

Abstract
Objective
To provide evidence that idiopathic inflammatory myopathy (IM) with myasthenia gravis (MG) frequently shows thymoma association and polymyositis (PM) pathology and shares clinicopathologic characteristics with IM induced by immune checkpoint inhibitors (ICIs).
Methods
We analyzed the clinicopathologic features of 10 patients with idiopathic IM and MG identified in 970 consecutive patients with biopsy-proven IM.
Results
Seven patients (70%) had thymoma. IM and MG were diagnosed with more than 5-year time difference in 6 thymomatous patients and within 1 year in 1 thymomatous and 3 nonthymomatous patients. Seven thymomatous patients showed rhabdomyolysis-like features with respiratory failure (4/7), dropped head (3/7), cardiac involvement (2/7), and positive anti–acetylcholine receptor (anti-AChR) and anti-titin antibodies (7/7 and 4/6, respectively) but rarely showed ocular symptoms (2/7) or decremental repetitive nerve stimulation (RNS) responses (1/7) at IM diagnosis. Three nonthymomatous patients showed acute cardiorespiratory failure with rhabdomyolysis-like features (1/3), positive anti-AChR and anti-titin antibodies (3/2 and 2/2, respectively), and fluctuating weakness of the skeletal muscle without ocular symptoms (3/3). Muscle pathology showed a PM pathology with infiltration of CD8-positive CD45RA-negative T-lymphocytes (9/9), scattered endomysial programmed cell death 1 (PD-1)–positive cells (9/9), and overexpression of programmed cell death ligand 1 (PD-L1) on the sarcolemma of muscle fibers around the infiltrating PD-1–positive cells (7/9).
Conclusion
Rhabdomyolysis-like features, positive anti-AChR antibody without decremental RNS responses, and PD-L1 overexpression are possible characteristics shared by ICI-induced IM. Frequent thymoma association in patients with idiopathic IM and MG may suggest thymoma-related immunopathogenic mechanisms, including dysregulation of the immune checkpoint pathway.
Idiopathic inflammatory myopathies (IMs) are a heterogeneous group of muscle disorders. Myasthenia gravis (MG) is rarely associated with IM. The reported clinical features of patients with both idiopathic IM and MG included brachio-cervical weakness or dropped head,1–5 respiratory decompensation,3–10 muscle swelling with pain,4,11 cardiac involvement,4,5,9 and markedly elevated serum creatine kinase (CK) levels.4,6,8 In addition, patients with anti–acetylcholine receptor (anti-AChR) antibody (Ab)-positive thymomatous IM without MG symptoms showing rapidly progressive weakness and respiratory failure with markedly elevated serum CK levels12,13 have been described. Although previous reports suggested some characteristics of idiopathic IM patients with MG and/or thymoma, clinical features, including the temporal relationship between the onset of IM and MG and the prevalence of clinical characteristics, including thymoma association, have not been well known because of the lack of systematical study conducted in a series of patients with IM. Furthermore, pathologic findings have not been studied systematically.
The rare combination of IM and MG has been recently reported in patients with immune-related adverse events induced by immune checkpoint inhibitors (ICIs) targeting programmed cell death 1 (PD-1) or cytotoxic T-lymphocyte–associated protein 4 (CTLA-4).14–17 The reported clinical features of these patients include cardiac involvement14,15 and rhabdomyolysis-like features with markedly elevated serum CK levels,14,15 suggesting similarities of the clinical features of IM between the 2 groups: ICI induced and idiopathic. Therefore, we analyzed the clinicopathologic characteristics of IM associated with MG in a series of patients with biopsy-proven IM to determine whether some characteristic features are shared by ICI-induced and idiopathic patients with both IM and MG.
