

Abstract
Scope:
The bioactive constituents in ginger extract responsible for their anti-hyperglycemic effect and the underlying mechanisms are incompletely understood. Gingerenone A (Gin A) has been identified as an inhibitor of p70 S6 (S6K1), a kinase that plays a critical role in the pathogenesis of insulin resistance. Our study aims to evaluate if Gin A can sensitizes the insulin receptor by inhibiting S6K1 activity.
Methods and results:
Western blot analysis revealed that Gin A induced phosphatidylinositide-3 kinase (PI3K) feedback activation in murine 3T3-L1 adipocytes and rat L6 myotubes, as evidenced by increased AKTS473 and S6K1T389 but decreased S6S235/236 and insulin receptor substrate 1 (IRS-1)S1101 phosphorylation. Western blot and immunoprecipitation analysis revealed that Gin A increased insulin receptor tyrosine phosphorylation in L6 myotubes and IRS-1 binding to the PI3K in 3T3-L1 adipocytes. Confocal microscopy revealed that Gin A enhanced insulin-induced translocation of glucose transporter 4 (GLUT4) into the cell membrane in L6 cells. 2-NBDG(2-N-(Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)−2-deoxyglucose) fluorescent assay revealed that Gin A enhanced insulin-stimulated glucose uptake in 3T3-L1 adipocytes and L6 myotubes.
Conclusions:
Gin A overcomes insulin resistance and increases glucose uptake by inhibiting S6K1 activity. Gin A or other plant-derived S6K1 inhibitors could be developed as novel anti-diabetic agents.
Keywords: Gingerenone A, p70 S6 kinase, Insulin resistance, Insulin receptor, Glucose uptake
Introduction
More than one-third of the American adults have the metabolic syndrome, a condition characterized by the presence of insulin resistance, obesity, dyslipidemia, and hypertension [1, 2]. Insulin resistance is the primary force driving the development of type 2 diabetes, a chronic disease with an unprecedented prevalence. Currently, more than a dozen anti-diabetic drugs are available. However, the declining therapeutic efficacy or intolerable side-effects after long-term patient use have often prevented the effective control of hyperglycemia [3]. Patients with inadequate or poor control of hyperglycemia often develop diabetic complications such as blindness, frequent limb infections, diabetic nephropathy, and cardiovascular disease. These complications are responsible for most diabetes-related morbidity and mortality [3]. There has been increasing interest in screening chemical compounds from the herb and plant extracts to develop safer and more effective anti-diabetic drugs.
Insulin activates insulin receptor signaling by first phosphorylating the receptor itself. Activated insulin receptor in return phosphorylates other intracellular adaptor proteins such as insulin receptor substrates (IRS) [4, 5]. IRS interact with the p85 subunit of PI3K and activates PI3K, a kinase that phosphorylates and activates protein kinase B (AKT) (Fig. 1A) [4, 5]. AKT regulates glucose metabolism by multiple mechanisms [6, 7]. It inhibits gluconeogenesis, stimulates glycogen synthesis, and enhances glucose uptake in the insulin-sensitive tissues by triggering glucose transporter type 4 (GLUT4) translocation to the cell membrane (Fig. 1A) [4, 5]. The mechanistic target of rapamycin (mTOR), a kinase downstream of AKT, is constitutively activated by nutrient overload, subsequently leading to hyperactivation of p70 S6 kinase (S6K1) (Fig. 1A). S6K1 phosphorylates IRS-1 at serine 1101 [7, 8] and disrupts its interaction with PI3K, leading to poor AKT activation upon insulin stimulation [9]. S6K1 deficiency in S6K1−/− mice fed a high-fat diet (HFD) ameliorates obesity and hyperglycemia [10]. S6K1 is a key kinase that drives insulin resistance and induces obesity under nutrient overload conditions [6].
Fig. 1. The mechanisms of S6K1 inhibitors-induced feedback activation of the PI3K pathway and insulin receptor sensitization.
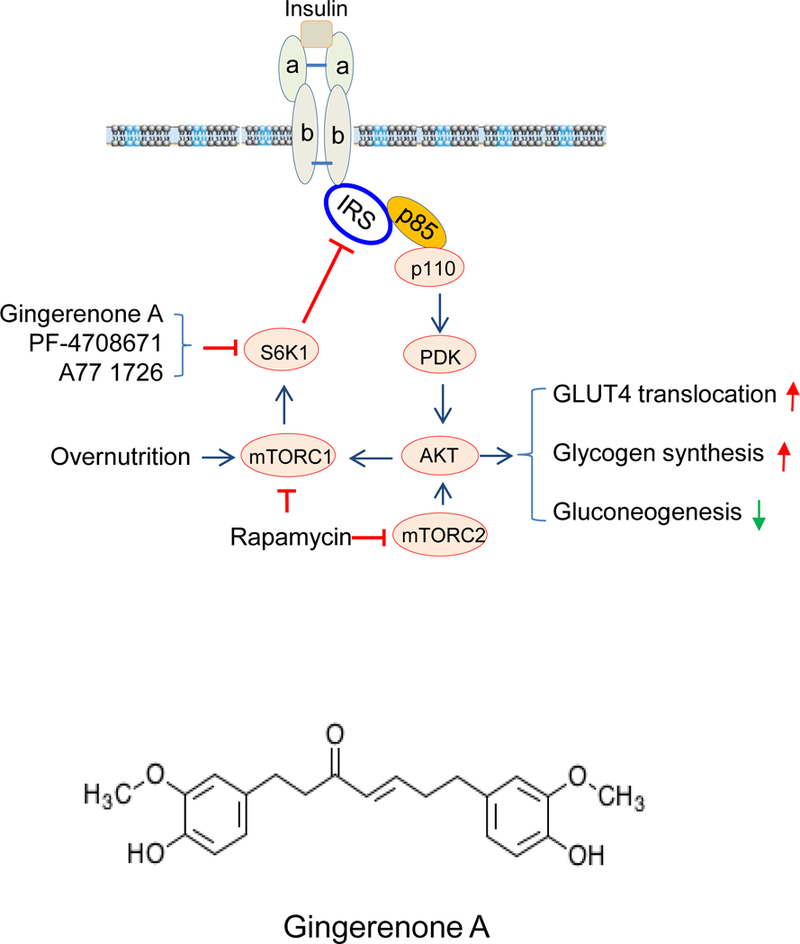
(A) Mode of action of Gin A and two other S6K1 inhibitors, PF-4708671 and A77 1726. Overnutrition with high concentrations of fatty acids and amino acids leads to constitutive S6K1 activation, which phosphorylates IRS-1S1101, leading to poor AKT activation. S6K1 inhibitors such as Gin A inhibit S6K1 activity and subsequently inhibit IRS-1S1101 phosphorylation, resulting in better interaction with the p85 subunit of PI3K and AKT activation. Activated AKT stimulates GLUT4 membrane translocation and glycogen synthesis but decreases gluconeogenesis. Chronic use of rapamycin leads to inhibition of both mTORC1 and mTORC2, thus exacerbating hyperglycemia. (B) Chemical structure of Gin A.
