured 4 h postdose did not indicate an effect of GSK2618960 on cytokine release. IL‐8 was increased at apparently random time points in some subjects, as was IFN‐γ. Throughout the study, there were no consistent changes from baseline in either of the T cell differentiation markers (IL‐17A or IL‐17F) or in the other cytokines measured (levels remained undetectable).
Immunogenicity
Five of six subjects receiving GSK2618960 0.6 mg kg–1 developed ADAs (titre range: 4–2560) and all six subjects receiving GSK2618960 2.0 mg kg–1 developed ADAs (titre range: 4–5120). Among the ADA‐positive subjects, 2/5 subjects receiving GSK2618960 0.6 mg kg developed NAbs (titre range: 2–10) and 5/6 subjects receiving GSK2618960 2.0 mg kg–1 had NAbs (titre range: 4–40). ADAs were persistent to day 169.
Discussion
There were neither safety nor tolerability concerns following a 1‐h IV infusion of either a single 0.6 mg kg–1 or 2.0 mg kg–1 dose of GSK2618960 in healthy subjects. From both a safety and mechanistic viewpoint, there was neither a short‐ nor long‐term impact on T‐lymphocyte counts or subpopulations throughout the 24‐week study period. There was an unexpected transient small decrease in B cells that could not easily be explained but may have been due to a redistribution phenomenon. Overall, there were no meaningful changes to inflammatory cytokines or differentiation markers (e.g. IL‐17A and IL‐17F).
GSK2618960 successfully blocked IL‐7R signalling above the threshold concentration (1 μg ml–1) required for full target engagement, as evidenced by total inhibition of STAT5 phosphorylation (in an ex vivo assay), and this corroborated previous in vitro findings (data on file). Thus, GSK2618960 may prove effective in modulating the autoinflammatory activity of pathogenic T cells in localized diseased tissue (e.g. salivary glands in patients with Sjögren's syndrome). These observations are in agreement with a PK/PD study reported by Kern et al. in cynomolgus monkeys 38, showing that treatment with an IL‐7Rα mAb prevents the ability of IL‐7 to initiate downstream signalling proximal from the IL‐7R, as evidenced by inhibition of STAT5 phosphorylation.
Importantly, GSK2618960 inhibited IL‐7‐induced phosphorylation of STAT5 in a manner that was in concordance with drug exposure, target engagement and its proposed mechanism of action.
The duration at which GSK2618960 exposures were maintained (in vivo) above the in vitro ‘threshold’ of 1 μg ml–1 concentration and the understanding of consequential drug PD were central to both the study rationale and to the importance of defining a starting dose and regimen for future clinical studies in patients. The actual duration of exposure above the threshold concentration believed to achieve full RO was 7 days and 21 days (0.6 mg kg–1 and 2.0 mg kg–1 doses, respectively), which was shorter than originally predicted (11 days and 30 days). The half‐life of the 0.6 mg kg–1 and 2.0 mg kg–1 single doses administered by IV infusion was 57.4 (±7.6) and 123.0 (±26.8) h, respectively, which is relatively short when compared with that of a typical mAb against soluble targets (21 days) 39. Clearance by target‐mediated drug disposition (TMDD) provides an additional, but saturable, clearance pathway for some antibodies 40. Thus, it appears that the level of target expression and, consequently, the extent of TMDD were higher than originally anticipated. Despite the low level of target, the TMDD phenomena can still play a relevant role in explaining nonlinear PK, and this is mainly because of the high turnover of the target that contributes to the saturable elimination of the antibody. The decreased clearance rate of GSK2618960 at the higher dose level would support this.
Target engagement (>95% RO) was achieved at a threshold concentration of >1 μg ml–1, which corroborated the in vivo PK and target engagement findings. The relationship between PK and RO is illustrated in Figure 6 , with the fitted LOESS curve superimposed. As was expected, the extent of duration of target engagement (RO) correlated with the inhibition of phosphorylation of STAT5.
Figure 6.