Methods
Patients
Clinical records and biopsy reports were reviewed for 970 consecutive patients, who were referred to our department for pathologic diagnosis between April 1986 and December 2017. IM diagnosis was based on the criteria proposed by Bohan and Peter18,19; in addition, both (1) elevated serum CK levels and (2) muscle biopsy findings of inflammatory changes with major histocompatibility complex (MHC) class I expression on non-necrotic muscle fibers and sometimes with necrotic and/or regenerating fibers18–20 were required. Exclusion of muscular dystrophy by immunohistochemistry and clinical features was also required. Inclusion body myositis (IBM) was excluded using the 188th European Neuromuscular Centre IBM criteria.21 Sarcoid myopathy was excluded on the basis of clinical and pathologic findings.22 Patients who developed IM as an adverse effect of drugs, including ICIs, were excluded from this study. MG diagnosis was based on clinical features of weakness with increased fatigability of skeletal muscles and one or more of the following 3 criteria: (1) a positive edrophonium infusion test, (2) decremental repetitive nerve stimulation (RNS) responses, and (3) increased jitter or blocking on a single-fiber electromyogram (SFEMG). Thymoma diagnosis was made on the basis of CT of the anterior mediastinum tumor and confirmed by pathologic examination.
In recruiting patients with both IM and MG, patients with IM who had a past or subsequent history of MG were included. Because the patients with thymoma sometimes have MG or subclinical MG or may develop MG during follow-up periods, the patients with IM who had a past or subsequent history of thymoma were also included. By these methods, among 970 consecutive patients with IM, we found 10 patients with both IM and MG (1.0%) and 1 non-MG thymomatous patient with IM (figure 1). The 11 patients with both IM and MG included 3 patients who have been previously reported in separate reports (no. 11, Yamaguchi et al., 2000; no. 5, Maekawa et al., 2014; and no. 9, Suzuki et al., 2014).23–25
Figure 1. Flowchart of patient inclusion.
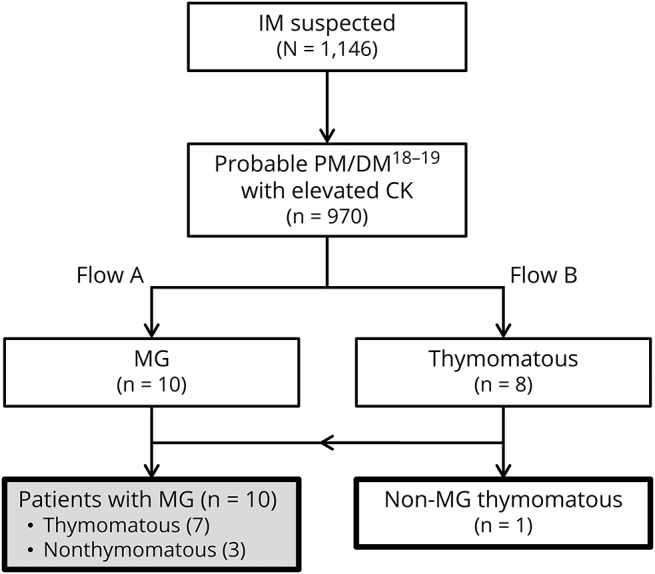
Clinical and pathologic features of 1,146 patients were evaluated based on Bohan and Peter's myositis criteria and serum CK levels, and then 970 patients were identified as more than possible polymyositis or dermatomyositis with an elevated serum CK level. Among these patients, 10 patients with a history of MG in flow A and 8 patients with a history of thymoma in flow B were identified. Finally, 10 patients with MG were recruited excluding the 7 overlapping patients between flow A and flow B. In addition, 1 non-MG patient with a history of thymoma was also recruited. CK = creatine kinase; DM = dermatomyositis; PM = polymyositis.
Definition of onset of IM and MG
IM onset was defined as the time of development of sustained skeletal muscle weakness with elevated serum CK levels. MG onset was defined as the earlier of 2 dates: (1) the date of development of weakness with increased fatigability of skeletal muscles (e.g., fluctuating diplopia, ptosis, or skeletal muscle power) or (2) the date of detection of abnormality on RNS or an SFEMG in weakened muscles.
Histochemical and immunohistochemical analysis
Stored frozen muscle samples were processed into 10-μm sections for routine histochemistry and immunohistochemistry. Immunohistochemistry were performed with a conventional method, using the substrate 3,3'-diaminobenzidine tetrahydrochloride (DAB). Primary antibodies against MHC class I (Dako, Agilent Technologies, CA), MHC class II (Dako), CD4 (Dako), CD8 (Nichirei Biosciences, Tokyo, Japan), CD20 (Abcam, Cambridge, UK), CD21 (Dako), CD45/leukocyte common antigen (Dako), CD45RA (Dako), CD68 (Dako), myxovirus resistance protein A (M x A; Abcam), PD-1 (Abcam), programmed cell death ligand 1 (PD-L1; Abcam), and CTLA-4 (Abcam) were used.
