

Abstract
Background/Purpose:
Lichen planus is a T-cell-mediated mucocutaneous disorder characterized histopathologically by a band of chronic inflammatory cells in the subepithelial zone and degeneration of basal layer. The present study was aimed to evaluate the distribution and quantitative assessment of cluster of differentiation 1a (CD1a)-positive Langerhans cells (LCs) in oral lichen planus (OLP), thus to determine the role of LCs pertaining to the changes occurring in OLP.
Materials and Methods:
Five cases of normal oral mucosa and 20 cases of OLP were immunostained with CD1a antibody; the positive cells were counted manually in the photomicrographs and statistically analyzed using t-test, Mann–Whitney test, and Wilcoxon signed-rank test.
Results:
The average percentage of CD1a-positive LCs in normal subjects was 0.9%, and in the OLP cases higher percentage was observed (3.93%). The statistical comparison of these two parameters was significant (P=0.018). The degree of basal cell degeneration and density of subepithelial infiltrate on statistical comparison with the concentration of CD1a-positive LCs showed significant results.
Conclusion:
LCs play a pivotal role in the recruitment of CD4+ and CD8+ cells to the subepithelial region and basal keratinocytes apoptosis. A small number of study subjects, assessment of only CD1a molecule and LCs in the epidermis only were a few of the drawbacks of the study.
KEY WORDS: Antigen-presenting cells, langerhans cells, oral epithelium, oral lichen planus
Introduction
Oral lichen planus (OLP) is a common T-cell mediated chronic mucocutaneous disorder affecting the oral mucosa.[1]
Several studies on immune-mediated lesions have shown that there is a difference in the number and distribution of the Langerhans cells (LCs). Their results have highlighted that LCs have a crucial role in their pathogenesis.[2] The role of cluster of differentiation 1a (CD1a) expressing LCs pertaining to OLP changes such as basal cell degeneration and the density of the subepithelial infiltrate are not much explored.
The current study was undertaken to evaluate the distribution and quantitative assessment of CD1a-positive LCs in OLP lesions pertaining to the changes observed in OLP.
Materials and Methods
The material for the present study included 20 cases of OLP and five normal oral mucosae. The study and control samples were subjected to immunohistochemical marker CD1a and to evaluate the expression of LCs in the epithelium.
Two to three serial sections of 3 μm thickness were made, taken in aminopropyltriethoxysilane (APES) pre-coated slides for immunohistochemistry. The sections in the APES coated slides were de-paraffinized by heating, and then, de-waxing was done with xylene for 1 h. The sections were rehydrated following a routine protocol and kept in tap water for 10 min. Heat antigen retrieval procedure was done under citrate buffer of pH-6 using pressure cooker.
After cooling, the sections were treated with 3% hydrogen peroxide and power block for 10 min in each to reduce nonspecific binding. The sections were incubated with CD1a (Novocastra, USA) for 30 min. Then, slides were washed in phosphate-buffered saline and treated with polyhorseradish peroxidase enzyme (Scytek, USA) for 30 min. Immunostaining was developed by treating the sections with freshly prepared diaminobenzidine (3,3’-diaminobenzidine tetrahydrochloride) solution for 5 min, and the excess chromogen was removed by rinsing. The slides were stained in Mayer's Hematoxylin for 1 min, then bluing was carried out in running tap water for 10 min. The sections were dehydrated in graded alcohol, cleared with xylene, and mounted using DePeX.
The LCs positive for CD1a appeared as brown-colored end product in the dendritic process, cytoplasm, and cell border of LCs. The areas that exhibited numerous CD1a-positive cells with good intensity of staining were recorded with Nikon digital camera. Photomicrographs were taken from three representative areas in the immunohistochemical-stained OLP sections and the sections from the normal subjects under ×40. The number of positive LCs and negative epithelial cells was counted manually both in OLP and in normal epithelium for the percentage. The distribution of CDIa-positive LCs in different epithelial layers, i.e., upper, mid-spinous, and suprabasal layers, as well as the basal cell degeneration and density of inflammatory cells in the subepithelial zone, were graded into mild (+), moderate (++), and intense (+++) by two experienced oral pathologists. The results were compared statistically using t-test, Mann–Whitney test, and Wilcoxon signed-rank test.
