

The recent discovery of Orsay virus, the only known natural virus of Caenorhabditis elegans, provides a unique opportunity to study virus-host interactions that mediate infection in a genetically tractable multicellular model organism. As viruses remain a global threat to human health, better insights into cellular components that enable virus infection and replication can ultimately lead to the development of new targets for antiviral therapeutics.
KEYWORDS: Caenorhabditis elegans, Orsay virus, drl-1
ABSTRACT
Orsay virus is the only known natural virus pathogen of Caenorhabditis elegans, and its discovery has enabled virus-host interaction studies in this model organism. Host genes required for viral infection remain understudied. We previously established a forward genetic screen based on a virus-inducible green fluorescent protein transcriptional reporter to identify novel host factors essential for virus infection. Here, we report the essential role in Orsay virus infection of the dietary restriction-like (drl-1) gene, which encodes a serine/threonine kinase similar to the mammalian MEKK3 kinase. Ablation of drl-1 led to a >10,000-fold reduction in Orsay virus RNA levels, which could be rescued by ectopic expression of DRL-1. DRL-1 was dispensable for Orsay replication from an endogenous transgene replicon, suggesting that DRL-1 affects a prereplication stage of the Orsay life cycle. Thus, this study demonstrates the power of C. elegans as a model to identify novel virus-host interactions essential for virus infection.
IMPORTANCE The recent discovery of Orsay virus, the only known natural virus of Caenorhabditis elegans, provides a unique opportunity to study virus-host interactions that mediate infection in a genetically tractable multicellular model organism. As viruses remain a global threat to human health, better insights into cellular components that enable virus infection and replication can ultimately lead to the development of new targets for antiviral therapeutics.
INTRODUCTION
Orsay virus is the only known natural virus pathogen of Caenorhabditis elegans (1). The recent discovery of Orsay virus has enabled identification of novel host factors essential for infection and immunity in this classic model organism (2–7). As many C. elegans genes and fundamental biological processes are evolutionarily conserved in higher eukaryotes, further dissection of virus-host interactions using this novel platform has the potential to identify novel insights into basic cellular biology and targets for antiviral therapeutics.
Orsay virus is a nonenveloped, positive-sense, single-stranded RNA virus most closely related to members of the Nodaviridae family (1). Orsay virus has a bipartite genome encoding an RNA-dependent RNA polymerase (RdRp) in the RNA1 segment, which is ∼3.4 kb. The viral capsid protein and a novel capsid-delta fusion protein whose function has been implicated in viral egress are encoded in the RNA2 segment, which is ∼2.5 kb (1, 5).
We previously described an unbiased forward genetic screen to define host factors that mediate Orsay virus infection in C. elegans (3). Central to the screen is the jyls8;rde-1(ne219) reporter strain, which is hypersensitive to Orsay virus and carries an integrated transcriptional green fluorescent protein (GFP) reporter under the virus-inducible pals-5 promoter (2). In our previous study, following ethyl methanesulfonate (EMS) mutagenesis, two mutants, sid-3(vir9) and viro-2(vir10), were isolated that fail to express GFP when challenged with Orsay virus and show a significant defect in viral RNA accumulation. sid-3 encodes a nonreceptor tyrosine kinase, and viro-2 is an ortholog of human Wiskott Aldrich syndrome proteins (N-WASP). These represent the first host genes identified to be necessary for Orsay virus infection in C. elegans. Both of these genes are required for early, prereplication steps of the virus life cycle. As the EMS screen in the previous study was not saturating, here we expanded the genetic screen to identify another novel host gene, drl-1, that is also critical for Orsay virus infection of C. elegans. As with sid-3 and viro-2, we determined that drl-1 acts at a step preceding viral RNA replication. drl-1 encodes a putative serine-threonine kinase that shares similarity with human mitogen-activated protein kinases (MAPK), implicating, for the first time in C. elegans, a role for a MAPK pathway in viral infection.
RESULTS
A forward genetic screen identified a novel host factor critical for Orsay virus infection in C. elegans.
A chemical mutagenesis screen was performed using the previously described jyls8;rde-1(ne219) strain to identify additional host factors critical for Orsay virus infection in C. elegans (3). The goal of the screen was to isolate mutants that failed to express GFP upon virus infection, referred to as Viro (for virus-induced reporter off) mutants.
Approximately 2,000 haploid genomes were mutagenized with EMS. Initially 127 Viro hits were isolated, and after several rounds of validation (i.e., bleaching and reinfection), two independent Viro mutants remained. Mating with the previously identified sid-3(vir9) and viro-2(vir10) mutants demonstrated that one of these Viro mutants belonged to the same genetic complementation group as sid-3(vir9), while the other mutant, Viro-4, represented an independent complementation group. Viro-4 reproducibly failed to express GFP following Orsay virus infection (Fig. 1A). As the microsporidial pathogen Nematocida parisii can also activate GFP expression in jyls8;rde-1(ne219), we challenged Viro-4 with N. parisii. The mutant displayed intestinal GFP expression, demonstrating the integrity of the GFP transgene and that the failure to induce GFP was virus specific (Fig. 1A).
FIG 1.