Detection of autoantibodies
Myositis-specific autoantibodies (MSAs), including anti–Jo-1, anti–PL-7, anti–PL-12, anti–Mi-2, and anti–signal recognition particle 54-kDa protein Abs, were detected by dot-blotting using recombinant proteins (Diarect AG, Freiburg, Germany). Anti–transcriptional intermediary factor 1 gamma and anti–melanoma differentiation-associated gene 5 Abs were detected by immunoprecipitation, and anti–3-hydroxy-3-methylglutaryl-CoA reductase Ab was detected by ELISA, as described in our earlier study.26 Anti-titin Ab was also detected by ELISA (DLD Diagnostika, Hamburg, Germany).
Standard protocol approvals, registrations, and patient consents
The study was approved by the institutional ethics committees of the University of Tokyo Hospital. Written informed consent was obtained from each patient at the time of biopsy and serum sampling.
Data availability
Anonymized data will be shared by request from any qualified investigators.
Results
Clinical features of patients at IM diagnosis
Table 1 shows the clinical features and histopathologic findings of the skeletal muscles of 10 patients with both IM and MG; the patients' detailed clinical features are described in appendix e-1 links.lww.com/NXI/A95.
Table 1.
Clinical and pathologic features of IM in patients with both IM and MG



The mean age at IM diagnosis was 60 years (range: 47–78 years). Six patients were women. None of the patients had skin rash or interstitial lung disease. Seven patients were associated with invasive thymoma, and the other 3 had no thymoma. The temporal relationship between the onset of IM and MG are as follows: (1) 5 thymomatous patients (nos. 1, 2, and 4–6) had preexisting MG (diagnosed more than 9 years before their IM diagnosis); (2) 1 thymomatous patient (no. 7) had preexisting IM (diagnosed with MG 5 years after the IM diagnosis); and (3) 1 thymomatous patient (no. 3) and 3 nonthymomatous patients (nos. 8–10) had concurrent IM and MG (diagnosed with IM and MG within 1 year). Clinical features related to MG at IM diagnosis included fluctuating ocular symptoms (2/10), abnormal RNS responses on weakened extremity muscles (4/10), positive edrophonium infusion test (2/3), and positive anti–AChR Ab (9/10).
Thymomatous IM patients rarely showed fluctuating ocular symptoms (2/7) and abnormal RNS responses on extremities (1/7). By contrast, 3 nonthymomatous IM patients showed abnormal RNS responses (3/3) and positive edrophonium infusion test (2/2) but showed no ocular symptoms (0/3).
Before IM diagnosis, 2 patients had a history of transient elevation of serum CK levels without limb weakness (4,209 IU/L in no. 1 and 518 IU/L in no. 8). Progression of weakness in IM was rapid in 5 patients. Four thymomatous patients with normal RNS tests on extremities presented with rapid progression of weakness with acute markedly elevated serum CK levels (1,008–10,226 IU/L) at IM development (rhabdomyolysis-like features) and required ventilator support. One nonthymomatous patient (no. 9) developed respiratory muscle weakness with relatively preserved limb muscle strength, cardiac failure, and acute elevation of serum CK levels (1,140 IU/L).
The clinical features of the 10 patients included respiratory failure requiring ventilator support (5/10), severe weakness of extremities (4/10), dropped head (4/10), myalgia (4/10), dysphagia (5/9), and cardiac involvement (3/10). One thymomatous patient (no. 7) had initial symptoms of muscle swelling and pain in the forearms.
The mean serum CK level was 2,712 IU/L. The serum testing for autoantibodies demonstrated anti-AChR Ab (9/10), MSAs (0/9), and anti-titin Ab (6/8). A needle EMG, performed for 9 patients, showed spontaneous activities at rest in most of them (8/9). Three patients (3/10) showed cardiac involvements. A cardiac muscle biopsy performed in 1 patient (no. 6) showed abundant T-lymphocyte infiltration consistent with lymphocytic myocarditis.