Results
Most of the cases which exhibited severe basal cell degeneration demonstrated intense inflammatory infiltrate in the subepithelial zone. In contrast, the inflammatory infiltrate was present in milder degree in OLP cases that exhibited basal cell degeneration in focal areas.
Both the study and control samples subjected to immunohistochemical marker CD1a demonstrated LCs in suprabasal, mid-spinous, and upper spinous layers of the epithelium. The distribution was predominantly confined to the mid-spinous layers in both the groups [Figures 1 and 2]. The LCs located in suprabasal layer showed one or two dendritic processes and exhibited round morphology whereas the cells in the upper and mid-spinous layer demonstrated more dendritic processes [Figure 3]. However, the LCs observed in the OLP epithelium demonstrated numerous dendritic processes, especially in mid-spinous and suprabasal layers, and few cells presented as round cells. The percentage of LCs count in the normal oral epithelium ranged from 0.62% to 4.5%, and the average was 0.9%. The CD1a expressing LCs average percentage in OLP epithelium was 3.93%; however, the highest percentage was 11.85%. In OLP, presence of LCs was graded into normal (1%–3%), mild (3%–6%), moderate (6%–9%), and intense (above 9%). The presence of CD1a-positive LCs in OLP was in normal grade in seven cases, mild in 10 cases, and it was moderate in two cases and intense in one case.
Figure 1.

The normal oral epithelium exhibits a very few CD1a-positive Langerhans cells (Immunohistochemistry, ×40)
Figure 2.

Histopathology of oral lichen planus shows CD1a-positive Langerhans cells in the upper spinous, mid spinous, and suprabasal layers (Immunohistochemistry, ×10)
Figure 3.

The histopathology of oral lichen planus showing intensely stained dendritic Langerhans cells both in the epithelium and in upper dermis (immunohistochemistry, ×40)
The difference of means of LCs present in the epithelium of normal oral mucosa and OLP was statistically evaluated using Student's t-test and found to be significant (P=0.018).
The distribution of CD1a-positive LCs in the normal oral epithelium and in the OLP epithelium is given in the Figures 4 and 5.
Figure 4.
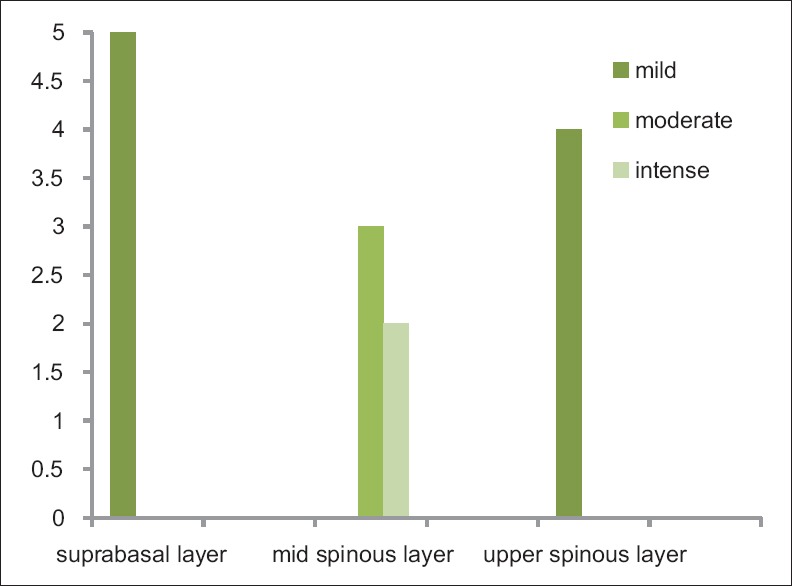
Distribution of Langerhans cells in the suprabasal, mid-spinous, upper spinous layers in normal oral mucosa
Figure 5.