While S6K1 appears to be a novel therapeutic target for the control of hyperglycemia, whether plant-derived S6K1 inhibitors can sensitize the insulin receptor, improve glucose metabolism, and control hyperglycemia has not been studied. Zingiber species belong to the Zingiberaceae (ginger) family and have been widely used as spice additives and plant medicines [11]. Ginger extracts and its pungent phenolic components can increase glucose uptake and reduce blood glucose level [12–15]. Gingerols and shogaols in ginger extract have been identified as two types of the bioactive ingredients mainly responsible for their anti-hyperglycemia and anti-obesity effects [16–18]. These compounds regulate glucose metabolism by activating AMPK through an unresolved mechanism [16–18]. Recently, another compound extracted from ginger, Gin A, was found to inhibit S6K1 and Janus Kinase 2 (JAK2) activities, and to induce apoptosis in JAK-activated cancer cell lines [19]. More recently, Suk et al. reported that Gin A activates AMPK in adipocytes in vitro and in vivo, leading to the suppression of obesity and inflammation in HFD-fed mice [20]. However, this compound has not been studied in the context of diabetes. Here we report that Gin A sensitized the insulin receptor by AKT feedback activation, leading to increased GLUT4 translocation to the cell membrane, and increased glucose uptake in 3T3-L1 adipocytes and L6 myotubes.
2. Materials and methods
2.1. Reagents.
Gingerenone A was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). PF-4708671 and dexamethasone were obtained from Sigma Aldrich (St. Louis, MO). Rosiglitazone was obtained from Calbiochem (EMD Millipore, Billerica, MA). 3-Isobutyl-1-methylxanthine (IBMX). Insulin and 2-NBDG were purchased from Invitrogen (Life Technologies, Grand Island, NY). Antibodies against AKT, S6K1, S6, IRS-1, p85 of the PI-3 kinase and phospho-antibodies (AKTS473, pERKT202/204, S6K1T389, S6S235/236, IRS-1S1101, and IRY1146) were purchased from Cell Signaling Technology (Danvers, MA). A GLUT4 expression vector (mCherry-Glut4-myc) was obtained from Dr. Amira Klip (The Hospital for Sick Children, Toronto, Ontario).
2.2. Cell lines and differentiation.
L6 cells (a rat myoblast cell line) were grown in complete DMEM containing 10% fetal bovine serum. L6 myoblasts were differentiated into myotubes in DMEM by switching with the media containing 2% calf serum for two weeks. 3T3-L1 adipocytes (<15 passages) were differentiated as described by Zebisch et al. [21]. An adipocyte-like phenotypic change in >95% of the cells was considered to successfully induced cell differentiation. Both L6 and 3T3-L1 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA).
2.3. Western blot.
3T3-L1 adipocytes were cultured in serum-free medium overnight, whereas L6 myotubes were cultured in serum-free medium for 2h. Gin A at indicated concentrations (2.5–40 μM) or PF-4708671 (10 μM) was added and incubated for the indicated time, followed by stimulation with 100 nM insulin for the indicated time. Unstimulated cells were included as a negative control. Cells were harvested and lysed in NP-40 lysis buffer containing 1% NP-40; 50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 5 mM EDTA; 10 μg/ml aprotinin; 10 μg/ml leupeptin; 1 mM phenylmethylsulfonyl fluoride; 2 mM sodium pervanadate. Protein phosphorylation and total amounts of proteins were analyzed by Western blot with the indicated antibodies. NIH Image-J software was used to quantify the density of the bands followed by normalization with the arbitrary units of their corresponding total proteins. Results were presented as the mean ± standard deviation (SD) from three independent experiments in bar graphs.
2.4. IRS-1 binding to the PI-3 kinase.
3T3-L1 adipocytes were grown in serum-free medium overnight and then incubated either in essential balanced salt solution (EBSS) or EBSS containing 2X amino acids (AA) in the absence or presence of Gin A or PF-4708671 (20 μM) for 4 hr. Cells were stimulated with insulin (100 nM) for 10 min. Cells were harvested and lysed in NP-40 buffer, followed by immunoprecipitation with an anti-p85 rabbit monoclonal antibody. Immunoprecipiates were analyzed by electrophoresis, followed by Western blot with anti-p85 and anti-IRS-1 antibodies.
2.5. Visualization of GLUT4 membrane translocation.
L6 cells were seeded on coverslips and then transiently transfected with mCherry-Glut4-myc expression vector DNA using TurboFect Reagent according to the manufacturer’s instruction. After incubation for 24 hr, the cells were treated with Gin A 10 μM for 4 hr in EBSS containing 10% FBS with or without 2X AA. Cells were left unstimulated or stimulated with 100 nM insulin for 30 min. The coverslips were fixed in 4% paraformaldehyde at room temperature for 10 min. The coverslips were mounted with 50% glycerin in PBS containing 4,6-diamidino-2-phenylindole (0.5 μg/ml; Sigma Chemical Co.). mCherry-tagged Glut4 fluorescence was visualized under a Leica LP8 confocal microscope. The numbers of cells with GLUT4 membrane staining in 10 randomly selected fields from each treatment were divided by the total numbers of mCherry-GLUT4-positive cells. Percents of the GLUT4 membrane-positive cells were calculated and presented in a bar graph. The results represent the mean ± standard deviation from one of three independent experiments in triplicate.
2.6. Glucose uptake.
3T3-L1 adipocytes were first incubated in serum- and glucose-free DMEM overnight, then incubated either in EBSS or EBSS containing 2X AA in the absence or presence of Gin A or PF-4708671 (20 μM) for 2 hr. 2-NBDG (50 μM) was added at last 1 hr. L6 myotubes were incubated in serum- and glucose-free DMEM for 2 hr, Gin A or PF-4708671 and 2-NBDG were added and then incubated for 1 hr. Cells were stimulated with 50 nM insulin. Subsequently, the culture media were removed. The cell monolayers were washed with PBS three times. The fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm using a fluorescence microplate reader.
2.7. Dosage information.
L6 and 3T3-L1 cells were cultured in the absence or presence of Gin A at concentrations of 10–40 μM for 4 hr. No visible cytotoxicity such as cell floating, rounding or ruffling etc. was observed. Gin A causes cytotoxicity only in cancer cells that are addicted to an oncogene and does not have cytotoxicity in untransformed cells such as L6 and 3T3-L1 cells [19] [20]. Suk et al. [20] reported that Gin A at 40 μM does not cause 3T3-L1 adipocytes after incubation for 6 days. Likewise, there was no cytotoxicity in HEK293 cells after incubation with Gin A (50 μM) for 24 or 48 hr (X. Xu, unpublished observations).
