

Abstract
Mass spectrometry of intact proteins and protein complexes has the potential to provide a transformative level of information on biological systems, ranging from sequence and post-translational modification analysis to the structures of whole protein assemblies. This ambitious goal requires the efficient fragmentation of both intact proteins and the macromolecular, multi-component machines they collaborate to create through non-covalent interactions. Improving technologies in an effort to achieve such fragmentation remains perhaps the greatest challenge facing current efforts to comprehensively analyze cellular protein composition and is essential to realizing the full potential of proteomics. In this work, we describe the use of a trimethyl pyrylium (TMP) fixed-charge covalent labeling strategy aimed at enhancing fragmentation for challenging intact proteins and intact protein complexes. Combining analysis of TMP-modified and unmodified protein complexes results in a greater diversity of regions within the protein that give rise to fragments, and results in an up to 2.5-fold increase in sequence coverage when compared to unmodified protein alone, for protein complexes up to 148 kDa. TMP modification offers a simple and powerful platform to expand the capabilities of existing mass spectrometric instrumentation for the complete characterization of intact protein assemblies.
GRAPHICAL ABSTRACT
Rapid characterization of proteins in increasingly complex mixtures by mass spectrometry (MS) has emerged as a powerful platform for understanding the details of biology and biochemistry at an unprecedented level of detail. Conventional proteomics workflows utilize enzymatic digestion prior to separation and MS to sequence the peptides excised from proteins in mixtures as complex as whole cell lysates1. This rapid characterization of exceptionally complex protein mixtures has enabled a revolution in biological analysis2, from determining the proteins and interactions involved in disease processes to aiding in solving the three-dimensional structures of proteins and protein complexes. Despite these advances, current ‘bottom-up’ proteomics approaches are typically unable to completely identify all the post-translational modifications and proteoforms that are crucially important for biological function3. In response to such deficiencies, ‘top-down’ MS technologies have been developed that are capable of sequencing intact proteins without enzymatic digestion4–6. Such tools are typically capable of capturing a wide-ranging snapshot of proteoform composition, but typically lack the ability to completely characterize even medium-sized protein sequences7,8. This deficiency is amplified by the fact that most proteins require the actions or presence of other proteins to accomplish their biological function9,10. Detecting and analyzing these interactions requires characterization of not just individual intact proteins, but the preservation and analysis of the non-covalent complexes they form in the cell. Both bottom-up and typical top-down proteomics require denaturation and/or enzymatic digestion of proteins, often precluding any analysis of structure and dynamics of these complex assemblies.
Direct characterization of intact protein assemblies using native MS11 offers a promising alternative to determine protein interactions, along with significant structural and mechanistic insights into the function of multiprotein machines12–14. Furthermore, native MS has the potential to provide protein sequence and structural information in the context of the same experiment, making it a promising tool for the study of protein complexes. However, sequencing technology coupled to native MS experiments lags far behind complementary bottom-up and top-down approaches targeting small, monomeric proteins. Incomplete fragmentation of proteins limits the ability of native MS to identify unknown proteins within complexes and prevents detailed analysis of the proteoforms incorporated within such assemblies. While collision induced dissociation (CID) is a widely available and effective technology for peptide sequencing, CID information extracted from large proteins and protein complexes analyzed under native conditions is often limited by the low charge density observed for analytes under such conditions15. In many cases, sequence coverage is concentrated into a few labile regions, e.g. flexible or terminal loop areas8. Achieving full sequence coverage for such large protein systems is one of the key challenges facing top-down proteomics, as well as the establishment of native MS workflows for wide-ranging structural proteomics.
Approaches to improve sequence coverage in top-down proteomics have focused largely on the development of ion activation paradigms other than CID. Electron transfer and capture dissociation (ETD16,17 and ECD18–21), infrared multiphoton dissociation (IRMPD22,23), electron ioniziation dissociation (EID24), and ultraviolet photodissociation (UVPD7,25) have demonstrated significant improvements in sequence coverage relative to and in combination with CID. Current state of the art fragmentation is typically achieved by employing several activation methods in concert, but often still provides only modest coverage of large proteins and protein complexes7,8. Implementing multiple activation techniques often requires instrument modification beyond the capability of many laboratories and is expensive, and many promising new techniques remain limited to a small subset of available MS platforms. CID remains the most widely available fragmentation technique and is by far the most common technique to be used in combination with others, making further development of CID techniques an attractive target for improving sequencing technology.
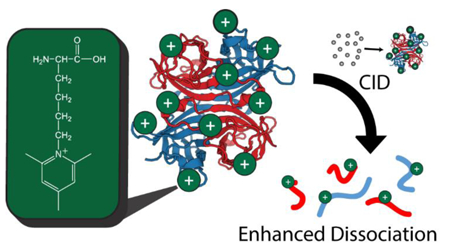
Chemical modification of protein complexes offers the potential for a new set of complementary methods to expand intact protein characterization by MS. Derivatization of single peptides with reagents that bear intrinsic positive charge has previously been shown to alter the dissociation pathways accessed in CID, improving sequencing for bottom-up proteomics experiments26–29. Several reports have used fixed charge derivatization to alter the charge states of electrosprayed protein ions30,31, but expanding this concept to sequencing of large proteins and protein complexes has proven highly challenging due to the difficulty of maintaining a fixed charge at the energies required to cause backbone fragmentation in such systems32. In this report, we present the use of trimethyl pyrylium33,34 (TMP) to covalently tether a stable positive charge to protein lysine side chains, altering the energy of various dissociation pathways to enable improved sequencing for large protein complex ions. Fixed charge modification by TMP provides orthogonal sequence coverage to other forms of biomolecular activation in the gas phase, opening a new pathway for improved sequence coverage of challenging protein targets using a simple derivatization that relies upon a commercially available and inexpensive reagent.
