

Abstract
Aims
The aim of the present study was to quantitate the hypoglycaemic effects of dipeptidyl peptidase‐4 inhibitors (DPP‐4i), glucagon‐like peptide‐1 receptor agonists (GLP‐1r) and sodium glucose cotransporter 2 inhibitors (SGLT2i) as add‐on treatments to metformin monotherapy in patients with type 2 diabetes mellitus (T2DM) using a model‐based meta‐analysis (MBMA).
Methods
A systematic literature search of public databases was conducted to develop models that describe the time courses of the fasting plasma glucose (FPG)‐ and haemoglobin A1c (HbA1c)‐lowering effects of three antidiabetic classes using NONMEM 7.3.0.
Results
Seventy‐six publications were eligible for this study, and 873 FPG and 1086 HbA1c values were collected. We developed a physiological indirect response model that described the time courses of FPG and HbA1c and simulated reductions in these values 90 days after the initiation of add‐on treatments. FPG and HbA1c reductions with once weekly exenatide, liraglutide and dulaglutide were greater than those with other drugs. Mean changes from baseline FPG and HbA1c with these drugs were as follows: exenatide (−22.5 and −16.6%), liraglutide (−22.1 and −16.3%), and dulaglutide (−19.3 and −14.3%). The hypoglycaemic effects of DPP‐4i and SGLT2i were similar.
Conclusions
Once weekly exenatide, liraglutide and dulaglutide provided better hypoglycaemic effects among the antidiabetic drugs analysed. Long‐acting GLP‐1r appears to be more useful for T2DM patients inadequately controlled with metformin monotherapy.
Keywords: metformin, model‐based meta‐analysis, population pharmacodynamics, type 2 diabetes mellitus
What is Already Known about this Subject
Clinical practice guidelines recommend metformin as first‐line therapy; however, we sometimes encounter patients who do not respond well to this treatment. Although some classes of antidiabetic drugs have been identified as candidates for adjunctive treatments to metformin monotherapy, there is no consistent consensus regarding add‐on second‐line therapy.
What this Study Adds
The FPG‐ and HbA1c‐lowering effects of three classes of antidiabetics, DPP‐4i, GLP‐1r and SGLT2i, as add‐ons to metformin may be evaluated using an MBMA approach.
Long‐acting GLP‐1r appears to be more useful than other drugs for T2DM patients inadequately controlled with metformin monotherapy.
Introduction
Type 2 diabetes mellitus (T2DM) is characterized by a chronic hyperglycaemic state due to decreases in insulin secretion and sensitivity 1, 2. The estimated prevalence of diabetes worldwide is more than 400 million, and the total number of patients with diabetes is predicted to increase to 629 million by 2045 3. Appropriate glycaemic control based on haemoglobin A1c (HbA1c) and fasting plasma glucose (FPG) is required in order to prevent various complications, such as retinopathy, nephropathy, and neuropathy 4, 5. The American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) recommend metformin as first‐line monotherapy for most T2DM patients 6. Metformin is a biguanide that decreases blood glucose concentrations by inhibiting gluconeogenesis in the liver 7. Secondary failure may occur after long‐term metformin therapy 8, 9, 10, 11. Therefore, the ADA and EASD recommend dual therapy with metformin and other antidiabetic drugs if glycaemic control is not achieved. There is an extensive list of pharmacological therapies available for the second‐line adjunctive treatment of T2DM, including sulfonylureas (SU), thiazolidines (TZD), dipeptidyl peptidase‐4 inhibitors (DPP‐4i), glucagon‐like peptide‐1 receptor agonists (GLP‐1r), sodium glucose cotransporter 2 inhibitors (SGLT2i), and basal insulins. DPP‐4i, GLP‐1r and SGLT2i, which have novel mechanisms of action, are less likely to cause weight gain and hypoglycaemia 12, 13, 14; therefore, the use of these drugs is increasing. In Japan, the share of DPP‐4i in the total oral antidiabetic drugs market reached 69% in 2015 15. In terms of dual therapy with metformin monotherapy, since few randomized controlled trials (RCTs) have directly compared the efficacy of these drugs, a consistent consensus regarding the most appropriate drugs as add‐ons is lacking.
A model‐based meta‐analysis (MBMA) is an extension of a traditional meta‐analysis. A traditional meta‐analysis has the following limitations: (1) it may only be applicable when direct head‐to‐head RCTs exist, and (2) observation periods and doses are limited to specific ranges. In contrast, MBMA, which involves a meta‐analysis using mathematical models, has the capacity to perform indirect comparisons even though head‐to‐head RCTs are lacking. In addition, MBMA may incorporate longitudinal and dose–response data, thereby allowing for the quantification of dose–response relationships and time courses of effects. Therefore, MBMA is more flexible than a traditional meta‐analysis and is expected to provide more information 16, 17.
The aim of the present study was to develop a population pharmacodynamic (PPD) model that quantitates the FPG‐ and HbA1c‐lowering effects of DPP‐4i, GLP‐1r and SGLT2i as add‐ons to metformin monotherapy in T2DM patients using an MBMA approach.