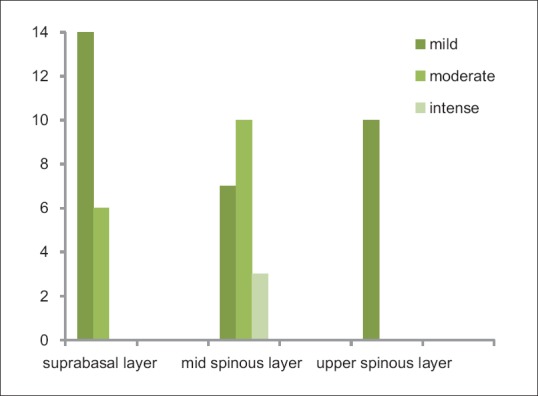
Distribution of Langerhans cells in the suprabasal, mid-spinous, upper spinous layers in oral lichen planus
The degree of basal cell degeneration observed in OLP cases and the density of chronic inflammatory cell infiltration was compared with the concentration of CD1a-positive LCs in OLP using Wilcoxon signed-rank test and found to be highly significant at P=0.001 and 0.002, respectively [Figure 6].
Figure 6.
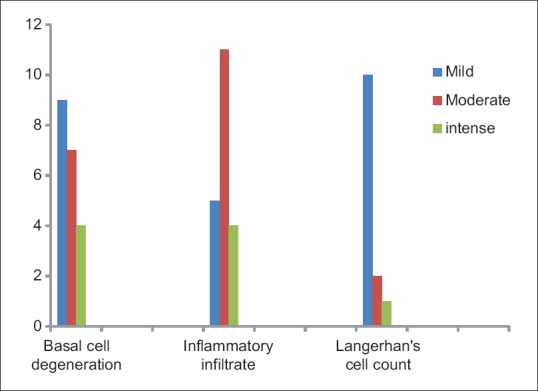
Comparison of various grades of basal cell degeneration, inflammatory cell infiltrate, and Langerhans cells in oral lichen planus cases
Discussion
OLP affects 1% to 2% of the general adult population, and the female-male ratio is 1.4:1. It affects the individuals in the fifth and sixth decades of life.[3,4] The characteristic microscopic features observed in the OLP are band of chronic inflammatory cells in the subepithelial zone and liquefaction degeneration of the basal cell layer.[5]
The LCs in OLP can express major histocompatibility complex (MHC) Class II molecules which present the antigen to the CD4+ T cells. The interleukin (IL)-12 secreted from these two cell types may induce CD4+ T cells to release cytokines such as IL-2 and interferon-gamma (IFN-γ).[6,7,8] In addition, the keratinocytes present the antigen to the CD8+ T helper-cells through MHC Class 1 molecules, these cytotoxic cells together with the cytokines secreted by CD4+ T-helper cells may induce the apoptosis of the basal keratinocytes.[9]
LCs are dendritic cells, commonly positioned in the upper spinous layer of the skin, mucosa of the upper gastrointestinal tract and female genital tract.[10] The special structure and markers used to distinguish LCs from other dendritic cells are Birbeck granules, CD1a, E-cadherin, MHC Class II molecules, Langerin, and S-100.[10] Among these various markers, the CD1a reaction is considered as a reliable and the gold standard method for the demonstration of LCs. The CD1a have a similar function-like MHC class 1 molecules in antigen presentation. The CD1 isoforms such as CD1a, CD1b, CD1c expressed on antigen presenting cells (APCs) present various forms of self and microbial lipid antigen to CD4+, CD8+, and double-negative T cell populations.[11]
In spite of active search on the pathogenesis of OLP, the antigen that induces the immune response is yet to be identified. The triggering agent may be an exogenous antigen or self peptide. The first antigen might be MHC Class II-restricted, virus-derived polypeptide, that may be processed through an endosomal compartment within the dendritic cells. The APC derived pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), IL-1 and IL-12 result in the generation of IL-2 and IFN-γ. These two cytokines in addition with locally derived IFN-α induce chemokine (C-X-C motif) ligand (CXCL) 9 – CXCL11 might result in the recruitment and activation of CXCR3 (+) cytotoxic CD8+ T cells.[12]
The second antigen may be a self-peptide that is heat shock protein (HSP). This autoantigen is developed by the basal keratinocytes after exposure to the exogenous agents such as stress, genetic predisposition, hypertension, malignant neoplasms, exposure to dental materials, drugs, infectious agents such as bacterial and viral infections, autoimmune mechanism, immunodeficiency, food allergies, habits, trauma, and diabetes.[1,12,13] The susceptibility to develop OLP may be due to dysregulated HSP gene expression or an inability to inhibit the immune response, occurring after self HSP recognition. Many reports showed an increased expression of HSP in OLP. The second antigen is presented to CD8+ T cells directly by the basal keratinocytes through MHC Class 1 molecules. The cytotoxic and pro-apoptotic mediators expressed by the CD8+ T cells may mediate the basal keratinocytes apoptosis.[1,14]
