

Abstract
The RBP associated with lethal yellow mutation (RALY) is a member of the heterogeneous nuclear ribonucleoprotein family whose transcriptome and interactome have been recently characterized. RALY binds poly-U rich elements within several RNAs and regulates the expression as well as the stability of specific transcripts. Here we show that RALY binds PRMT1 mRNA and regulates its expression. PRMT1 catalyzes the arginine methylation of Fused in Sarcoma (FUS), an RNA-binding protein that interacts with RALY. We demonstrate that RALY down-regulation decreases protein arginine N-methyltransferase 1 levels, thus reducing FUS methylation. It is known that mutations in the FUS nuclear localization signal (NLS) retain the protein to the cytosol, promote aggregate formation, and are associated with amyotrophic lateral sclerosis. Confirming that inhibiting FUS methylation increases its nuclear import, we report that RALY knockout enhances FUS NLS mutants’ nuclear translocation, hence decreasing aggregate formation. Furthermore, we characterize the RNA-dependent interaction of RALY with FUS in motor neurons. We show that mutations in FUS NLS as well as in RALY NLS reciprocally alter their localization and interaction with target mRNAs. These data indicate that RALY’s activity is impaired in FUS pathology models, raising the possibility that RALY might modulate disease onset and/or progression.
INTRODUCTION
The RNA-binding protein (RBP) RALY (RBP associated with lethal yellow mutation) is a ubiquitously expressed member of the heterogeneous nuclear ribonucleoprotein (hnRNP) group (Michaud et al., 1993; Rhodes et al., 1997; Jiang et al., 1998; Busch and Hertel, 2012). RALY forms ribonucleoparticle complexes (RNPs) on interaction with RNA, and it has been identified as a component of both Barentsz and Staufen 2 transport RNPs in rat brain (Fritzsche et al., 2013). RALY contains an RNA recognition motif (RRM) that has been recently validated (Rossi et al., 2017), a predicted consensus sequence for a bipartite nuclear localization signal (NLS), and a peculiar C-terminal domain that contains several embedded glycine-arginine motifs, referred to as the GRR region (Tenzer et al., 2013). In cell lines, RALY localizes not only to the nucleus, with the exclusion of the nucleoli, but also to the cytoplasm, where it associates to polyribosomes (Rossi et al., 2017). Recently, we identified the RALY-associated RNAs in human cell lines, and we found that RALY preferentially binds poly-U stretches within the 3′ untranslated region (UTR) of transcripts (Rossi et al., 2017).
By comparing the set of differentially expressed genes in response to RALY silencing with the list of RALY interacting RNAs, the transcript coding for the protein arginine N-methyltransferase 1 (PRMT1) was found enriched in RALY-containing RNPs and down-regulated on RALY silencing (Cornella et al., 2017; Rossi et al., 2017). PRMT1 belongs to the PRMT family of enzymes responsible for protein arginine methylation, a common posttranslational modification of nuclear RBPs that modulates their localization (Pahlich et al., 2006; Bedford and Clarke, 2009). A well-studied target of PRMT1 is the Fused in Sarcoma protein (FUS) (Dormann et al., 2012; Tradewell et al., 2012; Yamaguchi and Kitajo, 2012; Scaramuzzino et al., 2013).
FUS is an ubiquitously expressed hnRNP and belongs to the FET (FUS, EWS, and TAF-15) family of RBPs that includes also Ewing’s sarcoma RNA binding protein 1 (EWSR1) and TATA-binding protein-associated factor 2N (TAF-15) (Tan and Manley, 2009). FUS mediates a wide range of cellular processes, such as transcription regulation, RNA splicing, DNA repair, and damage response (Deng et al., 2014). In neurons, FUS has been described to play crucial roles in dendritic spine formation and stability, RNA transport, mRNA stability, and synaptic homeostasis (Fujii et al., 2005; Groen et al., 2013; Sephton et al., 2014; Udagawa et al., 2015).
FUS contains an N-terminal low complexity Q/G/S/Y domain, followed by a Gly-rich region, an RRM, two arginine-glycine-glycine (RGG) repeat regions interrupted by a Zinc-finger domain, and a nonconventional proline-tyrosine NLS (PY-NLS) at the C-terminal (Shang and Huang, 2016). The PY-NLS domain interacts with the nuclear import receptor Transportin 1, which translocates PY-NLS–containing proteins to the nucleus (Chook and Süel, 2011). By methylating arginine residues close to the PY-NLS region, PRMT1 decreases FUS binding affinity to Transportin 1 and reduces FUS nuclear translocation (Dormann et al., 2012). The regulation of FUS intracellular localization is particularly relevant since mutations that delocalize the protein to the cytoplasm are associated to Mendelian forms of amyotrophic lateral sclerosis (ALS), a progressive and fatal neurodegenerative disorder that affects both upper and lower motor neurons (MNs) (Bosco et al., 2010; Dormann et al., 2010; Gal et al., 2011; Ito et al., 2011; Kino et al., 2011; Zhang and Chook, 2012).
ALS-linked FUS mutants can form nuclear and cytoplasmic aggregates, sequestering different proteins and RNAs and altering the normal protein–protein and protein–RNA interactions (Kwiatkowski et al., 2009; Vance et al., 2009; Groen et al., 2010; Nomura et al., 2014; Schwartz et al., 2014; Jun et al., 2017). In the cytoplasm, the formation of FUS mutants’ inclusions can alter the dynamics of the stress granules (SGs) (Baron et al., 2013; Vance et al., 2013). SGs are cytoplasmic foci that contain RBPs including the poly-A binding protein 1 (PABP1) and translationally stalled mRNAs (Buchan, 2014). SG marker proteins have been detected in both neuronal and glial cytoplasmic FUS-positive protein inclusions, hypothesizing that SG dysregulation exerts a pathogenic role in motor neurodegeneration (Baron et al., 2013; Vance et al., 2013). Thus, by regulating FUS intracellular localization, PRMT1 is a pivotal determinant of FUS SG formation, interaction with proteins and RNAs, and, therefore, ALS-causative mutants’ cytotoxicity (Dormann et al., 2012; Tradewell et al., 2012; Yamaguchi and Kitajo, 2012; Scaramuzzino et al., 2013). Interestingly, FUS has been detected by mass spectrometry in immunoprecipitated RALY RNPs from HeLa cells (Tenzer et al., 2013).
In this study, we characterize the interaction between RALY and FUS. We show that RALY binds PRMT1 mRNA and regulates its expression in HeLa cells. By down-regulating PRMT1 expression, RALY knockout (KO) decreases FUS arginine methylation, thus increasing FUS mutants’ nuclear import and reducing the number of aggregates in the cytoplasm. We also show that RALY and FUS interact in MNs by means of their RNA-binding domains. More importantly, mutations in FUS alter RALY intracellular localization and its interaction with target mRNAs. These data indicate that RALY’s activity is impaired in cellular models of FUS neurodegenerative pathologies, hence possibly contributing to the disease progression. Moreover, our results suggest that mutations in RALY-encoding gene can alter FUS localization and functionality, therefore potentially inducing a pathological state.
RESULTS
RALY down-regulation impacts FUS methylation and its intracellular localization
We have recently found that PRMT1 mRNA is enriched in RALY RNPs by RNA immunoprecipitation-sequencing (RIP-seq) (Rossi et al., 2017). In addition, PRMT1 was found down-regulated in RALY-silenced cells (Cornella et al., 2017). To validate these RIP-seq data, we performed RIP from HeLa cell extract. The obtained results confirmed that PRMT1 mRNA was significantly enriched in RALY immunoprecipitates compared with normal rabbit immunoglobulin G (IgG) (Figure 1A). We also analyzed GAPDH mRNA as negative control since it was not enriched in RALY RNPs (Rossi et al., 2017; Cornella et al., 2017). We then verified the decrease of PRMT1 mRNA and protein expression in RALY KO HeLa cells by quantitative reverse transcription-PCR (qRT-PCR) and Western blot analysis, respectively. The results showed that PRMT1 was down-regulated in RALY KO cells at both the mRNA and protein levels (Figure 1, B and C). These findings are also in agreement with recent results published by another group (Bondy-Chorney et al., 2017). These data not only confirm the interaction between RALY and PRMT1 mRNA but also show that RALY down-regulation leads to a reduction of the levels of PRMT1 mRNA and protein.
FIGURE 1:

RALY regulates PRMT1 expression, and ensuing FUS arginine methylation. (A) PRMT1 mRNA is enriched in RALY-containing RNPs. The RNA, purified after RALY or normal rabbit IgG IP in HeLa cell extract, was analyzed by qRT-PCR. The mRNA enrichment was calculated relatively to the 10% of RNA input. The scatter plot represents results from three independent experiments. The p value was calculated comparing PRMT1 with GAPDH using an unpaired two-tailed Student’s t test (*p < 0.05). (B) PRMT1 mRNA is down-regulated in RALY KO HeLa cells compared with control. PRMT1 mRNA was analyzed by qRT-PCR and normalized on GAPDH. The scatter plot represents results from three independent experiments. The p value was calculated with unpaired two-tailed Student’s t test (*p < 0.05). (C) PRMT1 protein is down-regulated in RALY KO HeLa cells compared with control cells. PRMT1 protein was analyzed by Western blot and normalized on ACTININ. The column graph represents means ± SEM results from five independent experiments. The p value was calculated with unpaired two-tailed Student’s t test (***p < 0.001). (D) FUS arginine methylation is decreased on RALY KO in HeLa cells. FUS protein was immunoprecipitated from RALY KO and control HeLa cells and analyzed by Western blot. The me-FUS band optical density was quantified and normalized on corresponding immunoprecipitated and input FUS bands. The scatter plot represents results from three independent experiments. The p value was calculated with unpaired two-tailed Student’s t test (*p < 0.05).
A well-described target of PRMT1 is FUS and methylated FUS (me-FUS) is present in cytosolic inclusions (Jun et al., 2017). Since FUS has been identified as a component of RALY RNP complexes (Tenzer et al., 2013), we decided to study the effect of RALY down-regulation on FUS activity. To this aim, we started by assessing whether a down-regulation of RALY could result in a diminished FUS arginine methylation. We first performed FUS immunoprecipitation (IP) from RALY KO and control HeLa cell protein extracts and then measured mono- and dimethylarginine by immunoblotting with a specific antibody (Figure 1D). The results confirmed that RALY KO HeLa cells had lower levels of me-FUS (Figure 1D). Thus, PRMT1 expression was decreased on RALY KO and, as a consequence, FUS was less methylated at arginine residues.
