

Abstract
AIM: Huntington’s disease (HD) is an inherited disease caused by an expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin gene (HTT) that ultimately leads to neurodegeneration. To study the molecular basis of this disease, induced pluripotent stem cells (iPSCs) generated from patients’ fibroblasts were used to investigate axonal mitochondrial trafficking and the nature of nuclear indentations.
METHODS: Pathological and control iPSCs generated from patients with a low number of repeats were differentiated in striatal neurons of the brain. Mitochondrial density was measured along the axon using tubulin beta 3 co-staining in pathological and control neurons. To investigate the connection of nuclear roundness with calcium dysregulation, several calcium inhibitors were used. Proteasome system inhibition was applied to mimic premature neuronal ageing.
RESULTS: We found that the mitochondrial density was approximately 7.6 ± 0.2 in neurites in control neurons but was only 5.3 ± 0.2 in mutant neurons with 40-44 CAG repeats (p-value <0.005). Neuronal ageing induced by proteasome inhibitor MG132 significantly decreased the mitochondrial density by 15% and 25% in control and mutant neurons to 6.5 ± 0.1 (p-value < 0.005) and 4.0 ± 0.3 (p-value < 0.005), respectively. Thus, for the first time, an impairment of mitochondrial trafficking in pathological neurons with endogenous mutant huntingtin was demonstrated. We found that inhibiting the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the ryanodine-receptor (RyR) or the inositol 1,4,5-trisphosphate receptor (IP3R) by specific inhibitors did not specifically affect the nuclear roundness or survival of pathological neurons differentiated from patient iPSCs. Therefore, nuclear calcium homeostasis is not directly associated with HD pathology.
CONCLUSION: Identifying HD iPSCs and differentiating from them neurons provide a unique system for modelling the disease in vitro. Impairments of mitochondrial trafficking and nuclear roundness manifest long before the disease onset, while premature neuronal ageing enhances differences in mitochondrial distribution.
Keywords: Reprogramming, Differentiation, Huntingtin, Mitochondria trafficking, Calcium Homeostasis
Introduction
Huntington's disease (HD) is an incurable hereditary neurodegenerative disease that occurs at a frequency of 1-20 per 100,000 people and typically manifests between 35–55 years of age. Clinically, HD causes a combination of steadily progressing movement, cognitive and psychiatric disorders with a wide spectrum of signs and symptoms. HD is characterized by extensive neurodegeneration, primarily affecting the striatal region of the brain. There is no cure that can stop or reverse the disease. HD is caused by an expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin gene (HTT) that leads to a pathological elongation of polyglutamine repeats in the huntingtin protein[1]. Normal alleles of the HTT gene contain 10-35 CAG repeats, and when the number of repeats exceeds 35, the disease develops. In most clinical cases, the number of CAG repeats does not exceed 50. However, a higher number of CAG repeats leads to the juvenile form of HD, which begins in childhood or adolescence and tends to progress more quickly than the adult-onset form [2-4]. Despite its monogenic nature, HD pathogenesis is incredibly complex, and despite a large number of studies using various transgenic models, the mechanisms leading to neuronal loss in HD patients are still unknown.
First introduced in 2006, genetic reprogramming of somatic cells to pluripotency provides new possibilities for modelling hereditary diseases using personalized human cells carrying pathological alleles. Easily accessible adult somatic cells, such as skin fibroblasts or blood cells, are genetically reprogrammed to induced pluripotent stem cells (iPSCs) that closely resemble embryonic stem cells (ESCs). Similar to ESCs, iPSCs can grow unlimitedly in culture and differentiate into any somatic cell type. The latter is particularly valuable for studying human neurodegenerative diseases due to the difficulties of obtaining neurons from patients with neurodegenerative diseases and the limitations of transgenic models. Using this technology, recent findings have enhanced our understanding of the ways in which cells regulate and respond to expanded polyglutamine proteins such as mutant huntingtin. The HD iPSC Consortium generated and studied a set of 14 iPSC lines from three HD patients with a significant number of CAG repeats, including the juvenile form of the disease (60, 109, 180), and from healthy donors. In mutant neurons differentiated from iPSCs, pathological changes associated with cell electrophysiology, metabolism and adhesion were detected. Mutant neurons with 180 CAG repeats had an increased level of cell death in comparison with the normal controls[5]. Aggregates of mutant huntingtin in neurons differentiated from patient iPSCs were observed after prolonged cultivation in vitro or transplantation into the rat brain[6].
