

Primary VZV infection results in the widespread dissemination of the virus throughout the host. Viral transportation is known to be directly influenced by susceptible immune cells in the circulation. Moreover, infection of immune cells by VZV results in attenuation of the antiviral mechanisms used to control infection and limit spread. Here, we provide evidence that human monocytes, which are highly abundant in the circulation, are permissive to productive VZV infection. Furthermore, monocyte-derived macrophages were also highly permissive to VZV infection, although VZV-infected monocytes were unable to differentiate into macrophages. Exploring the relationships between VZV and permissive immune cells, such as human monocytes and macrophages, elucidates novel immune evasion strategies and provides further insight into the control that VZV has over the immune system.
KEYWORDS: varicella-zoster virus
ABSTRACT
Varicella-zoster virus (VZV) is associated with viremia during primary infection that is presumed to stem from infection of circulating immune cells. While VZV has been shown to be capable of infecting a number of different subsets of circulating immune cells, such as T cells, dendritic cells, and NK cells, less is known about the interaction between VZV and monocytes. Here, we demonstrate that blood-derived human monocytes are permissive to VZV replication in vitro. VZV-infected monocytes exhibited each temporal class of VZV gene expression, as evidenced by immunofluorescent staining. VZV virions were observed on the cell surface and viral nucleocapsids were observed in the nucleus of VZV-infected monocytes by scanning electron microscopy. In addition, VZV-infected monocytes were able to transfer infectious virus to human fibroblasts. Infected monocytes displayed impaired dextran-mediated endocytosis, and cell surface immunophenotyping revealed the downregulation of CD14, HLA-DR, CD11b, and the macrophage colony-stimulating factor (M-CSF) receptor. Analysis of the impact of VZV infection on M-CSF-stimulated monocyte-to-macrophage differentiation demonstrated the loss of cell viability, indicating that VZV-infected monocytes were unable to differentiate into viable macrophages. In contrast, macrophages differentiated from monocytes prior to exposure to VZV were highly permissive to infection. This study defines the permissiveness of these myeloid cell types to productive VZV infection and identifies the functional impairment of VZV-infected monocytes.
IMPORTANCE Primary VZV infection results in the widespread dissemination of the virus throughout the host. Viral transportation is known to be directly influenced by susceptible immune cells in the circulation. Moreover, infection of immune cells by VZV results in attenuation of the antiviral mechanisms used to control infection and limit spread. Here, we provide evidence that human monocytes, which are highly abundant in the circulation, are permissive to productive VZV infection. Furthermore, monocyte-derived macrophages were also highly permissive to VZV infection, although VZV-infected monocytes were unable to differentiate into macrophages. Exploring the relationships between VZV and permissive immune cells, such as human monocytes and macrophages, elucidates novel immune evasion strategies and provides further insight into the control that VZV has over the immune system.
INTRODUCTION
Varicella-zoster virus (VZV) is a highly species-specific human alphaherpesvirus that results in two clinically distinct diseases. Primary varicella (chickenpox) presents as a disseminated cutaneous rash which is cleared by the host immune response. During primary infection, the virus establishes lifelong latency in dorsal root ganglia (DRG) (1). Reactivation of the virus leads to herpes zoster (shingles), which manifests as a dermatomal rash of vesicular lesions, typically at sites innervated by the latently infected ganglia from which the virus reactivates (2). Both primary and reactivated VZV infection results in viremia, which is presumed to be facilitated by infection of host immune cells (3, 4), and VZV is detectable in circulating peripheral blood mononuclear cells (PBMC) of patients with varicella and herpes zoster (5–7). A number of studies have reported the susceptibility of several PBMC subsets to VZV infection, including T lymphocytes, dendritic cells (DCs), and natural killer (NK) cells (8–10), and direct trafficking of the virus via infected immune cells, particularly DCs and T lymphocytes, is thought to contribute to the dissemination of the virus during primary infection (8, 11, 12).
Monocytes contribute between 15% and 30% of circulating lymphocytes and have the capacity to enter nonlymphoid organs under steady-state conditions to replenish tissue-resident macrophage and DC populations (13, 14). Monocytes play important roles in immune surveillance and pathogen clearance (15), and during microbial inflammation, migrating circulatory monocytes are also able to replace skin-resident Langerhans cells and dermal DCs with inflammatory DC populations (16, 17). Few investigations have focused exclusively on the susceptibility of monocytes to infection by VZV. Two studies initially described the exposure of human monocytes to VZV in vitro and reported that VZV infection of monocytes was abortive (18, 19). These studies did, however, highlight the potential of human macrophages to support productive VZV infection. In contrast, further studies observed VZV glycoproteins on monocytes by immunofluorescence staining following exposure to the virus, suggesting that these cells may have been productively infected (20, 21). A more recent study exposed isolated monocytes to cell-associated VZV but did not report the proportion of VZV-infected monocytes (22). Direct demonstration of productive infection of monocytes has yet to be reported, and the functional impact of any productive infection of monocytes has also not been clarified.
We have previously provided evidence that VZV can productively infect human monocyte-derived DCs, plasmacytoid DCs, and NK cells in vitro (9, 10, 12, 23). In the current study, we sought to determine the susceptibility of human monocytes derived from PBMC to VZV infection. To this end, freshly isolated human monocytes from PBMC were exposed to VZV and assessed for infection and consequent virus-specific modulation. We report that primary human monocytes are fully permissive to productive VZV infection and are capable of transmitting infectious virus to other cells. Monocyte viability and longevity were reduced following infection, and VZV selectively targeted cell surface CD14, HLA-DR, CD11b, and the macrophage colony-stimulating factor receptor (M-CSFR) for downregulation. Furthermore, VZV-infected monocytes were impaired in their capacity to undergo endocytosis. We also demonstrate that VZV-infected monocytes are unable to differentiate into macrophages, whereas macrophages are highly susceptible to VZV infection. Our study is the first to clearly demonstrate, using multiple approaches, that human monocytes are permissive to productive VZV infection and identify the impact of VZV infection on monocyte function and differentiation.
RESULTS
Isolation and infection of human CD14+ monocytes by VZV.
Human CD14+ monocytes were obtained from human blood by positive selection of freshly isolated PBMC with anti-human CD14 magnetic beads. Preisolation PBMC and postisolation CD14+ fractions were assessed by flow cytometry for the proportions of CD14+ CD11b+ monocytes, as CD11b is a pan-monocyte marker (24). Monocytes were enriched from approximately 30% of PBMC to >95% of isolated cells (Fig. 1A). For VZV infection, mock- or VZV-infected human foreskin fibroblasts (HFF) were carboxyl fluorescein succinimidyl (CFSE) labeled (to allow their exclusion from subsequent analysis) and cocultured with monocytes at a ratio of one HFF to two monocytes (1:2) in a cell-associated infection. At 24, 48, and 72 h postinfection (hpi), monocytes were assessed for VZV antigen, a complex of glycoproteins E and I (gE:gI), which is expressed late in the virus replicative cycle. VZV gE:gI was readily detected on the surface of monocytes exposed to VZV-infected HFF (Fig. 1B). On average, 25% of monocytes were VZV gE:gI positive (gE:gI+) with a range of 7% to 62% between 24 and 72 hpi (Fig. 1C).
FIG 1.