PRMT1-mediated arginine methylation is relevant for regulating FUS intracellular localization. In fact, PRMT1 silencing can restore the nuclear translocation of ALS-linked FUS mutants, reducing the cytoplasmic aggregation (Dormann et al., 2012). For this reason, we analyzed in RALY KO cells the intracellular localization of hemagglutinin (HA)-tagged FUS carrying ALS-linked mutations within the NLS, namely R521C, R521H, and P525L, as well as of wild-type (WT) FUS. We validated FUS mutants by expressing them in HeLa cells and analyzing the intracellular localization and aggregate formation by immunofluorescence and high-content image assay. Our data show that FUS P525L is strongly retained in the cytoplasm and also creates aggregates (Supplemental Figure S1, A and B). To promote aggregate formation, we treated the cells with arsenite and then stained them with anti-HA and anti-PABP1 antibodies, a well-known marker of SGs. As expected, arsenite treatment significantly delocalized to the cytoplasm all the analyzed mutants and recruited them to SGs (Supplemental Figure S1, B and C). In arsenite-treated RALY KO HeLa cells, we observed that the nuclear translocation of FUS mutants was strongly increased compared with control cells, while cytosolic inclusions were markedly reduced (Figure 2A).
FIGURE 2:

Nuclear translocation of wild-type and mutant FUS is increased in RALY knockout cells. (A) Immunofluorescence of RALY KO and control HeLa cells transfected with HA-tagged FUS constructs and treated with arsenite. Cells were stained with anti-HA and anti-PABP1 antibodies and then detected with Alexa Fluor 594- and Alexa Fluor 488–conjugated secondary antibodies, respectively. The scale bar corresponds to 40 μm. (B) The graphs report the quantification of nucleus/cytoplasm signal of HA staining (corresponding to transfected FUS constructs), obtained by high-content image analysis, in untreated (top graph) and arsenite-treated HeLa cells (bottom graph). Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare RALY KO to control cells (*p < 0.05; **p < 0.01; ***p < 0.001). (C) The graph reports the quantification of FUS-HA number of spots per cell, obtained by high-content image analysis. Spots, induced by arsenite treatment, were detected with PABP1 staining and then analyzed for HA-positive staining. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare RALY KO to control cells (**p < 0.01; ***p < 0.001).
To obtain an unbiased quantitative analysis, we performed high-content image analysis. Interestingly, the ratio of the nuclear/cytoplasmic signal for both WT and mutated FUS increased in RALY KO HeLa cells compared with control cells, in untreated as well as arsenite-treated cells (Figure 2B). Consistently, both WT and mutated FUS proteins were less recruited to SGs on arsenite treatment in RALY KO cells compared with controls (Figure 2C). The same results were obtained when HeLa cells were transfected with small interfering RNA (siRNA) against RALY (si-RALY) or control nontargeting siRNA (si-CTRL), confirming that the observed effects were specifically caused by RALY down-regulation (Supplemental Figure S2, A–C). To define whether the reduced recruitment of FUS into SGs in RALY KO cells was due to a general impairment of SG formation, we measured the number of SGs in RALY KO cells by high-content image analysis. We observed that RALY KO increased the cellular susceptibility to metabolic stress since more SGs were detected in comparison to control cells (Supplemental Figure S2D). Thus, the reduction of FUS recruitment to SGs in RALY KO cells is not related to a general decrease of SG formation.
To verify the functional link between RALY knockout, PRMT1 down-regulation and decreased formation of FUS mutants’ cytoplasmic aggregates in RALY KO cells, we overexpressed either green fluorescent protein (GFP)-tagged RALY or GFP-tagged PRMT1 and analyzed WT and mutant FUS cytoplasmic aggregate formation. To this aim, we performed two high-content image assays: in the first assay, we compared the number of aggregates measured in RALY KO cells expressing RALY-GFP with that measured in untransfected RALY KO HeLa and untransfected HeLa cells (Figure 3A); in the second assay, we compared the number of aggregates measured in RALY KO cells expressing PRMT1-GFP with that measured in untransfected RALY KO HeLa cells and in untransfected HeLa cells (Figure 3B). We found that the overexpression of both RALY and PRMT1 significantly increased the formation of FUS mutants’ cytoplasmic aggregates in comparison to RALY KO cells. We found no differences in the intracellular localization of endogenous FUS on RALY silencing compared with control cells (unpublished data). In fact, RGG arginine methylation does not alter the nuclear import of endogenous FUS, because the PY-NLS binds to Transportin 1 tightly enough to allow FUS translocation into the nucleus (Dormann et al., 2012). Taken together, these data propose RALY as a regulator of FUS methylation. However, since PRMT1 overexpression only partially rescued the RALY KO phenotype, other mechanisms regulating FUS trafficking might be affected on RALY down-regulation.
FIGURE 3:
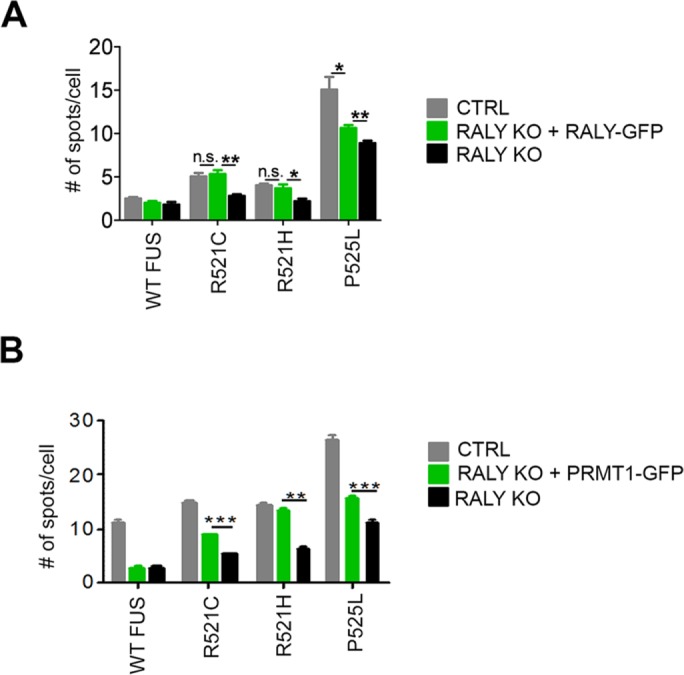
RALY and PRMT1 overexpression partially retrieves FUS mutants’ aggregate formation. (A) The graph reports the quantification of FUS-HA number of spots per cell, obtained by high-content image analysis of control, RALY KO, and RALY KO + RALY-GFP HeLa cells. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare RALY KO + RALY-GFP to either control or RALY KO cells (*p < 0.05; **p < 0.01). n.s., not significant. (B) The graph reports the quantification of FUS-HA number of spots per cell, obtained by high-content image analysis of control, RALY KO, and RALY KO + PRMT1-GFP HeLa cells. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare RALY KO + PRMT1-GFP to either control or RALY KO cells (**p < 0.01; ***p < 0.001). n.s., not significant.
Development-dependent regulation of RALY and FUS expression in nerve cells
Considering the above results, we asked whether FUS and RALY could physically interact in nerve cells. Since RALY was not characterized in neuronal tissues, we started analyzing its expression in mouse cortex and spinal cord at different stages of development. Western blot analysis of protein extracts revealed that RALY expression increased during development in both mouse cortex and spinal cord (Figure 4A). In the spinal cord, we compared the expression pattern of RALY with FUS, TAF-15, EWSR1, and TAR DNA-binding protein 43 (TDP-43), all RNA-binding proteins whose alterations have been also correlated with either familial or sporadic ALS (Kabashi et al., 2008; Neumann et al., 2009). Similarly to RALY, TDP-43 and FUS proteins showed an increase in their expression during development (Figure 4A). In contrast, the levels of EWSR1 and TAF-15 proteins remained unchanged, indicating that their expression was not development dependent (Figure 4A). We successively analyzed RALY intracellular expression pattern in primary cultures of murine MNs. RALY was detected not only within the nuclear compartment but also along axonal processes. Moreover, distinct particles were clearly present in growth cones (Figure 4B). Together, these data show that the expression of RALY increases during neuron and MN aging, suggesting an intriguing role during development and that RALY is present along the axons of cultured MNs.
FIGURE 4:

FUS and RALY display similar expression profiles and are components of the same RNPs in MNs. (A) RALY expression increases with age in the mouse nervous system. Protein expression was analyzed by Western blot in mouse tissues at embryo-day 18 (E18) and postnatal days 1 (P1), P3, P7, P15, P30, and P90. In the cortex (top panel) RALY was analyzed; in the spinal cord (bottom panel) RALY, EWSR1, TAF-15, FUS, and TDP-43 were analyzed. As protein loading control, β-III TUBULIN was detected. (B) RALY localizes to MN axons and growth cones. Immunofluorescence assay displaying RALY in green and Tau in red. The scale bar corresponds to 10 μm. (C) Size fractionation of NSC-34 cell line RNA granules. Each fraction was processed for Western blot and probed with the indicated antibodies indicated. One-tenth of the cell extract was loaded in the first well as input control. Cell extract was either untreated (top panel) or RNase treated (bottom panel). RALY- and FUS-containing RNPs were enriched in fractions 4–9 (RALY) or fractions 2–6 (FUS), respectively. PABP1 bands indicate that the isolated RNPs were still intact. RPL26 serves as a marker to follow ribosomal fractionation. RNase treatment (bottom panel) shifted all proteins, including the RNP marker PABP1, toward lighter fractions on top of the gradient, meaning that RALY- and FUS-containing complexes were true RNA granules. (D) EWSR1, TAF-15, FUS, PABP1, and TDP-43 coimmunoprecipitate with RALY in an RNA-dependent manner. RALY was immunoprecipitated from either untreated or RNase-treated NSC-34 protein extracts. Normal rabbit IgG IP was performed as a control. Immunoprecipitated proteins were processed for Western blot and probed with the indicated antibodies. (E) RALY and FUS colocalize in nucleus and cytoplasm. PLA was performed in HeLa and NSC-34 cells to analyze RALY and FUS localization. Fluorescent signals identified colocalization spots in both nucleus (labeled by DAPI staining in blue) and cytoplasm (outlined by bright field acquisition). The scale bar corresponds to 40 μm. (F) RALY and FUS colocalize in MN axon. PLA was performed on MN primary cells and combined with SMI32 staining. Fluorescent signals identified colocalization spots in both nucleus, cell soma, and axon. The scale bar corresponds to 40 μm.