Methods
Literature search
The ‘targeted drugs’ in the present study included DPP‐4i (sitagliptin, vildagliptin, alogliptin, linagliptin, teneligliptin, anagliptin, saxagliptin, trelagliptin and omarigliptin), GLP‐1r (liraglutide, exenatide, lixisenatide and dulaglutide), and SGLT2i (ipragliflozin, dapagliflozin, luseogliflozin, tofogliflozin, canagliflozin and empagliflozin), which are all approved in Japan. A systematic literature search of PubMed, the Cochrane library (CENTRAL/CCTR: Cochrane Central Register of Controlled Trials), and ClinicalTrials.gov (https://clinicaltrials.gov/) was conducted on 3 March 2016. The words used in the search were (‘metformin’ OR ‘targeted drugs’) AND (‘diabetes’ OR ‘diabetic’). Details of the search terms are provided in Supplementary Table S1. Only clinical trials satisfying the following inclusion criteria were included in the analysis: (1) randomized double‐blind clinical trials, (2) patients diagnosed with T2DM, (3) targeted drugs added to metformin monotherapy because of inadequate glycaemic control, (4) HbA1c or FPG values used as clinical indicators, and (5) published in English. We excluded trials focusing on specific populations, such as renal failure and paediatric subjects. MBMA was conducted according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) 18.
We extracted the following information from eligible studies: mean and median values of HbA1c and FPG at each time point, sample size, dosage, duration of T2DM, duration of metformin therapy, age, sex, race (e.g., Caucasians, Asians, and others), body mass index (BMI) and body weight. Graphical data were converted to numerical data using GetData Graph Digitizer® version 2.26 (http://getdata‐graph‐digitizer.com).
Model development
A PPD analysis was performed using NONMEM 7.3.0 (Icon Development Solutions, Ellicott City, Maryland) with first‐order conditional estimation with interaction method (FOCE‐INTER). Graphical processing of the NONMEM output was performed with R (version 3.3.2).
In the present study, a physiological indirect response model was established to describe the time courses of FPG and HbA1c 19. Overall model structures for FPG and HbA1c are shown as follows:
FPG (DPP‐4i and GLP‐1r):
FPG (SGLT2i):
HbA1c:
where Kin,FPG and Kout,FPG are the FPG production rate constant and FPG elimination rate constant, respectively. Changes in HbA1c were modelled as secondary changes dependent on the baseline ratio of FPG (FPG/BaselineFPG), with the HbA1c production rate constant (Kin,HbA1c) and HbA1c elimination rate constant (Kout,HbA1c). The description of HbA1c production also included the use of the power function λ 20, 21, 22. Kin,HbA1c is defined by Kin,HbA1c = BaselineHbA1c × Kout,HbA1c. BaselineFPG and BaselineHbA1c represent FPG and HbA1c levels before the initiation of dual therapies, respectively. BaselineFPG and BaselineHbA1c in the x th biomarker of the j th arm of the i th study are given by:
where TVBx is the estimated typical baseline FPG and HbA1c. ηB and κ are the random effects of inter‐study variabilities (ISV) and inter‐arm variabilities (IAV) 23, respectively. ISV and IAV were assumed to be symmetrically distributed as random variables with mean zero and variance ωISV 2 and ωIAV 2. IAV was weighted by the inverse of the square root of the number of patients in the study arm (Nij) normalized to 100 patients. The reason why IAV was included in this analysis was that IAV is purely the product of a small sample size, because in a randomized trial with an infinite sample size, there are no random differences across arms. Disease progression for FPG was assumed to be a proportional increase with a slope parameter (DPFPG) relative to the baseline. An exponential error model was used to describe ISV on DPFPG. The placebo effect to FPG levels (Eplacebo) was described by a constant model. An exponential error model was used to describe ISV on Eplacebo. The drug effect (Edrug) to FPG levels was as follows:
where Emax is the maximum treatment effect ranging between 0 and 1; ED50 is the dose resulting in 50% of Emax. The model included the individual potency (ED50) of each drug, but assumed the same Emax for drugs with the same mechanism of action. The drug effect was assumed to be constant across studies, i.e., ISV on Edrug was not estimated. These models indicate that DPP‐4i and GLP‐1r inhibit FPG production, and SGLT2i stimulates FPG elimination. An additive error model was used to describe residual error variability (RUV). RUV was weighted by the inverse of the square root of the number of patients in the study arm normalized to 100 patients. Ideally for mean data, residual variability needs to be weighted by the precision of the mean (the inverse of squared standard errors). However, since we did not obtain standard errors in many studies, residuals were weighted by the sample size.
After establishing the basic models, covariate modelling was conducted. Age, sex, race, BMI and body weight were selected as candidates for the covariate. Covariate selection was conducted based on clinical plausibility and differences in the objective function value (OFV) estimated by NONMEM between hierarchical models. Forward inclusion and backward exclusion were used to develop the covariate model. Significance levels for forward inclusion and backward exclusion were set at 0.01 and 0.001, respectively.
Model validation
During model building, changes in OFV, Akaike information criterion (AIC), relative standard errors and goodness‐of‐fit (GOF) plots were used for model evaluation. GOF was investigated using plots of the observation vs. population prediction (PRED) and individual predictions (IPRED), conditional‐weighted residuals (CWRES) vs. the treatment duration 24, CWRES vs. PRED, and absolute individual weighted residuals (IWRES) vs. IPRED. In order to assess the robustness of the final PD model, a prediction‐corrected visual predictive check (pcVPC) was conducted. An 80% prediction interval (PI) was defined for pcVPC from the 10th and 90th percentiles of simulated dependent data at each time point and was then compared with original data. One thousand simulations were performed for pcVPC. pcVPC was performed with the software package Perl‐speaks‐NONMEM version 4.8.1.