Pathologic findings
We observed various amounts of necrotic fibers and inflammatory cells in both the perimysium and the endomysium. All patients showed different levels of overexpression of MHC class I on muscle fibers with various extent levels: 1+, n = 2; 2+, n = 2; and 3+, n = 6 (table 1). Overexpression of MHC class II on muscle fibers was observed in 9 patients (9/10). All patients showed the findings of endomysial CD8+ T-lymphocytes surrounding (10/10) and sometimes invading (5/10) non-necrotic muscle fibers expressing MHC class I antigen and therefore met the pathologic criteria of polymyositis (PM)20 (definite PM, n = 5; probable PM, n = 5; figure 2A).
Figure 2. Histopathologic findings of patients with both inflammatory myopathy and myasthenia gravis.
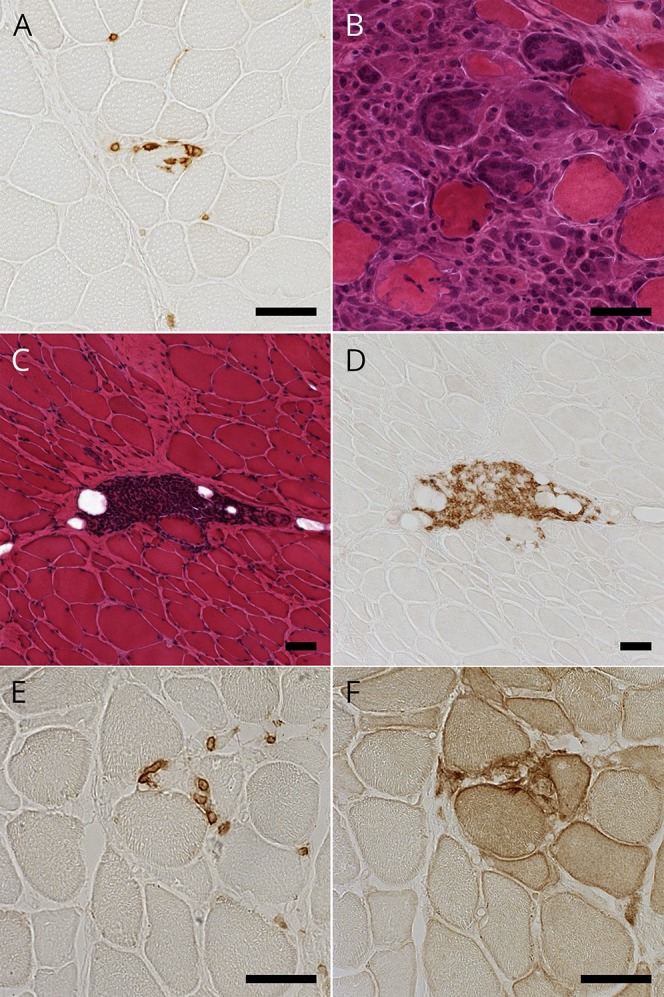
(A) Immunohistochemical staining for CD8 showing CD8-positive cells invading non-necrotic muscle fibers in patient 1. (B) Hematoxylin-eosin (HE) staining showing granulomatous lesion with multinucleated giant cells in patient 7. (C and D) Serial sections with HE staining and immunohistochemical staining for CD20 showing perimysial aggregation of CD20-positive cells in patient 8. (E and F) Serial sections with immunohistochemical staining for PD-1 and PD-L1 showing endomysial PD-1–positive cells and PD-L1 upregulation on non-necrotic fibers in patient 9. Scale bar = 50 μm. PD-1 = programmed cell death 1; PD-L1 = programmed cell death ligand 1.
Immunohistochemistry for CD45RA, which was performed in 9 frozen sample available patients, showed no endomysial CD45RA+ cells, indicating that the endomysial CD8+ T-lymphocytes surrounding and sometimes invading non-necrotic muscle fibers were all CD45RA− cells (9/9). Two patients (nos. 3 and 8) showed perimysial accumulation of CD45RA+ cells (figure e-1 links.lww.com/NXI/A95).
One thymomatous patient (no. 7) showed granulomatous inflammatory cell invasion with multinucleated giant cells (figure 2B). The granulomas and giant cells were positive for CD68. Two patients (nos. 3 and 8) showed focal perimysial accumulation of mononuclear cells, many of which were positive for CD20 (figure 2, C and D).