RALY and FUS are components of the same RNPs
To characterize RALY and FUS RNP complexes at biochemical level, we determined their sedimentation profile on density sucrose gradient, an assay that was previously employed to isolate endogenous RNPs (Kiebler et al., 1999; Fritzsche et al., 2013). We prepared cell lysates from the MN-like cell line NSC-34, a hybrid cell line produced by fusion of neuroblastoma with mouse MN-enriched primary spinal cord cells (Cashman et al., 1992). Importantly, NSC-34 cells expressed RALY and FUS proteins (Supplemental Figure S3). The fractionation pattern of RALY and FUS was compared with PABP1 and with the ribosomal marker RPL26. By adding RNase inhibitors, we observed a distinct fractionation pattern for each analyzed protein. In addition, the presence of PABP1 in discrete fractions suggested that RNPs were still intact and contained polyadenylated mRNAs. Interestingly, we observed the cosedimentation of FUS and RALY in at least three fractions (Figure 4C). Sample treatment with RNase shifted a significant portion of all analyzed proteins toward lighter fractions, thus confirming that the signal corresponded to intact RNP complexes (Figure 4C). To analyze whether the cofractionation reflected the presence of RALY and FUS in the same complexes, we carried out coimmunoprecipitation assay (co-IP) in NSC-34 cell protein extract using an anti-RALY antibody. Confirming RALY proteomic results in HeLa cells (Tenzer et al., 2013), we found that FUS protein, as well as PABP1, was present in the endogenous RALY immunoprecipitate (Figure 4D). In addition, though not detected by proteomic approach in HeLa cells (Tenzer et al., 2013), EWSR1, TAF-15, and TDP-43 proteins also coprecipitated with RALY in NSC-34 cells (Figure 4D). To assess the role of RNA in FUS-RALY interaction, the cell lysate was treated with RNase to disassemble RNP complexes prior to RALY immunoprecipitation. The treatment with RNase strongly reduced the association of RALY with FUS, TAF-15, TDP-43, and PABP1. Notably, the association of RALY with EWSR1 was not completely abolished after RNase treatment, suggesting the presence of an additional protein–protein interaction (Figure 4D). Reciprocal co-IP using an anti-FUS antibody confirmed the interaction (unpublished data). Taken together, these data suggest that RALY interacts in an RNA-dependent manner with FUS, TAF-15, EWSR1, PABP1, and TDP-43 in NCS-34 cells.
To visualize RALY-FUS interaction within the cell, we performed the proximity ligation assay (PLA) (Fredriksson et al., 2002). This technique leads to the detection of specific fluorescent dots linked to close proximity protein interactions (40 nm or below). Two different primary antibodies specific for RALY and FUS were used to visualize their interaction in HeLa, NSC-34 cells, and primary MNs (Figure 4, E and F). In HeLa cells, FUS and RALY PLA signal was detected in both nucleus and cytoplasm (Figure 4E). In NSC-34 cells, the signal was detected in the nucleus, cytoplasm, and, interestingly, also along processes indicating that RALY and FUS interact in all these compartments (Figure 4E). The specificity of PLA was confirmed in HeLa cells as well as in HeLa cells lacking RALY expression by using either only one primary antibody in the presence of both secondary antibodies or only secondary antibodies (Supplemental Figure S3, B and C). In both cases PLA signal was not detected, confirming the specificity of the assay. In primary MN cultures, PLA was combined with SMI32 staining to assure cell identity and visualize all the processes. As for NSC-34, RALY and FUS PLA signals were detected in the nucleus but also in cell body and processes of MNs (Figure 4F). In conclusion, RALY and FUS are components of the same RNP complexes in MNs not only in nuclear and cytosolic compartments but also along neuronal processes.
We then assessed which domain of FUS was required for forming RNP complexes with RALY. We used HeLa cells expressing deleted mutants of FUS tagged with GFP (Supplemental Figure S4A) (Groen et al., 2013). Western blot analysis showed that all FUS-GFP constructs have a stable expression at the expected molecular weight and without affecting RALY protein levels (Figure 5A). Moreover, all FUS-GFP constructs localized to the nuclear compartment, since none of the deletions compromised the NLS (Supplemental Figure S4B). The interaction of the constructs with endogenous RALY was determined by IP using an anti-GFP antibody followed by Western blot detection. The results revealed that only Δ360-501 FUS-GFP was undetectable in the co-IP with endogenous RALY, indicating that FUS might interact through its portion bearing the RGG regions and the Zn finger (Figure 5B). Since RGG regions and the Zn finger motif are required for the binding of FUS to nucleic acids (Iko et al., 2004; Schwartz et al., 2013), these findings support the RNA dependence of FUS-RALY interaction.
FIGURE 5:

RALY and FUS interact through their RNA-binding domains. (A) Western blot of protein extracts from HeLa cells, transfected with FUS-GFP constructs (Supplemental Figure S4), and probed with anti-GFP, anti-RALY, and anti-TUBULIN antibodies. (B) RALY coimmunoprecipitates with all FUS-GFP constructs but not with the one missing the RRM (Δ360-501). Co-IP experiment was performed by immunoprecipitating with anti-GFP antibody and probing the Western blot with anti-RALY antibody. (C–E) FUS-HA coimmunoprecipitates with all RALY-MYC/DDK constructs (Supplemental Figure S5) except the two missing the RRM (E). Co-IP experiment was performed by immunoprecipitating with anti-FLAG M2 affinity gel and probing the Western blots with anti-HA antibody and anti-Myc antibody. UT, untransfected; *, light IgG chain; **, heavy IgG chain.
Successively, we analyzed the regions of RALY needed for the interaction with FUS. We cloned different myelocytomatosis (MYC)/DDK-tagged deleted constructs of RALY and transfected them in HeLa cells to assess the intracellular localization (Supplemental Figure S5, A and B). All the deleted recombinant RALY proteins accumulate in the nucleus except full-length RALY and RALY(Δ1-142), whose signal was detected also in the cytoplasm (Supplemental Figure S5B). To analyze the interaction between FUS and RALY domains, we cotransfected RALY-MYC/DDK and FUS-HA constructs and performed IP with the anti-FLAG M2 affinity gel. FUS-HA was clearly detected in IPs of full-length RALY and RALY(Δ144-306) (Figure 5, C and D). A reduced FUS-HA signal was observed with RALY(Δ226-306) (Figure 5D). In contrast, we failed to immunoprecipitate FUS-HA with both RALY(Δ1-142) and RALY(Δ1-224) (Figure 5E). Both these constructs were missing the N-terminal region containing the RRM domain. Taken together, these data show that the N-terminal domain of RALY containing the RRM is required for the interaction with FUS.
ALS-linked FUS mutants maintain their interaction with RALY
Point mutations in FUS-coding gene have been linked to familial ALS (Kwiatkowski et al., 2009; Vance et al., 2009). In particular, mutations occurring in the PY-NLS cause the mislocalization of FUS that remains in the cytoplasm forming aggregates (Dormann et al., 2010; Lagier-Tourenne et al., 2010; Vance et al., 2013; Schwartz et al., 2014). Ultimately, this altered nucleus/cytoplasm equilibrium of FUS might change the composition and function of RNPs. Therefore, we asked whether ALS-linked point mutations might affect FUS-RALY interaction. We analyzed the interaction using HA-tagged FUS carrying ALS-linked mutations within the NLS, namely R521C, R521H, and P525L, stably expressed by NSC-34 cells. By immunofluorescence with anti-HA antibody, we verified FUS mutants’ expression and localization in NSC-34 cells (Figure 6A). Images showed that FUS R521C, R521H, and especially P525L are retained in the cytoplasm more than WT. Moreover, arsenite treatment confirmed that FUS NLS mutants are recruited to SGs (Figure 6A). These results corroborate the NSC-34 model in comparison with previously published data (Bosco et al., 2010; Dormann et al., 2010; Lenzi et al., 2015). We detected the coprecipitation of endogenous RALY with all FUS mutants, meaning that ALS-linked FUS mutants maintain the interaction with RALY (Figure 6B).
FIGURE 6:

ALS-linked FUS mutants interact with RALY. (A) FUS mutants are retained in the cytoplasm and recruited to SGs in differentiated NSC-34 cells. NSC-34 cells were induced for 48 h, treated with arsenite (or control), and processed for immunofluorescence. The staining was performed with anti-HA and anti-PABP1 antibodies and revealed with Alexa Fluor 594 and 488, respectively, DAPI and CellMask Deep Red plasma membrane stain. The scale bar corresponds to 40 μm. (B) RALY coimmunoprecipitates with WT and mutant FUS-HA. In the top panel, a co-IP assay was performed by immunoprecipitating WT and mutant FUS-HA with anti-HA antibody from doxycycline-induced NSC-34 cell extracts. Normal rabbit IgG were used as negative control. Immunoprecipitated proteins were processed for Western blot and probed with anti-HA and anti-RALY antibodies. In the bottom panel, Western blot analysis of input cell extracts probed with anti-HA antibody to detect FUS-HA construct’s expression, anti-RALY, and anti-TUBULIN antibody to verify homogeneous loading.
These results prompted us to investigate whether RALY could be mislocalized and recruited to cytoplasmic inclusions by FUS mutants.
ALS-linked FUS mutants alter RALY interaction with its targets mRNAs
Having assessed the interaction between RALY and FUS in NSC-34 cells, we tested whether ALS-linked FUS mutants could change RALY’s interaction with its target mRNAs. To address this point, we performed RALY RIP in NSC-34 cells stably expressing either WT or mutated FUS and analyzed the coimmunoprecipitated RNA by qRT-PCR. The presence of the following transcripts was assessed: H1fx and Raly as mRNAs bound by RALY; Dctn1, Sod1, and Sncb as mRNAs bound by both RALY and FUS; and Gapdh and B2m as negative controls (Lagier-Tourenne et al., 2012; Rossi et al., 2017). By normalizing on Gapdh, we did not observe any significant variation concerning the selected target mRNA expression levels in NSC-34 cells expressing FUS mutants compared with WT (Supplemental Figure S6A). After confirming the presence of selected mRNAs in RALY RNPs isolated from NSC-34 cells expressing WT or FUS NLS mutants (Supplemental Figure S6B), we compared the enrichment obtained from cells expressing each FUS mutant with the corresponding result obtained from cells expressing WT FUS (Figure 7A). The statistical analysis showed that the expression of FUS NLS mutants significantly decreased the interaction of RALY with all the analyzed target mRNAs (Figure 7A).