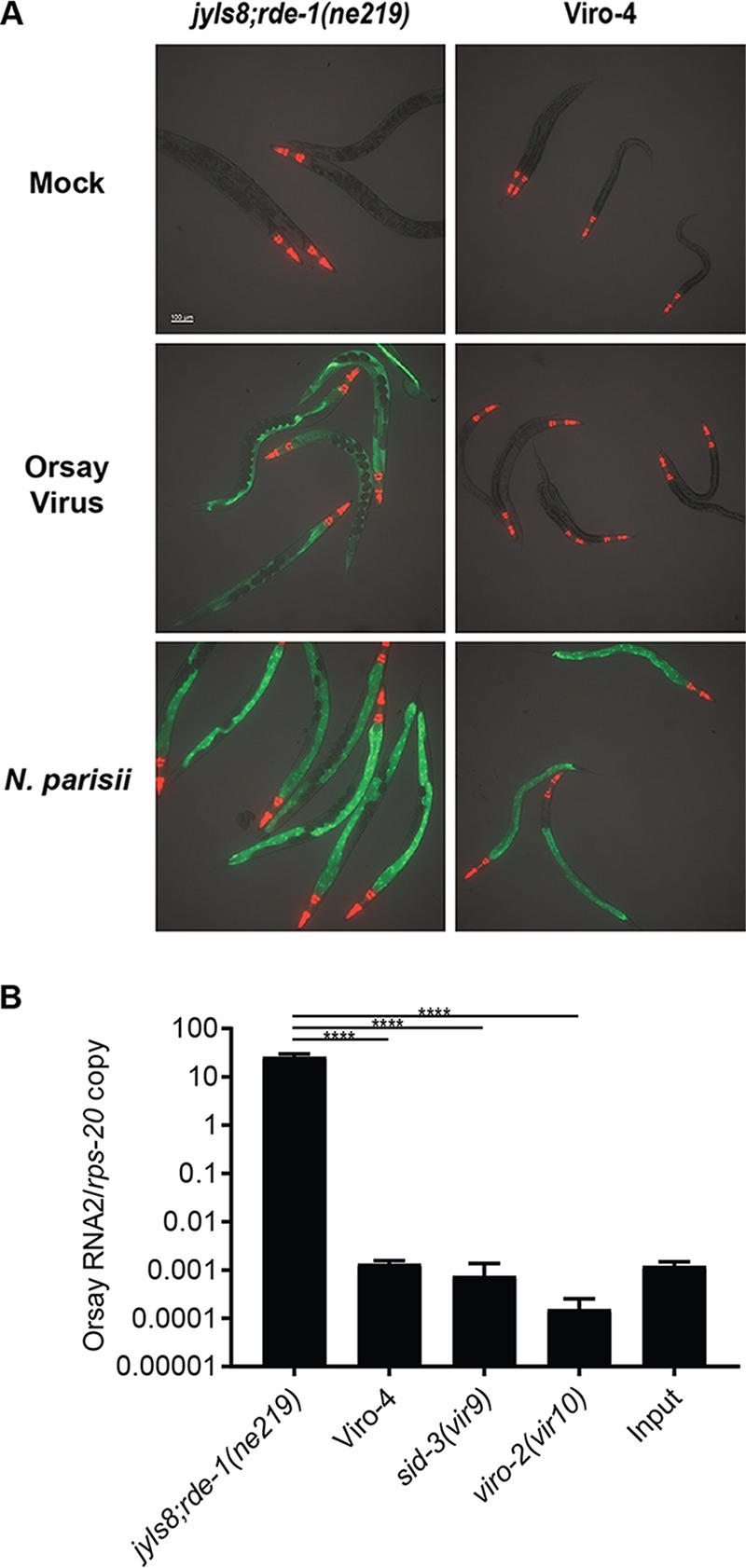
Chemical mutagenesis screen identified a mutant strain with a defect in GFP expression and viral RNA replication. (A) GFP reporter expression of Viro-4 mutants infected with Orsay virus or N. parisii at 3 days postinfection. (B) Orsay virus RNA2 levels quantified by real-time qRT-PCR at 3 days postinfection. Values are means plus standard deviations for three replicate wells. ****, P < 0.0005.
Orsay virus RNA levels in Viro-4 were measured by real-time quantitative reverse transcription-PCR (qRT-PCR) 3 days after infection. Compared to the unmutagenized reporter strain, Viro-4 showed a >10,000-fold reduction in Orsay viral RNA at 3 days postinfection, demonstrating a significant defect in virus RNA production (Fig. 1B). The Orsay virus RNA levels in Viro-4 were comparable to those seen in the previously identified sid-3(vir9) and viro-2(vir10) mutants (Fig. 1B).
Mutation in the drl-1 gene is responsible for the Viro-4 phenotype.
To identify the mutation responsible for the virus-resistant phenotype in the Viro-4 mutant, F2 bulk segregant analysis was performed by outcrossing the strain with the rde-1(ne219) mutant strain, which is also highly susceptible to Orsay virus infection, as previously described (3). Thirty-eight independent F2 cross-progenies that had the GFP and virus-resistant phenotype (data not shown) were isolated. Their F3 progenies were pooled and subjected to whole-genome sequencing and analyzed using the CloudMap bioinformatic pipeline (8).