2.8. Statistical analysis.
An unpaired Student t test was used to evaluate if there were significant differences in glucose uptake in 3T3-L1 adipocytes and L6 myotubes in various treatment groups. A p value <0.05 was considered statistically significant. All statistics analyses were conducted using a GraphPad Prism 6 software.
3. Results
3.1. Gin A induces PI3K feedback activation under insulin-sensitive conditions.
A prior study has shown that Gin A inhibits the activity of S6K1 and induces AKT feedback activation in the HCT116 colorectal cancer cell line [19]. We first evaluated the ability of Gin A to inhibit S6K1 activity and to induce the feedback activation of the PI3K pathway in 3T3-L1 adipocytes and in L6 myotubes. As shown in Fig. 2A, insulin induced AKTS473, S6K1T389, ERKT202/204, and S6S235/236 phosphorylation in 3T3-L1 adipocytes. Gin A enhanced insulin-induced phosphorylation of AKTS473 at 10, 20 and 40 μM in a dose-dependent manner. Interestingly, S6K1T389 phosphorylation was significantly increased by Gin A at 2.5 and 5 μM but started to decline at 10, 20, and 40μM. Consistently, Gin A enhanced insulin-induced AKTS473 phosphorylation in L6 myotubes in a dose-independent manner (Fig. 2B). S6K1T389 phosphorylation levels were lower in L6 myotubes treated with Gin A at high concentrations (20 and 40 μM) than that at low concentrations (2.5–10 μM). Gin A increased ERKT202/204 phosphorylation but decreased S6S235/236 phosphorylation in both 3T3-L1 adipocytes and L6 myotubes in a dose-dependent manner. PF-4708671 was included as a positive control. It significantly increased AKTS473, S6K1T389, and ERKT202/204 phosphorylation but decreased S6S235/236 phosphorylation in both 3T3-L1 adipocytes and L6 myotubes (Fig. 2). The observations of increased S6K1T389 phosphorylation by Gin A and PF-4708671 is consistent with numerous studies showing that inhibition of S6K1 activity leads to increased S6K1 phosphorylation [22, 23].
Fig. 2. Gin A induces feedback activation of the PI3K and MAPK pathways.
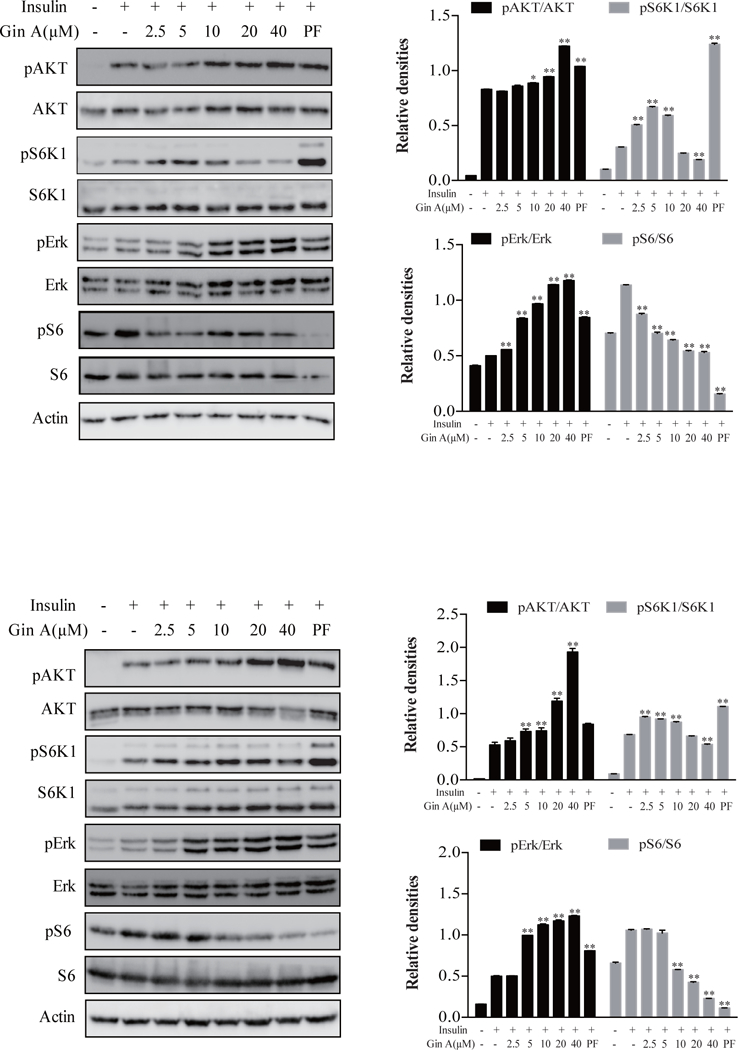
(A) 3T3-L1 adipocytes were starved in serum-free media overnight and then treated with Gin A or PF-4708671 (10 μM) for 5 hr. The cells were left unstimulated or stimulated with insulin (100 nM) for 10 min. (B) L6 myotubes were starved in serum-free media for 2 hr and then treated with Gin A or PF-4708671 (10 μM) for 2 hr. The cells were left unstimulated or stimulated with insulin (100 nM) for 5 min. Cells were harvested and analyzed for the phosphorylation of S6K1T389, AKTS473, ERKT202/204, and S6S235/236, followed by reprobing with their specific antibodies for total protein. β-actin was included as a loading control. Relative protein phosphorylation was determined by analyzing the density of the bands and presented as bar graphs. The results are the mean ± standard deviation (SD) from three experiments. *p<0.05; **p<0.01, compared to insulin-stimulated control.
3.2. Gin A induces PI3K feedback activation in 3T3-L1 adipocytes under insulin-resistant conditions.
We next determined the effect of Gin A on insulin signaling in these two cell lines cultured in the presence of 2X amino acids (AA), a condition of insulin resistance. As shown in Fig. 3A, Gin A at 10 μM enhanced insulin-induced S6K1T389 and AKTS473 phosphorylation more effectively in the presence of 2X AA than in its absence. Likewise, Gin A more effectively inhibited S6S235/236 and IRS-1S1101 phosphorylation in the presence of 2X AA than in its absence. Dose response revealed that Gin A at concentrations of 20 and 40 μM was less effective in inducing S6K1 phosphorylation than that with Gin A at 10 μM (Fig. 3B). In contrast, Gin A increased AKTS473 but decreased IRS1S1101 phosphorylation in a dose-dependent manner. Gin A did decrease S6S235/236 phosphorylation but not in a dose-dependent manner. PF-4708671 was included as a positive control. Again, it increased AKTS473 and S6K1T389 but decreased S6S235/236 and IRS1S1101 very effectively in 3T3-L1 adipocytes in the presence of 2X AA (Fig. 3B).