Simulation
Using the final models, we simulated reductions in FPG and HbA1c 90 days after the initiation of add‐on therapy. The dosage was set to the recommended dose of each drug in Japan. Parameter uncertainty, obtained from the variance–covariance matrix of the final model, was implemented in the simulations. The typical time courses of FPG and HbA1c for the three drugs (vildagliptin, exenatide and canagliflozin), which were selected from each drug class, were simulated.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 25, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 26, 27, 28.
Results
Data analysis
A total of 2397 publications were found in the initial literature search. After screening and eligibility evaluations, 76 studies (31 585 patients) were eligible for the analysis, including 55 studies on DPP‐4i (eight drugs), 10 on GLP‐1r (four drugs), and 18 on SGLT2i (four drugs). However, some targeted drugs (e.g., trelagliptin, luseogliflozin and tofogliflozin) were not included in the analysis because trial data were not available. Detailed literature search results are shown in the PRISMA flow diagram (Figure S1), and a study design summary is provided in Supplementary Table S2. The total numbers of FPG and HbA1c were 873 and 1086, respectively. The medians (ranges) for the baseline values of FPG and HbA1c were 165 mg dl−1 (138–244 mg dl−1) and 8.0% (7.0–9.3%), respectively. The characteristics of each targeted drug are summarized in Table 1.
Table 1.
Study characteristics for each drug
| Drug | Number of trials | Dose (mg) | Number of points | Age a (year) | Male a (%) | Caucasian a (%) | BMI a (kg m −2 ) | HbA1c a (%) | FPG a (mg dl −1 ) | Weight a (kg) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FPG | HbA1c | ||||||||||
| Placebo | 50 | 0 | 250 | 274 | 55.9 | 53.4 | 72.7 | 31.1 | 8.0 | 165 | 87.4 |
| DPP‐4 inhibitors | |||||||||||
| Sitagliptin | 24 | 50/100 | 132 | 134 | 55.1 | 54.1 | 57.6 | 30.9 | 8.0 | 160 | 86.7 |
| Vildagliptin | 10 | 50/100 | 52 | 74 | 55.8 | 52.8 | 76.5 | 30.3 | 8.1 | 173 | 81.8 |
| Alogliptin | 4 | 12.5/25 | 36 | 64 | 53.9 | 52.7 | 63.0 | 31.3 | 7.9 | 177 | 83.9 |
| Linagliptin | 6 | 1/5/10 | 26 | 48 | 58.7 | 54.0 | 75.1 | 30.2 | 8.0 | 166 | 85.0 |
| Teneligliptin | 1 | 20 | 5 | 5 | 55.7 | 55.1 | 0 | NA | 7.8 | 151 | NA |
| Anagliptin | 1 | 200 | 3 | 9 | 56.8 | 51.7 | 0 | 24.9 | 7.7 | 146 | 66.1 |
| Saxagliptin | 10 | 2.5/5/10 | 47 | 73 | 54.7 | 53.0 | 81.0 | 31.4 | 8.0 | 164 | 87.8 |
| Omarigliptin | 2 | 25 | 4 | 4 | 57.5 | 50.5 | NA | NA | 7.5 | 158 | 87.5 |
| GLP‐1 receptor agonists | |||||||||||
| Liraglutide | 2 | 0.5/0.6/1/1.2/ 1.5/1.8/2 | 11 | 15 | 53.5 | 54.0 | 0 | 25.9 | 8.6 | 177 | 68.4 |
| Exenatide | 3 | 0.01/0.02/2 | 18 | 30 | 52.7 | 53.7 | 78.5 | 33.0 | 8.2 | 167 | 94.6 |
| Lixisenatide | 4 | 0.005/0.01/0.02 /0.03/0.04/0.06 | 38 | 60 | 55.4 | 44.7 | 81.8 | 32.3 | 7.6 | 162 | 89.0 |
| Dulaglutide | 1 | 0.75/1.5 | 24 | 22 | 54.0 | 46.2 | 52.6 | 31.3 | 8.2 | 175 | 86.4 |
| SGLT2 inhibitors | |||||||||||
| Ipragliflozin | 2 | 12.5/50/150/300 | 27 | 27 | 56.6 | 50.0 | 91.3 | 31.8 | 7.8 | 157 | 89.3 |
| Dapagliflozin | 6 | 2.5/5/9.2/10 | 93 | 105 | 55.0 | 50.4 | 81.8 | 31.6 | 7.9 | 161 | 86.3 |
| Canagliflozin | 5 | 50/100/150/200/300 | 60 | 62 | 55.3 | 50.8 | 68.6 | 31.6 | 7.8 | 164 | 87.7 |
| Empagliflozin | 5 | 1/5/10/25/50 | 47 | 80 | 57.3 | 55.3 | 84.1 | 31.5 | 7.9 | 162 | 88.1 |
Values are given as medians.