Three candidate mutations were identified in the linked region on chromosome IV: a G-to-A nonsense mutation, which leads to a loss of the splice acceptor site of the ninth exon of the drl-1 gene, a G-to-A missense mutation, which results in a D522N amino acid substitution in the same drl-1 gene, and a G-to-A missense mutation in the cogc-8 gene, which results in an R360Q amino acid substitution. drl-1 encodes a putative serine/threonine kinase; RNA interference (RNAi) knockdown of drl-1 is reported to lead to an increase in lifespan, decrease in fat storage, and lower levels of oxidizing agents (9). By BLASTp analysis, the kinase DRL-1 has 26% sequence identity (44% sequence similarity) to the human MAP kinase kinase kinase (MEKK3), which has been shown to function in signaling cascades regulating cellular proliferation, differentiation, and inflammatory responses (10–14). cogc-8 encodes a subunit of lobe B of the conserved oligomeric Golgi complex and has been reported as a mediator of gonad morphogenesis and gonadal cell migration (15).
To determine which of these genes was responsible for the GFP and virus-resistant phenotype in the Viro-4 mutant, we first performed RNAi knockdown of drl-1 and cogc-8 in the drh-1 mutant background, which is hypersensitive to Orsay virus infection and has a functional feeding RNAi pathway. RNAi knockdown of drl-1 showed an approximately 10-fold reduction in Orsay virus RNA at 2 days postinfection compared to that of the empty RNAi feeding vector, cogc-8, or an irrelevant host gene, dpy-3 (Fig. 2). These results suggested that drl-1, not cogc-8, was the causal gene. To provide independent validation, we ectopically expressed wild-type DRL-1 from either a fosmid encompassing the drl-1 locus or a plasmid driven by the ubiquitous sur-5 gene promoter in the Viro-4 mutant. Orsay virus infection of both strains led to wild-type levels of both GFP expression and Orsay virus RNA at 3 days postinfection (Fig. 3A and B). These results demonstrated mutation of the gene drl-1 is responsible for the Viro-4 phenotype; this mutant will be referred to here as the drl-1(vir11) mutant strain.
FIG 2.
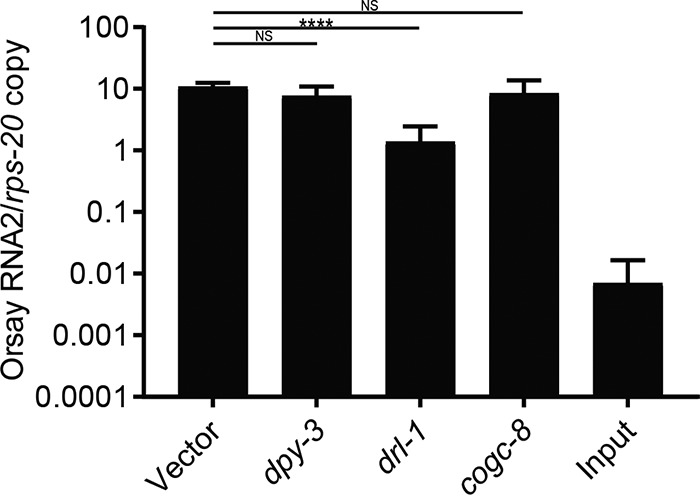
RNAi knockdown of candidate causal genes in the drh-1 mutant strain. Shown are results of real-time qRT-PCR of Orsay virus RNA2 levels 2 days after Orsay virus infection of animals fed RNAi against independent genes. Values are means plus standard deviations (error bars) for three independent biological experiments, each with three replicate wells. Statistically significant differences were determined by one-way analysis of variance (ANOVA) [F(3,32) = 14.47; P < 0.0001], with statistical difference identified between empty vector and drl-1 (P = 0.0001 by Dunnett’s multiple-comparison test). ****, P < 0.0005; NS, not significant (P > 0.05).
FIG 3.