Experimental
Chemical modification.
Avidin from chicken egg white, Alcohol dehydrogenase (ADH) from Saccharomyces cerevisiae, Ovalbumin from chicken (all from Sigma Aldrich, St. Louis, MO), were dissolved in 100mM triethylammonium bicarbonate (TEAB, Sigma Aldrich), pH 8.5, to make solutions containing 25uM protein for chemical modification. 2,4,6-Trimethyl pyrylium (TMP) tetrafluoroborate (Alfa Aesar, Haverhill, MA) was dissolved in 100mM TEAB, pH 8.5, vortexed for ten seconds to dissolve, and quickly added to protein solutions to 10- to 25-fold molar excess relative to the reactive Lysine residues present. Reaction solutions were briefly vortexed and allowed to react for 24 hours at room temperature. Following modification, proteins were buffer exchanged sequentially into 1M ammonium acetate, then 200mM ammonium acetate, both pH 7.4 (Sigma Aldrich), with P6 (Avidin) or P30 (ADH, Ovalbumin) microspin columns (BioRad Laboratories, Hercules, CA) according to manufacturer instructions. Buffer exchanged samples were either analyzed immediately or flash frozen with liquid nitrogen and stored at −80C prior to analysis.
Ion Mobility-Mass Spectrometry.
A quadrupole ion mobility-time of flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA) was used for all ion mobility experiments. 5uL of buffer-exchanged protein solution (20uM) was transferred to a gold-coated borosilicate capillary (0.78mm i.d., Harvard Apparatus, Holliston, MA) for direct infusion. Instrumental settings were optimized to preserve intact protein complexes prior to activation: capillary voltage 1.5 kV, sample cone 40 V, extraction cone 0 V. Gas flows (mL/min): source: 50, trap: 6 (Avidin) or 8 (ADH, Ovalbumin), helium cell: 200, IM separation: 90. IMS traveling wave settings were the same for all proteins: wave velocity: 150 m/s, wave height: 20 V, IMS bias: 5 V. Backing pressure was set to 5.5 mbar (Avidin), or 8.0 mbar (ADH, Ovalbumin). A single charge state of each protein complex was selected and collisionally activated in the trap cell (trap collision voltage: Avidin, 140 V; Ovalbumin, 160 V; ADH, 200 V) prior to ion mobility separation. Trap (collision cell) pressures were 3.8 E-2 mbar (Avidin, Ovalbumin) or 4.4e-2 mbar (ADH). Time of flight pressure was 1.8 E-6 mbar for all analyses. Scans were combined for 30 seconds (Avidin) or 10 minutes (Avidin, ADH, Ovalbumin) to obtain sufficient signal to noise ratios.
FT-ICR mass spectrometry.
After buffer exchange, protein solutions (diluted to 10 μM in 100 mM aqueous ammonium acetate) were transferred to a metal-coated borosilicate capillary (Au/Pd coated, 1 μm i.d., Thermo Fisher Scientific, West Palm Beach, FL, USA) and mounted in the nanospray ion source. Mass spectrometry experiments were performed using a 15T SolariX FTICR instrument equipped with an infinity cell (Bruker Daltonics, Bremen, Germany). The following instrument settings were used: ESI voltage: 1.2 – 1.3 kV, dry gas temperature: 180 °C, flow rate: 2.0 L/min, RF amplitude of ion funnels: 200 Vpp, Funnel 1 voltage: 200 V, Funnel 2 voltage: 6 V, Skimmer 1 voltage: 60 – 180 V (longer for larger precursor masses; not enough to induce fragmentation), Skimmer 2 voltage: 5 V, multipole 1 RF: 2 MHz, quadrupole RF: 1.4 MHz, transfer hexapole RF: 2 MHz, time-of-flight: 1 – 2 ms (higher for larger proteins). CID experiments were performed at collision cell voltages from 30–100V at a collision cell gas flow of 35%. Ions were accumulated for 500 ms in the collision cell before entering the infinity ICR cell. Source, quadrupole, and UHF pressures were 2.5e0, 3.7e-6, and 1.8e-9 mbar, respectively. At least 200 scans were combined to obtain a sufficiently high signal-to-noise ratio. IRMPD was performed using a 30 W CO2 laser (Synrad, Mukilteo, WA, USA) interfaced to the back of the instrument. Laser power was held at 95% with an irradiation time of 1 s.
Data Analysis.