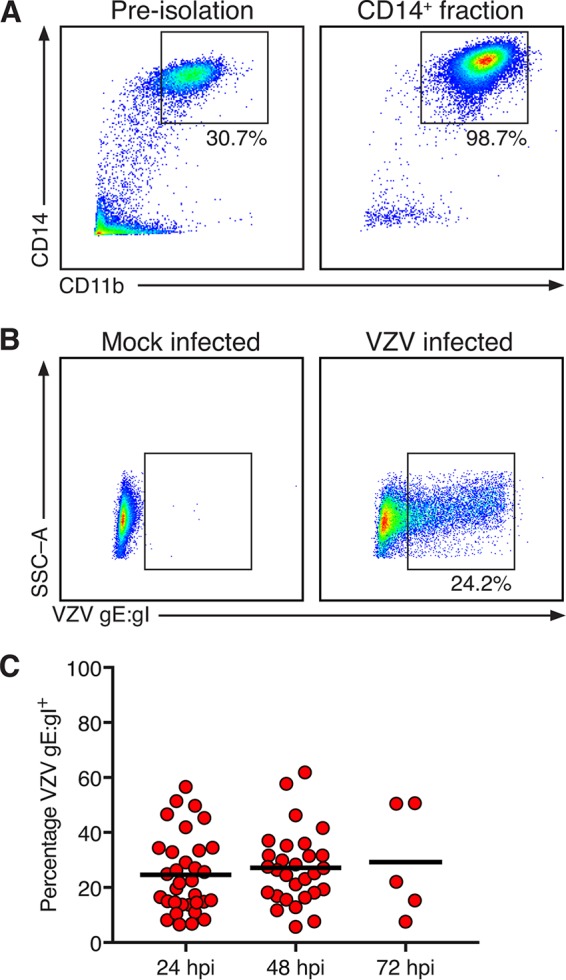
Isolation and infection of human CD14+ monocytes by VZV. (A) Preisolation PBMC and the positive fraction of anti-CD14 microbead isolation. The boxes display the proportion of live and single-cell monocytes expressing CD14 and CD11b, and the results are representative of those for 33 biologically independent donors. (B) CD14+ monocytes were cultured with mock- or VZV-infected HFF and assessed after 24, 48, and 72 h of culture. Representative plots indicate expression of VZV gE:gI on mock-infected (left) and VZV-infected (right) monocytes. Gating was performed on live, single cells, to the exclusion of the CFSE-labeled HFF inoculum. SSC-A, side scatter area. (C) VZV-infected monocytes exhibited cell surface VZV gE:gI+ at 24 hpi (n = 33), 48 hpi (n = 29), and 72 hpi (n = 5). The bars indicate the mean proportion at each time point.
VZV-infected monocytes display each temporal class of viral antigen.
Productive VZV infection requires expression of three temporally distinct classes of viral gene products: the immediate early (IE), early (E), and late (L) gene products (25). At 48 h after exposure of monocytes to the CSFE-labeled VZV-infected inoculum, cells were examined by immunofluorescence assay (IFA) for the presence and subcellular localization of viral proteins indicative of each temporal class in the VZV replicative cycle: IE protein IE62, E protein ORF29 (pORF29), and L protein gE. CSFE was used to label the VZV-infected inoculum, as this provided a reliable means to readily distinguish inoculating cells from monocytes. In VZV-exposed monocytes, all three temporal classes of viral proteins were readily detected (Fig. 2). The localization of each viral antigen was consistent with that of VZV replication, specifically, diffuse late-stage IE62, punctate nuclear pORF29, and diffuse plasma membrane localization of gE (26–28). No VZV antigens were detectable following mock infection or when VZV-exposed monocytes were stained with isotype antibodies (Fig. 2).
FIG 2.

VZV-infected monocytes display hallmarks of productive viral infection. VZV-infected monocytes were examined at 48 hpi by immunofluorescence staining for the presence and localization of immediate early protein IE62, early protein pORF29, and late glycoprotein gE (red staining) and DAPI nuclei (blue staining). Representative images of merged channels and individual viral antigen staining, mock-infected monocytes, and isotype control antibody-stained VZV-infected cells are shown. All images were acquired at a ×63 magnification and are representative of those from 5 independent experiments.
Detection of VZV virions by backscattered electron microscopy.
To determine whether virions could be visualized in VZV-infected monocytes, VZV-exposed monocytes at 24 hpi underwent fluorescence-activated cell sorting (FACS) to isolate VZV gE:gI+ monocytes and to exclude any CSFE-labeled infecting HFF inoculum. Purified infected monocytes were then fixed and processed for backscattered scanning electron microscopy (BSEM). Virions were readily observable on the cell surface of VZV gE:gI+ monocytes (Fig. 3A). Higher magnification revealed multiple virions along the periphery of infected monocytes (Fig. 3A, arrows). Higher magnification also revealed the internal capsid structure of virions on VZV gE:gI+ monocytes (Fig. 3A, arrowhead). Within the nucleus of many VZV gE:gI+ monocytes, viral nucleocapsids of the expected size of approximately 130 nm were observed (Fig. 3B). Most of these nucleocapsids in the nucleus of infected monocytes exhibited densely contrasting cores, presumed to be viral genetic material and consistent with previous reports of VZV-infected lymphocytes assessed through electron microscopy (EM) (29). The detection of VZV antigens representing each temporal class of replicating VZV, the detection of virions associated on the surface of VZV-infected monocytes, and the identification of viral nucleocapsids in the nucleus of infected monocytes indicate that human monocytes are permissive to fully productive VZV infection.
FIG 3.

Backscattered scanning electron microscopy of VZV-infected human monocytes. VZV-infected monocytes were collected and sorted by FACS. VZV gE:gI+ monocytes were fixed and processed for serial block-face backscattered scanning electron microscopy. (A) Images depict a representative VZV gE:gI+ monocyte, with the two insets (i and ii) being magnified on the right. Arrows, VZV virions; arrowheads, virions with observable capsid structures. (B) Images depict a representative VZV gE:gI+ monocyte, with the inset, magnified on the right, showing VZV capsid structures within the nucleus (open arrowheads).
VZV-infected monocytes transfer infectious virus to HFF.
Propagation of VZV in vitro results in highly cell-associated virus which relies on cell-to-cell contact to transfer infectious virus (30, 31). As an initial step, we determined whether VZV-infected monocytes released cell-free virus. Supernatants from VZV-infected monocytes at 48 hpi were centrifuged to remove cellular debris, before being used to inoculate HFF monolayers. HFF monolayers were monitored for the appearance of a cytopathic effect over a period of 5 days. No cytopathic effect was observed in HFF at any stage, indicating that infected monocytes do not release detectable cell-free VZV. To determine whether VZV-infected monocytes could transfer infectious virus to HFF, we conducted an infectious center assay (ICA). VZV-infected monocytes at 48 hpi were isolated by FACS to exclude any CSFE-labeled infecting HFF inoculum, before being inoculated onto uninfected HFF monolayers and incubated for 5 days. The monolayers were then stained by IFA for VZV pORF29 and VZV gE to detect infectious centers (Fig. 4A). This analysis revealed the development of distinct VZV infectious centers (Fig. 4A). No viral antigens were detected when mock-infected monocytes were inoculated onto HFF monolayers. To eliminate the possibility of plaque formation through passive transfer of extracellular virions attached to the surface of monocytes, we also performed the infectious center assay with monocytes pretreated with low-pH citrate buffer, which is known to inactivate and detach surface-bound virions (32–35), prior to inoculation of HFF monolayers. Infectious centers with observable VZV antigen staining were detected following inoculation of citrate buffer-treated VZV-infected monocytes (Fig. 4B). When enumerated, fewer infectious centers were observed following inoculation of citrate buffer-treated VZV-infected monocytes than following inoculation of VZV-infected monocytes that had not been treated with citrate buffer, suggesting that externally adherent infectious particles partially contribute to the transmission of virus (Fig. 4C). These experiments demonstrate that VZV-infected monocytes are able to transmit VZV to HFF and do so in the absence of cell-free VZV production.
FIG 4.

Transfer of infectious virus to HFF. (A) VZV-infected monocytes were isolated by FACS and seeded onto uninfected HFF monolayers for 5 days. The results of immunofluorescence assay (IFA) staining for early VZV pORF29 and late VZV gE shows infectious centers are presented. A merged IFA image of VZV-infected monocytes inoculated on HFF monolayers is shown. Individual channels show pORF29 (green), gE (red), DAPI (blue), and a merged image of mock-infected monocyte inoculation. All images were taken at a ×20 magnification and are representative of those from 3 independent experiments. (B) VZV-infected monocytes were pretreated with citrate buffer prior to inoculation onto uninfected HFF monolayers. A merged IFA image of citrate buffer-treated VZV-infected monocytes inoculated on HFF monolayers is shown. Individual channels of VZV gE (red) and DAPI (blue) are depicted. All images were taken at a ×20 magnification and are representative of those from 3 independent experiments. (C) Enumeration of infectious centers observed following inoculation of untreated and citrate buffer-treated, VZV-infected monocytes onto uninfected HFF. Each symbol represents an independent donor.