FIGURE 7:

ALS-linked FUS mutants retain RALY in the cytoplasm, recruit it to cytosolic aggregates, and alter its interaction with mRNAs. (A) FUS mutants impair RALY interaction with Dctn1, Sod1, Sncb, Hifx, and Raly mRNAs. Doxycycline-induced NSC-34 cell extracts were processed for RIP analysis. The graph shows the statistical analysis of five independent experiments, normalized on Gapdh. To compare all the experiments, the yield was set equal to 1 for RALY RIPs in cells expressing WT FUS; hence the yield for RALY RIPs in cells expressing FUS mutants was calculated proportionately. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare FUS mutant with WT FUS expressing cells (*p < 0.05; **p < 0.01). (B) FUS mutants R521C, R521H, and P525L retain RALY in the cytoplasm. Doxycycline-induced NSC-34 cell extracts were processed for nucleus/cytoplasm separation and Western blot. On the right, the graph represents the mean of five independent experiments. Bars indicate means ± SEM, and p values were calculated with unpaired two-tailed Student’s t test to compare FUS mutant with WT FUS-overexpressing cells (*p < 0.05). (C) FUS mutants significantly retain RALY in the cytoplasm. The graph, obtained by high-content image analysis, reports the quantification of RALY nucleus/cytoplasm signal, in NSC-34 cells expressing WT or mutated FUS-HA, detected by immunostaining with anti-RALY and anti-HA antibody, respectively. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare FUS mutants with WT FUS-expressing cells (*p < 0.05; ***p < 0.001). (D) FUS mutants can recruit endogenous RALY to cytosolic aggregate triggered by arsenite treatment. Immunofluorescence images show FUS-HA staining (red) and endogenous RALY (green) in cytosolic aggregates. HeLa cells were transfected for 24 h before fixation. The scale bar corresponds to 10 μm.
To investigate which might be the possible outcome of the decreased RALY binding to mRNAs, we analyzed the protein levels of its targets DYNACTIN 1, H1X, SOD1, and BETA SYNUCLEIN. We also checked RALY protein levels that resulted unchanged on the expression of FUS mutants (Supplemental Figure S6, C and D). The Western blot results showed that, while BETA SYNUCLEIN expression was slightly lower but not statistically significant, DYNACTIN1, H1X, and SOD1 levels were decreased in NSC-34 cells expressing FUS mutants compared with WT (Supplemental Figure S6, C and D).
ALS-linked FUS mutants alter RALY intracellular localization
Having observed that FUS mutants alter RALY’s interaction with its target mRNAs, we hypothesized that ALS-linked FUS mutants could change RALY’s intracellular localization. We analyzed RALY by Western blot in nuclear and cytoplasmic extracts from NSC-34 cells expressing WT and mutant FUS-HA. Interestingly, we found that RALY became significantly more cytoplasmic on overexpression of FUS mutants, in particular on the overexpression of FUS P525L (Figure 7B). We then validated the Western blot data by performing a high-content image analysis. RALY nuclear/cytoplasmic signal analysis was performed on selection of FUS-HA expressing cells. The comparison with WT FUS cells confirmed that RALY is retained to the cytoplasm by FUS mutants’ expression (Figure 7C). To test whether endogenous RALY could also be recruited to FUS mutant cytoplasmic aggregates formed on arsenite treatment, we performed immunostaining on HeLa cells transfected with either WT or mutated FUS. The obtained images showed that endogenous RALY remained mainly nuclear when WT FUS was expressed while shifted to aggregates together with FUS NLS mutants (Figure 7D).
These data show that RALY interacts with FUS NLS mutants, consequently modifying its own intracellular distribution, recruitment to cytoplasmic aggregates, and interaction with specific target mRNAs. Moreover, our data indicate that the decrease of the binding of RALY to transcripts on expression of FUS mutants may result in a decrease of the corresponding encoded proteins. Since RALY associates to polyribosomes (Rossi et al., 2017), this effect may be related to an altered translational control. Alternatively, the reduction of RALY in the nucleus due to its cytoplasmic delocalization might lead to a diminished nucleus-cytoplasmic transport of the associated mRNAs.
RALY NLS mutant alters FUS intracellular localization and interaction with RNAs
Given the capability of FUS mutants to alter RALY’s intracellular localization and its interaction with RNAs, we investigated whether mutations in RALY could affect endogenous FUS. RALY contains a predicted consensus sequence for a potential bipartite nuclear localization signals (NLS1 and NLS2). We first validated the NLSs in HeLa cells by cloning either NLS1 or NLS2 or both at the N-terminus of an enhanced GFP (EGFP) and then measured the nuclear signal by high-content image assay (Supplemental Figure S7A). All constructs increased the ratio of the nucleus/cytosol GFP signal, with the full-length NLS showing the stronger nuclear translocation. NLS1 alone led to an intermediate accumulation and the NLS2 just a moderate effect (Supplemental Figure S7, B and C). Thus, the predicted consensus sequence in the middle region of RALY protein contains a functional NLS.
We then mutated the NLS to increase RALY’s cytoplasmic localization. Starting from RALY-GFP construct, two NLS1 and two NLS2 amino acids were mutated to l-alanines (Supplemental Figure S7E). After transfection of HeLa cells, we analyzed the nucleus/cytosol signal and found that the mutated protein (MUT RALY) was indeed more cytosolic compared with the WT (Supplemental Figure S7D). As the immunostaining images show, MUT RALY formed cytosolic aggregates that were likely SGs being positive for PABP1 staining, and the effect was even stronger on arsenite treatment (Supplemental Figure S7E). To analyze whether RALY mutants could delocalize endogenous FUS to the cytoplasm, we performed immunostaining on HeLa cells transfected with either WT or MUT RALY. Interestingly, we observed that MUT RALY retains FUS to the cytoplasm (Figure 8A). By inducing metabolic stress with arsenite treatment, we detected some cytoplasmic spots positive for both MUT RALY and FUS (Figure 8A). To quantify FUS delocalization to cytoplasm on MUT RALY expression, we carried out a high-content imaging assay on HeLa cells transfected with either WT or MUT RALY followed by immunostaining for endogenous FUS. The results confirmed that MUT RALY increased the cytosolic retention of endogenous FUS (i.e., decreased the nucleus/cytosol signal ratio), both with and without arsenite treatment (Figure 8B). Then, we tested whether RALY mutants were also able to alter FUS interaction with its target RNAs. To parallel the experiments describing FUS mutants’ effects on endogenous RALY, we transfected NSC-34 cells with either WT or MUT RALY for 24 h. Hence, we performed a RIP assay by immunoprecipitating FUS and analyzing with qRT-PCR four target RNAs: Dctn1, Sncb, Sod1, and Pink (Lagier-Tourenne et al., 2012). Data were normalized on Gapdh, which was not enriched in FUS RIP extracts. The results show that FUS binding to all the analyzed mRNAs was diminished on expression of MUT RALY (Figure 8C).
FIGURE 8:
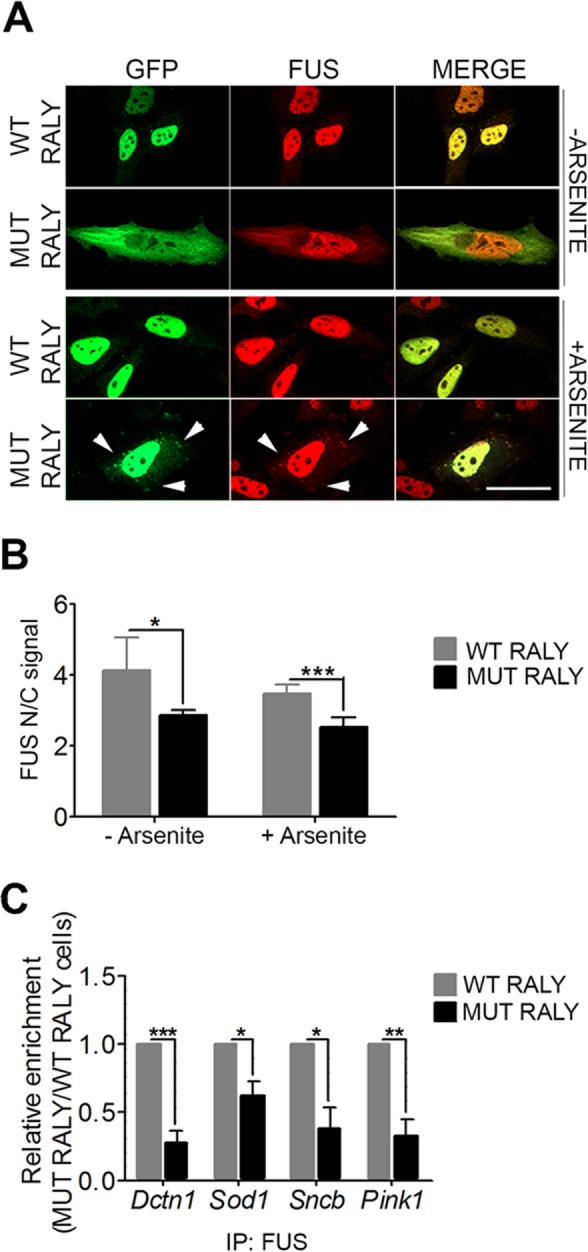
RALY mutant retains FUS in the cytoplasm and alters its interaction with mRNAs. (A) MUT RALY retains endogenous FUS in the cytoplasm and recruits it to cytosolic aggregates induced by arsenite treatment (arrowheads). Immunofluorescence images show WT or MUT RALY-GFP (green) and endogenous FUS (red) stained with anti-GFP and anti-FUS antibody, respectively. HeLa cells were transfected for 24 h before fixation. The scale bar corresponds to 20 μm. (B) MUT RALY significantly retains FUS in the cytoplasm. The graph reports the quantification of FUS nucleus/cytoplasm signal, obtained by high-content image analysis, in untreated and arsenite-treated HeLa cells. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare MUT RALY with WT RALY-overexpressing cells (*p < 0.05; ***p < 0.001). (C) RALY mutants impair FUS interaction with Dctn1, Sod1, Sncb, and Pink1 mRNAs. The graph shows the statistical analysis of five independent experiments, normalized on Gapdh. To compare all the experiments, the yield was set equal to 1 for FUS RIPs in cells expressing WT RALY; hence the yield for FUS RIPs in cells expressing MUT RALY was calculated proportionately. Bars indicate means ± SEM of five replicates, and p values were calculated with unpaired two-tailed Student’s t test to compare MUT RALY with WT RALY-overexpressing cells (*p < 0.05; **p < 0.01; ***p < 0.001).