For ion mobility-mass spectrometry data, slices of the 2D IM-MS data corresponding to peptide charge states were extracted from raw data to text format using TWIMExtract35, a data querying tool developed for processing Waters IM-MS data. Extracted data was smoothed (Savitsky-Golay, 0.2 m/z window size, 3 cycles), peak-picked, and de-isotoped (max charge 5, isotope mass tolerance 0.05 m/z, isotope intensity tolerance 100%) using mMass v5.5.036–38 to generate peak lists. Peaks were identified using an in-house single protein search script written in java to allow for identification of fragments containing variable numbers of intrinsically charged TMP modifications given the starting protein sequence. a, b, and y-type ions and neutral losses of water and ammonia were considered for identification of peaks after examination of the data revealed little contribution from other fragmentation pathways. Only terminal fragments were considered, as statistically confident identification of internal fragments was not possible given available resolution and mass accuracy. Mass tolerance of 10 ppm was used as the cutoff for peak identification. At least three replicates were used for all fragmentation data presented (Avidin n=5, ADH n=3, Ovalbumin n=3). Fragments identified in fewer than the majority of replicates were excluded from analyses. Error bars for sequence coverage plots were presented as two times the standard deviation in the number of cleavage sites observed for each replicate. For FT-ICR data, Data Analysis 4.0 (Bruker Daltonics, Billerica, MA) was used to extract and process raw data into peak lists. Peak lists were then processed using the same in-house search program and parameters as ion mobility-mass spectrometry data.
Results
CID-stable charge fixing chemical modification
In CID experiments, a large number of collisions with inert gas molecules are required to impart sufficient energy for the production of sequence informative fragment ions. This slow heating, along with the relatively rapid rate of intramolecular redistribution of the imparted vibrational energy39, results in fragmentation of the most labile bonds in the protein. Charge mobility plays a key role in the CID process, as “mobile protons”40–42 move along the peptide backbone, triggering b- and y-ion formation across a range of sites. Protein complexes preserved through the electrospray ionization (ESI) process (i.e. using native mass spectrometry conditions) typically have a low charge–to–mass ratio, and are furthermore stabilized by secondary, tertiary, and quaternary structural elements, making typical mobile-proton based CID an efficient process for only a small subset of the peptide bonds available. Fixing intrinsically-charged moieties has been shown to alter the fragmentation of peptides from primarily b- and y-type ions formed through mobile proton type fragmentation to primarily a-ions formed through charge-remote mechanisms26–29, but prior to this report, had not yet been extended to the CID of intact proteins and their assemblies.
The compound 2,4,6-trimethyl pyrylium (TMP) reacts with primary amines to produce an intrinsically charged pyridinium salt at the nitrogen of the original primary amine33,34 (Figure 1), resulting in the addition of C8H8+ with a mass shift of 104.06205 Da (Fig 1, Fig S1). The efficiency of labeling under native conditions is not perfect (Fig. S2, S3), resulting in a mixture of modification states. The TMP derivatization reaction proceeds under conditions that allow for native-like buffer conditions (see methods), resulting in labeling of intact protein complexes without significantly altering their structure (Figures S4-S7). IM profiles of the three protein complexes examined in this report before and after labeling are shown in supporting figures (S4-S6), demonstrating the preservation of a single conformation through the labeling procedure for all protein complexes examined. The drift times of the modified proteins increase slightly (5–10%), which can be attributed to the additional mass (up to ~10 kDa) added to the complexes by the reaction, which is consistent with previous work with native labeling of protein complexes43. Furthermore, collisional activation of TMP-modified Avidin tetramer results in an unfolding trajectory that mirrors that of the unmodified tetramer (Fig. S7), indicating preservation of the existing overall structure.
Figure 1.

Charge-fixing chemical modification scheme with TMP. At left, the cartoon structure of an example protein complex prior to modification with the structure of a lysine residue highlighted above. At right, the effect of TMP modification on primary amines present in the protein complex (lysine residues and the N-terminus). Reaction of the pyrylium with a primary amine results in a pyridinium derivative with a fixed positive charge on the nitrogen atom of the former amine. Plus signs indicate positive charges localized to lysine residues or N-termini throughout the protein complex.
Following ESI, both TMP fixed charges and mobile protons can influence the CID behavior of the complex. Unlike most commonly used, intrinsically charged modifications, such as sulfonium-based reagents44,45 and quaternary amines32,46–48, gas-phase decomposition of TMP-modified lysine is not energetically favorable under typical CID conditions for proteins and peptides, ensuring that the charges remain fixed throughout the process. This enables TMP-based fixed charges to alter the potential energy landscape associated with protein fragmentation and thus enhance the formation of sequence-informative product ions by CID.
Fixed-charge modification enhances sequence coverage in a model protein complex
Avidin, a 64 kDa homo-tetramer, has been studied extensively as a CID model for noncovalent protein assemblies in native mass spectrometry49,50. It exhibits some of the strongest noncovalent interactions between subunits of known protein complexes, making the tetramer a challenging target for top-down sequencing. TMP-modified Avidin tetramer was compared to unmodified Avidin using top-down ion mobility-mass spectrometry to determine the benefits of chemical modification for extracting protein sequence information directly from protein complex ions (Fig. 2). The ion mobility separation, specifically, was used to separate peptides of different charge states51, enhancing the ability of the time-of-flight mass analyzer to characterize the ion populations resulting from fragmentation of the complexes examined.
Figure 2.