Assessment of cell viability following VZV infection of monocytes.
VZV initiates programmed cell death of HFF; however, human sensory neurons and keratinocyte cell lines are protected from apoptosis following infection (36, 37). To determine the viability of VZV-infected monocytes, mock- and VZV-infected monocytes were assessed by multidiscriminant flow cytometry at 24 and 48 hpi. Isolated monocytes were cultured without the HFF inoculum in parallel. Mock- and VZV-infected monocytes and monocytes cultured without HFF were stained with live/dead dye (L/D) and VZV gE:gI, in conjunction with intracellular staining for cleaved caspase-3 (CC3). VZV-infected monocytes were identified as VZV gE:gI+ monocytes, whereas VZV exposed monocytes were identified as VZV gE:gI-negative (gE:gI–) monocytes from the same culture (Fig. 5A). CC3 is the terminal effector of cellular apoptosis, and discrimination between CC3 and L/D allows for classification of viable (CC3 negative [CC3–], L/D negative [L/D–]), early apoptotic (CC3 positive [CC3+], L/D–), late apoptotic (CC3+, L/D positive [L/D+]), and nonviable nonapoptotic (CC3–, L/D+) cells, as we previously established with VZV infection (37) (Fig. 5B). This analysis demonstrated a significant loss of viability of monocytes cultured without HFF in comparison to that of mock-infected and VZV-infected and exposed monocytes at 24 hpi (Fig. 5C), although no difference between VZV-infected monocytes and monocytes cultured without HFF was observed at 48 hpi. Moreover, there was a significant loss of viability of VZV-infected monocytes in comparison to that of mock-infected monocytes at 24 and 48 hpi and in comparison to that of VZV-exposed monocytes at 24 hpi (Fig. 5C). When cell death was examined, a greater proportion of early apoptotic cells was observed in monocytes cultured without HFF after 24 h than in mock-infected, VZV-infected, and VZV-exposed monocytes at 24 hpi (Fig. 5C). Greater proportions of late apoptotic monocytes were observed in monocytes cultured without HFF than in mock-infected monocytes and VZV-exposed monocytes at 24 and 48 hpi (Fig. 5C). There was no significant change in the examined forms of cell death between mock- and VZV-infected monocytes at 24 hpi; however, at 48 hpi there was a significant increase in the proportion of VZV-infected monocytes undergoing late apoptosis. These results indicate that coculture of monocytes with HFF improves viability during culture and that VZV-infected monocytes exhibit impaired viability with evidence of apoptosis in a minor proportion of monocytes at 48 hpi.
FIG 5.

Viability of VZV-infected monocytes. Monocytes without exposure to HFF and mock- and VZV-infected monocytes were stained with live/dead dye (L/D) and antibodies for VZV gE:gI and intracellular cleaved caspase-3 (CC3). (A) VZV-infected monocytes were identified as VZV gE:gI+ (red box), and VZV-exposed monocytes were identified as VZV gE:gI– (orange box). (B) Representative plot of VZV-infected monocytes stratified into viable cells (CC3–, L/D–) and cells with early apoptotic (CC3+, L/D–), late apoptotic (CC3+, L/D+), and nonapoptotic (CC3–, L/D+) cell death. (C) The proportions of monocytes (±SEM) at 24 hpi (top) and 48 hpi (bottom) are shown. Statistics were performed on the proportions of cells by repeated-measures two-way analysis of variance with Tukey’s multiple-comparison test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.00005; no symbol, an insignificant change.
VZV-infected monocytes exhibit impaired endocytosis.
A key function of monocytes is in aiding the adaptive immune system by acting as antigen-presenting cells (APCs) (38). To measure the capacity of VZV-infected monocytes to take up antigen, endocytosis was assessed by flow cytometry. Mock- and VZV-infected monocytes at 24 hpi were incubated with fluorescein isothiocyanate (FITC)-labeled dextran at either 37°C or 4°C to differentiate between internalized and surface-bound dextran, respectively. Dextran-FITC was readily detected on both mock- and VZV-infected (gE:gI+) monocytes, with a clear distinction in the median fluorescence intensity (MFI) of dextran between incubation at 37°C and that at 4°C (Fig. 6A). The MFI of surface-bound dextran at 4°C was subtracted from the MFI of dextran at 37°C to determine the proportion of internalized dextran. VZV-infected monocytes exhibited significantly less internalized dextran than mock-infected monocytes (Fig. 6B). These data demonstrate that VZV-infected monocytes are reduced in their ability to take up environmental antigens.
FIG 6.
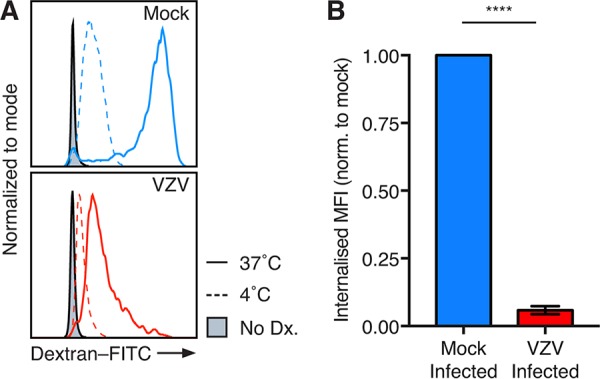
VZV-infected monocytes exhibit a decreased endocytic ability. Mock- and VZV-infected monocytes at 24 hpi were incubated with 1 mg/ml dextran-FITC for 1 h at 37°C or 4°C. Flow cytometry gating was performed on live, single cells, prior to assessment of VZV infection by VZV gE:gI staining. (A) Representative histogram of dextran-FITC (Dx) uptake at 37°C and 4°C and monocytes without dextran (filled). (B) The MFI of surface-bound dextran-FITC at 4°C subtracted from the MFI of surface and internalized dextran-FITC at 37°C to generate the internalized MFI (±SEM). The internalized MFI of VZV-infected monocytes was normalized (norm.) to that of the respective mock-infected monocytes in 4 independent experiments. Statistical analyses were performed by paired two-tailed Student's t test. ****, P < 0.00005.
Immunophenotyping of VZV-infected monocytes.
VZV is known to selectively downregulate a diverse range of immune markers on a variety of susceptible immune cells, including downregulation of major histocompatibility complex (MHC) class II on lipopolysaccharide (LPS)-matured monocyte-derived dendritic cells (MDDC) and gamma interferon (IFN-γ)-mediated MHC class II expression on fibroblasts (12, 23, 39). To determine whether VZV modulates the expression of immune markers during infection of monocytes, mock- and VZV-infected monocytes at 24 and 48 hpi were assessed by flow cytometry for the proportions of cells expressing six functionally important immune markers. These included HLA-ABC and HLA-DR as previously known targets of VZV infection; monocyte-specific markers CD14 and CD11b, which play key roles in establishing the functional phenotype of monocytes (24); and the macrophage colony stimulating factor receptor (M-CSFR), which is essential for macrophage colony-stimulating factor (M-CSF)-mediated macrophage differentiation. As there is uncertainty whether VZV-exposed monocytes (i.e., VZV gE:gI–) are truly uninfected, we chose to examine VZV-infected (VZV gE:gI+) monocytes in isolation.