Neurodegeneration may be connected to the increased caspase activity that was observed in the culture of mutant neurons when BDNF growth factor was removed from the culture medium. With the decreased expression of BDNF and reduced oxygen consumption that characterize mitochondria functioning, fragmentation and a decrease in the length of neurites were observed[7,8]. Neuronal metabolism was impaired in neurons that were differentiated in vitro from iPSCs established from patients with a low number of CAG repeats (fewer than 50); an increased lysosomal content in mutant neurons was shown compared with normal control neurons[9]. Recently, we established a number of iPSC lines from patients with HD with a low number of CAG repeats (42-44) showed their pluripotency in vitro and in vivo, and demonstrated that mutant neurons acquire pathological phenotypes in vitro, such as nuclear invaginations, increased expression of calcium-binding proteins and increased store-operated calcium entry. Premature ageing was associated with the formation of huntingtin aggregates and increased death of mutant neurons[10]. These results and other data support the calcium hypothesis of HD pathogenesis.
Neuronal calcium signalling involves several calcium channels: voltage-gated calcium channels of the plasma membrane (VGCC), NMDA receptors, AMPA receptors, TRP and store-operated calcium entry (SOC) channels. The main Ca2+-release channels in the endoplasmic reticulum (ER) belong to either the ryanodine-receptor (RyR)[11] or the inositol 1,4,5-trisphosphate (IP3)-receptor (IP3R)[12] families. Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the plasma membrane Ca2+-ATPase (PMCA) and the Na+/Ca2+ exchanger (NCE) control the calcium levels in the cytoplasm[13]. Similar to the ER, mitochondria can store Ca2+; however, mitochondrial Ca2+ storage is regulated by the mitochondrial calcium uniporter (MCU), a mechanism that is distinct from those used by the ER[14]. According to the calcium hypothesis of HD, destabilization of neuronal Ca2+ signalling is one of the toxic functions of the mutant HTT protein. In neurons, the mutant HTT sensitizes IP3R[15] and NMDA receptors[16] and increases SOC[17], resulting in an excess of calcium in the cytosol. The mechanism of toxicity is mediated by the activation of calpains and the excessive accumulation of calcium in the mitochondria. Mitochondrial function is impaired in HD transgenic cellular models[15, 18, 19]. However, mitochondrial function has not been investigated in human neurons differentiated from iPSCs with a low number of excessive CAG repeats.
Here, we utilized a previously established model system of medium spiny neurons differentiated from iPSCs[10] to investigate the possible effect that calcium homeostasis has on nuclear invaginations in pathological neurons and to study the mitochondria in the axons of normal and mutant neurons.
Materials and methods
Cell lines
Pluripotent stem cell lines from HD patients (iPSHD11, iPSHD22, iPSHD34) and healthy controls (iPSRG2L, endo-iPS12, hESM01) were cultured in mTeSR1 medium (Stemcell Technologies, Canada) on a Matrigel™ substrate (BD Biosciences, USA) as described by the manufacturer. Striatal neurons were differentiated and characterized as described previously[10]. Study was approved by local ethics committee.