BMI, body mass index; DPP‐4, dipeptidyl peptidase‐4; FPG, fasting plasma glucose; GLP‐1, glucagon‐like peptide‐1; HbA1c, haemoglobin A1c; NA, not applicable; SGLT2, sodium glucose cotransporter 2
PPD models
Figure 1 shows a constructed indirect response model that describes changes in FPG and HbA1c levels over time for all treatments. Table 2 shows the population PPD parameter estimates of FPG and Hb1Ac. Estimated typical BaselineFPG, Kout,FPG, Eplacebo, and DPFPG were 165 mg dl−1, 0.0936/day, 0.0168, and 0.0204/year, respectively. The dose–response relationship for each drug was characterized by the Emax model with a different Emax for each drug class and a different ED50 for every drug within each class. ED50 for teneligliptin was fixed to 0 in the final model because the estimate was close to 0, leading to convergence difficulties. The relationship between FPG and HbA1c was nonlinear and described by a power function with a different λ for the placebo and each drug class. Estimated typical BaselineHbA1c and Kout,HbAc were 7.96% and 0.0393/day, respectively. None of the covariates were found to significantly improve the PPD model.
Figure 1.
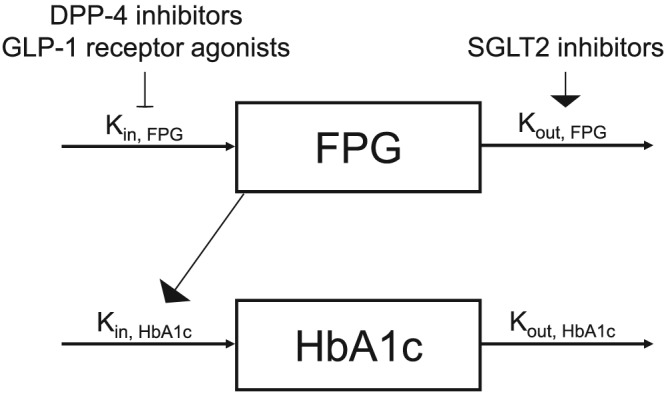
The final population pharmacodynamic model describing the time course of fasting plasma glucose (FPG) and haemoglobin A1c (HbA1c)
DPP‐4, dipeptidyl peptidase‐4; GLP‐1, glucagon‐like peptide‐1; Kin,FPG, FPG production rate constant; Kin,HbA1c, HbA1c production rate constant; Kout,FPG, FPG elimination rate constant; Kout,HbA1c; HbA1c elimination rate constant; SGLT2, sodium glucose cotransporter 2
Table 2.
Population PPD parameter estimates
| Parameter | Mean | RSE (%) |
|---|---|---|
| Baseline FPG (mg dl −1 ) | 165 | 1.0 |
| K out, FPG (/day) | 0.0936 | 33.9 |
| Baseline HbA1c (%) | 7.96 | 0.6 |
| K out, HbA1c (/day) | 0.0393 | 14.3 |
| E placebo | 0.0168 | 23.2 |
| λ placebo | 0.893 | 15.0 |
| DP FPG (/year) | 0.0204 | 17.9 |
| DPP‐4 inhibitors | ||
| E max, DPP‐4i | 0.116 | 5.2 |
| ED 50, sitagliptin (mg day −1 ) | 11.0 | 41.9 |
| ED 50, vildagliptin (mg day −1 ) | 9.51 | 55.6 |
| ED 50, alogliptin (mg day −1 ) | 2.36 | 52.1 |
| ED 50, linagliptin (mg day −1 ) | 1.80 | 26.7 |
| ED 50, teneligliptin (mg day −1 ) | 0 fixed | – |
| ED 50, anagliptin (mg day −1 ) | 31.4 | 33.8 |
| ED 50, saxagliptin (mg day −1 ) | 1.47 | 48.2 |
| ED 50, omarigliptin (mg day −1 ) | 3.15 | 66.7 |
| λ DPP‐4i | 0.831 | 3.7 |
| GLP‐1 receptor agonists | ||
| E max, GLP‐1r | 0.266 | 6.4 |
| ED 50, liraglutide (mg day −1 ) | 0.377 | 34.7 |
| ED 50, exenatide BID (mg day −1 ) | 0.0120 | 12.7 |
| ED 50 , exenatide QW (mg week −1 ) | 0.498 | 9.2 |
| ED 50, lixisenatide (mg day −1 ) | 0.0392 | 21.8 |
| ED 50, dulaglutide (mg week −1 ) | 0.328 | 5.4 |
| λ GLP‐1r | 0.777 | 8.7 |
| SGLT2 inhibitors | ||
| E max, SGLT2i | 0.199 | 13.6 |
| ED 50, ipragliflozin (mg day −1 ) | 14.3 | 52.5 |
| ED 50, dapagliflozin (mg day −1 ) | 4.06 | 35.5 |
| ED 50, canagliflozin (mg day −1 ) | 30.0 | 28.1 |
| ED 50, empagliflozin (mg day −1 ) | 4.54 | 62.3 |
| λ SGLT2i | 0.654 | 5.2 |
| Inter‐study variability and inter‐arm variability | ||
| ISV on Baseline FPG (%) | 8.4 | 13.2 |
| ISV on K out, FPG (%) | 105.4 | 19.0 |
| ISV on Baseline HbA1c (%) | 4.4 | 10.1 |
| ISV on K out, HbA1c (%) | 48.3 | 67.6 |
| ISV on E placebo (%) | 113.1 | 16.6 |
| ISV on DP FPG (%) | 92.1 | 22.4 |
| IAV on Baseline FPG (%) | 2.6 | 10.7 |
| IAV on Baseline HbA1c (%) | 1.2 | 15.8 |
| Residual error variability | ||
| Additive error FPG (mg dl −1 ) | 3.35 | 4.4 |
| Additive error HbA1c (%) | 0.0721 | 9.3 |
DPFPG, the coefficient of disease progression; DPP‐4i, dipeptidyl peptidase‐4 inhibitors; ED50, dose resulting in 50% of Emax; Emax, maximum drug effect; Eplacebo, placebo effect; FPG, fasting plasma glucose; GLP‐1r, glucagon‐like peptide‐1 receptor agonists; IAV, inter‐arm variability; ISV, inter‐study variability; Kout, FPG, FPG elimination rate constant; Kout, HbA1c, HbA1c elimination rate constant; RSE, relative standard error; SGLT2i, sodium glucose cotransporter 2 inhibitors
Model validation
GOF plots show the high predictive performance of the constructed models, and systematic deviations were not observed (Figure S2). pcVPCs for each drug class are shown in Figure 2. These models captured most of the observed data, indicating the good predictive performance of the models. These results suggest that the final models adequately describe the time courses for the FPG‐ and HbA1c‐lowering effects of the targeted drugs.