Immunohistochemistry for PD-1 and PD-L1 showed scattered endomysial PD-1+ cells (9/9) and overexpression of PD-L1 on the sarcolemma of muscle fibers near PD-1+ cells (7/9). Of interest, 2 patients (nos. 1 and 9) showed PD-L1 overexpression extending on non-necrotic fibers around the fibers invaded by the PD-1+ cells (figure 2, E and F). Immunohistochemistry for CTLA-4 demonstrated CTLA-4+ cells in endomysial infiltration (4/9) (figure e-2 links.lww.com/NXI/A95).
Associated thymoma
The World Health Organization classifications of invasive thymoma in 7 patients were B2 (n = 5), B3 (n = 1), and no data available (n = 1) (table 1). The mean age at thymoma diagnosis was 45 years (range: 36–56 years). Thymoma diagnosis was preceded to IM diagnosis more than 12 years (range: 12–17 years) in 5 patients (nos. 1, 2, and 4–6), and both thymoma and IM were codiagnosed in 2 patients (nos. 3 and 7). The disease statuses of thymoma at IM diagnosis were as follows: no recurrence or progression without therapy (n = 3), untreated (n = 2), recurrence (n = 1), and no progression under chemotherapy (n = 1).
Thymomatous IM patient without MG symptoms
Our series included 1 thymomatous IM patient without MG symptoms (no. 11, details in appendix e-1 links.lww.com/NXI/A95). This 51-year-old woman had moderate to severe weakness in her limbs and respiratory muscles requiring ventilator support. Laboratory data showed an elevated serum CK level (695 IU/L) and positive anti-AChR and anti-titin Abs. Needle EMG showed spontaneous activities, but RNS test was normal. Chest CT showed an anterior mediastinum tumor (1 cm in size), which was revealed to be necrotic tissues. Histopathologic diagnosis of muscle biopsy was definite PM.20 She was successfully treated with prednisolone (PSL). Her anterior mediastinum tumor increased in size without neurologic symptoms. Five years after the diagnosis of IM, the tumor was resected and pathologically confirmed as combined B2/B3 thymoma. Her 11-year follow-up showed no exacerbation of IM or thymoma or development of MG.
Clinical courses and response to treatments
One patient (no. 8) showed normalized serum CK levels during bed rest and refused immunotherapy. He showed no exacerbation of symptoms for 2.6 years until final follow-up. The remaining 9 patients, including 2 patients who showed normalization of serum CK levels during bed rest, were treated with immunotherapy (table 2). The treatments included immunosuppressants alone in 1 patient (no. 4) and increasing doses of oral PSL (20–60 mg) in the other 8 patients in combination with various immunosuppressive therapies: oral immunosuppressants (n = 4), high-dose IV methylprednisolone (n = 4), high-dose IV immunoglobulin (n = 2), and immunoabsorption (n = 1). Of interest, the elevated serum CK levels normalized within 3 weeks in all the treated patients, including the 3 patients with very high serum CK levels (nos. 1, 6, and 7).
Table 2.
Treatment of IM in patients with both IM and MG

The mean follow-up period was 3.8 years (range: 0.5–10.9 years). During the follow-up period, 3 patients died. The causes of death were sepsis following diffuse panbronchiolitis (n = 2, patients 5 and 7) and bronchopneumonitis complicated by cardiac dysfunction and pure red cell aplasia (n = 1, patient 9). The modified Rankin Scale scores at final follow-up of the 7 patients who were alive were grade 0 (n = 1), grade 1 (n = 4), and grade 3 (n = 2).
Discussion
The prevalence of thymoma association in patients with both IM and MG (70%) was much more than that in patients with only MG (10%).27 Thymoma is known to be associated with various autoimmune diseases. The frequent thymoma association in IM patients with both IM and MG should indicate that the association of these 2 diseases is not coincidental but induced by thymoma-related immunopathogenic mechanisms.