This analysis revealed a significant reduction in the proportion of CD14+ monocytes during VZV infection at both 24 and 48 hpi (Fig. 7A). In contrast, there was no significant modulation of the proportions of infected monocytes expressing HLA-ABC or HLA-DR at either time point. While there was a modest reduction in the proportion of CD11b+ infected monocytes at 24 hpi, this phenotype was no longer observed by 48 hpi. In contrast, there was a substantial reduction in the proportion of infected monocytes expressing M-CSFR at both 24 and 48 hpi (Fig. 7A).
FIG 7.

Cell surface immunophenotyping of VZV-infected monocytes. (A) Mock-infected monocytes (blue) and VZV-infected (red) monocytes were examined for the proportions of cells expressing the indicated cell surface markers at 24 and 48 hpi by flow cytometry. Gating was performed on live, single cells, prior to assessment of VZV infection by VZV gE:gI staining. Symbols depict the proportions of monocytes from multiple independent donors expressing the indicated markers, and bars represent the mean. (B) The MFI of immune markers on VZV-infected monocytes was normalized to that of the respective mock-infected monocytes at each time point. Bars represent the mean MFI (±SEM). Results are from multiple independent experiments examining CD11b (n = 8), HLA-ABC (n ≥ 8), HLA-DR (n ≥ 8), CD14 (n ≥ 18), and M-CSFR (n ≥ 9). Statistical analyses were performed for proportional and MFI analysis by paired two-tailed Student's t test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.00005; ns, not significant.
In conjunction with the modulation of monocyte proportions, the MFI of cells retaining expression of each immune marker was similarly assessed. The MFI of CD14 on VZV-infected monocytes was significantly reduced at 24 and 48 hpi (Fig. 7B). The MFI of HLA-ABC remained consistent following VZV infection; however, the MFI of HLA-DR was significantly diminished at 48 hpi (Fig. 7B). At both 24 and 48 hpi, the MFI of CD11b was significantly downmodulated by VZV infection, and there was a striking downregulation in the MFI of M-CSFR at 24 and 48 hpi. Taken together, these results indicate that VZV can manipulate infected monocytes by downregulating the proportions of cells expressing several key immune cell markers and by targeting further immune cell markers for downregulation at the protein level.
VZV infection impairs monocyte-to-macrophage differentiation.
Extravasation of circulating monocytes and differentiation into tissue-specific macrophages or dendritic cells are key functions of human monocytes in vivo (13), with stimulation of M-CSFR by M-CSF commonly being used to generate monocyte-derived macrophages in vitro (40). To assess the impact of VZV infection of monocytes on their capacity to differentiate into macrophages, an infection time course was undertaken. Monocytes isolated from four independent blood donors were mock and VZV infected and collected at 24 hpi prior to adherence for 2 h onto fresh culture plates. After 2 h, nonadherent monocytes were aspirated and adherent monocytes were cultured in the presence of M-CSF for 3 days to stimulate the generation of M-CSF-derived macrophages (M-Mφ). On each day, adherent monocytes were collected for flow cytometric assessment of viability and the proportion of cells expressing VZV gE:gI (Fig. 8A). Adherent monocytes remained viable in culture between day 0 and day 2, regardless of exposure to mock- or VZV-infected inoculum HFF, with a measurable reduction in the viability of monocytes exposed to VZV-infected HFF being seen at day 3 (Fig. 8B). This analysis also revealed a steady decline in the number of VZV-infected (gE:gI+) adherent monocytes (Fig. 8C). By day 3, two donors no longer had viable monocytes remaining in culture, and the remaining two donors had a >95% loss of adherent monocytes expressing VZV gE:gI (Fig. 8C). Mock-infected monocytes remained viable throughout the time course and exhibited the expected modulation of macrophage-specific markers CD206 and CD14 (data not shown). These results demonstrate that VZV-infected monocytes do not remain viable during culture for M-Mφ differentiation.
FIG 8.
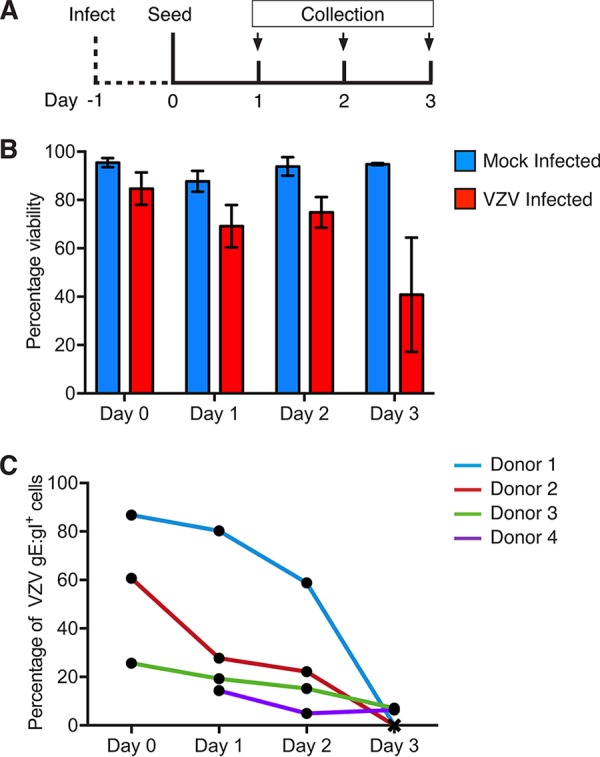
Differentiation of VZV-infected monocytes. Mock- and VZV-infected monocytes were collected at 24 hpi (day 0) and adhered into tissue culture plates in serum-free medium for 2 h at 37°C under 5% CO2. Nonadherent monocytes were aspirated, and the cultures were maintained for 3 days in medium supplemented with 25 ng/ml M-CSF (Peprotech). Adherent monocytes were collected by gentle scraping into the medium, stained for viability and VZV gE:gI, and analyzed by flow cytometry. (A) Timeline for infection and seeding. (B) Proportions of viable adherent monocytes cultured with mock-infected (blue) or VZV-infected (red) HFF. (C) Proportions of VZV gE:gI+ monocytes across each day measured for four independent donors.
As infected monocytes did not remain viable during M-CSF differentiation, we sought to determine whether monocytes that had already differentiated to become M-Mφ could be infected with VZV. Previous studies have reported that monocyte-derived macrophages are susceptible to productive VZV infection, although M-Mφ were not specifically examined (18, 19). M-Mφ were generated from freshly isolated monocytes and cocultured with CFSE-labeled VZV-infected HFF at a ratio of 1:5. At 48 hpi, VZV exposed M-Mφ were assessed by IFA for the presence and localization of macrophage-specific HLA-DR and VZV pORF29. Macrophages were identified by IFA by surface expression of HLA-DR, and VZV pORF29 was observed in the nucleus of VZV-exposed M-Mφ (Fig. 9A). Mock-infected M-Mφ did not exhibit pORF29 staining, whereas HLA-DR was readily detectable. No HLA-DR or pORF29 staining was observed when isotype control antibodies were used. Following IFA analyses, flow cytometry for VZV gE:gI was performed at 24 and 48 hpi using M-Mφ derived from 7 (24 hpi) and 8 (48 hpi) independent blood donors. VZV gE:gI was readily detected on the cell surface of VZV-exposed M-Mφ (Fig. 9B). VZV gE:gI was detected on an average of 24.5% of M-Mφ at 24 hpi (range, 9% to 52%) and an average of 42.8% of M-Mφ at 48 hpi (range, 19% to 74%) (Fig. 9C). These results demonstrate that both monocytes and monocyte-derived M-Mφ are permissive to VZV infection. Furthermore, these results suggest that the failure of VZV-infected monocytes to differentiate into M-Mφ is directly related to the diminished viability of infected monocytes rather than to any intrinsic lack of permissiveness of M-Mφ to VZV replication.
FIG 9.