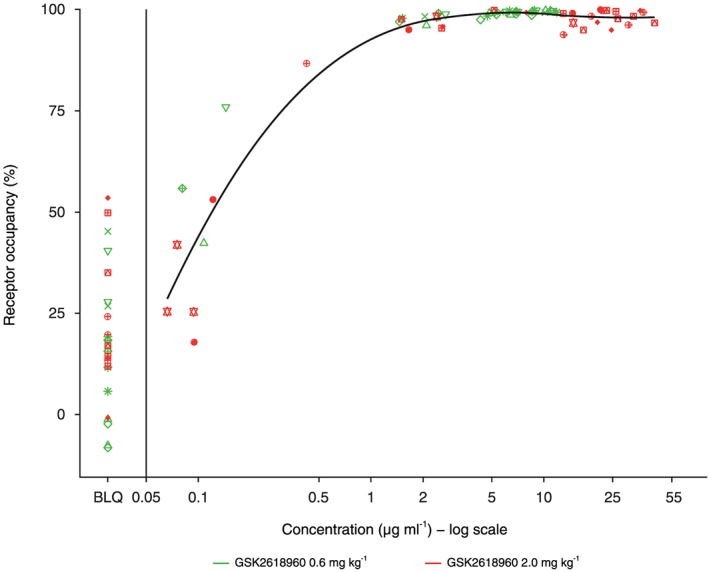
GSK2618960 plasma concentrations vs. receptor occupancy of interleukin‐7 receptor subunit‐α, for subjects in each treatment group with the fitted locally estimated scatterplot smoothing curve (post hoc analysis). Coloured symbols represent individual subjects. The vertical line represents the lower limit of quantification (50 ng ml–1). BLQ, below the lower limit of quantification
Although ADAs had not been observed in the first‐in‐human study (I7R116702; dose levels 0.001–0.15 mg kg–1) 34, 35, ADAs were first observed in 5/6 subjects administered a single IV infusion of GSK2618960 0.6 mg kg–1, and in all six subjects administered GSK2618960 2.0 mg kg–1, with higher titres generated in Cohort B (2.0 mg kg–1 dose) compared with Cohort A (0.6 mg kg–1 dose) suggesting a dose‐response relationship. Of note is that one subject generated a high titre (5120) by week 12 (assessed in the absence of circulating GSK2618960 and based on the titration results during method validation). Subjects who developed high and persistent titres of ADAs (7/12 across both cohorts) also developed NAbs). Furthermore, GSK2618960‐specific memory B cells were detected in a B cell enzyme‐linked immunospot assay (data not shown). Memory B cell activity developed as early as day 29, and was observed in all of those subjects who had developed ADAs by day 169. The frequency of such events seemed to be dose dependent. Following completion of Study 200 902, antidrug antibodies were observed in the 6‐month cynomolgus monkey study. ADAs to humanized antibodies are not unexpected in nonhuman primates and do not usually prevent progression to clinical development. It was noted that the results of clinical trial NCT02045732 (an anti‐IL7R mAb) listed that 3/3 subjects administered 0.25 mg kg–1 PF‐06342674 developed ADAs 41. Expression of target IL‐7 receptor on antigen presenting cells may increase the potential for immunogenicity, but there is no direct evidence. Host‐, disease‐ and product‐specific factors are known to influence immunogenicity – e.g. amino acid sequence, number and strength of T cell epitopes, post‐translational modification, structural alterations (aggregation, oxidation, deamidation, degradation), storage conditions, production and purification processes 42.
GSK2618960 exhibited a degree of (paradoxical) agonistic potential in the present study (assessed ex vivo), which corroborated a previous observation in the first‐in‐human study, I7R116702 34, 35. Agonistic activity could be exacerbated in vivo by an ADA response mediating crosslinking. However, the data indicated that there was no change to baseline agonism levels following a single‐dose IV infusion. Further evaluation of any impact of this phenomenon, using an optimized assay, could be incorporated into repeat‐dose studies in patients.