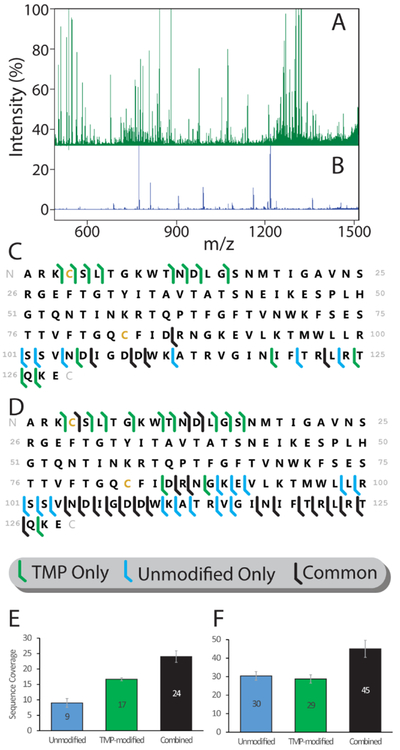
Enhanced sequencing of the Avidin tetramer following TMP modification. (A) Mass spectrum of fragments from CID of TMP-modified Avidin compared to (B), mass spectrum of fragments from unmodified Avidin presented on the same intensity axis. Sequencing information obtained from intact Avidin tetramer for thirty-second (C) and ten-minute (D) accumulation times (N=3). Cleavage sites unique to TMP-modified Avidin are highlighted in green, those unique to unmodified Avidin in blue, and common to both states in black. (E) Total sequence coverage obtained from thirty-second fragment accumulation, or (F) ten-minute fragment accumulation, for unmodified (blue), TMP-modified (green), and both datasets combined (black).
Top-down analyses often require significant signal accumulation times, as extensively fragmenting a single precursor results in many low intensity fragment ions52. The impact of TMP modification was examined on timescales that both mimic those utilized in the context of on-line separations coupled to MS detection (30 seconds total accumulation time) and an in-depth full coverage experiment, typically associated with separation tools coupled off-line to top-down MS (10 minutes total accumulation time). While significant accumulation times offer the most information about the impact of TMP on product ion populations and the future sequencing outcomes for this approach overall, the majority of applied top-down proteomics is typically done of the timescale of chromatographic separations, where elution of a species typically occurs on a timescale of seconds to tens of seconds. As such, the 30 second accumulation time data shown in Figure 2, where we observe the greatest enhancements to the number of product ion populations and their orthogonality to those produced from unmodified protein ions, represents the most practical assessment of the ability of TMP to serve as part of current top-down sequencing workflows.
The low mass region of the mass spectra acquired for unmodified and TMP-modified Avidin tetramers over the ten-minute time frame discussed above (Fig. 2A-B) exhibit a more even distribution of intensity amongst many fragment peaks when compared to equivalent spectra acquired for the unmodified protein. For example, CID of unmodified Avidin typically generates only approx. five fragment peaks that exceed 20% relative intensity (Fig. 2B), which is typical of CID of proteins having low, native-like charge states. The spectrum from TMP-modified Avidin, in contrast, shows intensity more evenly distributed across dozens of peaks (Fig. 2A). This fragment ion intensity distribution enables, in part, the substantial improvement in sequencing observed in our thirty-second accumulation runs, where TMP-modified Avidin generates nearly double the sequence coverage (17 cleavage sites vs 9) when compared to equivalent data for the unmodified protein tetramer (Fig. 2C, E). Most cleavage sites observed have numerous fragment ions corresponding to them (multiple charge states, neutral losses, and modification numbers), hence why the rich fragment spectra observed correspond to lower total numbers of cleavage sites. Modified Lysine residues are not explicitly labeled in Fig. 2 as the exact location of modification cannot be determined in all cases. Over the course of a full ten-minute accumulation, many peaks that are generated in very low abundance in the unmodified Avidin accumulate sufficient signal to be resolved and identified, reducing the difference in total coverage between modified and unmodified Avidin (Fig. 2D, F). The combined datasets, however, maintain a significant improvement in sequence coverage relative to any individual sequencing dataset due largely to the orthogonality of regions we observe to be covered within the Avidin sequence when using modified and unmodified precursor ions. We note that Avidin contains a highly heterogeneous glycosylation site at reside ASN-1753,54. Top-down fragmentation datasets from neither control nor TMP-modified Avidin reveal any evidence of this modification, despite extensive coverage of the n-terminus of the protein in our TMP-related data up to SER-15.
TMP modification acts to diversify the structural elements of the Avidin assembly from which sequence information can be obtained in addition to improving the total number of fragments observed. Figure 3 shows the X-ray structure of Avidin55 (PDB code: 1AVD), where highlighted residues indicate a detected fragmentation event for unmodified (Fig. 3B, E) and TMP-modified (Fig. 3C, F) tetramer. We use this surface map representation in order to visualize the locations of fragmentation events across the protein surface, taking into account the monomer and dimer substructures of the Avidin complex, which are shown in Figure 3 in place of the tetramer for simplicity. Fragmentation in the unmodified Avidin is confined to two main regions of the surface map with very little coverage of the total protein surface. In contrast, the regions of coverage in TMP-modified Avidin cover more of the protein surface and access regions left undetected in our datasets from unmodified Avidin, particularly at the n-terminus of the protein. The orthogonality of coverage is clearly demonstrated by projecting a combined fragmentation map of the Avidin complex (Fig. 3 D, G), where a few small regions are found in both modified and unmodified Avidin, but much of the observed coverage is unique to one experimental condition or the other.
Figure 3.