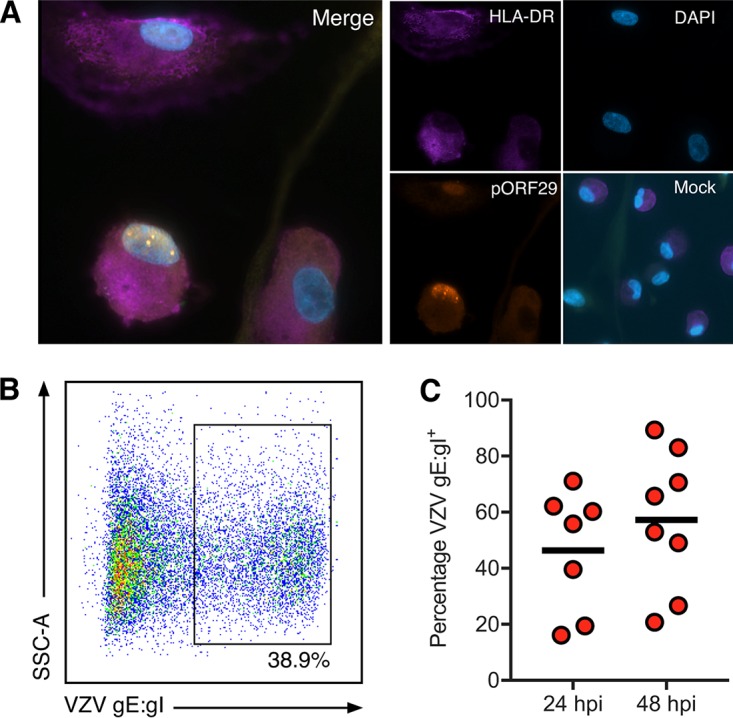
VZV infection of monocyte-derived macrophages. Freshly isolated monocytes were adhered under serum-free conditions as described in the text and cultured in 25 ng/ml M-CSF for 6 days to generate macrophages (M-Mφ). M-CSF macrophages were cocultured with VZV-infected CFSE-labeled HFF for 24 and 48 h. VZV exposed M-Mφ were examined by IFA for the presence of HLA-DR and VZV pOR29. (A) Merged IFA image of VZV-infected M-Mφ at 48 hpi. Individual channels of HLA-DR (purple), pORF29 (orange), and DAPI (blue) are shown. A merged image of mock-infected M-Mφ at the same time point is also shown. All images were taken at a ×20 magnification and are representative of those from 4 independent experiments. Macrophages were collected at 24 and 48 hpi and assessed for VZV gE:gI by flow cytometry. Gating was performed on live, single cells and excluded the CFSE-labeled HFF inoculum. (B) VZV-infected M-Mφ exhibited cell surface VZV gE:gI. (C) VZV-infected M-Mφ displayed surface VZV gE:gI at 24 hpi (n = 4) and 48 hpi (n = 5). Bars indicate the mean proportion at each time point.
DISCUSSION
This work identifies CD14+ monocytes as being fully permissive to VZV infection, with a consequence of this infection being functional impairment and an inability to remain viable during monocyte-macrophage differentiation. Previous studies that included examination of monocytes in the context of VZV exposure have yielded conflicting results. Studies of human monocytes isolated by glass adherence to cell-free VZV in vitro have suggested that VZV infection in monocytes was abortive (18, 19). In contrast, monocytes isolated by bead selection and infected with cell-free VZV exhibited VZV gene transcription and gE antigen expression (20, 21). The generation of cell-free VZV results in prohibitively low titers of VZV which require concentration, whereas cell-associated VZV infections are routinely used to transfer VZV to permissive populations in vitro (8, 12, 41, 42). In this respect, cell-associated infection has been utilized previously to examine infection of monocytes; however, this study exposed monocytes to a VZV-infected human embryonic lung fibroblast (HELF) inoculum and did not fully characterize productive infection of the exposed monocytes (22). Productive VZV infection of the monocytic-like THP-1 cell line, however, was confirmed by IFA and EM (43). Our characterization of VZV infection of primary monocytes therefore represents the most comprehensive investigation to date of the interaction of VZV with this cell type.
VZV-infected monocytes were identified on the basis of viral antigen expression, resulting in a population of VZV-exposed monocytes with undetectable VZV gE:gI expression. There is uncertainty regarding whether these cells constitute truly uninfected cells, whether they represent cells at early stages of infection undetectable by VZV gE:gI expression, or whether they may be abortively infected. Regardless, assessing the functional impact of VZV exposure on monocytes and, indeed, on other primary human cell types, such as T cells, DCs, and NK cells, would require a detailed characterization to properly attribute any functional impacts to VZV exposure. We believe that the field would be better served by a separate comprehensive study addressing this area and, as such, have only represented VZV-exposed monocytes in their viability following exposure.
The proportions of VZV gE:gI+ monocytes remained consistent across a 72-h time course with an absence of detectable cell-free VZV production by infected monocytes, which likely limited the spread of VZV between monocytes. In addition, infection of monocytes in suspension would render the continual cell-to-cell spread of VZV much less likely to occur. Virions were observed on the surface of infected monocytes and viral nucleocapsids were detected in the nucleus of VZV gE:gI+ monocytes by BSEM. The de novo synthesis of pORF29 was also observed in the nucleus of VZV-infected monocytes, and no internalized CFSE (which was used to label the infected HFF inoculum) was observed by IFA. These data demonstrate that monocytes did not simply take up the infecting inoculum but, rather, supported productive VZV infection. We have shown that exposure of human PBMC to cell-associated VZV results in approximately 8% of T cells becoming infected (10), corroborating previous reports (8). We also showed productive VZV infection of approximately 42% of NK cells (range, 17% to 65%) using the same inoculation method (10). The current study identified that, on average, 25% of monocytes (range, 7% to 62%) can be productively infected with VZV, which supports the notion that monocytes are relatively highly susceptible to infection. Moreover, transmission of VZV from monocytes to HFF was readily observed, including following citrate buffer pretreatment, although this treatment reduced the number of detectable infectious centers. The presence of free virions on the surface of infected monocytes, detected by BSEM, is consistent with this finding and suggests that virions may accumulate at this site, with the potential to infect bystander cells by cell-to-cell contact rather than by release of cell-free virus. Indeed, extracellularly adherent virions are presumed to facilitate the spread of VZV in a contact-dependent manner (27). Productively infected monocytes may therefore represent a means for viral dissemination in vivo.
Cell-free VZV-exposed monocytes were previously reported to exhibit annexin V staining at 12 hpi, suggesting that VZV infection results in cell death (20). However, cell death was not directly assessed in conjunction with markers of VZV infection in this study, and cell death remained unattributed to any specific form of cell death. In our current study, we observed a small proportion of VZV-infected monocytes undergoing apoptotic death at 48 hpi; however, the majority of VZV-infected monocytes remained viable at this time point. To ensure that subsequent analyses were not confounded by the presence of dying cells, we incorporated a live/dead dye in flow cytometry for functional evaluations and immune marker phenotyping. When VZV-infected monocytes were driven to macrophage differentiation with M-CSF, we observed the nearly complete cell death of these monocytes.
We observed high proportions of viable monocytes following coculture with uninfected HFF in comparison to the proportions of viable monocytes that had been cultured without HFF, which suggests that monocyte viability may be sustained through survival mechanisms induced by HFF coculture. Indeed, monocytes have been reported to undergo spontaneous apoptosis during in vitro culture (44), and this is consistent with our observations of monocytes cultured without HFF. The mechanism responsible for enhanced monocyte viability in the presence of HFF remains to be determined, although this may be due to specific interactions between these cell types. For example, CD11b binding extracellular matrix (ECM) component fibrinogen supports monocyte viability (45), as does engagement of the epidermal growth factor receptor (EGFR) by transforming growth factor α (TGF-α) (46), and both fibrinogen and TGF-α gene transcripts are highly expressed in HFF (47, 48). The observed downmodulation of CD11b MFI at 24 and 48 hpi on VZV-infected monocytes may contribute to impaired viability, although we cannot account entirely for the diminished proportions of inoculum HFF which succumbed to cell death and which would therefore be unable to further support cocultured monocytes.