It was envisaged that the binding of GSK2618960 to both sCD127 and membrane IL‐7Rα subunit (CD127) would set up a dynamic equilibrium – firstly, by preventing the binding of IL‐7 to both forms of the receptor and, secondly, by reducing IL‐7 clearance through receptor‐mediated internalization. Given that IL‐7 is constitutively expressed 43, 44, such a dynamic equilibrium might manifest as an increase in circulating levels of IL‐7 to a new threshold level. Indeed, the marked increase in the level of IL‐7 from baseline, the duration of that increase, and the onset and rate of decline were in concordance with drug dose, exposure and RO. These data indicate that GSK2618960 blocked clearance of IL‐7 through inhibition of receptor‐mediated internalization. Understanding the longer‐term impact of this (if any) will require repeat dose studies.
Similarly, sCD127 was speculated to increase upon complexing with GSK2618960 and have prolonged clearance until the complex was metabolized/excreted. Irrespective of the GSK2618960 dose, the rate of accumulation of sCD127 was slower than that of IL‐7 (increased levels of sCD127 only became apparent on day 2). Administration of the higher dose of GSK2618960 (2.0 mg kg–1 vs. 0.6 mg kg–1) and, consequentially, higher exposure, resulted in a relatively higher and longer duration of increase in circulating sCD127, as predicted. These data suggest that sCD127 (like IL‐7) accumulates upon engagement of IL‐7Rα by GSK2618960 and declines upon drug clearance. Circulating low TSLP concentrations were apparently unaffected following a single infusion of GSK2618960 at either dose level, which seemed to corroborate the notion that the binding of TSLP to its cognate heterodimeric receptor [cytokine receptor‐like factor 2 (CLFR2)] would be unlikely to be affected by GSK2618960, at least after a single infusion in the short term.
The main limitation of the present study was the inability to determine whether ADAs had an impact on GSK2618960 clearance. IgG antibodies, such as GSK2618960, typically have a half‐life of 21 days 39, which is unfortunately coincidental with the first experimental detection of ADAs, and so did not enable the determination of whether emergent ADAs had affected the extent and duration of drug exposure/clearance. Interpretation of PK data was yet further confounded by the single and relatively low dose levels tested and whereby TMDD was rationally believed to have shortened the half‐life. It is known that ADAs not only have the potential to increase the overall clearance rate, but also may interfere with the detection of free drug levels in PK assays 45. Further repeat‐dose clinical studies will be required to evaluate the impact of ADAs on GSK2618960 clearance.
To conclude, GSK2618960 was well tolerated when administered to healthy subjects as a single IV infusion at a dose capable of maintaining effective receptor blockade for up to 21 days. There were no SAEs, and no meaningful changes were observed in any haematological or immune cell populations or in the selected cytokine profiles. Single doses of GSK2618960 elicited ADAs with high incidence and persistent titres, and there was evidence of neutralizing capability. TMDD appeared to shorten the half‐life of GSK2618960 as compared with a typical IgG antibody. There was a clear PK/PD relationship between the duration of GSK2618960 exposure above the threshold concentration required to have an impact on important measures, such as the extent and duration of target engagement, and such PD measures as the extent and duration of inhibition of STAT5 phosphorylation and the duration of the increased levels of circulating IL‐7 and sCD127. Further repeat‐dose studies are warranted in patients, to properly assess the potential of GSK2618960 to have an impact on T cells and other cell populations in inflamed tissues and to evaluate any clinical improvement.
Competing Interests
J.C. is employed by Cambridge University Hospitals NHS Foundation Trust but is obligated to spend 50% of his time on clinical trial research with GSK Clinical Unit, Cambridge, via a secondment agreement with GSK (for which he receives no shares, stock or employee benefits). A.C. has received honoraria for talks, and support for travel to conferences, from Genzyme (a Sanofi company) and his institution has received research funding from the same source. S.B., K.B., K.C., B.D., J.E., D.F., L.F., S.G., J.‐J.O., J.P., S.S.F., A.S., A.v.M. and A.W. are employees of, and hold stocks/shares in, GSK. O.K.E. and D.S. were employees of GSK at the time the study was conducted. J.C., S.M. and J.Y. were employees of GSK at the time the study was conducted, and hold stocks/shares in GSK. F.G. and P.T. were employees of, and held stocks/shares in, GSK at the time the study was conducted.