In 7 thymomatous patients with both IM and MG, ocular symptoms or decremental RNS responses were rarely observed at IM diagnosis (2/7 or 1/7, respectively). Among them, 4 patients with normal RNS responses developed rapidly progressive muscle weakness and respiratory failure with acute elevation of serum CK to very high levels (rhabdomyolysis-like features). Previous studies reported patients with similar clinical features: IM and anti–AChR Ab-positive thymomatous MG presenting with normal RNS responses28 and rapidly progressive weakness and respiratory failure in conjunction with markedly elevated serum CK levels.7,12 Our findings demonstrated that thymomatous patients with both IM and MG frequently showed nonsimultaneous progression of IM and MG and often included those with severe progression and markedly elevated serum CK levels. The nonsimultaneous progression of IM and MG suggests that distinctly different thymoma-related immunopathogenic mechanisms on skeletal muscles are involved in the development of each disease.
Three nonthymomatous IM patients in our study shared the following characteristic features with thymomatous IM patients: none showed ocular symptoms, 1 patient showed acute respiratory and cardiac failure with acute serum CK level elevation, 2 patients (2/2) showed positive anti-titin Ab, and all 3 patients showed PM pattern pathology, characterized by CD8+ T-lymphocyte–mediated cellular autoimmunity. PM pattern pathology is a rare pathologic finding among patients with IM29,30 and is suggested as a secondary pathologic pattern associated with other systemic autoimmune diseases, including connective tissue diseases, viral infections, cancer, and drug toxicity.31 The overlapping of clinical, serologic, and pathologic characteristics between thymomatous and nonthymomatous IM patients may suggest a common immune mechanism underlying the IM development in patients with both IM and MG, irrespective of thymoma association. However, the immune mechanical background of nonthymomatous IM patients may be heterogeneous, considering that 1 nonthymomatous IM patient (no. 10) was seronegative for anti-AChR Ab.
We found 1 non-MG thymomatous IM patient (no. 11) with features similar to those of thymomatous patients with both IM and MG, except for the presentation of MG. A previous report described 1 non-MG thymomatous IM patient with similar clinical features: positive anti-AChR Ab, normal RNS responses, a very high serum CK level (7,271 IU/L), and a pathologic finding of endomysial CD8+ cells with CD68 + giant cells.13 The clinical features in both patients supported the idea that thymoma-related immunopathogenic mechanisms can affect skeletal muscle toward the development of IM, even without MG.
At the time of IM diagnosis, 5 patients had a preexisting thymoma, and among them, 2 patients showed no signs of recurrence. In the literature, we found a total of 4 patients with both IM and MG with preexisting thymoma for 4–17 years before IM diagnosis.4,9,32 The thymoma status of the 4 patients at IM diagnosis was not described, except in 1 patient who was autopsied and showed no thymoma recurrence. However, some studies have described thymoma recurrence long after thymectomy33 and microscopic thymoma (smaller than 1 mm), which cannot be detected by imaging studies.34 Considering the suggested thymoma-related immunopathogenic mechanisms in the development of IM, we recommend not only careful checkup for thymoma recurrence in patients with preexisting thymoma but also regular checkup for thymoma development in nonthymomatous patients with both IM and MG.
The presence of endomysial infiltration of T-lymphocytes (lymphorrhage) in the skeletal muscle tissue of patients with MG has been known for decades, particularly in patients with thymomatous MG.32,35 One suggestion is that the lymphocytic infiltration does not represent autoimmune IM, but rather, an infiltration consisting of CD8+CD45RA+-naive T-lymphocytes derived from lymphocyte-rich thymoma. However, in our series, CD8+CD45RA− cells but not CD8+CD45RA+ naive cells were the lymphocytes infiltrating the endomysium, and the extensive expression of MHC class I was observed in all patients (9/9 and 10/10, respectively), suggesting a true cell-mediated PM pattern of inflammation as a pathologic phenotype in our patients.
In addition to PM pattern pathology, massive collections of CD20+ B-lymphocytes were observed in 2 patients and CD68+ granulomas in 1 patient. These pathologic findings have already been reported in patients with both idiopathic IM and MG.3,9–11,13,28 Because the thymus gland is a central lymphatic organ that provides various immunologic functions, multifactorial autoimmune dysregulation in thymic disorders, including thymoma, has been reported.36 Further studies are needed to elucidate the overall immune mechanisms that cause these complicated pathologic features.