Immunocytochemistry
Cells were washed 2 times with PBS (PanEco, Russia), fixed with 4% paraformaldehyde (Sigma-Aldrich, USA) for 20 min at room temperature, and washed 3 times with PBS-0.1% Tween20 (Sigma-Aldrich, USA). The non-specific absorption of antibodies was blocked by 30 min of incubation in PBS containing 0.1% Tween20, 5% FBS (HyClone, USA), 2% goat serum (HyClone, USA), and 0.1% Triton X-100 (Sigma-Aldrich, USA) at room temperature. Primary antibodies were applied at the dilutions recommended by the manufacturer in PBS containing 0.1% Tween20, 5% FBS, and 2% goat serum and were incubated for 1 hour at room temperature before being washed 3 times for 5 min in PBS-0.1% Tween20. Secondary antibodies were applied at the dilutions recommended by the manufacturer, incubated for 30 min at room temperature in the dark, washed 3 times for 5 min in PBS-0.1% Tween20, incubated with DAPI (4',6-diamino-2-feniliindol dihydrochloride) (Sigma-Aldrich, USA) 0.1 µg/ml in PBS for 10 min, and washed 2 times in PBS-0.1% Tween20. The following antibodies were used in this study: primary antibody for TOMM20 (ab56783, Abcam), TUBB3 (ab18207, Abcam), and secondary antibody goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor® 555 conjugate (A-21428, Invitrogen), goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor® 488 conjugate (A-11001, Invitrogen). Images were acquired using a Axiovert 40 CFL fluorescence microscope (Zeiss AG, Germany) or Axio Imager A1 (Zeiss AG, Germany) and were processed using the Axiovision software.
Quantitative analysis of mitochondria distribution
After fixation, cells were immunostained with anti-TUBB3 and anti-TOMM20 antibodies. Images were captured with an Axio Imager A1 (Zeiss AG) microscope with 400x magnification. Axons were detected and measured using TUBB3 staining, followed by counting the number of TOMM20 stained spots along the same axon. At least seven fields of view were analysed for each cell line. Mitochondrial density was counted per 100 µm of axonal length.
Quantitative analysis of cell nuclear morphology
Neurons for the analysis were cultured in a 48-well plate and treated with the indicated chemical compounds for 24 h before fixation, as described in[10]. We used 50 μM 2-Aminoethyl diphenylborinate (2-ABP), 2 µM ruthenium red (RR), 500 nМ of Thapsigargin, or 100 μM of 1,4-Dithiothreitol (DTT), all from Sigma-Aldrich, for the treatments. Cells were then fixed with 4% paraformaldehyde (Sigma-Aldrich, USA) for 20 min at room temperature and stained with DAPI (Sigma-Aldrich). Images of stained cells were obtained using an Axiovert 40 CFL fluorescence microscope (Zeiss AG). To evaluate the roundness of the cellular nuclei, the following equation was employed: nuclear roundness = (nuclear perimeter)2 / (4 × π × area). The nuclear roundness of wild-type (WT) neurons was taken to be 100%. Calculations were performed using self-developed software, which is available on demand.
Quantitative analysis of cell death during premature cell ageing
Cell death was evaluated as previously described [10]. Briefly, after treatment, neuronal death was measured using a MultiTox-Fluor Multiplex Cytotoxicity Assay (Promega, USA) according to the manufacturer’s instructions. Fluorescence was detected using a DTX 880 Multimode Microplate Reader (Beckman Coulter, USA). To evaluate the level of cell death (LoCD), the following equation was employed: ([cytotoxicity in a well with cells] − [cytotoxicity in a well without cells]) / ([viability in a well with cells] − [viability in a well without cells]). The LoCD of MG132-treated WT neurons was considered 100%. To screen for chemical compounds in the MG132-induced cell ageing model, the following equation was used to determine the LoCD: ([LoCD in a well with 10 μM MG132 and drug] − [LoCD in a well with drug alone] − [LoCD in a well with 10 μM MG132 alone] + [LoCD in a well without MG132 or drug]). Chemical compounds were added 24 h prior to measuring at concentrations of 50 μM for 2-APB and 2 μM for RR.
Statistical analysis
Each experiment for neurons differentiated from each cell line was repeated at least three times. Quantifiable data are given as the mean ± the standard error of the mean. Comparisons of means were performed using a one-tailed Student’s t-test or a one-tailed Welch's t-test for unequal variances. In all cases, p-values <0.05 indicated statistically significant differences between means.