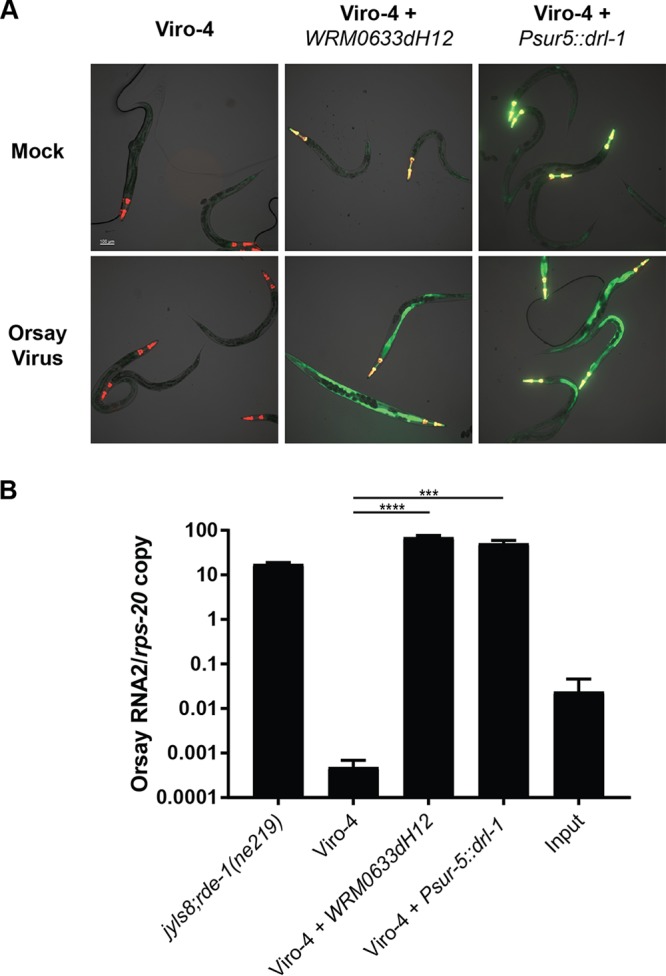
Orsay RNA replication and GFP expression patterns in Viro-4 mutants carrying a fosmid containing the endogenous drl-1 locus or overexpressing DRL-1. (A) GFP response at 3 days postinfection of Viro-4 mutants expressing the drl-1 locus or ectopically expressing DRL-1 infected with Orsay virus. The Pmyo2::YFP plasmid was coinjected as a transgenic marker. (B) Orsay virus RNA2 levels at 3 days postinfection of Viro-4 mutants expressing the drl-1 locus or overexpressing DRL-1 infected with Orsay virus. ***, P < 0.005; ****, P < 0.0005.
DRL-1 is necessary for a prereplication step of the Orsay virus life cycle.
To assess the stage of the Orsay life cycle upon which DRL-1 acts, we generated a transgenic drl-1(vir11) strain carrying an extrachromosomal array of plasmids encoding the Orsay virus RNA1 segment under the control of a heat shock promoter as previously described (3). As a control to determine the level of Orsay RNA generated by DNA templated transcription, a transgenic drl-1(vir11) strain carrying extrachromosomal arrays of a D601A polymerase-dead mutant of Orsay virus RNA1 was also generated (3). We previously determined that the wild-type RNA1, but not the D601A mutant, expressed in jyls8;rde-1(ne219) is sufficient to support replication of the Orsay virus RNA1 segment as a replicon system and activate GFP expression. Heat shock of drl-1(vir11) mutants carrying the wild-type Orsay RNA 1 segment induced intestinal GFP expression at 3 days after induction, whereas the D601A mutant-carrying animals did not (Fig. 4A). By qRT-PCR, ∼100-fold more viral RNA was detected in the wild-type RNA1-carrying line than the mutant expressing the polymerase-dead mutant Orsay virus RNA1 segment. The level of Orsay RNA1 replication was comparable to that observed in the analogous wild-type jyls8;rde-1(ne219) strain (Fig. 4B). These observations demonstrate that the absence of DRL-1 does not affect Orsay virus replication from an endogenous transgene. Therefore, DRL-1 is necessary for a prereplication step of the Orsay life cycle.
FIG 4.
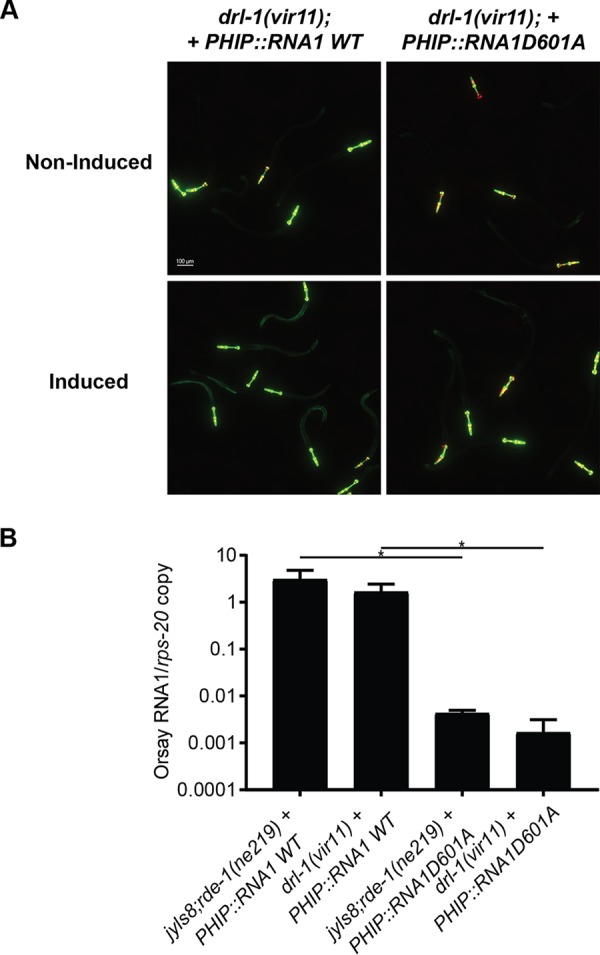
Impact of drl-1 on Orsay virus RNA replication induced from a transgene. (A) GFP expression of strains carrying either wild-type or polymerase-dead mutant Orsay virus RNA1 under a heat-inducible promoter in the drl-1(vir11) background 3 days after heat shock. The Pmyo2::YFP plasmid was coinjected as a transgenic marker. (B) Quantification of Orsay virus RNA1 replication 3 days after heat shock by real-time qRT-PCR. *, P < 0.05.