Fig. 3. Gin A induces feedback activation of the PI3K pathway in 3T3-L1 adipocytes under insulin resistance conditions.
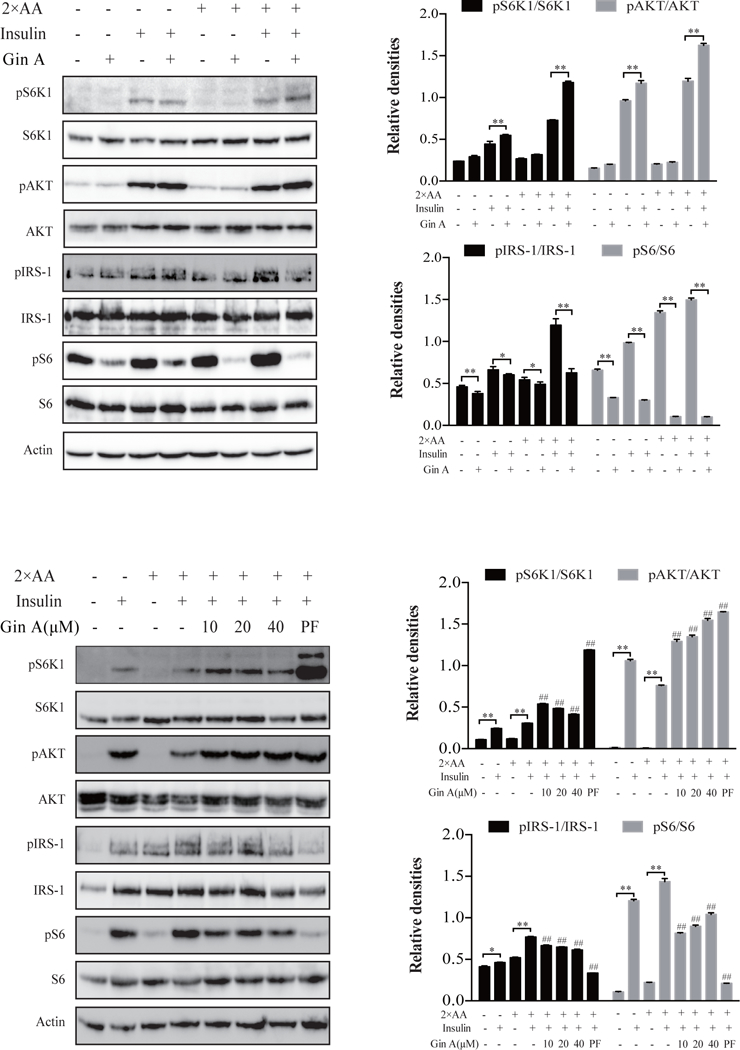
3T3-L1 adipocytes were starved in serum-free medium overnight and incubated in EBSS or EBSS containing 2X AA, then treated with or without Gin A (10 μM) (A) or with the indicated concentrations of Gin A or PF-4708671 (20 μM) (B) for 5 hr. The cells were left unstimulated or stimulated with insulin (100 nM) for 10 min. Cells were harvested and analyzed for the phosphorylation of S6K1T389, AKTS473, S6S235/236, and IRS-1S1101, followed by reprobing with their specific antibodies for total protein. β-actin was included as a loading control. Relative protein phosphorylation was determined by analyzing the density of the bands and presented as bar graphs. The results are the mean ± SD from three experiments. *p<0.05; **p<0.01; #p<0.05; ##p<0.01, compared to insulin-stimulated control.
3.3. Gin A induces PI3K feedback activation in L6 myotubes under insulin-resistant conditions.
We also assessed the effect of Gin A on insulin signaling in L6 myotubes cultured in the presence of 2X AA. As shown in Fig. 4A, Gin A at 10 μM enhanced insulin-induced S6K1T389 and AKTS473 phosphorylation more effectively in the presence of 2X AA than that in its absence. Gin A inhibited IRS-1S1101 and S6S235/236 phosphorylation in the absence or presence of 2X AA. Dose response analysis revealed that Gin A at 10 μM maximally increased S6K1 phosphorylation (Fig. 4B). Gin A increased AKTS473 but decreased IRS1S1101 phosphorylation in a dose-dependent manner. Gin A decreased S6S235/236 phosphorylation but not in a dose-dependent manner. PF-4708671 was included as a positive control. Again, it increased AKTS473 and S6K1T389 but decreased S6S235/236 and IRS1S1101 very effectively in L6 myotubes in the presence of 2X AA (Fig. 4B).
Fig. 4. Gin A induces feedback activation of the PI3K pathway in L6 myotubes under insulin resistance conditions.
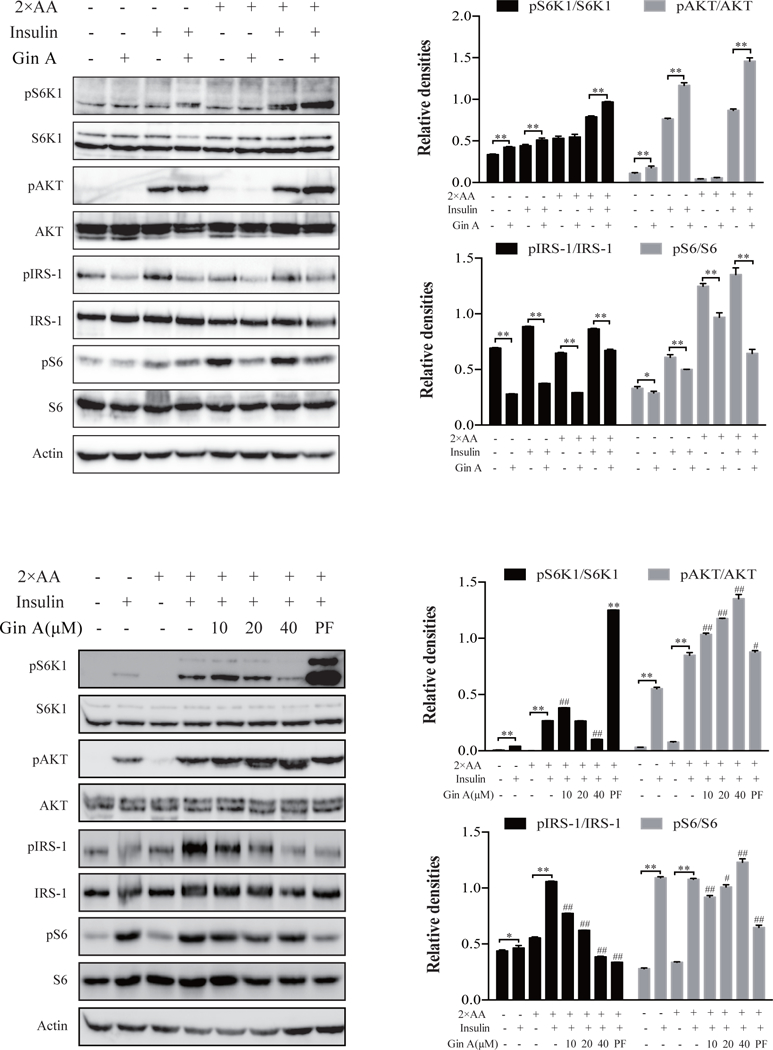
L6 myotubes were starved in serum-free media for 2 hr and incubated with EBSS or EBSS containing 2X AA, then treated with or without Gin A (10 μM) (A) or treated with the indicated concentrations of Gin A or PF-4708671 (20 μM) (B) for 2 hr. The cells were left unstimulated or stimulated with insulin (100 nM) for 5 min. Cells were harvested and analyzed for the phosphorylation of S6K1T389, S6S235/236, IRS-1S1101 and AKTS473, followed by reprobing with their specific antibodies for total protein. β-actin was included as a loading control. Relative protein phosphorylation was determined by analyzing the density of bands and presented as bar graphs. The results are the mean ± SD from three experiments. *p<0.05; **p<0.01, #p<0.05; ##p<0.01, compared to the control (with insulin and 2X AA but without drug).