Results
Previously, we found significant changes in calcium homeostasis and cell death of medium spiny neurons differentiated from HD iPSC lines[10]. However, the involvement of mitochondria in HD pathogenesis was obscured in our system because mitochondria are thought to be synthesized in the perinuclear region and must be trafficked to spines in response to cellular signals[20, 21], particularly by elevated intracellular calcium[22]. Thus, we decided to compare the number of mitochondria distributed along the length of the axons of normal and pathological neurons (mitochondrial density). To estimate the distribution of mitochondria, fixed cell cultures were stained with antibodies to microtubules (TUBB3), and mitochondria were considered to be colocalized upon detection with antibodies to the mitochondrial membrane receptor (TOMM20). We found that in control neurons, approximately 7.6 ± 0.2 mitochondria were distributed along 100 μm of neurite microtubules, whereas only 5.3 ± mitochondria were distributed in mutant neurons (p-value <0.005) (Figure 1). Thus, for the first time, a decrease in the density of mitochondria in neurites of human neurons with endogenous mutant huntingtin was demonstrated. It should be noted that neurons were differentiated in vitro for 2-3 months and that we did not observe any differences in cell death between WT and HD neurons, even though neuronal ageing made pathological neurons more susceptible to cell death[10]. To mimic cell ageing, we applied the proteasome inhibitor, MG132, to WT and HD neurons and calculated the mitochondrial density. The mitochondrial density was reduced in both WT (iPSRG2L, endo-iPS12) and HD (iPSHD11, iPSHD22) neurons: 6.5 ± 0.1 (p-value < 0.005) and 4.0 ± 0.3 (p-value < 0.005), respectively (Fig. 1). Thus, MG132 decreases the density of mitochondria in the neurites of WT and HD neurons, regardless of the presence of mutant HTT, indicating that at the periphery of the axons, the number of mitochondria can be decreased significantly. Previously, we found an increased level of nuclear indentation in human HD iPSC-derived neurons[10]. Importantly, nuclear impairment was also demonstrated in human post-mortem brain slices of HD patients[23]. Invaginations of the nuclear membrane found on neuronal cells contain a sufficiently large amount of calcium channels[24-26]. It is unclear whether the nuclear membrane impairment observed in HD neurons is due to improper calcium regulation in the nucleus or whether the cellular homeostasis of calcium and invagination of the nuclear membrane are unrelated manifestations of the HD phenotype. Cytoplasmic calcium can enter the cell nucleus via the nuclear pore complexes, but SERCA pumps may shield the nucleus against calcium entry. Ryanodine receptors and IP3 receptors are capable of amplifying nuclear calcium signals[27]. Therefore, we decided to block SERCA to investigate the effects that the calcium level has on nuclear morphology. We applied thapsigargin on WT and HD neurons. We found that thapsigargin caused nuclear invaginations in control neurons (Figure 2A) and increased the mean value of cell nuclei roundness from 100 ± 3% to 113 ± 3% (p-value <0.005). In pathological neurons, thapsigargin also increased the mean value of nuclei roundness from 123 ± 4% to 138 ± 4% (p-value <0.05) (Figure 2B). Because thapsigargin is an inducer of ER stress[28], we decided to investigate whether the increase in the nuclear roundness is due to ER stress. Pathologic and normal neurons were treated with another ER stress inducer, DTT[28]. We did not observe any changes in nuclear envelope roundness in either WT or HD neurons (Figure 2B). Therefore, nuclear invaginations may be caused by a violation of calcium homeostasis mediated by SERCA inhibition, but this pathway is not directly associated with HD pathology. The increased content of IP3R and RyR on the surface of the invaginated nuclear membranes of neurons[29] can also lead to increased calcium entry into the nucleus and morphological changes. To investigate this issue, we used 2-APB as an inhibitor of IP3R and ruthenium red (RR) as an inhibitor of RYR [30] that also inhibits MCU[31]. Neurons that were differentiated from WT and HD iPSCs were treated with 2-APB or RR, and the mean value of the nuclear roundness was analysed.