VZV infection selectively reduced the proportion of CD14+ monocytes while simultaneously downregulating the cell surface expression of CD14. Infection of human monocytes by VZV has been shown to activate Toll-like receptor 2 (TLR2) through a CD14-dependent mechanism to produce interleukin-6 (IL-6) and IL-8 (22). It remains to be determined if the downregulation of CD14 impacts proinflammatory cytokine production. Moreover, TLR9 requires constitutive CD14 interaction and is required for plasmacytoid DC secretion of IFN-α, tumor necrosis factor, and IL-6 (49, 50). We have previously shown that VZV infection in the skin results in an influx of peripheral DCs and that VZV can abrogate TLR9-mediated IFN-α production (9). It has been suggested, however, that TLR9-independent mechanisms of IFN-α production may be induced by VZV (51). As only a few TLRs mediate a wide response to a large number of stimuli, it is unsurprising that ancillary proteins, such as CD14, are required for these pathways.
We observed unaltered endogenous levels of cell surface MHC class I on VZV-infected monocytes. This contrasts with the VZV-specific downregulation of MHC class I on VZV-infected, LPS-matured human CD14+ MDDC (23). Moreover, the selective downregulation of cell surface MHC class I has been reported in VZV-infected HFF and T cells (39, 52). This result may imply a cell type specificity of MHC class I downregulation following VZV infection or may be a result of the greater expression and/or stability of MHC class I on monocytes, the downmodulation of which was not detectable during the infection time course. Interestingly, MHC class II was selectively downregulated on VZV-infected monocytes at 48 hpi. Previously, only IFN-γ-induced MHC class II was reported to be modulated by VZV in HFF and keratinocytes, whereas endogenous levels remained unaffected (53, 54). Our observation therefore represents the first instance of endogenous MHC class II modulation by VZV. A reduction of cell surface MHC class II may suggest the altered susceptibility of VZV-infected monocytes to CD4+ T cells. However, as T cells (11, 12) are permissive to VZV infection, any change in CD4+ T cell activity in response to VZV-infected monocytes may be complicated if VZV infection of these effector cells themselves may influence their function.
VZV-infected monocytes were additionally impaired in their ability to take up dextran particles. Uptake of environmental antigen and presentation on MHC molecules are necessary for the action of conventional APCs, such as DCs and macrophages, and there is increasing evidence that monocytes can directly contribute to antigen presentation, in addition to being progenitors for conventional APCs (reviewed in reference 55). VZV infection may therefore impair the ability of monocytes to take up antigen for processing and present antigen in the context of MHC class II.
VZV infection selectively impaired the cell surface expression of M-CSFR at both 24 and 48 hpi, which implies that VZV-infected monocytes may be less capable of responding to M-CSF. In the case of M-CSF driving monocytes to macrophage differentiation (M-Mφ), M-CSF engagement with the M-CSFR promotes prosurvival signaling and induces differentiation into M1- and M2-like macrophages (reviewed in reference 56). The experiments that we undertook to explore this possibility resulted in the sequential and almost complete loss of cell viability when VZV-infected monocytes were stimulated to differentiate to macrophages with M-CSF. M-Mφ were, however, highly susceptible to VZV infection, suggesting that infected cell viability was the determinant for impaired differentiation and that this was not an inherent factor resulting from the loss of permissiveness of M-Mφ to VZV infection.
Human monocytes are highly abundant in the circulation, are capable of sensing and defending against infection, and are instrumental in resolving inflammation (57). The ability of VZV to infect and modulate the function of this cell type may enhance the capacity of this virus to establish infection in the human host. The capacity of VZV to infect monocytes may also have implications for macrophages in combatting infection, as VZV-infected monocytes were unable to differentiate into macrophages. This work furthers our understanding of the diverse array of immune-modulatory strategies that herpesviruses such as VZV undertake during infection.
MATERIALS AND METHODS
Cell isolation and tissue culture.
Peripheral blood mononuclear cells (PBMC) were derived from healthy human blood (with ethics approval from the University of Sydney) by gradient centrifugation with Ficoll-Hypaque Plus (GE Healthcare). CD14+ monocytes were isolated from PBMC via positive selection with anti-human CD14 magnetic beads (Miltenyi Biotec) and cultured in RPMI 1640 medium (Lonza) supplemented with 10% fetal calf serum (FCS; Sigma) and 50 IU/ml penicillin and streptomycin (pen-strep; Gibco). Human foreskin fibroblasts (HFF; ATCC) were cultured in Dulbecco’s modified Eagle’s medium (Lonza) supplemented with 10% FCS and 50 IU/ml penicillin and streptomycin. All cultures were maintained at 37°C under 5% CO2 unless indicated otherwise.
Macrophage generation.
To generate M-CSF-derived macrophages (M-Mφ), freshly isolated monocytes were collected and adhered to tissue culture plates in serum-free RPMI 1640 medium supplemented with 50 IU/ml pen-strep at a density of 5 × 105/ml for 2 h at 37°C under 5% CO2. Nonadherent cells were aspirated, and the medium was replaced with RPMI 1640 medium supplemented with 10% FCS, 50 IU/ml pen-strep, and 25 ng/ml recombinant human M-CSF (Miltenyi). The cultures were maintained at 37°C under 5% CO2 for 6 days with a medium change on day 3. For immunofluorescent staining of VZV-infected M-Mφ, monocytes were seeded onto glass coverslips and cultured as described above.
Viruses and infection.
Low-passage-number clinical strain VZV-S (58) (kindly provided by A. Arvin, Stanford University) was propagated in HFF. For infections, VZV-infected HFF were labeled with carboxyl fluorescein succinimidyl (CFSE; Thermo Fisher Scientific) or cell-trace violet (CTV; Thermo Fisher Scientific) per the manufacturer’s instructions where indicated and used to inoculate human CD14+ monocytes at a ratio of 1:2 HFF to monocytes and human M-Mφ at a ratio of 1:5. HFF and monocyte cultures were centrifuged at 150 × g to induce contact.
Flow cytometry.
Cells were washed with phosphate-buffered saline (PBS), stained with live/dead Zombie Dye (BioLegend) per the manufacturer’s instruction, and immunostained in flow cytometry staining buffer (PBS with 1% FCS and 10 mM EDTA). Prior to antibody staining, cells were incubated with the Fc receptor blocking solution human TruStain FcX (BioLegend) per the manufacturer’s instructions. The antibodies used were anti-CD14 (clone MPhiP-9), anti-cleaved caspase-3 (clone C92-605), anti-HLA-DR (clone G46-6), and anti-M-CSFR (clone 9-4D2-1E4) (all from BD Biosciences), anti-CD11b (clone ICRF44) and anti-HLA-ABC (clone W6/32) (both from BioLegend), and anti-gE:gI (clone SG1; Meridian Life Sciences) conjugated in-house to r-phycoerythrin (RPE; Bio-Rad). Respective isotype control antibodies were also used (BD Biosciences, BioLegend). Staining was for 30 min at 4°C prior to fixation with BD Cytofix (BD Biosciences) per the manufacturer’s instructions. For intracellular staining, cells were permeabilized with BD Perm/Wash buffer (BD Biosciences) prior to staining. For the cell viability assay, monocytes were stained with anti-cleaved caspase-3 overnight. Flow cytometry was performed on a BD LSR Fortessa-X20 flow cytometer, and data analysis was performed with FlowJo software (Tree Star).
Immunofluorescence assay (IFA).
Monocytes were spotted onto microscope slides and left to dry before fixation with 4% paraformaldehyde (BD Biosciences). The slides were blocked with 20% normal donkey serum (NDS; Sigma-Aldrich) and stained with primary antibodies in 10% NDS for 1 h at room temperature (RT). The primary antibodies used were anti-IE62 (clone IE62; Meridian Life Sciences), anti-pORF29 (kindly provided by P. R. Kinchington), anti-gE (clone vN-20; Santa Cruz Biotechnology), and anti-HLA-DR (clone LN-3; Novocastra). All primary antibody incubations were followed by incubation with 1:200 Alexa Fluor 488-, Alexa Fluor 546-, or Alexa Fluor 594-conjugated secondary antibodies (Thermo Fisher) at RT, in conjunction with ProLong Gold antifade mountant with DAPI (4′,6-diamidino-2-phenylindole; Thermo Fisher). Images were obtained on a Zeiss deconvolution microscope (Zeiss).