DISCUSSION
The recently established Orsay virus-C. elegans model provides a robust system to define host-virus interactions. Initial studies in this nascent field have begun to identify novel proviral and antiviral factors (2–7). We previously described a forward genetic screening strategy to identify proviral host factors which identified sid-3 and viro-2 (3). As that pilot screen was not saturated, as evidenced by the absence of multiple alleles of the same gene, we extended the screen to another 2,000 haploid genomes. Here, we identified drl-1 as a novel host gene essential for Orsay virus infection of C. elegans.
Only one prior publication has characterized drl-1. It is reported to be a negative regulator of life span and health, likely through nutrient sensing and metabolic shifts (9). In that study RNAi knock down of drl-1 led to an extension in longevity, an increase in fatty acid degradation, and a decrease in cellular reactive oxygen species (ROS). Whether drl-1 mediates virus infection through these pathways or functions in a novel pathway remains to be explored.
The phenotypes we observed in drl-1(vir11) with respect to Orsay virus infection phenocopied our previously reported sid-3(vir9) and viro-2(vir10) mutants. Specifically, in addition to the failure to activate GFP following Orsay infection, Orsay RNA levels were reduced to comparable levels by >10,000-fold. In addition, DRL-1, like SID-3 and VIRO-2, was dispensable for Orsay replication from an endogenous transgene, suggesting that all 3 of these genes act at a prereplication step of the Orsay virus life cycle. Whether they act together to affect the same step or distinct steps is currently unknown. While there is no experimental data directly linking DRL-1 to SID-3 or VIRO-2, given that DRL-1 has reported kinase activity (9), it is tempting to speculate that DRL-1 functions in a signaling pathway linked to SID-3 and/or VIRO-2.
In contrast to sid-3 and viro-2, which have clear mammalian orthologs, there is no clear mammalian ortholog of drl-1. By BLASTp analysis, the human gene with greatest similarity is that encoding MEKK3, with 26% sequence identity (44% sequence similarity). MEKK3 is a serine/threonine kinase and a member of the MAPK family. MEKK3 has been reported to regulate signaling pathways, such as ERK, JNK, and p38, involved in various cellular processes, such as proliferation, differentiation, apoptosis, and inflammatory responses in response to extracellular signals and stresses (10–14). In C. elegans, the p38 MAPK pathway plays a role in the response to bacterial pathogens, but there have been no reports linking MAPK pathways to viruses in C. elegans (17).
Increasing evidence demonstrates the crucial role of MAPK signaling pathways in viral infection in mammals. For example, influenza A virus (IAV) infection requires activation of the MAPK/ERK pathway for efficient viral replication, as inhibition of this pathway reduces IAV titers in human lung carcinoma cells and mouse lung tissues (18, 19). Conversely, constitutive activation of this pathway yielded higher virus titers in cell culture and enhanced mortality in vivo (18, 20). Hepatitis C virus (HCV) and coxsackievirus B3 (CVB3) also upregulate activity of the MAPK cascade upon infection, and inhibiting this pathway suppresses both HCV and CVB3 viral replication in cell culture (21–24). The amino acid similarity, albeit somewhat limited, between C. elegans DRL-1 and human MEKK3 raises the possibility that the MAPK pathway’s function in mediating viral infection is evolutionarily conserved from C. elegans to humans. Future studies will dissect whether DRL-1 interacts with C. elegans MAPK pathways as well as whether human MEKK3 or related kinases are important for infection by mammalian viruses.
MATERIALS AND METHODS
C. elegans culture and maintenance.
C. elegans strains WM27 rde-1(ne219) and RB2519 drh-1(ok3495) were obtained from the Caenorhabditis Genetics Center (CGC) and maintained under standard laboratory culture conditions unless otherwise specified (25) (Table 1). In brief, animals were fed Escherichia coli OP50 on nematode growth medium (NGM) plates in a 20°C incubator and chunked every 3 to 4 days to a new NGM plate seeded with E. coli OP50.
TABLE 1.