Figure 1:
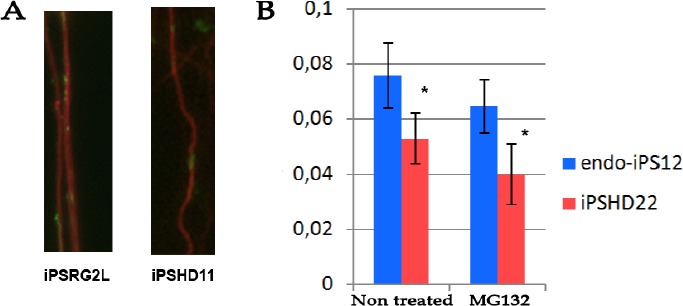
Axonal mitochondria density in wild type and HD spiny neurons differentiated from respective iPSC lines. (A) Representative immunochistochemical micrograph of axonal fragment stained with antibodies to tubulin beta-3 (TUBB3 shown in red) and mitochondrial marker TOMM20 (shown in green). (B) Number of mitochondria per 1 μm of axonal length (y axis) in neurons differentiated from WT (endo-iPS12) cell line and pathological HD iPSCs (iPSHD22) without treatment and after application of 10 μM MG132 for 24 h (p-value <0.005). Scale bar 100μm.
Figure 2:
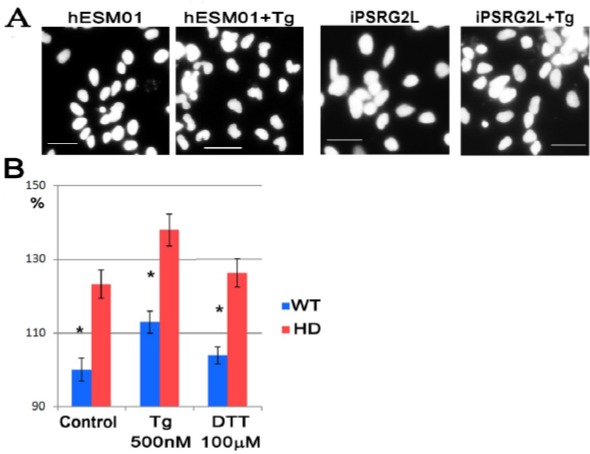
Thapsigargin induces nuclear indentations in spiny neurons independently from ER stress. (A) Representative micrograph of neuronal nuclei differentiated from WT cell line hESM01 before and after treatment with 250 μM of Tg for 24 h. Stained with DAPI, scale bar 50 μm. C shaped nuclei in Tg treated neurons are more pronounced. (B) Evaluation of nuclear roundness in not treated neurons and cell after treatment with thapsigargin (Tg) and dithiothrietol (DTT) for 24 h. WT is a mean of nuclear roundness in neurons differentiated from three control cell lines (hESM01, iPSRG2L, and endo-iPS12); HD is a mean of nuclear roundness in neurons differentiated from three pathological cell lines (iPSHD11, iPSHD22, and iPSHD34) p-value <0.05. From 2408 up to 4059 nuclei were scored in each sample.
We found that neither 2-APB nor RR had a significant influence on nucleus roundness in either WT or HD neurons (Figure 3A). Since it was previously shown that IP3R and RYR are involved in the HD pathology, including neuronal death in vitro mediated by the increased activity of these calcium channels[15, 30], we also decided to study the effect of the receptors’ inhibitors on in vitro differentiated neurons. Previously we did not observe any differences in cell death between WT and HD neurons only during ageing caused by proteasomal system inhibition the premature death of pathological neurons was detected. The SOC entry inhibitor EVP4593 rescued neurons from the death[10]. However, unlike EVP4593, neither 2-APB nor RR exhibited neuroprotective properties in our model reducing LoCD (Figure 3B). Therefore, it is unlikely that nuclear invaginations are directly connected to the intacellular calcium level or are directly involved in neurodegeneration during HD.