In conclusion, RALY mutant with a higher cytosolic retention is able to alter endogenous FUS localization and the interaction with its target mRNAs. These findings hint at the possibility that mutations occurring in RALY NLS induce phenotype similar to FUS-linked neurodegenerative disorders.
RALY and FUS do not cooperatively regulate the expression levels of target mRNAs
We observed that the expression of ALS-linked FUS mutants alters RALY binding to the target mRNAs which are also bound by FUS (Figure 7A) and diminishes their protein level (Supplemental Figure S6, C and D). Therefore, we investigated whether RALY and FUS cooperatively regulate gene expression. To this aim, we transiently silenced FUS in WT and RALY KO HeLa cells, and measured mRNA and protein levels of DCTN1, SOD1 and FUS. First, we checked FUS expression and found that FUS expression is down-regulated in RALY KO cells at both protein and mRNA levels (Supplemental Figure S8). Second, we confirmed the effectiveness of the silencing in both WT and RALY KO cells by comparing FUS protein and mRNA levels in si-FUS transfected cells with si-CTRL transfected ones and found that FUS expression was almost abolished after 72 h of si-FUS treatment (Supplemental Figure S8).
Interestingly, our results show that, in RALY KO cells compared with WT, DYNACTIN1, SOD1 and BETA SYNUCLEIN were strongly down-regulated at protein level, as well as at mRNA level for DYNACTIN1 and SOD1 (Supplemental Figure S8, B and C). The decreased expression of H1X protein and mRNA confirmed what we have previously published (Rossi et al., 2017). Our data report that FUS silencing does not change the expression of the analyzed targets both in WT and in RALY KO cells, with exception of a slight increase of SOD1 protein in WT RALY/si-FUS cells (Supplemental Figure S8, A and B). These results obtained in HeLa cells are in agreement with what previously reported by RNA-seq analysis on FUS KO mouse brain, where DCTN1, SOD1, and SNCB are bound by FUS, but their expression levels are not influenced by FUS (Lagier-Tourenne et al., 2012). Our experiments, however, show no alterations for H1X expression on FUS silencing, but this divergence may be due to the different experimental models used.
DISCUSSION
The transport of RNA by RBPs toward dendrites and axons is a fundamental process for the correct development of neurons and the maintenance of their physiology (Smith et al., 2014; Glock et al., 2017). Underlying the importance of RBPs, an increasing number of mutations hitting their coding genes have been identified as causative drivers or associated with neurological pathologies. In this study, we characterized the interaction between FUS, an RBP deeply involved in the onset of ALS, and the hnRNP RALY. We described the behavior of different mutated FUS forms on RALY down-regulation and mutation and vice versa. Arginine methylation and the binding to Transportin1 control FUS localization, condensation, and dissociation in the form of fibrils, gels, or droplets (Guo et al., 2018; Hofweber et al., 2018; Mikhaleva and Lemke, 2018; Qamar et al., 2018; Yoshizawa et al., 2018).
ALS-linked FUS mutants are characterized by the formation of protein and RNA containing aggregates in the cytoplasm. Arginine methylation by PRMT1 is a key determinant of PY-NLS FUS mutant localization, where RGG methylation by PMRT1 abrogates nuclear translocation and determines cytosolic retention with aggregates formation. Abolishing RGG methylation is sufficient to overcome PY-NLS mutations and restore the nuclear localization of the mutated protein (Dormann et al., 2012). In addition, it has been recently proposed that Transportin1 is not only implicated in nucleocytoplasmic transport but also acts as a chaperone that can regulate the condensation and/or dissociation of FUS assemblies (Guo et al., 2018; Hofweber et al. 2018; Qamar et al., 2018; Yoshizawa et al., 2018). Therefore, ALS mutations decreasing the affinity with Transportin1 impair not only the nuclear transport but also the chaperone activity that contributes to the formation of the pathologic aggregates (Guo et al., 2018; Hofweber et al., 2018).
After validating PRMT1 mRNA binding and expression regulation by RALY in our cellular model, we found that PRMT1 down-regulation on RALY KO results in diminished FUS methylation levels (Figure 1). As a consequence, PY-NLS mutated FUS is more translocated to the nucleus and less recruited to SGs (Figure 2). Interestingly, in patients with frontotemporal lobar degeneration (FTLD) with no identified genetic mutations, hypomethylated wild-type FUS has been detected in cytoplasmic inclusions (Neumann et al., 2009; Dormann et al., 2010, 2012; Mackenzie et al., 2010; Urwin et al., 2010; Snowden et al., 2011; Rainero et al., 2017). Indeed, emerging evidence has showed that hypomethylated FUS induces the formation of heterogeneous gel-like assemblies that disrupt RNP granule function (Hofweber et al., 2018; Qamar et al., 2018). Although no mutations in RALY encoding gene have been identified yet, the data we obtained from RALY KO cell model point out impairments in RALY function as a possible cause of hypomethylation.
By overexpressing either RALY-GFP or PRMT1-GFP in RALY KO HeLa cells, FUS mutant incorporation into aggregates is partially retrieved, thus corroborating that RALY participation in PRMT1 regulation could be considered part of the intricate mechanism causing pathological aggregates formation (Figure 3). Whether PRMT1 silencing boosts or rescues neurodegeneration induced by FUS mutants is still a matter of debate and seems to depend on the expressed FUS mutant for many reasons. First, FUS cytoplasmic inclusions can have methylated, such as in ALS, or hypomethylated residues, such as in FTLD, and can, by any means, lead to neuronal loss (Dormann et al., 2012). Second, the rescued nuclear localization and diminished aggregate formation of P525L FUS by PRMT1 down-regulation (Figure 2) does not imply that FUS toxicity is eliminated, its functionality retrieved, and neuronal survival reestablished. Third, Jun and colleagues showed that R521C FUS aggregate formation, and ensuing neurotoxicity, is enhanced by PRMT1 silencing, while PRMT1 overexpression rescues neurite degeneration (Jun et al., 2017). Our results indicate that tuning RALY expression represents an indirect path for modulating FUS methylation and aggregate formation. However, it is necessary to take into account that this molecular mechanism may either reduce or boost FUS cytotoxicity, depending on FUS mutants and cellular type.
We characterize RALY-FUS interaction and demonstrate that they bind to each other in an RNA-dependent manner (Figure 5). In addition, we show that RALY coimmunoprecitates with PABP1 and with other FUS interactors such as EWSR1, TAF-15, and TDP-43 (Figure 5). Interestingly, FUS and RALY proteomic studies reveal that they share a wide number of protein interactors (Tenzer et al., 2013; Kamelgarn et al., 2016). Notably, RALY, FUS, TDP-43, EWSR1 and TAF-15 bind chromatin and are all involved in transcription regulation (Lalmansingh et al., 2011; Yang et al., 2014; Izhar et al., 2015; Cornella et al., 2017).
Indeed, the ALS-linked FUS mutants interact with RALY and are able to delocalize RALY to the cytosol, recruit it to aggregates, and diminish its binding to RNAs (Figures 6 and 7).
These events lead to a reduction of DYNACTIN 1, H1X, and SOD1 proteins expression, suggesting processing alterations for RALY target mRNAs in cells expressing FUS ALS-linked mutants (Supplemental Figure S6, B and C). Dctn1 and Sod1 mRNAs are shared FUS and RALY targets (Lagier-Tourenne et al., 2012; Rossi et al., 2017), and therefore their diminished protein expression may be due to alterations in both FUS- and RALY-mediated processing in NSC-34 cells expressing FUS mutants. Interestingly, loss-of-function mutations in Dctn1 gene cause ALS and a slow progressive hereditary motor neuropathy (Puls et al., 2003; Münch et al., 2004; Puls et al., 2005). Furthermore, a decreased expression of Dctn1 has been reported in ALS murine models and patients (Ikenaka et al., 2012; Ikenaka et al., 2013; Kuz´ma-Kozakiewicz et al., 2013). SOD1 is also a well-known gene involved in genetic forms of ALS (Renton et al., 2014). In contrast, nothing is yet known about the function of H1X in neurons. Interestingly, we also show that these targets are down-regulated in RALY KO cells (Supplemental Figure S8). Although FUS knockdown has no additive effect in RALY KO cells, our results suggest that these two proteins associate in RNPs, and bind to common mRNAs, but exert different functions.
Further experiments are required to identify the mechanisms by which RALY regulates the expression level of the above-mentioned proteins. Recently, we showed that RALY can regulate RNA metabolism in HeLa cells transcriptionally and posttranscriptionally (Rossi et al., 2017). We reported that RALY is able to cosediment with ribosomes and polysomes, suggesting that it might have a potential role in the translational control of specific transcripts. Moreover, we demonstrated that RALY binds E2F1 mRNA within poly-U in 3′UTR and regulates its stability (Cornella et al., 2017). In addition, RALY associates to gene promoters and acts as a transcriptional cofactor, together with the long noncoding RNA Lexis, at the level of cholesterol biogenesis pathway in mouse liver (Sallam et al., 2016). We also characterized the association of RALY with chromatin and demonstrated that RALY interacts with transcriptionally active chromatin. This association is partially abrogated on RNA degradation (Cornella et al., 2017).
Several mechanisms can be responsible of the intracellular delocalization of RALY observed on expression of ALS-linked FUS mutants. For instance, we can hypothesize that RALY might be passively trapped in the gel-like/fibril aggregates generated by FUS mutants. Alternatively, we cannot exclude that nuclear/cytoplasmic shuttling alterations caused by nuclear pore dysfunction might also be the driving factor for RALY mislocalization.
In addition, we report that the expression of NLS-mutated RALY, which is delocalized to the cytoplasm, can retain FUS into the cytoplasm and decrease its binding to mRNAs (Figure 8). Notably, the analyzed mRNAs (i.e., Dctn1, Sod1, Pink1, and Sncb) that are FUS targets with decreased binding by NLS-mutated RALY expression are all known to play relevant roles in neuron physiology. Therefore, it would be of utmost interest to check for mutations in the RALY encoding gene in FUS-linked neurodegenerative disease patients.