Cell sorting.
Cultures of monocytes and HFF were washed with flow cytometry staining buffer and stained with anti-gE:gI and anti-HLA-DR or CD11b antibodies. Monocyte populations were isolated by selecting for CFSE- or CTV-negative, HLA-DR+, or CD11b+ monocytes on a FACSAria IIu flow cytometer. In some experiments, VZV gE:gI+ cells were further isolated from monocyte populations. Monocytes were maintained on ice in flow cytometry staining buffer prior to infectious center assays and preparation for electron microscopy.
BSEM.
VZV-infected monocytes underwent cell sorting at 24 hpi to isolate VZV gE:gI+ monocytes and to exclude the HFF inoculum. Monocytes were fixed in 2.5% glutaraldehyde (ProSciTech) prior to preparation for backscattered scanning electron microscopy (BSEM) by the Australian Centre for Microscopy & Microanalysis (ACMM) at the University of Sydney. Resin-embedded samples were sectioned for BSEM and imaged on a Sigma VP HD (Zeiss) scanning electron microscope.
Infectious center assay.
VZV-infected monocytes underwent cell sorting at 48 hpi to isolate monocytes from the HFF inoculum. Sorted monocytes were inoculated onto uninfected HFF monolayers on glass coverslips at a ratio of 1:5 HFF to monocytes. Cultures were centrifuged at 150 × g for 10 min to induce contact and cultured for 5 days prior to IFA staining. Coverslips were stained for immunofluorescent detection of VZV pORF29 and gE as described above. In some experiments, sorted monocytes prior to inoculation were treated with citrate buffer (40 mM C6H5O7Na3, 135 mM CaCl, 10 mM KCl [pH 3]) for 60 s at RT before washing in PBS.
Endocytosis assay.
Monocytes at 24 hpi were incubated with 1 mg/ml dextran fluorescein (molecular weight, 40,000; Thermo Fisher Scientific) for 1 h at 37°C or 4°C. Monocytes were kept on ice and stained for flow cytometry with anti-gE:gI. Flow cytometry was performed on monocytes incubated with dextran at 4°C or 37°C and monocytes incubated without dextran. The dextran MFI at 4°C was subtracted from the MFI of dextran at 37°C to generate an internalized MFI. Then, the VZV-infected monocyte MFI was normalized to the MFI of mock-infected monocytes.
Statistical analyses.
The statistical tests performed are indicated where applicable. All analyses were performed using GraphPad Prism software.
ACKNOWLEDGMENTS
We thank Paul Kinchington from the University of Pittsburgh for VZV pORF29-specific antibodies and Chelsea Gerada from the University of Sydney for assistance with cell viability assays. We acknowledge the facilities and the scientific and technical assistance of the Australian Centre for Microscopy & Microanalysis, University of Sydney; the Sydney Cytometry Facility; and the Bosch Institute Advanced Microscopy Facility, University of Sydney.
J.J.K. is supported by an Australian Postgraduate Award scholarship.
REFERENCES
- 1.Arvin A. 2001. Varicella-zoster virus In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Lungu O, Annunziato PW, Gershon A, Staugaitis SM, Josefson D, LaRussa P, Silverstein SJ. 1995. Reactivated and latent varicella-zoster virus in human dorsal root ganglia. Proc Natl Acad Sci U S A 92:10980–10984. doi: 10.1073/pnas.92.24.10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mainka C, Fuss B, Geiger H, Hofelmayr H, Wolff MH. 1998. Characterization of viremia at different stages of varicella-zoster virus infection. J Med Virol 56:91–98. doi:. [DOI] [PubMed] [Google Scholar]
- 4.Satyaprakash AK, Tremaine AM, Stelter AA, Creed R, Ravanfar P, Mendoza N, Mehta SK, Rady PL, Pierson DL, Tyring SK. 2009. Viremia in acute herpes zoster. J Infect Dis 200:26–32. doi: 10.1086/599381. [DOI] [PubMed] [Google Scholar]
- 5.Vafai A, Wellish M, Gilden DH. 1988. Expression of varicella-zoster virus in blood mononuclear cells of patients with postherpetic neuralgia. Proc Natl Acad Sci U S A 85:2767–2770. doi: 10.1073/pnas.85.8.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozaki T, Kajita Y, Asano Y, Aono T, Yamanishi K. 1994. Detection of varicella‐zoster virus DNA in blood of children with varicella. J Med Virol 44:263–265. doi: 10.1002/jmv.1890440309. [DOI] [PubMed] [Google Scholar]
- 7.Koropchak CM, Graham G, Palmer J, Winsberg M, Ting SF, Wallace M, Prober CG, Arvin AM. 1991. Investigation of varicella-zoster virus infection by polymerase chain reaction in the immunocompetent host with acute varicella. J Infect Dis 163:1016–1022. doi: 10.1093/infdis/163.5.1016. [DOI] [PubMed] [Google Scholar]
- 8.Ku CC, Padilla JA, Grose C, Butcher EC, Arvin AM. 2002. Tropism of varicella-zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers. J Virol 76:11425–11433. doi: 10.1128/JVI.76.22.11425-11433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huch JH, Cunningham AL, Arvin AM, Nasr N, Santegoets SJ, Slobedman E, Slobedman B, Abendroth A. 2010. Impact of varicella-zoster virus on dendritic cell subsets in human skin during natural infection. J Virol 84:4060–4072. doi: 10.1128/JVI.01450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell TM, McSharry BP, Steain M, Ashhurst TM, Slobedman B, Abendroth A. 2018. Varicella zoster virus productively infects human natural killer cells and manipulates phenotype. PLoS Pathog 14:e1006999. doi: 10.1371/journal.ppat.1006999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moffat JF, Stein MD, Kaneshima H, Arvin AM. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol 69:5236–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abendroth A, Morrow G, Cunningham AL, Slobedman B. 2001. Varicella-zoster virus infection of human dendritic cells and transmission to T cells: implications for virus dissemination in the host. J Virol 75:6183–6192. doi: 10.1128/JVI.75.13.6183-6192.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geissmann F, Jung S, Littman DR. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 14.Autissier P, Soulas C, Burdo TH, Williams KC. 2010. Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry A 77:410–419. doi: 10.1002/cyto.a.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell AJ, Roediger B, Weninger W. 2014. Monocyte homeostasis and the plasticity of inflammatory monocytes. Cell Immunol 291:22–31. doi: 10.1016/j.cellimm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Kupper TS, Fuhlbrigge RC. 2004. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol 4:211–222. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, Sieling PA, Liu YJ, Rea TH, Bloom BR, Modlin RL. 2005. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med 11:653–660. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arbeit RD, Zaia JA, Valerio MA, Levin MJ. 1982. Infection of human peripheral blood mononuclear cells by varicella-zoster virus. Intervirology 18:56–65. doi: 10.1159/000149304. [DOI] [PubMed] [Google Scholar]
- 19.Baba M, Shigeta S. 1983. Incomplete growth of varicella-zoster virus in human monocytes. Microbiol Immunol 27:767–777. doi: 10.1111/j.1348-0421.1983.tb00642.x. [DOI] [PubMed] [Google Scholar]
- 20.Konig A, Homme C, Hauroder B, Dietrich A, Wolff MH. 2003. The varicella-zoster virus induces apoptosis in vitro in subpopulations of primary human peripheral blood mononuclear cells. Microbes Infect 5:879–889. doi: 10.1016/S1286-4579(03)00177-1. [DOI] [PubMed] [Google Scholar]
- 21.Koenig A, Wolff MH. 2003. Infectibility of separated peripheral blood mononuclear cell subpopulations by varicella-zoster virus (VZV). J Med Virol 70:S59–S63. doi: 10.1002/jmv.10323. [DOI] [PubMed] [Google Scholar]