The LCs are involved both directly and indirectly in the recruitment of CD8+ T-cells to the subepithelial zone.[14] This dendritic cell has an active role in the epithelial immune mechanism, both in physiological and in disease status.[10] Its involvement in the pathogenesis of OLP is a well-established fact; however, this statement is contradicted by some investigators.[15,16]
The LCs are derived from the bone marrow progenitor cells and appear in the epithelium at about the 7th week of gestation. The myeloid progenitor gives rise to a monocyte which is the precursor for LCs.[17] In the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4, transforming growth factor (TGF)-β and activin A, the LCs develop from the monocytes. After exposure to various stimuli, there is an increase in the expression of Class II MHC and co-stimulatory molecules.[18] The CD1a molecules are a family of non-polymorphic transmembrane glycoprotein expressed on the cell surface in conjunction with β2-microglobulin. It is present on the APCs such as dendritic cells and B lymphocytes.[19]
Salamero et al. in 2001 observed CD1a molecules both in cell surface and intracellularly in association with MHC Class II molecules in LCs using confocal microscope. In the present study, the LCs observed in the normal and in the OLP epithelium exhibited CD1a positivity in the cytoplasm and cell membrane.[19]
The LCs were predominantly observed in the mid-spinous layer of normal oral epithelium. This observation was concurrent with the findings of de Witte et al.[20] The LCs constitute 2%–8% of the intra-epithelial cell content.[10] There was approximately one LC per 53 epidermal cells that represent 1.86%. It was suggested that LCs form territory for the epidermal cells and act as immune surveillance.[21] The present study observed that the percentage of LCs in normal oral mucosa was 0.9%, and it was less than the data given in the literature. Negishi et al. reported that the LC number was highest in the lining mucosa than in masticatory mucosa [Table 1].[22]
Table 1.
Langerhans cells count and the other morphological changes mentioned in the present study and literature

The quantitative assessment of CD1a-positive LCs in 20 cases of OLP showed a higher percentage than in normal oral mucosa. Comparing the density of CD1a-positive LCs between the normal subjects and the OLP, the result showed to be significant (P=0.018).
The results of the present study are concurrent with the study of Hasséus et al., Gueiros et al., and Mccartan et al.[15,16,24] However, a study on LCs density in OLP by Farthing et al. showed that no change in the number of LCs in lichen planus subjects. Their results showed the presence of more number of dendritic processes in the CD1a-positive LCs of OLP than in the control mucosa. Similar finding was observed in the current study. They suggested that the increase in the number of dendritic processes represent either an increase in the surface antigen or dendritic growth [Table 1].[23]
It is believed that the epidermal environment can promote the attraction of precursors and their differentiation into LCs. The keratinocytes secrete macrophage inflammatory protein 3a/chemokine (C-C motif) ligand 20 and TGF may direct CCR6+ expressing LC precursors towards the epithelium and enhance their entry into the LC pathway.[25] The information available in the literature and the results of the current study speculate that the altered epithelium in the OLP lesion may be the factor involved in the migration of LC precursors toward the epithelium. In addition, the cytokines liberated by the keratinocytes have a role in the maturation of LCs.