Funding for this study (NCT02293161) was provided by GSK. Medical writing support was provided by Clare Slater PhD, CMPP, Fishawack Indicia Ltd, UK, and was funded by GSK. All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. The authors wish to thank all subjects and staff of the GSK Clinical Unit, Cambridge, UK, and all past and present members of the GSK2681960 Project Team for their contributions. Special thanks to Jatin Patel, Kevin Page, Thomas Lee, Jerry Page and Karen Liao (Study 200902).
Contributors
J.E., A.v.M., K.C., D.F., S.M., D.S., K.B., J.Y. and J.Cr. contributed to the 200 902 study concept and/or design; B.D., J.‐J.O., F.G., P.W.T., J.Y., J.Ch. and A.C. contributed to the I7R116702 study supplement concept and/or design; B.D., D.F., A.W., S.S.F., J.‐J.O., O.K.‐E. and J.P. were involved in the acquisition of 200 902 data; J.E., A.v.M., L.F., S.G., K.C., A.S., D.F., B.D., S.M., A.W., S.S.F. and K.B. provided data analysis or interpretation. All authors critically revised this manuscript.
Supporting information
Data S1 Study design: Dosing rationale
Data S2 Study assessments and procedures: Pharmacodynamics/biomarker assessments
Data S3 Pharmacodynamics: Phosphorylation of STAT5
Ellis, J. , van Maurik, A. , Fortunato, L. , Gisbert, S. , Chen, K. , Schwartz, A. , McHugh, S. , Want, A. , Santos Franco, S. , Oliveira, J.‐J. , Price, J. , Coles, A. , Brown, K. , Su, D. , Craigen, J. L. , Yang, J. , Brett, S. , Davis, B. , Cheriyan, J. , Kousin‐Ezewu, O. , Gray, F. , Thompson, P. W. , and Fernando, D. (2019) Anti‐IL‐7 receptor α monoclonal antibody (GSK2618960) in healthy subjects – a randomized, double‐blind, placebo‐controlled study. Br J Clin Pharmacol, 85: 304–315. 10.1111/bcp.13748.
Clinicaltrials.gov identifier: NCT02293161.
References
- 1. Hartgring SA, Willis CR, Alcorn D, Nelson LJ, Bijlsma JW, Lafeber FP, et al Blockade of the interleukin‐7 receptor inhibits collagen‐induced arthritis and is associated with reduction of T cell activity and proinflammatory mediators. Arthritis Rheum 2010; 62: 2716–2725. [DOI] [PubMed] [Google Scholar]
- 2. van Roon JA, Glaudemans KA, Bijlsma JW, Lafeber FP. Interleukin 7 stimulates tumour necrosis factor alpha and Th1 cytokine production in joints of patients with rheumatoid arthritis. Ann Rheum Dis 2003; 62: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Roon JA, Verweij MC, Wijk MW, Jacobs KM, Bijlsma JW, Lafeber FP. Increased intraarticular interleukin‐7 in rheumatoid arthritis patients stimulates cell contact‐dependent activation of CD4(+) T cells and macrophages. Arthritis Rheum 2005; 52: 1700–1710. [DOI] [PubMed] [Google Scholar]
- 4. De Benedetti F, Massa M, Pignatti P, Kelley M, Faltynek CR, Martini A. Elevated circulating interleukin‐7 levels in patients with systemic juvenile rheumatoid arthritis. J Rheumatol 1995; 22: 1581–1585. [PubMed] [Google Scholar]
- 5. Rihl M, Kellner H, Kellner W, Barthel C, Yu DT, Tak PP, et al Identification of interleukin‐7 as a candidate disease mediator in spondylarthritis. Arthritis Rheum 2008; 58: 3430–3435. [DOI] [PubMed] [Google Scholar]
- 6. Bonifati C, Trento E, Cordiali‐Fei P, Carducci M, Mussi A, D'Auria L, et al Increased interleukin‐7 concentrations in lesional skin and in the sera of patients with plaque‐type psoriasis. Clin Immunol Immunopathol 1997; 83: 41–44. [DOI] [PubMed] [Google Scholar]
- 7. Bikker A, Kruize AA, Wenting M, Versnel MA, Bijlsma JW, Lafeber FP, et al Increased interleukin (IL)‐7Ralpha expression in salivary glands of patients with primary Sjogren's syndrome is restricted to T cells and correlates with IL‐7 expression, lymphocyte numbers and activity. Ann Rheum Dis 2012; 71: 1027–1033. [DOI] [PubMed] [Google Scholar]
- 8. Bikker A, Moret FM, Kruize AA, Bijlsma JW, Lafeber FP, van Roon JA. IL‐7 drives Th1 and Th17 cytokine production in patients with primary SS despite an increase in CD4 T cells lacking the IL‐7Ralpha. Rheumatology (Oxford) 2012; 51: 996–1005. [DOI] [PubMed] [Google Scholar]