Some immune-related adverse events induced by antitumor therapy with ICIs have been recently reported to cause the association of MG with IM.14,16,17 A recent study of 12 nonthymomatous patients who developed MG after antitumor therapy with nivolumab (an anti–PD-1 monoclonal Ab) reported that 4 patients had IM and 3 patients had myocarditis, including 1 patient with both IM and myocarditis.15 This study also showed the following clinical features of patients with nivolumab-induced MG: markedly elevated CK levels (10/12, 83%), positive anti-AChR Ab (10/12, 83%), positive edrophonium infusion test (4/7, 57%), and abnormal decremental RNS responses (2/7, 29%). Patients with ICI-induced IM have been often reported to have rhabdomyolysis-like clinical features with myocarditis or respiratory failure.37 Therefore, the assumption that patients with ICI-induced IM share similar immunopathogenic mechanisms with patients with both idiopathic IM and MG should be reasonable. Moreover, we recently found PD-1+ cell infiltration and extended PD-L1 (a ligand of PD-1) overexpression on non-necrotic fibers in patients with anti–PD-1 Ab-induced IM.16 The similarity of clinical features between patients with ICI-induced IM and those with idiopathic IM and MG prompted us to examine PD-1, PD-L1, and CTLA-4 expressions in our patients. Subsequently, we found PD-L1 overexpression on non-necrotic muscle fibers around PD-1+ cells in 7 patients, including 2 patients with extended PD-L1 overexpression, and endomysial infiltration of CTLA-4+ cells in 4 patients. CTLA-4+ cells in endomysial infiltrates with invasion of muscle fibers have been reported in patients with idiopathic PM or IBM.38 Both PD-1/PD-L1 and CTLA-4/B7 pathways are involved in the induction of immune tolerance on activated cytotoxic T-lymphocytes, although the 2 pathways operate at different stages of an immune response.39 The expression of PD-1, PD-L1, and CTLA-4 in IM pathology may suggest dysregulation of immune checkpoint molecules in the microenvironment of IM in patients with IM and MG. The backgrounds of the dysregulation of the immune checkpoint molecules in our patients remain unclear. However, a previous report has shown that genetic polymorphisms of CTLA-4 are associated with thymomatous MG patients.40 The finding indicates involvement of the immune checkpoint pathway in the development of autoimmune diseases in thymomatous patients.
Our study has some limitations. First, the number of patients with both IM and MG is small because of the rarity of simultaneous occurrence of these 2 diseases. Second, we did not always follow the clinical courses in patients with IM for a long time enough to detect MG association. Third, MG activity may be underestimated because SFEMG or edrophonium infusion test had not been performed in all of our patients. Despite these limitations, our study showed that idiopathic IM with MG frequently shows thymoma association and PM pathology. In addition, this study demonstrated that idiopathic IM with MG shares clinicopathologic characteristics with IM induced by ICIs. Further studies should be conducted to determine the essential mechanisms of thymoma-related autoimmune dysregulation in patients with both IM and MG, particularly focusing on the roles of immune checkpoint molecules.
Acknowledgment
The authors thank Naoko Tokimura, Masako Nishizawa, and Yuki Ogiwara for technical assistance.
Glossary
- anti-AChR
anti–acetylcholine receptor
- CK
creatine kinase
- CTLA-4
cytotoxic T-lymphocyte–associated protein 4
- IBM
inclusion body myositis
- ICI
immune checkpoint inhibitor
- IM
inflammatory myopathy
- MG
myasthenia gravis
- MHC
major histocompatibility complex
- MSA
myositis-specific autoantibody
- PD-1
programmed cell death 1
- PD-L1
programmed cell death ligand 1
- PM
polymyositis
- PSL
prednisolone
- RNS
repetitive nerve stimulation
- SFEMG
single-fiber electromyogram
Author contributions
N. Uchio: study design and concept, acquisition of data, analysis and interpretation, and drafting of the manuscript. K. Taira, C. Ikenaga, A. Unuma, M. Kadoya, and K. Yoshida: acquisition of data. S. Nakatani-Enomoto, Y. Hatanaka, Y. Sakurai, Y. Shiio, and K. Kaida: acquisition of data and critical revision of the manuscript. A. Kubota: critical revision of the manuscript. T. Toda: study supervision. J. Shimizu: study design and concept, acquisition of data, analysis and interpretation, and critical revision of the manuscript.