Figure 2.
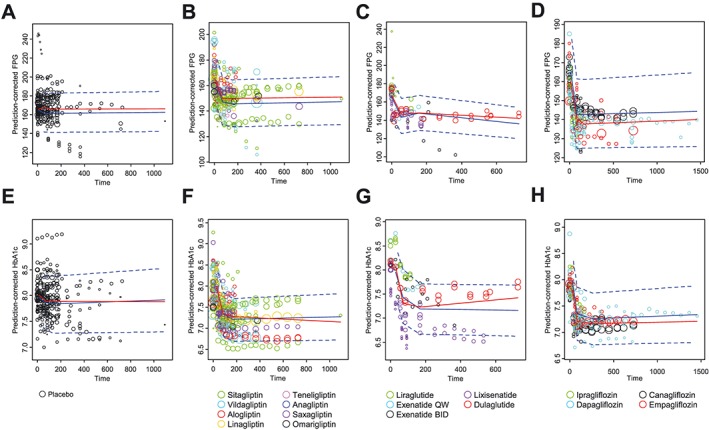
Prediction‐corrected visual predictive check plots for fasting plasma glucose (FPG) of the placebo (A), dipeptidyl peptidase‐4 inhibitors (DPP‐4i) (B), glucagon‐like peptide‐1 receptor agonists (GLP‐1r) (C), and sodium glucose cotransporter 2 inhibitors (SGLT2i) (D) as well as haemoglobin A1c (HbA1c) of the placebo (E), DPP‐4i (F), GLP‐1r (G) and SGLT2i (H). Red solid lines represent the observed median. Blue solid and dashed lines represent the predicted median and 80% prediction intervals, respectively. Open circles represent observed data, and the symbol size is proportional to the number of subjects in each study
QW, once weekly; BID, twice daily
Simulation
Based on the final models, we simulated reductions in FPG and HbA1c 90 days after the initiation of add‐on therapy (Figure 3). The dosage was set to the recommended dose of each drug in Japan. Parameter uncertainty was implemented in the simulations because some parameters (e.g., ED50 for omarigliptin and empagliflozin, Table 2) were estimated with poor precision. Among these drugs, GLP‐1r (exenatide QW, liraglutide and dulaglutide) showed superior FPG‐ and HbA1c‐lowering effects (−22.2, −19.7 and −19.4% for FPG, and −16.8, −14.7 and −14.7% for HbA1c, respectively). FPG‐ and HbA1c‐lowering effects were similar between DPP‐4i and SGLT2i. Median reductions in FPG were −10.9 to −14.4% for SGLT2i and −9.6 to −12.6% for DPP‐4i. Median reductions in HbA1c were −6.9 to −9.2% for SGLT2i and −7.6 to −10.0% for DPP‐4i. The typical time courses of FPG and HbA1c of the three drugs, which were selected from each drug class, were simulated and are shown in Figure 4.
Figure 3.
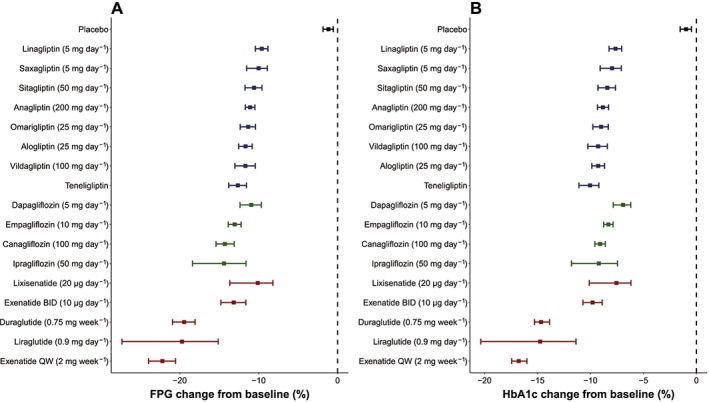
Reductions in FPG (A) and HbA1c (B) 90 days after the initiation of add‐on therapy. Each square and bar represent the median and 90% confidence interval from model simulation (n = 1000) for each drug. Red, green, and blue squares and bars represent changes from the baseline in FPG for GLP‐1r, SGLT2 and DPP‐4, respectively. The dosage was set to the recommended dose of each drug in Japan
QW, once weekly; BID, twice daily
Figure 4.