In our study, the number of dendritic processes observed in LCs in the normal subjects was comparatively less than in the LCs observed in OLP [Figure 1]. According to Walsh et al., the signals for CD1a expression could be mediated by cytokines such as IL-1, GM-CSF, TNF-α, and IL-6 and were liberated by activated keratinocytes and monocytes.[26] There was an upregulation of these cytokines in OLP and was proved by various studies. Therefore, these cytokines may have a role in an increased expression of dendritic process in OLP. Thus, it reflects the key role of LCs in the OLP pathogenesis.[27]
The present study showed the distribution of LCs mainly in the suprabasal and mid-spinous layers of OLP [Figures 2 and 3]. However, Chou and Daniels found a higher position of LCs in OLP epithelium than in the normal tissue and suggested it might be due to an increased turnover of epithelium in OLP. The difference in the distribution of LCs in the upper spinous layer might be due to the migration toward the basal layer.[28]
The binding and localization of LCs in the epithelial cells are mediated by E-cadherin which is expressed on the surface. After exposure to the allergen, the LCs migrate from the epithelium as a result of downregulation of E-cadherin.[29] The LCs seen in the suprabasal layer in OLP epithelium appeared as round cells with few dendritic processes. This finding speculates that the possible reason for such an appearance may be detachment of LCs from the adjacent keratinocytes.
The basal cell degeneration is triggered by CD8+ cytotoxic T-cells, and the possible mechanisms involved are, T-cell-secreted TNF-α binding TNF-α receptor 1 on the keratinocyte surface, T-cell surface CD95 L (Fas ligand) binding CD95 (Fas) on the keratinocyte surface, and T-cell-secreted granzyme B entering the keratinocyte through perforin-induced membrane pores.[1]
The density of LCs was observed in an increased level in cases which exhibited moderate and severe amount of basal cell degeneration and dense subepithelial inflammatory infiltrate. On statistical evaluation, these two factors were highly significant (P=0.001 and 0.002 respectively).
The LCs recruit CD8+ T-cells to the subepithelial region through the CD4+ T-cells by presenting antigen through MHC Class II molecules and secreting cytokines such as TNF-α, IL-1, and IL-12. The activated CD4+ T-cells secrete IL-2 and INF-γ which in turn recruits CD8+ T-cells to the subepithelial region and trigger the basal cell apoptosis.[30] The basal keratinocytes undergoing apoptosis may be phagocytosed by LCs. The auto-antigen present in keratinocytes can be processed and presented to the CD4+ T-cells. Another mechanism is the CD1a expressing APCs are capable of presenting the antigen to both CD4+ T-cells and CD8+ T-cells directly.[11]
Conclusion
With all the scientific background in conjunction with the results of the present study, it is emphasized at this juncture that the LCs have a pivotal role in the recruitment of CD4+ and CD8+ cells to the subepithelial region and in the keratinocytes apoptosis. The limitation of the present study was that it evaluated only the LCs in the epithelium and not in other areas like lamina propria. Also it dealt with CD1a expression only and the other forms of CD1 and their role in OLP were not studied. The number of subjects in the study was also small.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Sugerman PB, Savage NW, Walsh LJ, Zhao ZZ, Zhou XJ, Khan A, et al. The pathogenesis of oral lichen planus. Crit Rev Oral Biol Med. 2002;13:350–65. doi: 10.1177/154411130201300405. [DOI] [PubMed] [Google Scholar]