3.4. Gin A increases the interaction of the PI3K with IRS-1 and sensitizes the insulin receptor.
We next determined whether AKT feedback activation resulted from increased interaction between IRS-1 and the PI3K. As shown in Fig. 5A, insulin increased the binding of the p85 subunit of the PI3K to IRS-1 in 3T3-L1 adipocytes. Gin A further increased the interaction of IRS-1 with the PI3K in a dose-dependent manner. PF-4708671 included as a positive control also increased the binding of p85 subunit of the PI3K to IRS-1 (Fig. 5A). Total IRS-1 and p85 levels were not changed in 3T3-L1 adipocytes incubated in the absence or presence of Gin A for 4 hr. Gin A increased insulin-stimulated tyrosine phosphorylation of the insulin receptor slightly more effectively in L6 myotubes in the presence of 2X AA than that in its absence (Fig. 5B).
Fig. 5. GinA sensitizes insulin receptor signaling.
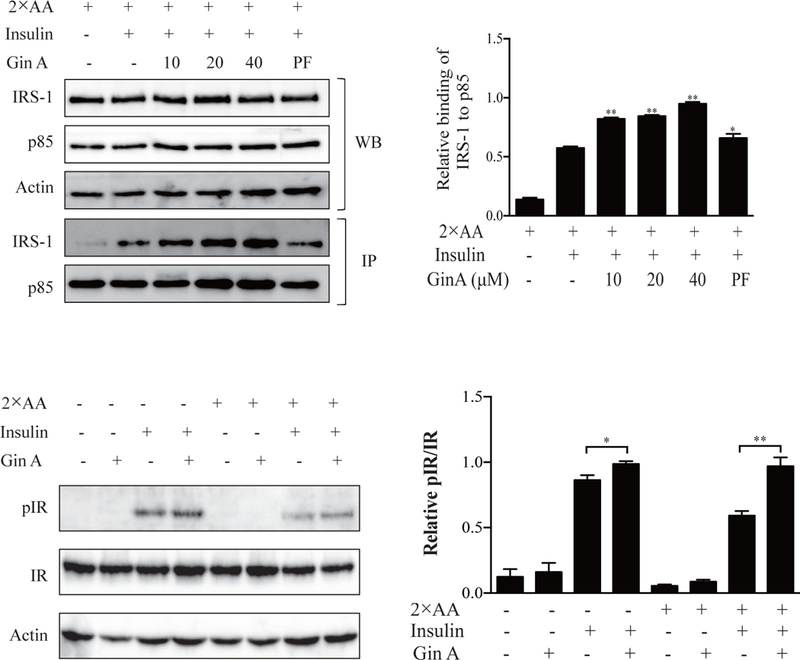
(A) 3T3-L1 adipocytes were starved in serum-free media overnight and incubated with EBSS or EBSS containing 2X AA, then treated with the indicated concentrations of Gin A or PF-4708671 (20 μM) for 4 hr. The cells were left unstimulated or stimulated with insulin (100 nM) for 10 min. Cells lysates were immunoprecipitated with an anti-p85 antibody followed by probing with anti-p85 and anti-IRS-1 antibodies. (B) 3T3-L1 adipocytes were treated as (A). Cell lysates were analyzed for IRY1146 phosphorylation, followed by reprobing with an antibody against total IR protein. β-actin was included as a loading control. Relative IRS-1 binding to the p85 subunit of PI3K (A) or relative IR phosphorylation levels (B) were determined by analyzing the density of the bands and presented as bar graphs. The results are the mean ± SD from three experiments. *p<0.05; **p<0.01; #p<0.05; ##p<0.01, compared to the control (with insulin and 2X AA but without drug).
3.5. Gin A increases GLUT4 translocation to cell membrane.
It is well established that AKT activation triggers the translocation of GLUT4 from cytoplasmic vesicles to the cell membrane [7, 24]. Here we tested if AKT feedback activation by Gin A led to GLUT4 translocation from the cytoplasm to the cell membrane. As shown in Fig. 6A, GLUT4 was present mostly in the cytoplasm of DMSO-treated L6 cells but was translocated to the membrane of L6 cells upon insulin stimulation (Fig. 6A). Gin A slightly increased GLUT4 translocation from the cytoplasm into the cell membrane in the absence of 2X AA. In contrast, Gin A significantly enhanced insulin-stimulated GLUT4 translocation from the cytoplasm to the cell membrane in the presence of 2X AA (Fig. 6A & B).
Fig. 6. Gin A enhances insulin-stimulated GLUT4 translocation to the cell membrane.