Mapping locations of peptide bond cleavage on Avidin. (A) Depiction of the monomer surface map used for comparison of cleavage locations, generated from Avidin crystal structure (PDB code: 1AVD). Monomer and dimer surfaces from the complex were used for ease of view. (B-D) Cleavage site map of fragments from thirty-second accumulation for unmodified, TMP-modified, and both combined, respectively. (E-G) Cleavage site maps from (B-D) presented on dimer surfaces to highlight interface locations in the complete Avidin tetrameric complex. In the combined views (D, G), blue coloring indicates cleavage sites unique to unmodified Avidin, green coloring indicates those unique to TMP-modified Avidin, and black coloring indicates sites common to both.
Modification extends proteomic sequencing to large protein complexes
Many proteins of critical biological importance assemble into complexes with masses that extend to hundreds of kilodaltons and beyond56,57. Such sizes are sufficient to be highly challenging for current top-down sequencing technologies, leaving important post-translational modifications located deep within these sequences inaccessible. In order to investigate the ability of TMP derivatization to bridge this technology gap, we modified two large protein complexes and carried out top-down sequencing experiments in a mode similar to those described above. Alcohol dehydrogenase (ADH) from yeast, a 148 kDa tetramer containing several phosphorylation sites, and Ovalbumin from chicken, a 170 kDa tetramer, were modified with TMP and subjected to CID for sequencing (Fig. 4).
Figure 4.

Enhanced sequencing of large protein complexes ADH and Ovalbumin. (A, D) Total sequence coverage (number of unique peptide bond cleavage sites) obtained from modified and unmodified ADH (a) and Ovalbumin (d) (N=3). (B, E) Sequence map of cleavage sites obtained from ADH (B) and Ovalbumin (E). Black dots indicate the middle 150 (b) or 200 (e) residues of the protein sequence, from which no coverage was obtained for any condition. (C, F) Cleavage location maps for ADH (C) and Ovalbumin (F). As in figure 3, only a monomer is shown of the tetrameric structure to allow view of all sides. Coverage unique to unmodified protein is colored blue, TMP-modified protein is colored green, and sites common to both states are colored black
Sequencing data obtained for unmodified ADH results in relatively few fragment ions that originate primarily (>90%) from the n-terminal region of the protein. Such results are common for typical top-down sequencing efforts involving ADH, in which fragmentation rarely penetrates past reside 30 of the protein sequence17,21, though concentrated efforts with multiple activation methods have achieved additional coverage23. In contrast, TMP modification of ADH yielded a dramatic improvement in CID sequence coverage compared to unmodified ADH, with more than twice as many sequence-informative fragments detected for the TMP-modified protein (Fig. 4 A, C). Furthermore, while TMP-modification led to enhanced coverage at the ADH n-terminus, on par with previous ETD17 and ECD21 datasets, it also unlocked significant fragmentation from the c-terminus, including 12 new c-terminal fragment sites that cover an additional 57 residues not typically accessed in unmodified ADH by CID, ECD, or ETD data. Similar to Avidin, TMP modification resulted in orthogonal coverage information and the combined analysis of both modified and unmodified data resulted in the most total coverage, with nearly two and a half times the coverage of the unmodified ADH alone. Our TMP-modified sequencing data also covers a phosphorylation site at SER-31658–60, which was previously shown to be up-regulated in the presence of a mating pheromone58. Phosphorylation at this site was not detected in our dataset, likely indicating the ADH standard used is not phosphorylated at this site, although we cannot rule out loss of the attached phosphate during CID.
Unlike ADH and Avidin, both n- and c-termini of unmodified Ovalbumin exhibited significant sequence coverage values in our experiments. The compact, low-charge monomer of Ovalbumin was analyzed, rather than the intact tetramer due to low amounts of tetramer in both unmodified and modified spectra. Despite the significant coverage already present in unmodified Ovalbumin, TMP modification still resulted in a substantial improvement in coverage (Fig. 4B, D), with an average over three replicates of nearly 50% more fragmentation sites than unmodified Ovalbumin. Like ADH and Avidin, combining the modified and unmodified data resulted in the best overall sequence coverage for Ovalbumin, with an 80% improvement over unmodified Ovalbumin alone, on average (n=3). Ovalbumin contains several PTM sites; however, coverage of these regions of the protein sequence was not significantly extended through TMP modification. As might be expected given the similarity of the coverage maps in Fig. 4D, a superposition of fragmentation data with Ovalbumin monomer structure shows an incremental, yet significant, improvement in the diversity of regions of the protein surface covered through CID fragmentation of the TMP modified protein (Fig. 4F).
Modification of ADH and Ovalbumin demonstrates the potential of fixed charge derivatization with TMP to expand the capabilities of intact sequencing for large proteins and protein complexes. The dramatic improvement in sequencing of the ADH tetramer, and particularly the generation of fragments from the previously intractable c-terminal region represent the potential of TMP modification for enabling sequencing of previously inaccessible regions of large protein complexes. TMP modification compares favorably to state of the art fragmentation methods such as ECD and ETD without the advanced instrumentation requirements of those techniques, and provides complementary coverage in many cases. Furthermore, modification by TMP enabled coverage of a PTM site near the c-terminus of ADH that has not been previously accessed before by any top-down analysis to our knowledge. In the case of Ovalbumin, where essentially the same regions of the protein are fragmented in both the modified and unmodified cases, the improvement to total sequence coverage remains substantial.