- 2.Cury PR, Furuse C, Rodrigues AE, Barbuto JA, Araújo VC, Araújo NS, et al. Interstitial and Langerhans’ dendritic cells in chronic periodontitis and gingivitis. Braz Oral Res. 2008;22:258–63. doi: 10.1590/s1806-83242008000300012. [DOI] [PubMed] [Google Scholar]
- 3.Sugerman PB, Savage NW. Oral lichen planus: Causes, diagnosis and management. Aust Dent J. 2002;47:290–7. doi: 10.1111/j.1834-7819.2002.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 4.Sousa FA, Rosa LE. Oral lichen planus: Clinical and histopathological considerations. Braz J Otorhinolaryngol. 2008;74:284–92. doi: 10.1016/S1808-8694(15)31102-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shklar G. Lichen planus as an oral ulcerative disease. Oral Surg Oral Med Oral Pathol. 1972;33:376–88. doi: 10.1016/0030-4220(72)90467-7. [DOI] [PubMed] [Google Scholar]
- 6.Farthing PM, Cruchley AT. Expression of MHC class II antigens (HLA DR, DP and DQ) by keratinocytes in oral lichen planus. J Oral Pathol Med. 1989;18:305–9. doi: 10.1111/j.1600-0714.1989.tb00402.x. [DOI] [PubMed] [Google Scholar]
- 7.Kang K, Kubin M, Cooper KD, Lessin SR, Trinchieri G, Rook AH, et al. IL-12 synthesis by human Langerhans cells. J Immunol. 1996;156:1402–7. [PubMed] [Google Scholar]
- 8.Simark Mattsson C, Jontell M, Bergenholtz G, Heyden M, Dahlgren UI. Distribution of interferon-gamma mRNA-positive cells in oral lichen planus lesions. J Oral Pathol Med. 1998;27:483–8. doi: 10.1111/j.1600-0714.1998.tb01917.x. [DOI] [PubMed] [Google Scholar]
- 9.Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: Facts and controversies. Clin Dermatol. 2010;28:100–8. doi: 10.1016/j.clindermatol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Barrett AW, Cruchley AT, Williams DM. Oral mucosal Langerhans’ cells. Crit Rev Oral Biol Med. 1996;7:36–58. doi: 10.1177/10454411960070010301. [DOI] [PubMed] [Google Scholar]
- 11.Mizumoto N, Takashima A. CD1a and langerin: Acting as more than Langerhans cell markers. J Clin Invest. 2004;113:658–60. doi: 10.1172/JCI21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lodi G, Scully C, Carrozzo M, Griffiths M, Sugerman PB, Thongprasom K, et al. Current controversies in oral lichen planus: Report of an international consensus meeting. Part 1. Viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40–51. doi: 10.1016/j.tripleo.2004.06.077. [DOI] [PubMed] [Google Scholar]
- 13.Ismail SB, Kumar SK, Zain RB. Oral lichen planus and lichenoid reactions: Etiopathogenesis, diagnosis, management and malignant transformation. J Oral Sci. 2007;49:89–106. doi: 10.2334/josnusd.49.89. [DOI] [PubMed] [Google Scholar]
- 14.Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: Clinical and histological perspectives. J Invest Dermatol. 2009;129:1088–99. doi: 10.1038/sj.jid.2009.42. [DOI] [PubMed] [Google Scholar]
- 15.Hasséus B, Jontell M, Brune M, Johansson P, Dahlgren UI. Langerhans cells and T cells in oral graft versus host disease and oral lichen planus. Scand J Immunol. 2001;54:516–24. doi: 10.1046/j.1365-3083.2001.00988.x. [DOI] [PubMed] [Google Scholar]