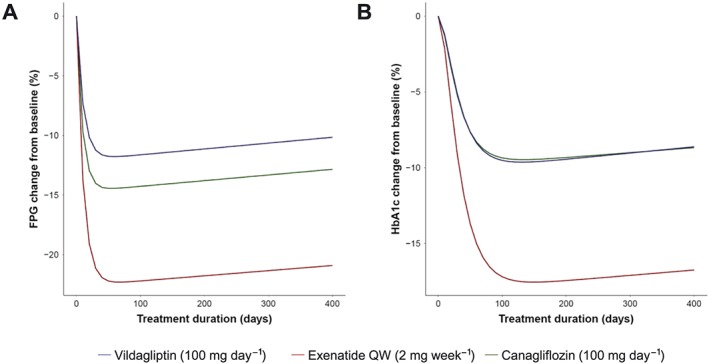
Typical time courses of fasting plasma glucose (FPG) (A) and haemoglobin A1c (HbA1c) (B)
Discussion
The aim of the present study was to quantitate the hypoglycaemic effects of DPP‐4i, GLP‐1r and SGLT2i as add‐on treatments to metformin monotherapy in T2DM patients using MBMA.
We demonstrated that GLP‐1r was associated with greater reductions in FPG and HbA1c than the other treatments tested within the approved dosages in Japan (Figure 3). The significant superiority of GLP‐1r to DPP‐4i as add‐on therapy to metformin has been suggested in most RCTs. For example, exenatide (2 mg week−1) resulted in significantly greater improvements in HbA1c than sitagliptin (100 mg day−1) in the DURATION‐2 study 29. Furthermore, the network meta‐analysis (NMA) conducted by Zintzaras et al. indicated a higher proportion achieving the HbA1c goal with GLP‐1r than other combination therapies with metformin 30. The DURATION‐8 study, which compared the efficacy and safety of exenatide (2 mg week−1) vs. dapagliflozin (10 mg day−1), showed that reductions in HbA1c at week 12 were greater in patients given exenatide 31.
In comparisons between long‐ and short‐acting GLP‐1r, treatments with long‐acting GLP‐1r (i.e., exenatide QW, dulaglutide and liraglutide) have been associated with greater reductions in FPG and HbA1c (Figure 3); long‐acting GLP‐1r provide relatively stable drug concentration–time profiles in the long term, leading to stable glycaemic control 32, 33, 34. The NMA conducted by Kayaniyil et al. showed that the administration of exenatide QW led to a slightly higher proportion of patients achieving the glycaemic target than exenatide BID and lixisenatide 35.
The present study developed a PPD model that combined with the physiological relationship between FPG and HbA1c. During the model building process, we combined the mechanism of action of each drug class into the model: DPP‐4i and GLP‐1r inhibit FPG production, and SGLT2i stimulates FPG elimination. A large number of physiological models have been developed to describe the relationship between FPG and HbA1c. Our PPD parameters were similar to those reported previously 21, 22. The FPG progression rate was estimated to be 2.04%/year and was similar to that reported by Stringer et al. (1.7%/year) 22. In the present study, disease progression for FPG was assumed to be a proportional increase with DPFPG. Several different disease progression models have been investigated (e.g., log‐linear and exponential), but were not found to be superior.
The relationship between FPG and HbA1c was found to be nonlinear and was described by a power function with a different λ for the placebo and each drug class. Our estimated λ was similar to values reported in previous studies (0.74 and 0.71) 20, 21. The nonlinear relationship between FPG and HbA1c may result from the contribution of postprandial glucose because the value for HbA1c is the result of FPG and postprandial glucose 36. In addition, previous studies demonstrated that mean plasma glucose (the arithmetic mean of FPG and postprandial glucose) correlated better with HbA1c than FPG alone 37, 38.
This MBMA has several limitations. For example, covariate information obtained from the literature was limited. Some information, such as the metformin dose and durations of T2DM and metformin monotherapy, was not consistently reported; therefore, we were unable to include these as candidates for the covariate analysis. Since this information may contribute to patient heterogeneity, the results of the covariate analysis need to be interpreted with caution.
In conclusion, this MBMA quantified the hypoglycaemic effects of DPP‐4i, GLP‐1r and SGLT2i when they were added to metformin monotherapy. The simulations based on PPD models suggested that long‐acting GLP‐1r (i.e., exenatide QW, liraglutide and dulaglutide) were more effective than other drugs for T2DM patients inadequately controlled with metformin monotherapy.
Competing Interests
There are no competing interests to declare.