High resolution MS analysis of large complexes
Ion mobility-mass spectrometry (IM-MS) analysis of ADH and Ovalbumin revealed significant portions of the protein sequences, despite being generally unable to isotopically resolve large (mass greater than approx. 8000 Da), highly charged (z > 5) fragment ions. To resolve these large fragments and further confirm the sequencing improvements offered by TMP, high resolution FT-ICR tandem MS analysis was performed on modified and unmodified ADH and Ovalbumin.
A comparison of CID fragmentation spectra acquired from the same ADH samples on both the FT-ICR and IM-MS platforms reveals clear trade-offs between the two instruments for top-down sequencing experiments. On the IM-MS platform, high energy CID and control of pressure and gas flow enabled extensive fragmentation of the large protein complex, but the mass resolution of the time of flight mass analyzer is insufficient to resolve isotopes for the largest fragments. The FT-ICR platform, in contrast, readily resolves any fragments generated, but did not bring about the degree of fragmentation observed on the IM-MS instrument used (Fig. 5D). Fragments identified for CID of modified and unmodified Ovalbumin precursors were nearly identical between the two platforms, with the exception of several high mass fragments identified on the FT-ICR platform that could not be isotopically resolved on the IM-MS instrument.
Figure 5.

FT-ICR MS sequencing of large protein complexes with chemical modification. (A) Comparison of fragment mass spectra from IRMPD of intact ADH tetramer unmodified (top) and TMP-modified (bottom). (B) Unique cleavage sites obtained from IRMPD of ADH unmodified (blue) and TMP-modified (green). (C) FT-ICR sequencing of Ovalbumin using both CID (left) and IRMPD (right) for unmodified (blue), TMP-modified (green), and combined (black) analyses. (D) Comparison of sequence information obtained for CID of Ovalbumin on FT-ICR (gray), IM-MS (beige), and common to both instruments (brown).
Infrared multiphoton photodissociation (IRMPD) was performed in addition to CID for both ADH and Ovalbumin on the FT-ICR platform. As is the case with our CID datasets discussed above, TMP modified ADH and Ovalbumin exhibited dramatic increases in fragmentation when compared to unmodified proteins. In the case of ADH, only a single fragment could be observed at low intensity in the IRMPD spectrum of the unmodified complex, while the TMP-modified complex had nearly a dozen peaks corresponding to five sequence informative cleavage sites (Fig. 5A, B). Analysis of IRMPD data acquired for TMP-modified Ovalbumin revealed a similar trend, with activation of the protein producing 18 sites of coverage compared to just three for the unmodified Ovalbumin (Fig. 5C).
Like CID, IRMPD requires step-wise heating of the protein in order to deposit sufficient energy to elicit sequence-informative fragmentation61. TMP modification results in improved sequence coverage for both the CID and IRMPD experiments, presumably due to the fact that both methods typically cleave the weakest bonds present in the protein sequence, and that this landscape of bond energies is significantly altered in TMP-bound protein ions. Modification by TMP thus represents a strategy for enhancing sequence coverage of intact proteins and protein complexes across slow-heating fragmentation techniques available on a great many MS instrument platforms. Large protein complexes often require many activation techniques operating together to achieve sufficient sequence coverage for post-translational modification (PTM) analysis and identification purposes, and it is clear from the data reported here that TMP modification, or similar strategies based on the principles discussed here, have the potential to enable an increased role for CID/IRMPD tools within such workflows.
Conclusion
Top-down and native MS have emerged as valuable techniques for the analysis of intact proteins and protein complexes, but have been limited by incomplete sequence coverage, particularly for large proteins and complexes. Chemical modification using intrinsically-charged moieties such as TMP provides a simple and effective method to substantially enhance sequence coverage of intact proteins and protein complexes. Using this approach, we demonstrate enhancements in sequence coverage of three challenging model systems of 50–150% over analysis of the unmodified protein complexes by CID alone. We show that TMP modification provides these benefits across multiple instrument platforms that utilize multiple activation techniques, with enhancements to coverage in both CID and IRMPD datasets. TMP-enhanced CID compares favorably with standard top-down activation techniques like ECD, demonstrating both comparable coverage in ECD-accessible regions and providing coverage of previously intractable regions of the ADH tetramer.
Despite fixing many positive charges to our model proteins through TMP modification, the fragmentation we observe remains dominated by the b- and y-type ions characteristic of mobile proton-induced fragmentation, as is typical for CID of unmodified proteins. The absence of a shift to primarily a-type ions, contrasting to experiments with charge-derivatized peptides, is a novel and somewhat surprising result that demonstrates the persistence of mobile-proton behavior for high mass ions capable of intramolecular charge pairing. Clearly, however, the fragmentation pattern of intact proteins can be altered by fixed charges, likely through changing the relative ordering of bond strengths throughout the molecule as a result of the new locations of fixed charges imparted by TMP. We cannot rule out that some of the observed alterations to fragmentation are due to pathways associated with charge remote fragmentation events, which was the ultimate aim of the TMP modification chemistry described here. Further data collection will be necessary to verify and quantify such channels within TMP modified proteins.