- 16.Gueiros LA, Gondak R, Jorge Júnior J, Coletta RD, Carvalho Ade A, Leão JC, et al. Increased number of Langerhans cells in oral lichen planus and oral lichenoid lesions. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:661–6. doi: 10.1016/j.oooo.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Jaitley S, Saraswathi T. Pathophysiology of Langerhans cells. J Oral Maxillofac Pathol. 2012;16:239–44. doi: 10.4103/0973-029X.99077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borkowski TA, Letterio JJ, Farr AG, Udey MC. A role for endogenous transforming growth factor beta 1 in Langerhans cell biology: The skin of transforming growth factor beta 1 null mice is devoid of epidermal Langerhans cells. J Exp Med. 1996;184:2417–22. doi: 10.1084/jem.184.6.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salamero J, Bausinger H, Mommaas AM, Lipsker D, Proamer F, Cazenave JP, et al. CD1a molecules traffic through the early recycling endosomal pathway in human Langerhans cells. J Invest Dermatol. 2001;116:401–8. doi: 10.1046/j.1523-1747.2001.01264.x. [DOI] [PubMed] [Google Scholar]
- 20.de Witte L, Nabatov A, Pion M, Fluitsma D, de Jong MA, de Gruijl T, et al. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med. 2007;13:367–71. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 21.Bauer J, Bahmer FA, Wörl J, Neuhuber W, Schuler G, Fartasch M, et al. A strikingly constant ratio exists between Langerhans cells and other epidermal cells in human skin. A stereologic study using the optical disector method and the confocal laser scanning microscope. J Invest Dermatol. 2001;116:313–8. doi: 10.1046/j.1523-1747.2001.01247.x. [DOI] [PubMed] [Google Scholar]
- 22.Negishi A, Mogi K, Yamaguchi T. Histopathological study on the distribution of Langerhans cells in oral mucosa. Kitakondu Med J. 2007;5:307–10. [Google Scholar]
- 23.Farthing PM, Matear P, Cruchley AT. The activation of Langerhans cells in oral lichen planus. J Oral Pathol Med. 1990;19:81–5. doi: 10.1111/j.1600-0714.1990.tb00801.x. [DOI] [PubMed] [Google Scholar]
- 24.McCartan BE, Lamey PJ. Expression of CD1 and HLA-DR by Langerhans cells (LC) in oral lichenoid drug eruptions (LDE) and idiopathic oral lichen planus (LP) J Oral Pathol Med. 1997;26:176–80. doi: 10.1111/j.1600-0714.1997.tb00454.x. [DOI] [PubMed] [Google Scholar]
- 25.Musso T, Scutera S, Vermi W, Daniele R, Fornaro M, Castagnoli C, et al. Activin A induces Langerhans cell differentiation in vitro and in human skin explants. PLoS One. 2008;3:e3271. doi: 10.1371/journal.pone.0003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh LJ, Seymour GJ, Powell RN. The regulation of Langerhans cell T6, DR and DQ antigen expression: An hypothesis. J Oral Pathol. 1988;17:43–6. doi: 10.1111/j.1600-0714.1988.tb01504.x. [DOI] [PubMed] [Google Scholar]
- 27.Jungell P. Oral lichen planus. A review. Int J Oral Maxillofac Surg. 1991;20:129–35. doi: 10.1016/s0901-5027(05)80001-3. [DOI] [PubMed] [Google Scholar]
- 28.Chou MJ, Daniels TE. Langerhans cells expressing HLA-DQ, HLA-DR and T6 antigens in normal oral mucosa and lichen planus. J Oral Pathol Med. 1989;18:573–6. doi: 10.1111/j.1600-0714.1989.tb01554.x. [DOI] [PubMed] [Google Scholar]
- 29.Jakob T, Brown MJ, Udey MC. Characterization of E-cadherin-containing junctions involving skin-derived dendritic cells. J Invest Dermatol. 1999;112:102–8. doi: 10.1046/j.1523-1747.1999.00475.x. [DOI] [PubMed] [Google Scholar]
- 30.Ivanovski K, Nakova M, Warburton G, Pesevska S, Filipovska A, Nares S, et al. Psychological profile in oral lichen planus. J Clin Periodontol. 2005;32:1034–40. doi: 10.1111/j.1600-051X.2005.00829.x. [DOI] [PubMed] [Google Scholar]