Strains used in this study
| Laboratory name | Strain name | Relevant genotype |
|---|---|---|
| WM27 | rde-1(ne219) | [rde-1(ne219)V] |
| ERT54 | jyls8 | jyIs8[Ppals-5::GFP; Pmyo-2::mCherry] |
| RB2519 | drh-1(ok3495) | [drh-1(ok3495)IV] |
| WUM32 | jyIs8;rde-1(ne219); virEx20[PHIP::RNA1WT-1] | {virEx20[PHIP::OrsayRNA1WT-1; Pmyo-2::YFP]; jyIs8[Ppals-5::GFP;Pmyo-2::mCherry];rde-1(ne219) V} |
| WUM33 | jyIs8;rde-1(ne219); virEx21[PHIP::RNA1D601A-1] | {virEx21[PHIP::OrsayRNA1D601A-1; Pmyo-2::YFP]; jyIs8[Ppals-5::GFP;Pmyo-2::mCherry];rde-1(ne219) V} |
| WUM45 | sid-3(vir9) | {jyIs8[Ppals-5::GFP; Pmyo-2::mCherry]; rde-1(ne219) V; sid-3(vir9)X} |
| WUM46 | viro-2(vir10) | {jyIs8[Ppals-5::GFP; Pmyo-2::mCherry]; rde-1(ne219) V; viro-2(vir10)III} |
| WUM73 | drl-1(vir11) | {jyIs8[Ppals-5::GFP; Pmyo-2::mCherry]; rde-1(ne219) V; drl-1(vir11)IV} |
| WUM74 | drl-1(vir11); virEx37[Psur-5::drl-1] | {virEx37[Psur-5::drl-1;Pmyo-2::YFP]; jyIs8[Ppals-5::GFP;Pmyo-2::mCherry];rde-1(ne219)V; drl-1(vir11)IV} |
| WUM75 | drl-1(vir11); virEx38[PHIP::RNA1WT-1] | {virEx38[PHIP::OrsayRNA1WT-1; Pmyo-2::YFP]; jyIs8[Ppals-5::GFP;Pmyo-2::mCherry];rde-1(ne219) V; drl-1(vir11)} |
| WUM76 | drl-1(vir11); virEx39[PHIP::RNA1D601A-1] | {virEx39[PHIP::OrsayRNA1D601A-1; Pmyo-2::YFP]; jyIs8[pals-5::GFP;Pmyo-2::mCherry];rde-1(ne219) V; drl-1(vir11)} |
| WUM77 | drl-1(vir11); virEx40[WRM0633dH12] | {virEx40[WRM0633dH12;Pmyo-2::YFP]; jyIs8[Ppals-5::GFP; Pmyo-2::mCherry];rde-1(ne219)V; drl-1(vir11)IV} |
Orsay virus preparation, infection, and RNA extraction.
Orsay virus was prepared by liquid culture as described previously (26), filtered through a 0.22-µm filter, and stored at −80°C. For all infection experiments, animals were bleached and then synchronized in M9 buffer (27) in 15-ml conical tubes with constant rotation at room temperature for 16 h. Five hundred arrested larval stage 1 (L1) larvae were seeded onto each well of a six-well plate with 20 µl of an E. coli OP50 lawn. L1 larvae were allowed to recover for 20 h at 20°C prior to infection. Orsay virus filtrate was thawed at room temperature and then diluted 1:10 with M9 buffer. For each well, 20 µl of virus filtrate was added into the middle of the bacterial lawn and incubated at 20°C. Three days after infection, animals were collected into 1.5-ml Eppendorf tubes by washing each well with 1 ml of M9 buffer and then pelleted by spinning for 1 min at 3,000 rpm in a benchtop centrifuge. M9 supernatant was removed and 300 µl TRIzol reagent (Invitrogen) was added to the tubes, and then the tubes were frozen in liquid nitrogen. For each experiment, three replicate wells were used for each infection condition unless otherwise indicated. Total RNA from infected animals was extracted using Direct-zol RNA miniprep (Zymo Research) purification according to the manufacturer’s protocol and eluted into 30 µl of RNase/DNase-free water.
C. elegans feeding RNAi knockdown.
RNAi feeding was used for gene knockdown as described previously (28). E. coli strain HT115, carrying double-stranded RNA expression cassettes for genes of interest, was induced using established conditions and then seeded into six-well NGM plates. dpy-3 and cogc-8 RNAi clones were from the Ahringer RNAi library (29). To construct the drl-1 feeding RNAi clone, a 1-kb genomic fragment of the gene was amplified by single-animal PCR using primers LS86 (TAAGCAGGATCCATTCTGCTACCAATTATCATTTTGCT) and LS87 (ATTCGTTGCTTATCATGCAATCTTATCAACACCATAACC). The PCR product was digested with BamHI, cloned into pL4440, and transformed in E. coli HT115. Twenty arrested L1 drh-1 mutant animals were seeded into each well of a six-well plate. After 72 h of RNAi feeding, Orsay virus was added to the plates as described above. At 48 h postinfection the infected C. elegans animals were collected, and 300 µl of TRIzol (Invitrogen) was added to each well for RNA extraction.
Orsay virus quantification by real-time qRT-PCR.
RNA extracted from infected animals was subjected to one-step real-time quantitative reverse transcription-PCR (qRT-PCR) to quantify Orsay virus replication as previously described (16). Briefly, the extracted viral RNA was diluted 1:100, and 5 µl was used in a TaqMan fast virus one-step qRT-PCR with primers and probe (Orsay_RNA2) that target the Orsay RNA2 segment. Absolute Orsay virus RNA2 copy number was determined by comparison to a standard curve generated using serial dilutions of Orsay virus RNA2 in vitro transcripts. Primers GW314 and GW315 and probe Orsay_RNA1, which target the Orsay virus RNA1 segment, were used to quantify Orsay virus RNA1 abundance (26). To control for variation in the number of animals, Orsay virus RNA levels were normalized to an internal control gene, rps-20, which encodes a small ribosomal subunit S20 protein required for translation in C. elegans (30). The means and standard deviations for three replicate wells are shown for all experiments. Each experiment was performed in two or more biologically independent experiments. Statistical testing was done by Student's t test, unless otherwise stated. Statistically significant comparisons are indicated: ****, P < 0.0005; ***, P < 0.005; **, P < 0.01; *, P < 0.05; NS, not significant (P < 0.05).