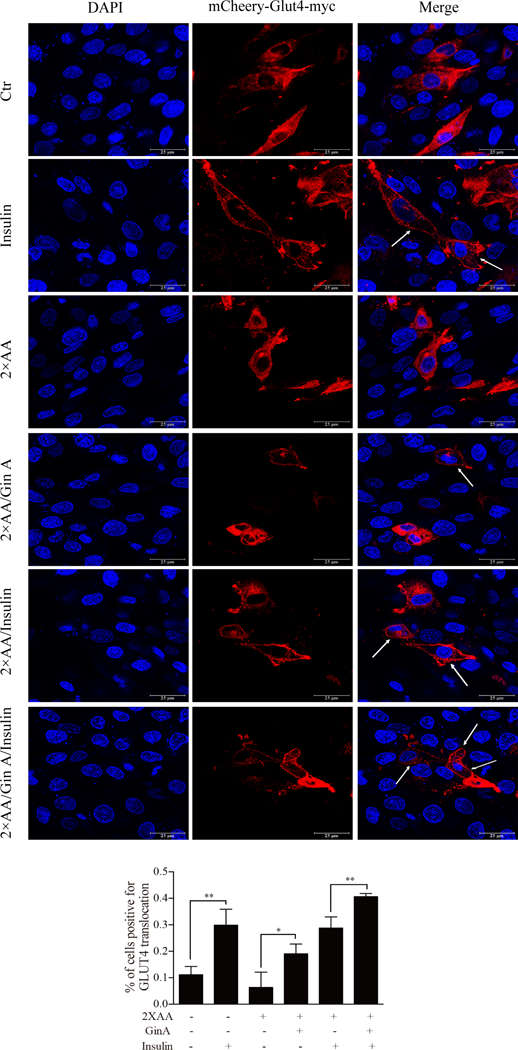
(A) L6 cells transfected with mCherry-Glut4-myc were treated with Gin A (10 μM) for 4 hr in EBSS containing 10% FBS with or without 2X AA. Cells were left unstimulated or stimulated with insulin (100 nM) for 30 min. After fixation, mCherry-tagged GLUT4 fluorescence was visualized under a Leica LP8 confocal microscope. Arrows denote the mCherry-tagged GLUT4 translocation to the cell membrane. (B) Quantification of GLUT4 translocation to the plasma membrane. The data represent the mean ± SD from one of three experiments with similar results. *p<0.05; **p<0.01
3.6. Gin A enhances insulin-stimulated glucose uptake.
Having shown that Gin A increased GLUT4 membrane translocation, we next determined the ability of Gin A to increase glucose uptake. As shown in Fig. 7A, intracellular glucose levels were significantly increased by insulin stimulation in 3T3-L1 adipocytes and L6 myotubes in the absence of 2X AA. The ability of insulin to stimulate glucose uptake was slightly weakened in 3T3-L1 adipocytes and L6 myotubes by 2X AA. Gin A further increased intracellular glucose levels in insulin-stimulated 3T3-L1 adipocytes and L6 myotubes in a dose-dependent manner (Fig. 7A) and in L6 myotubes (Fig. 7B). PF-4708671 was included as a positive control. It also increased intracellular glucose levels in 3T3-L1 adipocytes (Fig. 7A) and L6 myotubes (Fig. 7B). These results collectively suggest that elevated GLUT4 membrane translocation by Gin A due to insulin receptor sensitization is responsible for increased glucose uptake.
Fig. 7. Gin A increases glucose uptake.
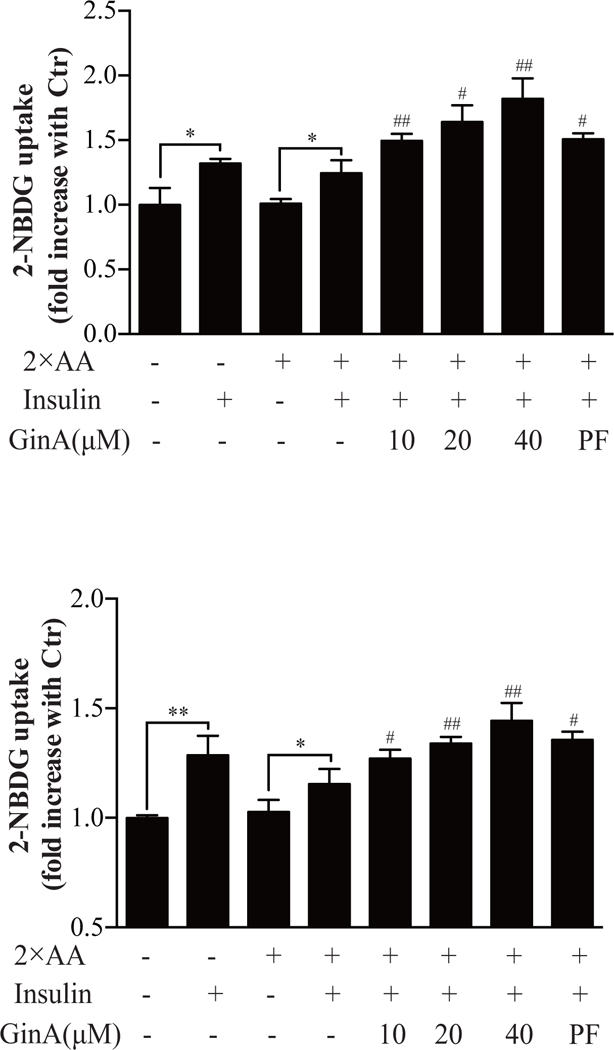
3T3-L1 adipocytes (A) and L6 myotubes (B) were starved in serum- and glucose-free media overnight and incubated with the indicated concentrations of Gin A or PF-4708671 in EBSS containing 2X AA for 1 hr. 2-NBDG (50 μM) was added for another 1 hr. The cells were left unstimulated or stimulated with insulin (50 nM) for 30 min. The cultured media were removed, followed by rinsing three times with PBS. The fluorescence intensity was read in a plate reader with the excitation wavelength of 485 nm and an emission wavelength of 535 nm. Data represents the mean ± SD of one experiment in triplicate. The experiments were repeated three times with similar results. *p<0.05; **p<0.01; #p<0.05; ##p<0.01, compared to the control (with insulin and 2X AA but without drug).
Discussion
It has been long recognized that ginger and ginger extract have beneficial effects on glucose and lipid metabolism [25]. Most studies have focused on the anti-diabetic and anti-obesity effects of gingerols and shogaols extracted from ginger [25–28]. These compounds exert their biological activities largely by activating AMPK, an energy sensor that regulates glucose and lipid metabolism [25–28]. Though S6K1 plays a central role in the metabolic syndrome and type 2 diabetes, whether targeting S6K1 with natural products can improve glucose metabolism remains unexplored. In the present study, we report that Gin A, a constituent in ginger extract and an inhibitor of S6K1, was able to sensitize the insulin receptor, to increase GLUT4 translocation into the plasma membrane, and to stimulate glucose uptake in adipocytes and myotubes. Our study is the first to report that a plant-derived S6K1 inhibitor can improve glucose metabolism by directly targeting a crucial enzyme involved in insulin resistance. Since the relative amount of Gin A and other bioactive agents such as gingerols and shogaols in ginger is unknown, how significantly Gin A contributes to the anti-hyperglycemic effect of Gin A is not clear, exploring S6K1 inhibitors from natural products could be a vital approach to discovering novel anti-diabetic drugs or dietary supplements for the control of hyperglycemia.