- 9. Bikker A, van Woerkom JM, Kruize AA, Wenting‐van Wijk M, de Jager W, Bijlsma JW, et al Increased expression of interleukin‐7 in labial salivary glands of patients with primary Sjogren's syndrome correlates with increased inflammation. Arthritis Rheum 2010; 62: 969–977. [DOI] [PubMed] [Google Scholar]
- 10. Jin JO, Kawai T, Cha S, Yu Q. Interleukin‐7 enhances the Th1 response to promote the development of Sjogren's syndrome‐like autoimmune exocrinopathy in mice. Arthritis Rheum 2013; 65: 2132–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willis CR, Seamons A, Maxwell J, Treuting PM, Nelson L, Chen G, et al Interleukin‐7 receptor blockade suppresses adaptive and innate inflammatory responses in experimental colitis. J Inflamm 2012; 9: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee LF, Logronio K, Tu GH, Zhai W, Ni I, Mei L, et al Anti‐IL‐7 receptor‐alpha reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T‐cell function. Proc Natl Acad Sci U S A 2012; 109: 12674–12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, et al IL‐7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci USA 2012; 109: 12668–12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kreft KL, Verbraak E, Wierenga‐Wolf AF, van Meurs M, Oostra BA, Laman JD, et al The IL‐7Ralpha pathway is quantitatively and functionally altered in CD8 T cells in multiple sclerosis. J Immunol 2012; 188: 1874–1883. [DOI] [PubMed] [Google Scholar]
- 15. Lee LF, Axtell R, Tu GH, Logronio K, Dilley J, Yu J, et al IL‐7 promotes T (H)1 development and serum IL‐7 predicts clinical response to interferon‐beta in multiple sclerosis. Sci Transl Med 2011; 3: 93ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. International Multiple Sclerosis Genetics Consortium , Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, et al Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357: 851–862. [DOI] [PubMed] [Google Scholar]
- 17. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al Robust associations of four new chromosome regions from genome‐wide analyses of type 1 diabetes. Nat Genet 2007; 39: 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carrette F, Surh CD. IL‐7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin Immunol 2012; 24: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dooms H. Interleukin‐7: fuel for the autoimmune attack. J Autoimmun 2013; 45: 40–48. [DOI] [PubMed] [Google Scholar]
- 20. Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat Genet 1998; 20: 394–397. [DOI] [PubMed] [Google Scholar]
- 21. Lundstrom W, Highfill S, Walsh ST, Beq S, Morse E, Kockum I, et al Soluble IL7Ralpha potentiates IL‐7 bioactivity and promotes autoimmunity. Proc Natl Acad Sci USA 2013; 110: E1761–E1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cella M, Otero K, Colonna M. Expansion of human NK‐22 cells with IL‐7, IL‐2, and IL‐1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci USA 2010; 107: 10961–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al Ectopic lymphoid‐organ development occurs through interleukin 7‐mediated enhanced survival of lymphoid‐tissue‐inducer cells. Immunity 2007; 26: 643–654. [DOI] [PubMed] [Google Scholar]
- 24. Borger P, Kauffman HF, Postma DS, Vellenga E. IL‐7 differentially modulates the expression of IFN‐gamma and IL‐4 in activated human T lymphocytes by transcriptional and post‐transcriptional mechanisms. J Immunol 1996; 156: 1333–1338. [PubMed] [Google Scholar]
- 25. Wan Q, Kozhaya L, ElHed A, Ramesh R, Carlson TJ, Djuretic IM, et al Cytokine signals through PI‐3 kinase pathway modulate Th17 cytokine production by CCR6+ human memory T cells. J Exp Med 2011; 208: 1875–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mehrotra PT, Grant AJ, Siegel JP. Synergistic effects of IL‐7 and IL‐12 on human T cell activation. J Immunol 1995; 154: 5093–5102. [PubMed] [Google Scholar]