Forward genetic screen for mutants unable to induce GFP expression following Orsay virus infection.
The jyIs8;rde-1 reporter strain was maintained on 10-cm plates seeded with 1.5 ml of E. coli OP50. Animals were grown to a stage with a high proportion of L4 larvae and were then collected by washing the plates with water. Animals were then treated with 50 mM EMS for 4 h at 20°C with constant rotation. Animals were washed with M9 buffer and recovered on 10-cm plates for 6 h. Ten L4 P0 animals were transferred to 10 new NGM plates and were allowed to lay about 10 F1 eggs each before being removed. F1 animals were washed away from plates after each animal laid about 20 eggs. Hatched F2 animals were challenged with 800 µl of a 1:10 dilution of Orsay virus filtrate per plate and screened for Viro mutants. For complementation assays, eight males of either sid-3(vir9) or viro-2(vir10) mutant strain were crossed with two hermaphrodites of the Viro mutants to define the ability of their progeny to complement the Viro phenotype.
Genetic mapping through F2 bulk segregant and CloudMap analysis.
Viro-4 mutants were crossed with the WM27 rde-1(ne219) strain, and F1 progenies were chosen and transferred to six-well plates. After 3 days, the F2 progenies were challenged with Orsay virus. A total of 72 F2 Viro animals were picked off F1 plates and transferred to six-well plates. F2 plates were replicated after 1 day of laying eggs. The original F2 plates were challenged again with Orsay virus, and both virus-induced GFP fluorescence and Orsay virus RNA levels were assayed at 3 days postinfection. Thirty-eight wells that yielded exclusively Viro animals and that yielded low RNA levels from replicated F2 plates were pooled, and genomic DNA from pooled samples was extracted using a Qiagen genomic DNA preparation kit according to the manufacturer’s protocol. DNA libraries were prepared using a Nextera library preparation kit (New England Biolabs) and then sequenced using an Illumina MiSeq standard. The raw sequence data were analyzed using the pipeline CloudMap for C. elegans gene mapping (8).
Plasmid construction for gene overexpression.
To construct the drl-1 expression plasmid (Psur-5::drl-1), the entire drl-1 gene was amplified from fosmid WRM0633dH12. The PCR product was digested by SacII and NotI and was then ligated into the plasmid containing the sur-5 promoter.
Ectopic trans-complementation with plasmids encoding DRL-1.
For rescue of Viro-4, a fosmid (WRM0633dH12) or an expression plasmid containing the drl-1 gene was injected into the mutant strain at a concentration of 50 ng/µl or 5 ng/µl, respectively, along with 5 ng/µl of Pmyo-2::YFP as a transgenic marker and 100 ng/µl of DNA ladder (NEB) to help establish the array (31). For Orsay virus infection experiments, ten stable transgenic arrays containing F2 animals were seeded into each well of a six-well plate. After 72 h of egg laying, Orsay virus was added to the plates as described above. At 72 h postinfection the infected animals were assayed for GFP fluorescence and collected for RNA extraction and qRT-PCR to assay virus replication.
Epifluorescence microscopy.
The microscopic visual scanning analysis for Orsay virus infection was carried out using a Leica stereo fluorescence microscope. Images of C. elegans mutant infections were acquired using a Zeiss Axio Imager M2 inverted fluorescence microscope equipped with a Hamamatsu Flash4.0 CMOS camera for fluorescence. Briefly, young adult to adult C. elegans animals were anesthetized with 1 mM levimisol and then put on a 2% dry agarose pad with a coverslip (5 by 5 cm) on top. Images were acquired from both fluorescence channels and bright-field channels.
ACKNOWLEDGMENTS
We thank Michael Nonet for sharing the WRM0633dH12 fosmid. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). L.E.S. is supported in part by the NSF Graduate Research Fellowship Program (grant DGE-1745038). This project was supported in part by NIH grant AI134967 to D.W. and American Heart Association grant 18TPA34230015 to D.W.