Contributors
H.I., Y.K., T.H., and I.I. wrote the manuscript. H.I., Y.T., Y.K., S.M., M.K., T.H., and I.I. designed the research. H.I., Y.T., and Y.K. analysed the data. The authors confirm that the PI for this paper is Ichiro Ieiri and that he had direct clinical responsibility for patients.
Supporting information
Table S1 Name of drugs used in the systematic literature search
Table S2 Characteristics of studies included in the model‐based meta‐analysis dataset
Figure S1 Results of the literature search
CENTRAL/CCTR, Central Register of Controlled Trials; DPP‐4i, dipeptidyl peptidase‐4 inhibitors; FPG, fasting plasma glucose; GLP‐1r, glucagon‐like peptide‐1 receptor agonists; HbA1c, haemoglobin A1c; SGLT2i, sodium glucose cotransporter 2 inhibitors
Figure S2 Goodness‐of‐fit plots of the final pharmacodynamic model for fasting plasma glucose (FPG) (A–E) and haemoglobin A1c (HbA1c) (F–J)
Population predictions were made using population mean parameters. Individual predictions were obtained using individual empirical Bayesian estimated parameters. Delta (Δ) is defined as the difference between the baseline values and observed or predicted values of FPG (A–D) and HbA1c (F–I). Black lines represent the line of identify (A, B, F and G) and y = 0 (C, D, E, H, I and J). Red lines represent spline curves. DPP‐4, dipeptidyl peptidase‐4; GLP‐1, glucagon‐like peptide‐1; SGLT2, sodium glucose cotransporter 2
Inoue, H. , Tamaki, Y. , Kashihara, Y. , Muraki, S. , Kakara, M. , Hirota, T. , and Ieiri, I. (2019) Efficacy of DPP‐4 inhibitors, GLP‐1 analogues, and SGLT2 inhibitors as add‐ons to metformin monotherapy in T2DM patients: a model‐based meta‐analysis. Br J Clin Pharmacol, 85: 393–402. 10.1111/bcp.13807.
References
- 1. Seltzer HS, Allen EW, Herron AL, Brennan MT. Insulin secretion in response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. J Clin Invest 1967; 46: 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tabák AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimäki M, Witte DR. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 2009; 373: 2215–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. International Diabetes Federation . IDF Diabetes Atlas, 8th edn. Brussels, Belgium: International Diabetes Federation, 2017. Available at http://www.diabetesatlas.org (last accessed 21 November 2017). [Google Scholar]
- 4. The International Expert Committee . International expert committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 2009; 32: 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long‐term results of the Kumamoto study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23 (Suppl. 2): B21–B29. [PubMed] [Google Scholar]
- 6. Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al Management of hyperglycemia in type 2 diabetes, 2015: a patient‐centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38: 140–149. [DOI] [PubMed] [Google Scholar]
- 7. Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non‐insulin‐dependent diabetes mellitus. N Engl J Med 1995; 333: 550–554. [DOI] [PubMed] [Google Scholar]
- 8. Brown JB, Conner C, Nichols GA. Secondary failure of metformin monotherapy in clinical practice. Diabetes Care 2010; 33: 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eurich DT, Simpson SH, Majumdar SR, Johnson JA. Secondary failure rates associated with metformin and sulfonylurea therapy for type 2 diabetes mellitus. Pharmacotherapy 2005; 25: 810–816. [DOI] [PubMed] [Google Scholar]
- 10. Boccuzzi SJ, Wogen J, Fox J, Sung JCY, Shah AB, Kim J. Utilization of oral hypoglycemic agents in a drug‐insured US population. Diabetes Care 2001; 24: 1411–1415. [DOI] [PubMed] [Google Scholar]
- 11. Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 2006; 355: 2427–2443. [DOI] [PubMed] [Google Scholar]
- 12. Raz I, Hanefeld M, Xu L, Caria C, Williams‐Herman D, Khatami H, et al Efficacy and safety of the dipeptidyl peptidase‐4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49: 2564–2571. [DOI] [PubMed] [Google Scholar]
- 13. Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6‐week course of glucagon‐like peptide 1 on glycaemic control, insulin sensitivity, and beta‐cell function in type 2 diabetes: a parallel‐group study. Lancet 2002; 359: 824–830. [DOI] [PubMed] [Google Scholar]
- 14. Cefalu WT, Stenlöf K, Leiter LA, Wilding JPH, Blonde L, Polidori D, et al Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia 2015; 58: 1183–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. 5Tanabe M, Motonaga R, Terawaki Y, Nomiyama T, Yanase T. Prescription of oral hypoglycemic agents for patients with type 2 diabetes mellitus: a retrospective cohort study using a Japanese hospital database. J Diabetes Investig 2017; 8: 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mandema JW, Gibbs M, Boyd RA, Wada DR, Pfister M. Model‐based meta‐analysis for comparative efficacy and safety: application in drug development and beyond. Clin Pharmacol Ther 2011; 90: 766–769. [DOI] [PubMed] [Google Scholar]
- 17. Mould DR. Model‐based meta‐analysis: an important tool for making quantitative decisions during drug development. Clin Pharmacol Ther 2012; 92: 283–286. [DOI] [PubMed] [Google Scholar]