S6K1 has been recently pursued as a molecular target for sensitizing the insulin receptor and for controlling hyperglycemia [29]. Shum et al. [29] reported that PF-4708671, a specific inhibitor of S6K1, sensitizes the insulin receptor and enhances glucose uptake. We recently identified S6K1 as a molecular target of A77 1726, the active metabolite of anti-rheumatoid arthritis (RA) drug leflunomide [30]. A77 1726 induces feedback activation of the PI3K pathway in A375 melanoma cells [31], NSC34 motoneuron cells [32], 3T3-L1 adipocytes, and L6 myotubes [33]. Byun et al. [19] reported that Gin A inhibits the activity of S6K1, leading to increased AKT phosphorylation in the HCT116 colorectal cancer cell line. Consistent with these observations, our present study showed that Gin A inhibited S6S235/236 and IRSS1101 phosphorylation and induced feedback activation of the PI3K pathway in L6 myotubes and 3T3-L1 adipocytes under normal and insulin resistance conditions. Gin A enhanced insulin-induced insulin receptor tyrosine phosphorylation and increased IRS-1 binding to PI3K. In the presence of high amino acid concentrations, Gin A enhanced insulin-induced GLUT4 translocation to the plasma membrane in L6 cells and increased glucose uptake in 3T3-L1 adipocytes and L6 myotubes. These observations suggest that Gin A improves insulin receptor signaling by inhibiting IRS-1S1101 phosphorylation.
We noticed that Gin A did not increase S6K1 phosphorylation in a dose-dependent manner. S6K1 phosphorylation levels in 3T3-L1 adipocytes and L6 myotubes treated with Gin A at 20–40 μM were lower than when it was used at 10 μM (Fig. 2–4). Suk et al. [20] recently reported that Gin A at 40 μM induced AMP-activated protein K (AMPK) phosphorylation in 3T3-L1 adipocytes and in epidermal white adipose tissue in vivo. Since AMPK suppresses mTORC1 activity directly by phosphorylating Raptor and indirectly by phosphorylating TSC2 [34, 35], we speculate that AMPK activation by Gin A at high concentrations leads to the suppression of mTOR activity, thus restraining the maximal increase of S6K1 phosphorylation. However, declining S6K1 phosphorylation did not lead to better inhibition of S6 phosphorylation (Fig. 3B & 4B). Instead, S6 phosphorylation levels in 3T3-L1 adipocytes and L6 myotubes under insulin-resistant conditions treated with high concentrations of Gin A (20 and 40 μM) were not significantly lower than that treated with Gin A at 10 μM. We speculate that p90RSK activation by feedback-activated ERK phosphorylates S6 [36], thus preventing a dose-dependent inhibition of S6 phosphorylation (Fig. 3B & 4B).
There has been growing interest in finding natural products from herbs and plants to control the metabolic syndrome [37, 38]. Some phytochemicals in spice ingredients, herbal products, and dietary supplements can control hyperglycemia and obesity by improving glucose and lipid metabolism [38–41]. Ginger has been long considered a herbal medicine and has beneficial effects on a variety of ailments, including inflammation, infection, cancer, diabetes, obesity, and cardiovascular diseases [18, 26]. Several bioactive constituents in ginger extract have been identified and characterized, including gingerols, shogoals, and paradols. Gingerols and shogoals largely exert their anti-hyperglycemic and anti-obesity effects by activating AMPK [16–18]. 6-dihydroparadol increases cholesterol efflux in a human THP-1 macrophage cell line by inhibiting proteasomal activity and increasing the protein levels of ATP-binding cassette transporters, ABCA1 and ABCG1 [42]. This bioactivity may contribute to the anti-atherogenic effect of ginger [42]. Suk et al. recently reported that Gin A activates AMPK in vitro in 3T3-L1 adipocytes and in vivo in epidermal white adipose tissue [20]. Consistent with these observations, we found that Gin A induced AMPK phosphorylation in both 3T3-L1 adipocytes and in L6 myotubes (data not shown). How Gin A activates AMPK is not known. Our recent study showed that inhibition of S6K1 activity by A77 1726 and PF-4708671 activates AMPK through the TGF-β-activated kinase 1 (TAK1) in A375 melanoma cells and in NSC34 motoneuron cells [31, 32]. In addition, Gin A may also activate AMPK by elevating the AMP levels since S6K1 deficiency leads to increased AMP/ATP ratios and AMPK activation in the skeletal muscle tissues and myotubes of S6K1-deficient mice [43, 44]. Since S6K1 can bind and phosphorylate AMPK α2 at S491, leading to the inhibition of AMPK activity [45], Gin A may also activate AMPK by inhibiting S6K1-mediated AMPKS491 phosphorylation. Though we cannot rule out the possibility that Gin A may target other molecules to regulate glucose and lipid metabolism, Gin A may activate AMPK through multiple mechanisms, but all by inhibiting S6K1 activity. It remains to be determined if AMPK activation by other constituents such as gingerols or shogaols in ginger extract [16–18] is also mediated by inhibition of S6K1 activity.
S6K1 plays a critical role in the pathogenesis of type 2 diabetes [6]. We and others recently reported that S6K1 inhibitors such as PF-4708671 and leflunomide can control hyperglycemia in type 2 diabetic mouse models [29, 33]. The anti-hyperglycemic and anti-obesity effects of leflunomide have been observed in leflunomide-treated RA patients [46] [47]. Leflunomide could be a leading candidate for treating type 2 diabetes since the side-effects of this clinically approved drug have been well characterized [48, 49]. However, A77 1726 has other biochemical activities, including inhibition of protein tyrosine kinase activities and inhibition of pyrimidine nucleotide synthesis [48]. Leflunomide could be particularly useful for treating RA patients with diabetes, but its immunosuppressive effect could be a concern in most other patients [33, 48]. Food-derived S6K1 inhibitors or S6K1 inhibitor-enriched dietary supplements such as Gin A and ginger extract should have a relatively safe profile and could be developed as anti-diabetic drugs.
Acknowledgements:
We greatly appreciate the grant support from Natural Science Foundation of China (81672463) and the Priority Academic Program Development of Jiangsu Higher Education Institutions to X. Xu, and the grant support from the National Institutes of Health (R01 CA204926) to Y. Li. We would like to thank again Dr. Amira Klip (The Hospital for Sick Children, Toronto, Ontario) for the mCherry-GLUT4-myc expression vector.