- 27. Alderson MR, Tough TW, Ziegler SF, Grabstein KH. Interleukin 7 induces cytokine secretion and tumoricidal activity by human peripheral blood monocytes. J Exp Med 1991; 173: 923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bikker A, Kruize AA, van der Wurff‐Jacobs KM, Peters RP, Kleinjan M, Redegeld F, et al Interleukin‐7 and Toll‐like receptor 7 induce synergistic B cell and T cell activation. PLoS One 2014; 9: e94756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E, et al Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 2017; 174 (Suppl. 1): S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He R, Geha RS. Thymic stromal lymphopoietin. Ann NY Acad Sci 2010; 1183: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Birben E, Sahiner UM, Karaaslan C, Yavuz TS, Cosgun E, Kalayci O, et al The genetic variants of thymic stromal lymphopoietin protein in children with asthma and allergic rhinitis. Int Arch Allergy Immunol 2014; 163: 185–192. [DOI] [PubMed] [Google Scholar]
- 32. Gauvreau GM, O'Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al Effects of an anti‐TSLP antibody on allergen‐induced asthmatic responses. N Engl J Med 2014; 370: 2102–2110. [DOI] [PubMed] [Google Scholar]
- 33. Liu W, Putnam AL, Xu‐Yu Z, Szot GL, Lee MR, Zhu S, et al CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+T reg cells. J Exp Med 2006; 203: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clinical Trials.gov . A first time in human study exploring safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of GSK2618960 in healthy volunteers and patients with relapsing remitting multiple sclerosis (RRMS) [online]. Available at https://clinicaltrials.gov/ct2/show/NCT01808482 (last accessed 6 June 2017).
- 35. GlaxoSmithKline . A first time In human study exploring preliminary safety, tolerability, pharmacokinetics and pharmacodynamics of GSK2618960 in healthy volunteers and patients with relapsing remitting multiple sclerosis [online]. Available at https://www.gsk‐clinicalstudyregister.com/files2/116702‐Clinical‐Study‐Result‐Summary.pdf#page=1&zoom=auto,‐99,798 (last accessed 6 June 2017).
- 36. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al THE Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 2017; 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kern B, Li W, Bono C, Lee LF, Kraynov E. Receptor occupancy and blocking of STAT5 signaling by an anti‐IL‐7 receptor alpha antibody in cynomolgus monkeys. Cytometry B Clin Cytom 2016; 90: 191–198. [DOI] [PubMed] [Google Scholar]
- 39. Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty‐first century. Nat Rev Drug Discov 2003; 2: 52–62. [DOI] [PubMed] [Google Scholar]
- 40. Levi M, Grange S, Frey N. Exposure‐response relationship of tocilizumab, an anti‐IL‐6 receptor monoclonal antibody, in a large population of patients with rheumatoid arthritis. J Clin Pharmacol 2013; 53: 151–159. [DOI] [PubMed] [Google Scholar]
- 41. Clinical Trials.gov . A study to evaluate the safety and tolerability of PF‐06342674 (RN168) in subjects with multiple sclerosis (MS) [online]. Available at https://clinicaltrials.gov/ct2/show/results/NCT02045732?sect=Xc0156&term=NCT02045732&rank=1#outcome6 (last accessed 6 June 2017).
- 42. Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J 2012; 14: 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin‐7 mediates the homeostasis of naive and memory CD8 T cells in vivo . Nat Immunol 2000; 1: 426–432. [DOI] [PubMed] [Google Scholar]
- 44. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al IL‐7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci USA 2001; 98: 8732–8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Felis‐Giemza A, Moots RJ. Measurement of anti‐drug antibodies to biologic drugs. Rheumatology 2015; 54: 1941–1943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Study design: Dosing rationale
Data S2 Study assessments and procedures: Pharmacodynamics/biomarker assessments
Data S3 Pharmacodynamics: Phosphorylation of STAT5