Figure 3:
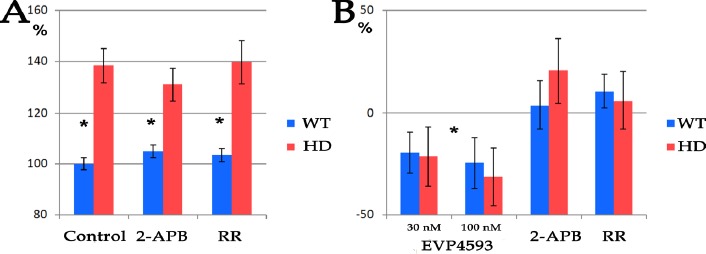
Inhibition of IP3R and RyR does not affect nuclear roundness and survival of pathological HD neurons. (A) Nuclear roundness of neurons differentiated from WT and HD iPSCs before and after 24 h treatment with 50μM 2-APB or 2 μM RR. WT is a mean of nuclear roundness in neurons differentiated from three control cell lines (hESM01, iPSRG2L, and endo-iPS12); HD is a mean of nuclear roundness in neurons differentiated from three pathological cell lines (iPSHD11, iPSHD22, and iPSHD34).From 1453 up to 2842 nuclei were scored in each sample (p-value <0.005). (B) Evaluation of IP3R and RyR inhibition on neuroprotection of pathological neurons during premature cell ageing caused by MG132 treatment. EVP4593 was used as a positive control. WT is a mean of three independent experiments performed on neurons differentiated from three control cell lines (hESM01, iPSRG2L, and endo-iPS12); HD is a mean of three independent experiments performed on neurons differentiated from three pathological cell lines (iPSHD11, iPSHD22, and iPSHD34).
Discussion
Mitochondria are the main sources of intracellular ATP and reactive oxygen species that form as a result of respiration. In addition, mitochondria actively participate in apoptosis and maintaining intracellular calcium homeostasis[32, 33]. Mitochondrial movement is dynamic in neurons and can vary between individual organelles and between axons and dendrites[34]. It is thought that an impairment of mitochondrial transport could result in inadequate distribution of ATP and sequestration of intracellular calcium. Accumulation of injured mitochondria due to inefficient removal could also have severe consequences, as damaged mitochondria may promote apoptosis by producing reactive oxygen species and releasing cytochrome c[35]. The decreased mitochondrial density observed in axons of human spiny neurons differentiated from HD patient iPSCs indicates that mitochondria trafficking was affected long before disease onset. Differentiated neurons in vitro correspond to roughly the last trimester of pregnancy and early postnatal period. Even the juvenile form of HD, with very high numbers of CAG repeats in the HTT gene, does not show clinical manifestations this early in development. There were some controversial reports on mitochondria isolated from patients or transgenic models investigating the effects of calcium concentration on mitochondria function[36-38]. However, it should be considered that isolated mitochondria are extracted from their physiological context, i.e., they have lost their native interactions with the ER and dynamics. Mitochondrial dynamics can be regulated on a genetic level. For example, Mitofusin-1 mediates mitochondrial fusion, whereas dynamin-related protein-1 controls fission. Mitochondrial fusion promotes a bioenergetically favourable state, while fission may contribute to both mitochondrial proliferation and apoptosis in neurons[39]. However, we did not find any statistically significant differences in the expression of mitochondria-associated genes between WT and HD neurons [10], thus indicating that impairment of mitochondria trafficking in HD neurons is a consequence of pathology. However, considering the significant inhibition of mitochondria trafficking that occurs during neuronal ageing, injured mitochondria may be the source of reactive oxygen species and cytochrome c and may lead to neurodegeneration, particularly in HD neurons.
The nuclear envelope is not simply a smooth-surfaced outer boundary but is interrupted by invaginations that extend within the nucleoplasm and morphologically resemble the ER[40]. Lamins are involved in nuclear reticulum proliferation[41] that could be directly connected with pathologies such as Hutchinson-Gilford progeria syndrome, Emery-Dreifuss muscular dystrophy and Dunnigan-type familial partial lipodystrophy[42,43]. However, no significant changes were found in the composition of lamins in HD neurons [10, 44]. Therefore, it is unlikely that the observed invaginations in HD are due to nuclear reticulum proliferation. The existence of nuclear reticulum continuous with ER offers a potential nuclear calcium-signalling source. Nuclear indentations contain a high calcium concentration and a range of calcium-releasing mechanisms, including the SERCA, IP3R, and RyR calcium channels[45]. In our model system of HD, inhibiting the receptors did not rescue pathological neurons from premature ageing and death. Therefore, it is unlikely that the observed nuclear indentations have a specific physiological function in these nuclear calcium-signalling pathways. This observation supports the hypothesis of the key role of the SOC entry in HD pathology. However, we cannot rule out the possibility that localized calcium release in the nucleus can elicit responses in gene expression changes that are unique and separate from those elicited by cytosolic calcium release.