REFERENCES
- 1.Felix MA, Ashe A, Piffaretti J, Wu G, Nuez I, Belicard T, Jiang Y, Zhao G, Franz CJ, Goldstein LD, Sanroman M, Miska EA, Wang D. 2011. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol 9:e1000586. doi: 10.1371/journal.pbio.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakowski MA, Desjardins CA, Smelkinson MG, Dunbar TL, Lopez-Moyado IF, Rifkin SA, Cuomo CA, Troemel ER. 2014. Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLoS Pathog 10:e1004200. doi: 10.1371/journal.ppat.1004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang H, Chen K, Sandoval LE, Leung C, Wang D. 2017. An evolutionarily conserved pathway essential for orsay virus infection of Caenorhabditis elegans. mBio 8:e00940-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanguy M, Veron L, Stempor P, Ahringer J, Sarkies P, Miska EA. 2017. An alternative STAT signaling pathway acts in viral immunity in Caenorhabditis elegans. mBio 8:e00924-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan W, Zhou Y, Fan Y, Tao YJ, Zhong W. 2018. Orsay delta protein is required for nonlytic viral egress. J Virol 92:e00745-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long T, Meng F, Lu R. 2018. Transgene-assisted genetic screen identifies rsd-6 and novel genes as key components of antiviral RNA interference in Caenorhabditis elegans. J Virol 92:e00416-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Pen J, Jiang H, Di Domenico T, Kneuss E, Kosalka J, Leung C, Morgan M, Much C, Rudolph KLM, Enright AJ, O'Carroll D, Wang D, Miska EA. 2018. Terminal uridylyltransferases target RNA viruses as part of the innate immune system. Nat Struct Mol Biol 25:778–786. doi: 10.1038/s41594-018-0106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. 2012. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192:1249–1269. doi: 10.1534/genetics.112.144204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chamoli M, Singh A, Malik Y, Mukhopadhyay A. 2014. A novel kinase regulates dietary restriction-mediated longevity in Caenorhabditis elegans. Aging Cell 13:641–655. doi: 10.1111/acel.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. 1996. Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3. Regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J Biol Chem 271:5361–5368. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 11.Chang X, Liu F, Wang X, Lin A, Zhao H, Su B. 2011. The kinases MEKK2 and MEKK3 regulate transforming growth factor-beta-mediated helper T cell differentiation. Immunity 34:201–212. doi: 10.1016/j.immuni.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG, Su B. 2004. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol 5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura K, Johnson GL. 2003. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J Biol Chem 278:36989–36992. doi: 10.1074/jbc.C300313200. [DOI] [PubMed] [Google Scholar]
- 14.Samanta AK, Huang HJ, Bast RC Jr, Liao WS. 2004. Overexpression of MEKK3 confers resistance to apoptosis through activation of NFkappaB. J Biol Chem 279:7576–7583. doi: 10.1074/jbc.M311659200. [DOI] [PubMed] [Google Scholar]
- 15.Kubota Y, Sano M, Goda S, Suzuki N, Nishiwaki K. 2005. The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development 133:263–273. doi: 10.1242/dev.02195. [DOI] [PubMed] [Google Scholar]
- 16.Jiang H, Franz CJ, Wang D. 2014. Engineering recombinant Orsay virus directly in the metazoan host Caenorhabditis elegans. J Virol 88:11774–11781. doi: 10.1128/JVI.01630-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, Ausubel FM. 2002. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 18.Droebner K, Pleschka S, Ludwig S, Planz O. 2011. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res 92:195–203. doi: 10.1016/j.antiviral.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Ludwig S, Wolff T, Ehrhardt C, Wurzer WJ, Reinhardt J, Planz O, Pleschka S. 2004. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett 561:37–43. doi: 10.1016/S0014-5793(04)00108-5. [DOI] [PubMed] [Google Scholar]
- 20.Olschlager V, Pleschka S, Fischer T, Rziha HJ, Wurzer W, Stitz L, Rapp UR, Ludwig S, Planz O. 2004. Lung-specific expression of active Raf kinase results in increased mortality of influenza A virus-infected mice. Oncogene 23:6639–6646. doi: 10.1038/sj.onc.1207883. [DOI] [PubMed] [Google Scholar]
- 21.Erhardt A, Hassan M, Heintges T, Haussinger D. 2002. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology 292:272–284. doi: 10.1006/viro.2001.1227. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Gong R, Qu J, Zhou Y, Liu W, Chen M, Liu Y, Zhu Y, Wu J. 2012. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. J Virol 86:1544–1554. doi: 10.1128/JVI.00688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber M, Watson KA, Selinka HC, Carthy CM, Klingel K, McManus BM, Kandolf R. 1999. Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. J Virol 73:3587–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo H, Yanagawa B, Zhang J, Luo Z, Zhang M, Esfandiarei M, Carthy C, Wilson JE, Yang D, McManus BM. 2002. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J Virol 76:3365–3373. doi: 10.1128/JVI.76.7.3365-3373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stiernagle T. 2006. Maintenance of C. elegans In The C. elegans Research Community, WormBook (ed), WormBook. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang H, Franz CJ, Wu G, Renshaw H, Zhao G, Firth AE, Wang D. 2014. Orsay virus utilizes ribosomal frameshifting to express a novel protein that is incorporated into virions. Virology 450-451:213–221. doi: 10.1016/j.virol.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahringer J. 2006. Reverse genetics In The C. elegans Research Community, WormBook (ed), WormBook. doi: 10.1895/wormbook.1.47.1. [DOI] [Google Scholar]
- 29.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 30.Melo JA, Ruvkun G. 2012. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149:452–466. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stinchcomb DT, Shaw JE, Carr SH, Hirsh D. 1985. Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol Cell Biol 5:3484–3496. doi: 10.1128/MCB.5.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]