- 22.Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, Finberg RW. 2005. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J Virol 79:12658–12666. doi: 10.1128/JVI.79.20.12658-12666.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrow G, Slobedman B, Cunningham AL, Abendroth A. 2003. Varicella-zoster virus productively infects mature dendritic cells and alters their immune function. J Virol 77:4950–4959. doi: 10.1128/JVI.77.8.4950-4959.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, Chalasani G, Taboas JM, Lakkis FG, Metes DM. 2017. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One 12:e0176460. doi: 10.1371/journal.pone.0176460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 14:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edson CM, Hosler BA, Poodry CA, Schooley RT, Waters DJ, Thorley-Lawson DA. 1985. Varicella-zoster virus envelope glycoproteins: biochemical characterization and identification in clinical material. Virology 145:62–71. doi: 10.1016/0042-6822(85)90201-6. [DOI] [PubMed] [Google Scholar]
- 27.Reichelt M, Brady J, Arvin AM. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J Virol 83:3904–3918. doi: 10.1128/JVI.02137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinchington PR, Turse SE. 1998. Regulated nuclear localization of the varicella-zoster virus major regulatory protein, IE62. J Infect Dis 178:S16–S21. doi: 10.1086/514263. [DOI] [PubMed] [Google Scholar]
- 29.Arvin AM, Moffat JF, Sommer M, Oliver S, Che X, Vleck S, Zerboni L, Ku CC. 2010. Varicella-zoster virus T cell tropism and the pathogenesis of skin infection. Curr Top Microbiol Immunol 342:189–209. doi: 10.1007/82_2010_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weller TH, Witton HM. 1953. Propagation in tissue cultures of cytopathogenic agents apparently derived from varicella vesicle fluids. AMA Am J Dis Child 86:644–645. [PubMed] [Google Scholar]
- 31.Cole NL, Grose C. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev Med Virol 13:207–222. doi: 10.1002/rmv.377. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Ali MA, Wang K, Sayre D, Hamel FG, Fischer ER, Bennett RG, Cohen JI. 2010. Insulin degrading enzyme induces a conformational change in varicella-zoster virus gE, and enhances virus infectivity and stability. PLoS One 5:e11327. doi: 10.1371/journal.pone.0011327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finnen RL, Mizokami KR, Banfield BW, Cai GY, Simpson SA, Pizer LI, Levin MJ. 2006. Postentry events are responsible for restriction of productive varicella-zoster virus infection in Chinese hamster ovary cells. J Virol 80:10325–10334. doi: 10.1128/JVI.00939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadaoka T, Depledge DP, Rajbhandari L, Venkatesan A, Breuer J, Cohen JI. 2016. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc Natl Acad Sci U S A 113:E2403–E2412. doi: 10.1073/pnas.1522575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Setas Pontes M, Devriendt B, Favoreel HW. 2015. Pseudorabies virus triggers glycoprotein gE-mediated ERK1/2 activation and ERK1/2-dependent migratory behavior in T cells. J Virol 89:2149–2156. doi: 10.1128/JVI.02549-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hood C, Cunningham AL, Slobedman B, Boadle RA, Abendroth A. 2003. Varicella-zoster virus-infected human sensory neurons are resistant to apoptosis, yet human foreskin fibroblasts are susceptible: evidence for a cell-type-specific apoptotic response. J Virol 77:12852–12864. doi: 10.1128/JVI.77.23.12852-12864.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerada C, Steain M, McSharry BP, Slobedman B, Abendroth A. 2018. Varicella-zoster virus ORF63 protects human neuronal and keratinocyte cell lines from apoptosis and changes its localization upon apoptosis induction. J Virol 92:e00338-18. 10.1128/JVI.00338-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DWH, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ. 2013. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abendroth A, Lin I, Slobedman B, Ploegh H, Arvin AM. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J Virol 75:4878–4888. doi: 10.1128/JVI.75.10.4878-4888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, Hamilton JA. 2012. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol 188:5752–5765. doi: 10.4049/jimmunol.1103426. [DOI] [PubMed] [Google Scholar]
- 41.Sloutskin A, Kinchington PR, Goldstein RS. 2013. Productive vs non-productive infection by cell-free varicella zoster virus of human neurons derived from embryonic stem cells is dependent upon infectious viral dose. Virology 443:285–293. doi: 10.1016/j.virol.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carpenter JE, Henderson EP, Grose C. 2009. Enumeration of an extremely high particle-to-PFU ratio for varicella-zoster virus. J Virol 83:6917–6921. doi: 10.1128/JVI.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nour AM, Reichelt M, Ku CC, Ho MY, Heineman TC, Arvin AM. 2011. Varicella-zoster virus infection triggers formation of an interleukin-1beta (IL-1beta)-processing inflammasome complex. J Biol Chem 286:17921–17933. doi: 10.1074/jbc.M110.210575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, Bigley V, Flavell RA, Gilroy DW, Asquith B, Macallan D, Yona S. 2017. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med 214:1913–1923. doi: 10.1084/jem.20170355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandor N, Lukacsi S, Ungai-Salanki R, Orgovan N, Szabo B, Horvath R, Erdei A, Bajtay Z. 2016. CD11c/CD18 dominates adhesion of human monocytes, macrophages and dendritic cells over CD11b/CD18. PLoS One 11:e0163120. doi: 10.1371/journal.pone.0163120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantley LC. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 47.Kueh J, Richards M, Ng SW, Chan WK, Bongso A. 2006. The search for factors in human feeders that support the derivation and propagation of human embryonic stem cells: preliminary studies using transcriptome profiling by serial analysis of gene expression. Fertil Steril 85:1843–1846. doi: 10.1016/j.fertnstert.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 48.Chen AC, Lee YL, Hou DY, Fong SW, Peng Q, Pang RT, Chiu PC, Ho PC, Lee KF, Yeung WS. 2012. Study of transforming growth factor alpha for the maintenance of human embryonic stem cells. Cell Tissue Res 350:289–303. doi: 10.1007/s00441-012-1476-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Bluml S, Grebien F, Bruckner M, Pasierbek P, Aumayr K, Planyavsky M, Bennett KL, Colinge J, Knapp S, Superti-Furga G. 2010. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med 207:2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, Lee KD, Coffman RL, Barrat FJ. 2006. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med 203:1999–2008. doi: 10.1084/jem.20060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu HR, Huang HC, Kuo HC, Sheen JM, Ou CY, Hsu TY, Yang KD. 2011. IFN-alpha production by human mononuclear cells infected with varicella-zoster virus through TLR9-dependent and -independent pathways. Cell Mol Immunol 8:181–188. doi: 10.1038/cmi.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen JI. 1998. Infection of cells with varicella-zoster virus down-regulates surface expression of class I major histocompatibility complex antigens. J Infect Dis 177:1390–1393. doi: 10.1086/517821. [DOI] [PubMed] [Google Scholar]
- 53.Abendroth A, Slobedman B, Lee E, Mellins E, Wallace M, Arvin AM. 2000. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J Virol 74:1900–1907. doi: 10.1128/JVI.74.4.1900-1907.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Black AP, Jones L, Malavige GN, Ogg GS. 2009. Immune evasion during varicella zoster virus infection of keratinocytes. Clin Exp Dermatol 34:e941–e944. doi: 10.1111/j.1365-2230.2009.03350.x. [DOI] [PubMed] [Google Scholar]
- 55.Jakubzick CV, Randolph GJ, Henson PM. 2017. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol 17:349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- 56.Martinez FO, Sica A, Mantovani A, Locati M. 2008. Macrophage activation and polarization. Front Biosci 13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 57.Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moffat JF, Zerboni L, Kinchington PR, Grose C, Kaneshima H, Arvin AM. 1998. Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J Virol 72:965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]