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Zimmet PZ, Magliano DJ, Herman WH, and Shaw JE, Lancet Diabetes Endocrinol, 2014, 2, 56. [DOI] [PubMed] [Google Scholar]
- 2.Zimmet P, Alberti KG, and Shaw J, Nature, 2001, 414, 782. [DOI] [PubMed] [Google Scholar]
- 3.Nathan DM, JAMA, 2015, 314, 1052. [DOI] [PubMed] [Google Scholar]
- 4.Guo S, J Endocrinol, 2014, 220, T1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo S, Drug Discov Today Dis Mech, 2013, 10, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dann SG, Selvaraj A, and Thomas G, Trends Mol Med, 2007, 13, 252. [DOI] [PubMed] [Google Scholar]
- 7.Copps KD and White MF, Diabetologia, 2012, 55, 2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boura-Halfon S and Zick Y, Am J Physiol Endocrinol Metab, 2009, 296, E581. [DOI] [PubMed] [Google Scholar]
- 9.Fenton TR and Gout IT, Int J Biochem Cell Biol, 2010, 43, 47. [DOI] [PubMed] [Google Scholar]
- 10.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, and Thomas G, Nature, 2004, 431, 200. [DOI] [PubMed] [Google Scholar]
- 11.Han JS, Lee S, Kim HY, and Lee CH, Molecules, 2015, 20, 16170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Tran VH, Duke CC, and Roufogalis BD, Planta Med, 2012, 78, 1549. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Tran VH, Kota BP, Nammi S, Duke CC, and Roufogalis BD, Basic Clin Pharmacol Toxicol, 2014, 115, 209. [DOI] [PubMed] [Google Scholar]
- 14.Rani MP, Padmakumari KP, Sankarikutty B, Cherian OL, Nisha VM, and Raghu KG, Int J Food Sci Nutr, 2011, 62, 106. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Yu H, Zhang X, Feng Q, Guo X, Li S, Li R, Chu D, and Ma Y, Nutrition, 2017, 36, 79. [DOI] [PubMed] [Google Scholar]
- 16.Lee JO, Kim N, Lee HJ, Moon JW, Lee SK, Kim SJ, Kim JK, Park SH, and Kim HS, J Cell Biochem, 2015, 116, 1401. [DOI] [PubMed] [Google Scholar]
- 17.Wei CK, Tsai YH, Korinek M, Hung PH, El-Shazly M, Cheng YB, Wu YC, Hsieh TJ, and Chang FR, Int J Mol Sci, 2017, 18, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semwal RB, Semwal DK, Combrinck S, and Viljoen AM, Phytochemistry, 2015, 117, 554. [DOI] [PubMed] [Google Scholar]
- 19.Byun S, Lim S, Mun JY, Kim KH, Ramadhar TR, Farrand L, Shin SH, Thimmegowda NR, Lee HJ, Frank DA, Clardy J, Lee SW, and Lee KW, J Biol Chem, 2015, 290, 23553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suk S, Kwon GT, Lee E, Jang WJ, Yang H, Kim JH, Thimmegowda NR, Chung MY, Kwon JY, Yang S, Kim JK, Park JHY, and Lee KW, Mol Nutr Food Res, 2017, 61, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zebisch K, Voigt V, Wabitsch M, and Brandsch M, Anal Biochem, 2012, 425, 88. [DOI] [PubMed] [Google Scholar]
- 22.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, and Alessi DR, Biochem J, 2010, 431, 245. [DOI] [PubMed] [Google Scholar]
- 23.Rosner M, Schipany K, and Hengstschlager M, Amino Acids, 2012, 42, 2251. [DOI] [PubMed] [Google Scholar]
- 24.Park JE, Lee JS, Lee HA, and Han JS, J Med Food, 2018, [Google Scholar]
- 25.Zhu J, Chen H, Song Z, Wang X, and Sun Z, Evid Based Complement Alternat Med, 2018, 2018, 5692962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Ke W, Bao R, Hu X, and Chen F, Ann N Y Acad Sci, 2017, 1398, 83. [DOI] [PubMed] [Google Scholar]
- 27.Sampath C, Rashid MR, Sang S, and Ahmedna M, Food Chem, 2017, 226, 79. [DOI] [PubMed] [Google Scholar]
- 28.Misawa K, Hashizume K, Yamamoto M, Minegishi Y, Hase T, and Shimotoyodome A, J Nutr Biochem, 2015, 26, 1058. [DOI] [PubMed] [Google Scholar]
- 29.Shum M, Bellmann K, St-Pierre P, and Marette A, Diabetologia, 2016, 59, 592. [DOI] [PubMed] [Google Scholar]
- 30.Doscas ME, Williamson AJ, Usha L, Bogachkov Y, Rao GS, Xiao F, Wang Y, Ruby C, Kaufman H, Zhou J, Williams JW, Li Y, and Xu X, Neoplasia, 2014, 16, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu X, Sun J, Song R, Doscas ME, Williamson AJ, Zhou J, Sun J, Jiao X, Liu X, and Li Y, Oncotarget, 2017, 8, 30438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun J, Mu Y, Jiang Y, Song R, Yi J, Zhou J, Sun J, Jiao X, Prinz RA, Li Y, and Xu X, Cell Death Dis, 2018, 9, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J, Sun J, Doscas ME, Ye J, Williamson AJ, Li Y, Li Y, Prinz RA, and Xu X, J Endocrinol, 2018, 237, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell RC, Yuan HX, and Guan KL, Cell Res, 2014, 24, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alers S, Loffler AS, Wesselborg S, and Stork B, Mol Cell Biol, 2012, 32, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anjum R and Blenis J, Nat Rev Mol Cell Biol, 2008, 9, 747. [DOI] [PubMed] [Google Scholar]
- 37.Lacroix IM and Li-Chan EC, Mol Nutr Food Res, 2014, 58, 61. [DOI] [PubMed] [Google Scholar]
- 38.Jung HS, Lim Y, and Kim EK, Int J Mol Sci, 2014, 15, 21505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu L, Li Y, Dai Y, and Peng J, Pharmacol Res, 2018, 130, 451. [DOI] [PubMed] [Google Scholar]
- 40.Ota A and Ulrih NP, Front Pharmacol, 2017, 8, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardie DG, Diabetes, 2013, 62, 2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D, Hiebl V, Ladurner A, Latkolik SL, Bucar F, Heiss EH, Dirsch VM, and Atanasov AG, Mol Nutr Food Res, 2018, e1800011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, and Withers DJ, Science, 2009, 326, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aguilar V, Alliouachene S, Sotiropoulos A, Sobering A, Athea Y, Djouadi F, Miraux S, Thiaudiere E, Foretz M, Viollet B, Diolez P, Bastin J, Benit P, Rustin P, Carling D, Sandri M, Ventura-Clapier R, and Pende M, Cell Metab, 2007, 5, 476. [DOI] [PubMed] [Google Scholar]
- 45.Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, and Kahn BB, Cell Metab, 2012, 16, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rho YH, Oeser A, Chung CP, Milne GL, and Stein CM, Arch Drug Inf, 2009, 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coblyn JS, Shadick N, and Helfgott S, Arthritis Rheum, 2001, 44, 1048. [DOI] [PubMed] [Google Scholar]
- 48.Breedveld FC and Dayer JM, Ann Rheum Dis, 2000, 59, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cannon GW and Kremer JM, Rheum Dis Clin North Am, 2004, 30, 295. [DOI] [PubMed] [Google Scholar]