Modification with TMP is a simple, single-step procedure that can be easily incorporated into an existing experimental workflow. Improving sequencing of intact proteins and complexes without the need for extensive instrument modifications has the potential to expand the capabilities of many laboratories to analyze intact proteins and complexes within top-down workflows. A comprehensive protein complex analysis workflow, utilizing TMP modification in conjunction with one or several activation techniques, holds the potential to provide both state-of-the-art sequencing and PTM information as well as structural and stoichiometric details for protein assemblies, enabling next generation experiments in structural proteomics.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the Nastional Institutes of Health (R01GM095832 to BTR and R01GM103479 and S10RR028893 to JAL) and the US Department of Energy (UCLA/DOE Institute for Genomics and Proteomics; DE-FC03–02ER63421). The authors thank M. W. Haskell for assistance with modeling of mass shift data. F.L. thanks the Research Foundation – Flanders (FWO) for funding a PhD fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Contents: Mass spectrum of TMP-modified Substance P peptide. Detailed analysis of modification efficiency for the Avidin tetramer and model used in associated computation. Arrival time profiles of modified and unmodified AvidinAvidin, ADH, and Ovalbumin. Collision induced unfolding fingerprints of Avidin. Table of assigned fragment ions for modified and unmodified AAvidin.
REFERENCES
- (1).Mayne J; Ning Z; Zhang X; Starr AE; Chen R; Deeke S; Chiang CK; Xu B; Wen M; Cheng K; Seebun D; Star A; Moore JI; Figeys D Anal. Chem 2016, 88, 95–121. [DOI] [PubMed] [Google Scholar]
- (2).Yates JR J. Am. Chem. Soc 2013, 135, 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Beck M; Claassen M; Aebersold R Curr. Opin. Biotechnol 2011, 22, 3–8. [DOI] [PubMed] [Google Scholar]
- (4).Catherman AD; Skinner OS; Kelleher NL Biochem. Biophys. Res. Commun 2014, 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Savaryn JP; Catherman AD; Thomas PM; Abecassis MM; Kelleher NL Genome Med. 2013, 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Siuti N; Kelleher NL Nat. Methods 2007, 4, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS J. Am. Chem. Soc 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Han X; Jin M; Breuker K; McLafferty FW Science (80-. ). 2006, 314, 109–112. [DOI] [PubMed] [Google Scholar]
- (9).Alberts B Cell 1998, 92, 291–294. [DOI] [PubMed] [Google Scholar]
- (10).Robinson CV; Sali A; Baumeister W Nature 2007, 450, 973–982. [DOI] [PubMed] [Google Scholar]
- (11).Leney AC; Heck AJ R.J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pukala TL; Ruotolo BT; Zhou M; Politis A; Stefanescu R; Leary JA; Robinson CV Structure 2009, 17, 1235–1243. [DOI] [PubMed] [Google Scholar]
- (13).Sharon M; Taverner T; Ambroggio XI; Deshaies RJ; Robinson CV PLoS Biol. 2006, 4, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chait BT; Cadene M; Olinares PD; Rout MP; Shi YJ Am. Soc. Mass Spectrom. 2016, 27, 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yin S; Loo JA Int. J. Mass Spectrom. 2011, 300, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Syka JEP; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Proc. Natl. Acad. Sci 2004, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lermyte F; Sobott F Proteomics 2015, 15, 2813–2822. [DOI] [PubMed] [Google Scholar]
- (18).McLafferty FW; Horn DM; Breuker K; Ge Y; Lewis MA; Cerda B; Zubarev RA; Carpenter BK J. Am. Soc. Mass Spectrom. 2001, 12, 245–249. [DOI] [PubMed] [Google Scholar]
- (19).Zubarev RA; Kelleher NL; McLafferty FW J. Am. Chem. Soc 1998, 120, 3265–3266. [Google Scholar]
- (20).Xie Y; Zhang J; Yin S; Loo JA J. Am. Chem. Soc 2006, 128, 14432–14433. [DOI] [PubMed] [Google Scholar]
- (21).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML Anal. Chem 2011, 83, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Little DP; Speir JP; Senko MW; O’Connor PB; McLafferty FW Anal. Chem 1994, 66, 2809–2815. [DOI] [PubMed] [Google Scholar]
- (23).Li H; Wongkongkathep P; Van Orden SL; Ogorzalek Loo RR; Loo JA J. Am. Soc. Mass Spectrom. 2014, 25, 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li H; Sheng Y; McGee W; Cammarata M; Holden D; Loo JA Anal. Chem 2017, 89, 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bowers WD; Delbert SS; Hunter RL; Mciver RT J. Am. Chem. Soc 1984, 106, 7288–7289. [Google Scholar]