Glossary
Abbreviations
- NMDA receptors
N-methyl-D-aspartate receptor
- AMPA receptors
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- TRP
Transient Receptor Potential
- PBS
Phosphate Buffered Saline
- FBS
Fetal Bovine Serum
Potential Conflicts of Interests
None
Supported by
FASO intramural research funding 0112-2016-0013 and Minobrnauki project RFMEFI60417X0184.
References
- 1.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72(6):971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65(4):267–77. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 3.Aziz NA, Jurgens CK, Landwehrmeyer GB, EHDN Registry Study Group. van Roon-Mom WM, van Ommen GJ, Stijnen T, Roos RA. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology. 2009;73(16):1280–5. doi: 10.1212/WNL.0b013e3181bd1121. [DOI] [PubMed] [Google Scholar]
- 4.Illarioshkin SN, Igarashi S, Onodera O, Markova ED, Nikolskaya NN, Tanaka H et al. Trinucleotide repeat length and rate of progression of Huntington’s disease. Ann Neurol. 1994;36(4):630–5. doi: 10.1002/ana.410360412. [DOI] [PubMed] [Google Scholar]
- 5.The HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11(2):264–78. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, Chang DJ, Kwon J, Oh SH, Shin DA, Kim HS, Do JT, Lee DR, Kim M, Kang KS, Daley GQ, Brundin P, Song J. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30(9):2054–62. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- 7.Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D. Qi X.Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest. 2013;123(12):5371–88. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11(2):253–63. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camnasio S, Delli Carri A, Lombardo A, Grad I, Mariotti C, Castucci A, Rozell B, Lo Riso P, Castiglioni V, Zuccato C, Rochon C, Takashima Y, Diaferia G, Biunno I, Gellera C, Jaconi M, Smith A, Hovatta O, Naldini L, Di Donato S, Feki A, Cattaneo E. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis. 2012;46(1):41–51. doi: 10.1016/j.nbd.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Nekrasov ED, Vigont VA, Klyushnikov SA, Lebedeva OS, Vassina EM, Bogomazova AN, Chestkov IV, Semashko TA, Kiseleva E5, Suldina LA, Bobrovsky PA, Zimina OA, Ryazantseva MA, Skopin AY, Illarioshkin SN, Kaznacheyeva EV, Lagarkova MA, Kiselev SL. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol Neurodegener. 2016;11:27–42. doi: 10.1186/s13024-016-0092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–85. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- 12.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87(2):593–58. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad V, Okunade GW, Miller ML, Shull GE. Phenotypes of SERCA and PMCA knockout mice. Biochem Biophys Res Commun. 2004;322(4):1192–203. doi: 10.1016/j.bbrc.2004.07.156. [DOI] [PubMed] [Google Scholar]
- 14.Bezprozvanny IB. Calcium signaling and neurodegeneration. Acta Naturae. 2010;2(1):72–82. [PMC free article] [PubMed] [Google Scholar]
- 15.Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinás R, Kristal BS, Hayden MR. Bezprozvanny I.. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc Natl Acad Sci. 2005;102(7):2602–7. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33(6):849–60. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem Biol. 2011;18(6):777–93. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H1, Li Q, Graham RK, Slow E, Hayden MR, Bezprozvanny I. Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington’s disease. Neurobiol Dis. 2008;31(1):80–8. doi: 10.1016/j.nbd.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, Kristal BS, Friedlander RM. Inhibitors of cytochrome c release with therapeutic potential for Huntington’s disease. J Neurosci. 2008;28(38):9473–85. doi: 10.1523/JNEUROSCI.1867-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis AF. Clayton DA In situ localization of mitochondrial DNA replication in intact mammalian cells. J Cell Biol. 1996;135(4):883–93. doi: 10.1083/jcb.135.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Okamoto K-I, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–87. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23(21):7881–8. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roos RA, Bots GT. Nuclear membrane indentations in Huntington's chorea. J Neurol Sci. 1983;61(1):37–47. doi: 10.1016/0022-510x(83)90053-9. [DOI] [PubMed] [Google Scholar]