As shown above, RALY interacts with TAF-15, EWSR1, and TDP-43, all proteins that harbor a prionlike domain and are therefore prone to form cytoplasmic inclusions in diverse neurodegenerative diseases (Ito et al., 2017). Given the link with these proteins, we investigated whether RALY could possess a similar domain through a bioinformatics analysis with the database PLAAC (prion-like amino acid composition) (Lancaster et al., 2014). Interestingly, the analysis detected the presence of a prionlike domain in the C-terminal region of RALY, more precisely in coincidence with the RGG region (amino acids 228–258) (Supplemental Figure S9). Together with the previous observations, this analysis enforces the possible involvement of RALY in the progression of neurodegenerative pathologies.
To support the importance of RALY-FUS interaction and interference when mutated, reciprocal alterations of protein localization in neurodegenerative models and patients’ samples have already been reported in literature. For instance, similarly to our findings, FUS has been described to interact with protein survival motor neuron (SMN), and ALS-linked FUS mutants alter SMN distribution by recruiting the protein to cytoplasmic inclusions (Groen et al., 2013). Another RBP, the RGNEF (Rho guanine nucleotide exchange factor), was detected in neuronal cytoplasmic inclusions of mutated FUS- and TDP-43-ALS patients’ spinal cord (Keller et al., 2012). In the same study, wild-type TDP-43 and FUS were detected in neuronal cytoplasmic inclusions of mutated FUS-ALS and TDP-43-ALS patients’ spinal cord, respectively.
In conclusion, we show that RALY and FUS interact and, if mutated, they can reciprocally alter their intracellular localization. This phenotype is likely driven by the RNA-mediated interaction but also by a modulation of PRMT1 expression operated by RALY that affects the nuclear/cytoplasmic distribution of FUS. Moreover, we report for the first time as far as we know that the altered distribution of both RALY and FUS impacts their functionality by decreasing their RNA-binding capacity. Our results propose RALY as a modulator of FUS and as a new possible player in neuronal physiology and pathological mechanisms.
MATERIALS AND METHODS
Cell cultures, transient transfections, and pharmacological treatments
NSC-34 (Tebu-BIO), HeLa (American Type Culture Collection), and SH-SY5Y cell lines were cultured in DMEM supplemented with 10% (vol/vol) fetal bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine (Lonza). Each cell line was tested for micoplasma by the Cell Tech Facility at CIBIO using the PlasmoTest–Mycoplasma Detection (InvivoGen). Cells were transiently transfected using the TransIT-LT1 Mirus according to the manufacturer’s protocol. For silencing, cells were transfected with ON-target plus Human RALY siRNA L-0123920 SMART-pool, siGENOME Non-Targeting siRNA pool D-00120613, and SMART pool ON-TARGET plus FUS siRNA L-009497-00-0005 (Dharmacon, GE Healthcare) using INTERFERin transfection reagent (Polyplus Transfection). Metabolic stress was induced using Na-arsenite (Sigma) for 1 h at the concentration of 0.5 mM in HeLa and 0.25 mM in NSC-34 cells as previously described (Vidalino et al., 2012) and then washed with phosphate-buffered saline (PBS) before fixation. HeLa cell line knockout for RALY (RALY KO) were generated with the CRISPR/Cas9 technology as previously described (Rossi et al., 2017).
MN primary cultures
Animal care and experimental procedures were conducted in accordance with the University of Trento ethics committee and were approved by the Italian Ministry of Health. Primary MNs from E13.5 mouse embryos were isolated and cultured as previously described (Conrad et al., 2011). Briefly, lumbar spinal cord tissues were carefully dissected under microscope, dissociated with trypsin, and transferred to a lectin-coated plate for 45 min to select MNs. After washing with HBSS to remove cells not attached to lectin, MNs were detached from lectin by treating for 1 min with depolarization solution (30 mM KCl, 0.8% NaCl) and collected in complete medium (Neurobasal, 1% B27, 5% horse serum, 0.5 mM Glutamax). Cells were then plated on 15-mm coverslips coated with 0.5 mg/ml polyornithine (Sigma) and 0.5 mg/ml laminin (Life Technologies) and cultured in prewarmed complete medium supplemented with 10 ng/ml BDNF (brain-derived neurotrophic factor), 10 ng/ml CNTF (ciliary neurotrophic factor), and 10 ng/ml GDNF (glial cell–derived neurotrophic factor) (Peprotech). After 5 d in vitro, MNs were fixed and processed for immunofluorescence.
Constructs
The sequences of the primers used in this study are listed in Supplemental Table S1. Retrotranscription was performed on total RNA isolated from HeLa cells using the TRIzol reagent (Invitrogen). RALY cDNA was amplified with the Phusion High-Fidelity DNA polymerase (New England BioLabs) and then cloned in frame with either EGFP (pEGFP-N1, Clontech, in BglII and AgeI restriction sites) or c-MYC (pCMV6-Entry, Origene, in NheI and NotI restriction sites). RALY mutants were created with the primers listed in Supplemental Table S1 and using the QuickChange site-directed mutagenesis kit (Stratagene), according to the manufacturer’s protocol, and cloned in pEGFP-N1. RALY domains were generated with the primers listed in Supplemental Table S1 and cloned in pCMV6-Entry.
The full-length human FUS cDNA was amplified from SK-N-BE cells with AccuPrime DNA polymerase (Invitrogen) and cloned in SgfI and MluI sites of pCMV6-AN-His-HA plasmid (OriGene) to generate the vector pCMV6-HIS-HA-hFUSwt, expressing the hFUS gene tagged at the N-terminus with polyhistidine (His) tag and HA. The mutants hFUSR521C, hFUSR521H, and hFUSP525L were obtained via PCR-directed mutagenesis by using plasmids listed in Supplemental Table S1. For lentiviruses preparation, the His-HA tagged genes were excised from pCMV6-HIS-HA plasmids and subcloned into the BamHI and XhoI sites of the vector pENTR1A (Addgene). The resulting vectors were then recombined with pLenti CMV/TO Puro DEST (Addgene) using Gateway LR-Clonase (Life Technologies) to get the lentiviral vectors expressing hFUSwt and its mutants under the control of a doxycycline-inducible promoter. Lentiviruses were then prepared in accordance with the protocols detailed by Campeau et al. (2009). The plasmids encoding human deleted FUS-GFP constructs were kindly provided by E. Groen (University of Edinburgh, Edinburgh, UK). The plasmids encoding human PRMT1-GFP was a kind gift from Frank Frank O. Fackelmayer (Institute of Molecular Biology and Biotechnology, Ioannina, Greece).
Transduction and generation of inducible cell lines
NSC-34 cells were transduced with the pLentiCMV_TetR_Blast vector (Addgene), constitutively expressing the tetracycline (Tet) repressor under the control of a CMV promoter, and selected for 7 d using 10 µg/ml Blasticidin (Sigma-Aldrich). The stable cells were infected with the lentiviral vectors in the presence of 4 µg/ml polybrene and selected by using 5 µg/ml puromycin. The overexpression of hFUSwt and mutants was induced with 2 µg/ml doxycycline (Clontech) for 48 h of culture.
Ribonucleoprotein immunoprecipitation
For RIP of endogenous RNP complexes, NSC-34 were UV-cross-linked, lysed in 10 mM HEPES, pH 7.4, 100 mM KCl, 5 mM MgCl2, 0.5% NP40, 1 mM dithiothreitol (DTT), protease, and RNase inhibitors for 3 h at –80°C and centrifuged at 10,000 × g for 20 min at 4°C. The protein concentration was measured by Bradford assay to use in the IP the same amount of sample. The supernatants were precleared and incubated overnight at 4°C with protein A magnetic beads (Invitrogen) coated with 2 μg of rabbit anti-RALY (Bethyl Laboratories), rabbit anti-FUS/TLS (Abcam) antibody, or normal rabbit IgG (Millipore). The beads were then washed four times with NT2 buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40, 1% Urea), and then RNA was isolated with TRIZOL (Life Technologies) and processed for qRT-PCR analysis.
Quantitative real-time PCR
Total RNA was isolated with TRIZOL reagent and retrotranscribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific Fermentas). The KAPA PROBE FAST qPCR Kit (KAPA Biosystems) was used for the qRT-PCR of mouse RALY and Dctn1. The KAPA SYBR FAST qPCR Kit was used for the qRT-PCR of mouse H1fx, Gapdh, B2m, Sncb and Sod1, Pink1, human PRMT1, and GAPDH. All primers and probes (listed in Supplemental Table S1) were purchased by IDT or Eurofins Genomics. The samples were incubated in the BioRad CFX96 Thermo Cycler for 40 cycles, and the results were analyzed with the Bio-Rad CFX Manager version 2.1.
Coimmunoprecipitation (coIP)
NSC-34 or HeLa cells were lysed in CHAPS buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% CHAPS, 10% glycerol, protease inhibitor cocktail, phosphatase inhibitor cocktail) and centrifuged at 14,000 × g for 10 min at 4°C. For RNase treatment, cell extracts were treated with RNaseA (100 μg/ml) for 15 min. For IP, protein extracts were incubated in a total volume of 1 ml at 4°C with either protein A for rabbit antibodies or protein G for mouse antibodies magnetic beads coated with 2 μg of primary antibody: rabbit anti-HA (A190-108A; Bethyl Laboratories), rabbit anti-RALY (A302-069; Bethyl Laboratories), and rabbit anti-FUS/TLS (ab23439, Abcam). For RALY-MYC/DDK and FUS-HA coIP, 30 μl of anti-FLAG M2 Affinity Gel (Sigma-Aldrich) were used. Beads were washed four times with CHAPS buffer, solubilized in reducing (DTT-containing) sample buffer, and analyzed by SDS–PAGE and Western blotting. After immunoblotting with primary antibodies, Western blot membranes were incubated with either mouse anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (211-032-171, Jackson Laboratories) or Clan-Blot IP detection kit HRP (21232, Thermoscientific) to minimize IgG heavy and light-chain signal interference.
Detection of arginine methylation
RALY KO or control HeLa cells were lysed in CHAPS buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% CHAPS, 10% glycerol, protease inhibitor cocktail, phosphatase inhibitor cocktail) and centrifuged at 14,000 × g for 10 min at 4°C. For IP, protein extracts were incubated O/N in a total volume of 1 ml at 4°C with protein A magnetic beads coated with 2 μg of rabbit anti-FUS/TLS (ab23439, Abcam). Beads were washed four times with CHAPS buffer, solubilized in reducing (DTT-containing) sample buffer, and analyzed by SDS–PAGE and Western blotting. The membrane was probed with rabbit anti-FUS/TLS to measure the amount of immunoprecipitated FUS, and with anti-mono and dimethyl arginine antibody (ab412, Abcam). To compare the levels of arginine methylation in RALY KO or control cell extracts, band optical density (OD) was measured and normalized as follows: [FUS IP mono and diMeArg OD/(FUS IP/FUS input)]. The experiment was repeated three times.