- (26).Vath JE; Biemann K Int. J. Mass Spectrom. Ion Process. 1990, 100, 287–299. [Google Scholar]
- (27).Adamczyk M; Gebler JC; Wu J Rapid Commun. Mass Spectrom. 1999, 13, 1413–1422. [DOI] [PubMed] [Google Scholar]
- (28).Chen W; Lee PJ; Shion H; Ellor N; Gebler JC Anal. Chem. 2007, 79, 1583–1590. [DOI] [PubMed] [Google Scholar]
- (29).Roth KDW; Huang Z-H; Sadagopan N; Watson JT Mass Spectrom. Rev 1998, 17, 255–274. [DOI] [PubMed] [Google Scholar]
- (30).Frey BL; Krusemark CJ; Ledvina AR; Coon JJ; Belshaw PJ; Smith LM Int. J. Mass Spectrom. 2008, 276, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Krusemark CJ; Frey BL; Belshaw PJ; Smith LM J. Am. Soc. Mass Spectrom. 2009, 20, 1617–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).He Y; Reilly JP Angew. Chemie - Int. Ed 2008, 47, 2463–2465. [DOI] [PubMed] [Google Scholar]
- (33).King LC; Ozog FJ J. Org. Chem 1955, 20, 448–454. [Google Scholar]
- (34).Li X; Cournoyer JJ; Lin C; O’Connor PB J. Am. Soc. Mass Spectrom. 2008, 19, 1514–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Haynes SE; Polasky DA; Dixit SM; Majmudar JD; Neeson K; Ruotolo BT; Martin BR Anal. Chem 2017, 89, 5669–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Strohalm M; Kavan D; Novák P; Volný M; Havlíček V Anal. Chem 2010, 82, 4648–4651. [DOI] [PubMed] [Google Scholar]
- (37).Strohalm M; Hassman M; Košata B; Kodíček M Rapid Commun. Mass Spectrom. 2008, 22, 905–908. [DOI] [PubMed] [Google Scholar]
- (38).Niedermeyer THJ; Strohalm M PLoS One 2012, 7, e44913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Baer T; Mayer PM J. Am. Soc. Mass Spectrom. 1997, 8, 103–115. [Google Scholar]
- (40).Cox KA; Gaskell SJ; Morris M; Whiting AJ Am. Soc. Mass Spectrom. 1996, 7, 522–531. [DOI] [PubMed] [Google Scholar]
- (41).Palzs B; Suhal S Mass Spectrom. Rev 2005, 24, 508–548. [DOI] [PubMed] [Google Scholar]
- (42).Dongré AR; Jones JL; Somogyi Á; Wysocki VH J. Am. Chem. Soc 1996, 118, 8365–8374. [Google Scholar]
- (43).Samulak BM; Niu S; Andrews PC; Ruotolo BT Anal. Chem 2016, 88, 5290–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhou X; Lu Y; Wang W; Borhan B; Reid GE J. Am. Soc. Mass Spectrom. 2010, 21, 1339–1351. [DOI] [PubMed] [Google Scholar]
- (45).Reid GE; Roberts KD; Simpson RJ; O’Hair RA J. J. Am. Soc. Mass Spectrom. 2005, 16, 1131–1150. [DOI] [PubMed] [Google Scholar]
- (46).Clifford-Nunn B; Showalter HDH; Andrews PC J. Am. Soc. Mass Spectrom. 2012, 23, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ko BJ; Brodbelt JS J. Am. Soc. Mass Spectrom. 2012, 23, 1991–2000. [DOI] [PubMed] [Google Scholar]
- (48).Krusemark CJ; Ferguson JT; Wenger CD; Kelleher NL; Belshaw PJ Anal. Chem 2008, 80, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Light-Wahl KJ; Schwartz BL; Smith RD J. Am. Chem. Soc 1994, 116, 5271–5278. [Google Scholar]
- (50).Schwartz BL; Light-Wahl KJ; Smith RD J. Am. Soc. Mass Spectrom. 1994, 5, 201–204. [DOI] [PubMed] [Google Scholar]
- (51).Zinnel NF; Pai PJ; Russell DH Anal. Chem 2012, 84, 3390–3397. [DOI] [PubMed] [Google Scholar]
- (52).Compton PD; Zamdborg L; Thomas PM; Kelleher NL Anal. Chem 2011, 83, 6868–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lee BS; Krishnanchettiar S; Lateef SS; Lateef NS; Gupta S Rapid Commun. Mass Spectrom. 2005, 19, 2629–2635. [DOI] [PubMed] [Google Scholar]
- (54).DeLange RJ; Huang T-SJ Biol. Chem 1971, 246, 698–709. [PubMed] [Google Scholar]
- (55).Pugliese L; Coda A; Malcovati M; Bolognesi MJ Mol. Biol 1993, 231, 698–710. [DOI] [PubMed] [Google Scholar]
- (56).Havugimana PC; Hart GT; Nepusz T; Yang H; Turinsky AL; Li Z; Wang PI; Boutz DR; Fong V; Phanse S; Babu M; Craig SA; Hu P; Wan C; Vlasblom J; Dar VUN; Bezginov A; Clark GW; Wu GC; Wodak SJ; Tillier ERM; Paccanaro A; Marcotte EM; Emili A Cell 2012, 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kirkwood KJ; Ahmad Y; Larance M; Lamond AI Mol. Cell. Proteomics 2013, 12, 3851–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gruhler A; Olsen JV; Mohammed S; Mortensen P; Faergeman NJ; Mann M; Jensen ON Mol. Cell. Proteomics 2005, 4, 310–327. [DOI] [PubMed] [Google Scholar]
- (59).Li X; Gerber SA; Rudner AD; Beausoleil SA; Haas W; Villén J; Elias JE; Gygi SP J. Proteome Res. 2007, 6, 1190–1197. [DOI] [PubMed] [Google Scholar]
- (60).Holt LJ; Tuch BB; Villen J; Johnson AD; Gygi SP; Morgan DO Science (80-. ). 2009, 325, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).McLuckey SA; Goeringer DE J. Mass Spectrom. 1997, 32, 461–474. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.