- 18. Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group . Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. PLoS Med 2009; 6: e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jusko WJ, Ko HC. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin Pharmacol Ther 1994; 56: 406–419. [DOI] [PubMed] [Google Scholar]
- 20. Hamrén B, Björk E, Sunzel M, Karlsson MO. Models for plasma glucose, HbA1c, and hemoglobin interrelationships in patients with type 2 diabetes following Tesaglitazar treatment. Clin Pharmacol Ther 2008; 84: 228–235. [DOI] [PubMed] [Google Scholar]
- 21. Stringer F, DeJongh J, Scott G, Danhof M. A model‐based approach to analyze the influence of UGT2B15 polymorphism driven pharmacokinetic differences on the pharmacodynamic response of the PPAR agonist sipoglitazar. J Clin Pharmacol 2014; 54: 453–461. [DOI] [PubMed] [Google Scholar]
- 22. Stringer F, DeJongh J, Enya K, Koumura E, Danhof M, Kaku K. Evaluation of the long‐term durability and glycemic control of fasting plasma glucose and glycosylated hemoglobin for pioglitazone in Japanese patients with type 2 diabetes. Diabetes Technol Ther 2015; 17: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahn JE, French JL. Longitudinal aggregate data model‐based meta‐analysis with NONMEM: approaches to handling within treatment arm correlation. J Pharmacokinet Pharmacodyn 2010; 37: 179–201. [DOI] [PubMed] [Google Scholar]
- 24. Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res 2007; 24: 2187–2197. [DOI] [PubMed] [Google Scholar]
- 25. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, et al The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 2017; 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bergenstal RM, Wysham C, Macconell L, Malloy J, Walsh B, Yan P, et al Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION‐2): a randomised trial. Lancet 2010; 376: 431–439. [DOI] [PubMed] [Google Scholar]
- 30. Zintzaras E, Miligkos M, Ziakas P, Balk EM, Mademtzoglou D, Doxani C, et al Assessment of the relative effectiveness and tolerability of treatments of type 2 diabetes mellitus: a network meta‐analysis. Clin Ther 2014; 36: 1443–1453. [DOI] [PubMed] [Google Scholar]
- 31. Frías JP, Guja C, Hardy E, Ahmed A, Dong F, Öhman P, et al Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION‐8): a 28 week, multicentre, double‐blind, phase 3, randomised control. Lancet Diabetes Endocrinol 2016; 4: 1004–1016. [DOI] [PubMed] [Google Scholar]
- 32. DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of exenatide in poly‐(d,l‐lactide‐co‐glycolide) microspheres produced an investigational long‐acting once‐weekly formulation for type 2 diabetes. Diabetes Technol Ther 2011; 13: 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Glaesner W, Vick AM, Millican R, Ellis B, Tschang S‐H, Tian Y, et al Engineering and characterization of the long‐acting glucagon‐like peptide‐1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev 2010; 26: 287–296. [DOI] [PubMed] [Google Scholar]
- 34. Lund A, Knop FK, Vilsbøll T. Glucagon‐like peptide‐1 receptor agonists for the treatment of type 2 diabetes: differences and similarities. Eur J Intern Med 2014; 25: 407–414. [DOI] [PubMed] [Google Scholar]
- 35. Kayaniyil S, Lozano‐Ortega G, Bennett HA, Johnsson K, Shaunik A, Grandy S, et al A network meta‐analysis comparing exenatide once weekly with other GLP‐1 receptor agonists for the treatment of type 2 diabetes mellitus. Diabetes Ther 2015; 7: 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA(1c). Diabetes Care 2003; 26: 881–885. [DOI] [PubMed] [Google Scholar]
- 37. Lledó‐García R, Mazer NA, Karlsson MO. A semi‐mechanistic model of the relationship between average glucose and HbA1c in healthy and diabetic subjects. J Pharmacokinet Pharmacodyn 2013; 40: 129–142. [DOI] [PubMed] [Google Scholar]
- 38. Ozmen S, Cil T, Atay AE, Tuzcu AK, Bahceci M. A simple way to estimate mean plasma glucose and to identify type 2 diabetic subjects with poor glycaemic control when a standardized HbA1c assay is not available. Diabet Med 2006; 23: 1151–1154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Name of drugs used in the systematic literature search
Table S2 Characteristics of studies included in the model‐based meta‐analysis dataset
Figure S1 Results of the literature search
CENTRAL/CCTR, Central Register of Controlled Trials; DPP‐4i, dipeptidyl peptidase‐4 inhibitors; FPG, fasting plasma glucose; GLP‐1r, glucagon‐like peptide‐1 receptor agonists; HbA1c, haemoglobin A1c; SGLT2i, sodium glucose cotransporter 2 inhibitors
Figure S2 Goodness‐of‐fit plots of the final pharmacodynamic model for fasting plasma glucose (FPG) (A–E) and haemoglobin A1c (HbA1c) (F–J)
Population predictions were made using population mean parameters. Individual predictions were obtained using individual empirical Bayesian estimated parameters. Delta (Δ) is defined as the difference between the baseline values and observed or predicted values of FPG (A–D) and HbA1c (F–I). Black lines represent the line of identify (A, B, F and G) and y = 0 (C, D, E, H, I and J). Red lines represent spline curves. DPP‐4, dipeptidyl peptidase‐4; GLP‐1, glucagon‐like peptide‐1; SGLT2, sodium glucose cotransporter 2