Preparation of cell extracts and Western blot
For total protein extracts, cells were washed with PBS, lysed in RIPA lysis buffer supplemented with protease inhibitors (Roche), and centrifuged at 14,000 × g for 10 min at 4°C. For nucleus/cytoplasm extract separation, cells were washed with PBS, lysed in 10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X-100, protease inhibitors, incubated 8 min on ice and centrifuged at 1300 × g for 10 min at 4°C. The supernatant, corresponding to the cytosolic fraction, was separated from the pellet, corresponding to the nucleus fraction. Nuclei were suspended in RIPA buffer and briefly sonicated. Equal amounts of proteins were separated by SDS–PAGE and blotted onto nitrocellulose (Schleicher and Schuell, Amersham).
Western blots were probed with the following antibodies: rabbit polyclonal anti-RALY (A302-070A; Bethyl), rabbit polyclonal anti-PRMT1 (ab3768; Abcam), rabbit polyclonal anti-SOD1 (NBP1-31204; Novus Biologicals), rabbit polyclonal anti-beta SYNUCLEIN (ab189276; Abcam), mouse monoclonal anti-GAPDH (SC-32233; Santa Cruz Biotechnology), mouse monoclonal anti-hnRNP A1 (NB 100-672; Novus Biologicals), rabbit polyclonal anti-Histone H1X (ab31272; Abcam), rabbit polyclonal anti-PABP1 (ab21060; Abcam), rabbit polyclonal anti-RPL26 (ab59567; Abcam), mouse monoclonal anti-TUBULIN (sc-53140; Santa Cruz Biotechnology), mouse monoclonal anti-ACTININ (sc-17829; Santa Cruz Biotechnology), rabbit polyclonal anti-FUS/TLS (ab23439; Abcam), rabbit polyclonal anti-HA (A190-108A; Bethyl), mouse monoclonal anti-MYC (M4439; Sigma-Aldrich), mouse monoclonal anti βIII-TUBULIN (T8578; Sigma-Aldrich), rabbit polyclonal anti-TAF15 (ab134916; Abcam), mouse anti-EWS (sc-28327; Santa Cruz Biotechnology), and goat anti-DYNACTIN1 (NB100-1110; Novus Biologicals). Primary antibodies were detected with HRP-conjugated goat anti-mouse, goat anti-rabbit, and mouse anti-goat secondary antibodies (1:5000; Santa Cruz Biotechnology) and Select ECL (GE Healthcare). Western blots were analyzed with the ChemiDoc XRS+ System (Bio-Rad).
Immunofluorescence microscopy
Immunofluorescence on cell cultures was carried out as previously described (Vidalino et al., 2012). Briefly, cells were washed in prewarmed PBS and then fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature. After permeabilization with 0.1% Triton X-100 for 5 min and incubation in blocking solution for 30 min, cells were incubated for 2 h at room temperature with the primary antibodies. The following antibodies were used: rabbit polyclonal anti-RALY (dilution 1:500, A302-070A; Bethyl), mouse anti-TAU (1:300, ab80579; Abcam), mouse anti-HA (1:500; H9658; Sigma-Aldrich), mouse anti-MYC (1:1000, M4439; Sigma-Aldrich), and rabbit anti-PABP1 (1:500, ab21060; Abcam). The following secondary antibodies (1:800; Invitrogen) were used: Alexa Fluor 488–conjugated goat anti-mouse (A11017) and anti-rabbit (A11008) IgGs, Alexa Fluor 594–conjugated goat anti-mouse (A11020) and anti-rabbit (A11012). The nucleus was stained with 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI) and cytoplasm with CellMask Deep Red plasma membrane stain (C10046, Molecular Probes). Microscopy analysis was performed using the Zeiss Observer Z.1 Microscope implemented with the Zeiss ApoTome device and with a PlanApo oil immersion lens (63×, NA = 1.4). Pictures were acquired using Zen imaging software package (Zeiss) and assembled with Adobe Photoshop 7.0. Images were not modified other than adjustments of levels, brightness, and magnification.
High-content analysis
Cells were plated (1 × 104 cells/well) in CellCarrier-96 Black (Perkin Elmer). RALY KO and control HeLa cells were produced as previously described (Rossi et al., 2017). The day after, cells were transfected with His-HA-hFUSwt or hFUSR521C, hFUSR521H or hFUSP525L and, after 24 h, treated with arsenite for 1 h, fixed, and processed for immunofluorescence. In rescue experiments, RALY KO cells were transfected with either RALY-GFP or PRMT1-GFP after 10 h from plating and then with His-HA-hFUSwt or hFUSR521C, hFUSR521H or hFUSP525L after 14 h. In RALY silencing experiment 2500 HeLa cells per well were plated, transfected after 24 h with either si-RALY or si-CTRL, and then transfected after further 48 h with His-HA-hFUSwt or hFUSR521C, hFUSR521H or hFUSP525L and fixed after 24 h before processing for immunofluorescence. Inducible NSC-34 cells were directly plated in doxycycline-containing medium to induce FUS-HA construct expression and after 48 h were fixed and processed for immunofluorescence. Plates were imaged on the High Content Screening System Operetta (PerkinElmer). In each well, images were acquired in 12 preselected fields with LWD 20× objective over four channels, with λ = 380 nm excitation/λ = 445 nm emission for DAPI, λ = 495 nm excitation/λ = 519 nm emission for Alexa Fluor 488, λ = 535 nm excitation/λ = 615 nm emission for Alexa Fluor 594, and λ = 647 nm excitation/λ = 681 nm emission for CellMasK Deep Red. For feature extraction, the images were analyzed by Harmony software version 4.1 (PerkinElmer). Briefly, individual cell nucleus and cytoplasm were segmented based on DAPI and CellMasK Deep Red plasma membrane stain, respectively. The Select Population algorithm allowed identifying the subpopulation of transfected cells by setting a fluorescence intensity threshold. Cells overexpressing His-HA-hFUSwt, hFUSR521C, hFUSR521H, and hFUSP525L were selected based on Alexa Fluor 594 fluorescence signal in the whole cell area, while cells overexpressing PRMT1-GFP or RALY-GFP were selected based on Alexa Fluor 488 fluorescence signal. In RALY silencing experiments, cells were further selected for RALY silencing based on the endogenous protein staining. HIS-HA-hFUSwt, hFUSR521C, hFUSR521H, hFUSP525L, or RALY mean intensity was quantified in the nuclear and cytoplasmic region. The Find Spots algorithm allowed localizing cytoplasmic protein aggregates and specifically select them by setting a spot area threshold ≥10 µm2. Each experiment was performed three times with five wells per experimental condition.
Optiprep density gradient centrifugation
NSC-34 cell were lysated in 25 mM HEPES, pH 7.4, 150 mM KCl, 8% glycerol, 0.1% NP40, 1 mM DTT, RNase and protease inhibitors, and the lysate was centrifuged at 20,000 × g for 10 min at 4°C. For RNase treatment, cell extracts were treated with RNaseA (100 μg/ml) for 15 min. The supernatant (Input) was layered on top of a 12-ml 15%–30% linear Optiprep (Sigma-Aldrich) density gradient (Fritzsche et al., 2013). Optiprep was diluted in 25 mM HEPES, pH 7.4, 150 mM KCl solution. After a centrifugation in a swinging bucket rotor (SW41; Beckman Coulter Genomics) at 280,000 × g at 4°C for 2.5 h, 12 fractions were collected from the top to the bottom of the gradient, and proteins were precipitated with trichloroacetic acid and acetone, solubilized in sample buffer, pH 8, and processed for Western blotting.
Proximity ligation assay
PLA was conducted according to manufacturer’s protocol (Sigma-Aldrich). Briefly, after fixation, permeabilization with 0.1% Triton X-100, and 40 min blocking, cells were incubated at room temperature for 2.5 h with primary antibodies: goat anti-FUS (1:300; Sigma-Aldrich) and rabbit anti-RALY (1:300; Bethyl). As negative controls, either no primary antibodies or goat anti-DYNACTIN1 (1:100; Novus Biologicals) and rabbit anti-RALY were incubated. After two 10-min washings with PBS, cells were incubated with Duolink In Situ PLA Probe anti-Rabbit PLUS and anti-Goat MINUS (Sigma-Aldrich), diluted 1:5 in blocking solution for 1 h at 37°C. After two 5-min washings in Wash Buffer A (Sigma-Aldrich), cells were incubated with ligase for 30 min at 37°C. After two 5-min washings in Wash Buffer A, cells were incubated with polymerase and amplification solution for 100 min at 37°C, washed twice for 10 min in Wash Buffer B (Sigma-Aldrich), once in 0.01% in Wash Buffer B, dried at room temperature in the dark, and mounted with Mounting Media with DAPI (Sigma-Aldrich). For MNs, after washings in Wash Buffer B, cells were incubated with mouse anti-SMI32 antibody (1:500, Abcam) and hence with Alexa Fluor 488–conjugated goat anti-mouse.
Data analysis
All quantifications were tested for significance with t tests and were considered significant if p < 0.05 and are expressed as standard error of the mean (SEM).
Supplementary Material
Acknowledgments
We thank E. Groen for kindly providing the plasmids to express deleted EGFP-tagged FUS. We thank Frank O. Fackelmayer for kindly providing the plasmid to express PRMT1-GFP. We are grateful to Valentina Adami and Michael Pancher (CIBIO-High Throughput Screening Core Facility) for the technical assistance during high-content image acquisition and analysis. We thank Alessandro Provenzani, Martin Hanczyc, and Elia Riciputi for their critical comments on the manuscript. This project was supported by the fund “Progetto Biotecnologie” from the University of Trento (to P.M.) and by the Project “Axonomix” (to P.M. and A.Q.).