- 24.Frotscher M, Kraft J, Zorn U. Fine structure of identified neurons in the primate hippocampus: a combined Golgi/EM study in the baboon. J Comp Neurol. 1988;275(2):254–70. doi: 10.1002/cne.902750207. [DOI] [PubMed] [Google Scholar]
- 25.Soriano E, Nitsch R, Frotscher M. Axo-axonic chandelier cells in the rat fascia dentata: Golgi-electron microscopy and immunocytochemical studies. J Comp Neurol. 1990;293(1):1–6. doi: 10.1002/cne.902930102. [DOI] [PubMed] [Google Scholar]
- 26.Kumar V, Jong YJI, O’Malley KL. Activated nuclear metabotropic glutamate receptor mGlu5 couples to nuclear Gq/11 proteins to generate inositol 1,4,5-trisphosphate-mediated nuclear Ca2+ release. J Biol Chem. 2008;283(20):14072–83. doi: 10.1074/jbc.M708551200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21(1):13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida H. ER stress and diseases. FEBS J. 2007;274(3):630–58. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 29.Malhas A, Goulbourne C, Vaux DJ. The nucleoplasmic reticulum: form and function. Trends Cell Biol. 2011;21(6):362–73. doi: 10.1016/j.tcb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki M, Nagai Y, Wada K, Koike T. Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem Biophys Res Commun. 2012;429((1-2)):18–23. doi: 10.1016/j.bbrc.2012.10.107. [DOI] [PubMed] [Google Scholar]
- 31.Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40((5-6)):553–60. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carafoli E. The fateful encounter of mitochondria with calcium: how did it happen? Biochim. Biophys. Acta. 2010;1797((6-7)):595–606. doi: 10.1016/j.bbabio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 33.Giacomello M, Drago I, Pizzo P, Pozzan T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 2007;14(7):1267–74. doi: 10.1038/sj.cdd.4402147. [DOI] [PubMed] [Google Scholar]
- 34.Ligon LA, Steward O. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol. 2000;427(3):340–50. doi: 10.1002/1096-9861(20001120)427:3<340::aid-cne2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 35.Lee HC, Wei YH. Mitochondrial role in life and death of the cell. J Biomed Sci. 2000;7(1):2–15. doi: 10.1007/BF02255913. [DOI] [PubMed] [Google Scholar]
- 36.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Cell. 2002;5(8):731–6. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 37.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13(14):1407–20. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 38.Brustovetsky N1, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s Disease. J Neurochem. 2005;93(6):1361–70. doi: 10.1111/j.1471-4159.2005.03036.x. [DOI] [PubMed] [Google Scholar]
- 39.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1(4):515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 40.Echevarría W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat Cell Biol. 2003;5(5):440–6. doi: 10.1038/ncb980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prüfert K, Vogel A, Krohne G. The lamin CxxM motif promotes nuclear membrane growth. J Cell Sci. 2004;117((Pt 25)) doi: 10.1242/jcs.01532. [DOI] [PubMed] [Google Scholar]
- 42.McClintock D, Gordon LB, Djabali K. Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc Natl Acad Sci U.S.A. 2004;103(7):2154–9. doi: 10.1073/pnas.0511133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roblek M, Schüchner S, Huber V, Ollram K, Vlcek-Vesely S, Foisner R, Wehnert M, Ogris E. Monoclonal antibodies specific for diseaseassociated point-mutants: lamin A/C R453W and R482W. PLoS ONE. 2010;5(5):e10604. doi: 10.1371/journal.pone.0010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bezprozvanny I, Kiselev SL. Neurons from skin mimic brain holes. Oncotarget. 2017;8(6):8997–8. doi: 10.18632/oncotarget.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bootman MD, Fearnley C, Smyrnias I, MacDonald F, Roderick HL. An update on nuclear calcium signalling. J Cell Sci. 2009;122((Pt 14)):2337–50. doi: 10.1242/jcs.028100. [DOI] [PubMed] [Google Scholar]