Abbreviations used:
- ALS
amyotrophic lateral sclerosis
- Co-IP
coimmunoprecipitation
- EWSR1
Ewing’s sarcoma RNA binding protein 1
- FTLD
frontotemporal lobar degeneration
- FUS
Fused in Sarcoma
- hnRNP
heterogeneous nuclear ribonucleoprotein
- IP
immunoprecipitation
- KO
knockout
- me-FUS
methylated FUS
- MN
motor neuron
- NLS
nuclear localization signal
- PLA
proximity ligation assay
- PRMT1
protein arginine N-methyltransferase 1
- PY-NLS
proline-tyrosine nuclear localization signal
- RBP
RNA-binding protein
- RGG
arginine-glycine-glycine
- RIP
RNA immunoprecipitation
- RNP
ribonucleoparticle
- RRM
RNA recognition motif
- SG
stress granule
- siCTRL
control nontargeting siRNA
- SMN
survival motor neuron
- TAF-15
TATA-binding protein–associated factor 2N
- TDP-43
TAR DNA-binding protein 43
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-02-0108) on October 24, 2018.
REFERENCES
- Baron DM, Kaushansky LJ, Ward CL, Sama RRK, Chian RJ, Boggio KJ, Quaresma AJC, Nickerson JA, Bosco DA. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol Neurodegener , 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG. (2009). Protein arginine methylation in mammals: who, what, and why. Mol Cell , 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Chorney E, Baldwin RM, Didillon A, Chabot B, Jasmin BJ, Côté J. (2017). RNA binding protein RALY promotes Protein Arginine Methyltransferase 1 alternatively spliced isoform v2 relative expression and metastatic potential in breast cancer cells. Int J Biochem Cell Biol , 124–135. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Sapp P, McKenna-Yasek D, Brown RH, Hayward LJ. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet , 4160–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR. (2014). mRNP granules. Assembly, function, and connections with disease. RNA Biol , 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Hertel KJ. (2012). Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA , 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One , e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. (1992). Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn , 209–221. [DOI] [PubMed] [Google Scholar]
- Chook YM, Süel KE. (2011). Nuclear import by karyopherin-βs: recognition and inhibition. Biochim Biophys Acta , 1593–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R, Jablonka S, Sczepan T, Sendtner M, Wiese S, Klausmeyer A. (2011). Lectin-based isolation and culture of mouse embryonic motoneurons. J Vis Exp JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornella N, Tebaldi T, Gasperini L, Singh J, Padgett RA, Rossi A, Macchi P. (2017). The hnRNP RALY regulates transcription and cell proliferation by modulating the expression of specific factors, including the proliferation-marker E2F1. J Biol Chem , 19674–19692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Gao K, Jankovic J. (2014). The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol , 337–348. [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J , 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, et al (2012). Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J , 4258–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Ostman A, Landegren U. (2002). Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol , 473–477. [DOI] [PubMed] [Google Scholar]
- Fritzsche R, et al (2013). Interactome of two diverse RNA granules links mRNA localization to translational repression in neurons. Cell Rep , 1749–1762. [DOI] [PubMed] [Google Scholar]
- Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. (2005). The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol , 587–593. [DOI] [PubMed] [Google Scholar]
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. (2011). Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging , 2323 e27–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glock C, Heumüller M, Schuman EM. (2017). mRNA transport & local translation in neurons. Curr Opin Neurobiol , 169–177. [DOI] [PubMed] [Google Scholar]
- Groen EJN, et al (2010). FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol , 224–230. [DOI] [PubMed] [Google Scholar]
- Groen EJN, et al (2013). ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet , 3690–3704. [DOI] [PubMed] [Google Scholar]
- Guo L, et al (2018). Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like comains. Cell , 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M, et al (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell , 706–719. [DOI] [PubMed] [Google Scholar]
- Ikenaka K, et al (2013). dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS One , e54511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenaka K, Katsuno M, Kawai K, Ishigaki S, Tanaka F, Sobue G. (2012). Disruption of axonal transport in motor neuron diseases. Int J Mol Sci , 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iko Y, et al (2004). Domain architectures and characterization of an RNA-binding protein, TLS. J Biol Chem , 44834–44840. [DOI] [PubMed] [Google Scholar]
- Ito D, Hatano M, Suzuki N. (2017). RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci Transl Med 9, eaah5436. [DOI] [PubMed] [Google Scholar]
- Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. (2011). Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol , 152–162. [DOI] [PubMed] [Google Scholar]
- Izhar L, Adamson B, Ciccia A, Lewis J, Pontano-Vaites L, Leng Y, Liang AC, Westbrook TF, Harper JW, Elledge SJ. (2015). A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep , 1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Guo X, Bhavanandan VP. (1998). Four distinct regions in the auxiliary domain of heterogeneous nuclear ribonucleoprotein C-related proteins. Biochim Biophys Acta , 229–233. [DOI] [PubMed] [Google Scholar]
- Jun MH, Ryu HH, Jun YW, Liu T, Li Y, Lim CS, Lee YS, Kaang BK, Jang DJ, Lee JA. (2017). Sequestration of PRMT1 and Nd1-L mRNA into ALS-linked FUS mutant R521C-positive aggregates contributes to neurite degeneration upon oxidative stress. Sci Rep , 40474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, et al (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet , 572–574. [DOI] [PubMed] [Google Scholar]
- Kamelgarn M, Chen J, Kuang L, Arenas A, Zhai J, Zhu H, Gal J. (2016). Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta , 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller BA, Volkening K, Droppelmann CA, Ang LC, Rademakers R, Strong MJ. (2012). Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol (Berl) , 733–747. [DOI] [PubMed] [Google Scholar]
- Kiebler MA, López-García JC, Leopold PL. (1999). Purification and characterization of rat hippocampal CA3-dendritic spines associated with mossy fiber terminals. FEBS Lett , 80–86. [DOI] [PubMed] [Google Scholar]
- Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N. (2011). Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res , 2781–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuz´ma-Kozakiewicz M, Chudy A, Kaz´mierczak B, Dziewulska D, Usarek E, Baran´czyk-Kuz´ma A. (2013). Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem Res, 2463–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, et al (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science , 1205–1208. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, et al (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci , 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet , R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalmansingh AS, Urekar CJ, Reddi PP. (2011). TDP-43 is a transcriptional repressor: the testis-specific mouse acrv1 gene is a TDP-43 target in vivo. J Biol Chem , 10970–10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster AK, Nutter-Upham A, Lindquist S, King OD. (2014). PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinforma Oxf Engl , 2501–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi J, De Santis R, de Turris V, Morlando M, Laneve P, Calvo A, Caliendo V, Chiò A, Rosa A, Bozzoni I. (2015). ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis Model Mech , 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IRA, et al (2010). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol (Berl) , 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud EJ, Bultman SJ, Stubbs LJ, Woychik RP. (1993). The embryonic lethality of homozygous lethal yellow mice (Ay/Ay) is associated with the disruption of a novel RNA-binding protein. Genes Dev , 1203–1213. [DOI] [PubMed] [Google Scholar]
- Mikhaleva S, Lemke EA. (2018). Beyond the transport function of import receptors: what’s all the FUS about? Cell , 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch C, et al (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology , 724–726. [DOI] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain J Neurol , 2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y. (2014). Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem , 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahlich S, Zakaryan RP, Gehring H. (2006). Protein arginine methylation: Cellular functions and methods of analysis. Biochim Biophys Acta , 1890–1903. [DOI] [PubMed] [Google Scholar]
- Puls I, et al (2003). Mutant dynactin in motor neuron disease. Nat Genet , 455–456. [DOI] [PubMed] [Google Scholar]
- Puls I, et al (2005). Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol , 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S, et al (2018). FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell , 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainero I, Rubino E, Michelerio A, D’Agata F, Gentile S, Pinessi L. (2017). Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct Neurol , 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci , 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes GH, Valbracht JR, Nguyen MD, Vaughan JH. (1997). The p542 gene encodes an autoantigen that cross-reacts with EBNA-1 of the Epstein Barr virus and which may be a heterogeneous nuclear ribonucleoprotein. J Autoimmun , 447–454. [DOI] [PubMed] [Google Scholar]
- Rossi A, Moro A, Tebaldi T, Cornella N, Gasperini L, Lunelli L, Quattrone A, Viero G, Macchi P. (2017). Identification and dynamic changes of RNAs isolated from RALY-containing ribonucleoprotein complexes. Nucleic Acids Res , 6775–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A, Sandhu J, Casero D, Vallim TQ, Hong C, et al (2016). Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature , 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaramuzzino C, Monaghan J, Milioto C, Lanson NA, Maltare A, Aggarwal T, Casci I, Fackelmayer FO, Pennuto M, Pandey UB. (2013). Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS One , e61576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Podell ER, Han SSW, Berry JD, Eggan KC, Cech TR. (2014). FUS is sequestered in nuclear aggregates in ALS patient fibroblasts. Mol Biol Cell , 2571–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, Cech TR. (2013). RNA seeds higher-order assembly of FUS protein. Cell Rep , 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, et al (2014). Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc Natl Acad Sci USA , E4769–E4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Huang EJ. (2016). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res , 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Rathod RJ, Rajkumar S, Kennedy D. (2014). Nervous translation, do you get the message? A review of mRNPs, mRNA-protein interactions and translational control within cells of the nervous system. Cell Mol Life Sci CMLS , 3917–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden JS, et al (2011). The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol (Berl) , 99–110. [DOI] [PubMed] [Google Scholar]
- Tan AY, Manley JL. (2009). The TET family of proteins: functions and roles in disease. J Mol Cell Biol , 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzer S, Moro A, Kuharev J, Francis AC, Vidalino L, Provenzani A, Macchi P. (2013). Proteome-wide characterization of the RNA-binding protein RALY-interactome using the in vivo-biotinylation-pulldown-quant (iBioPQ) approach. J Proteome Res , 2869–2884. [DOI] [PubMed] [Google Scholar]
- Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S. (2012). Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet , 136–149. [DOI] [PubMed] [Google Scholar]
- Udagawa T, et al (2015). FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat Commun , 7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin H, et al (2010). FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol (Berl) , 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, et al (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science , 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, et al (2013). ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet , 2676–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidalino L, Monti L, Haase A, Moro A, Acquati F, Taramelli R, Macchi P. (2012). Intracellular trafficking of RNASET2, a novel component of P-bodies. Biol Cell , 13–21. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A, Kitajo K. (2012). The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS One , e49267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Gal J, Chen J, Zhu H. (2014). Self-assembled FUS binds active chromatin and regulates gene transcription. Proc Natl Acad Sci USA , 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa T, et al (2018). Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell , 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZC, Chook YM. (2012). Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc Natl Acad Sci USA , 12017–12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.