Study funding
This work was supported by the Health and Labour Sciences Research Grant on Rare and Intractable Diseases (Validation of Evidence-based Diagnosis and Guidelines and Impact on QOL in Patients with Neuroimmunological Diseases) from the Ministry of Health, Labour and Welfare of Japan and a Grant-in-Aid for Scientific Research (KAKENHI; 15K09347) from JSPS.
Disclosure
N. Uchio, K. Taira, C. Ikenaga, M. Kadoya, A. Unuma, K. Yoshida, S. Nakatani-Enomoto, Y. Hatanaka, Y. Sakurai, and Y. Shiio report no disclosures. K. Kaida is an associate editor of Clinical and Experimental Neuroimmunology. A. Kubota and T. Toda report no disclosures. J. Shimizu received research support from Grants-in-Aid for Scientific Research. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NN.
References
- 1.Behan WM, Behan PO, Doyle D. Association of myasthenia gravis and polymyositis with neoplasia, infection and autoimmune disorders. Acta Neuropathol 1982;57:221–229. [DOI] [PubMed] [Google Scholar]
- 2.van de Warrenburg BP, Hengstman GJ, Vos PE, Boerman RH, ter Laak HJ, van Engelen BG. Concomitant dermatomyositis and myasthenia gravis presenting with respiratory insufficiency. Muscle Nerve 2002;25:293–296. [DOI] [PubMed] [Google Scholar]
- 3.Pestronk A, Kos K, Lopate G, Al-Lozi MT. Brachio-cervical inflammatory myopathies: clinical, immune, and myopathologic features. Arthritis Rheum 2006;54:1687–1696. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki S, Utsugisawa K, Yoshikawa H, et al. Autoimmune targets of heart and skeletal muscles in myasthenia gravis. Arch Neurol 2009;66:1334–1338. [DOI] [PubMed] [Google Scholar]
- 5.Venna N, Gonzalez RG, Zukerberg LR. Case 39-2011: a woman in her 90s with unilateral ptosis. N Engl J Med 2011;365:2413–2422. [DOI] [PubMed] [Google Scholar]
- 6.Hassel B, Gilhus NE, Aarli JA, Skogen OR. Fulminant myasthenia gravis and polymyositis after thymectomy for thymoma. Acta Neurol Scand 1992;85:63–65. [PubMed] [Google Scholar]
- 7.Otton SH, Standen GR, Ormerod IEC. T cell lymphocytosis associated with polymyositis, myasthenia gravis and thymoma. Clin Lab Haematol 2000;22:307–308. [DOI] [PubMed] [Google Scholar]
- 8.Antoine JC, Camdessanché JP, Absi L, Lassablière F, Féasson L. Devic disease and thymoma with anti-central nervous system and antithymus antibodies. Neurology 2004;62:978–980. [DOI] [PubMed] [Google Scholar]
- 9.Kon T, Mori F, Tanji K, Miki Y, Kimura T, Wakabayashi K. Giant cell polymyositis and myocarditis associated with myasthenia gravis and thymoma. Neuropathology 2013;33:281–287. [DOI] [PubMed] [Google Scholar]
- 10.Shah A, Pace A, Hilton D, Househam E, Weatherby S. Giant cell myositis responsive to combined corticosteroids and immunoglobulin. Pract Neurol 2015;15:456–459. [DOI] [PubMed] [Google Scholar]
- 11.Seton M, Wu CC, Louissaint A. Case 26-2013 : a 46-year-old woman with muscle pain and swelling. N Engl J Med 2013;369:764–773. [DOI] [PubMed] [Google Scholar]
- 12.Gidron A, Quadrini M, Dimov N, Argiris A. Malignant thymoma associated with fatal myocarditis and polymyositis in a 32-year-old woman with a history of hairy cell leukemia. Am J Clin Oncol 2006;29:213–214. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki H, Yano M, Kawano O, Hikosaka Y, Fujii Y. Thymoma associated with fatal myocarditis and polymyositis in a 58-year-old man following treatment with carboplatin and paclitaxel: a case report. Oncol Lett 2012;3:300–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura T, Fukushima S, Miyashita A, et al. Myasthenic crisis and polymyositis induced by one dose of nivolumab. Cancer Sci 2016;107:1055–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki S, Ishikawa N, Konoeda F, et al. Nivolumab-related myasthenia gravis with myositis and myocarditis in Japan. Neurology 2017;89:1–8. [DOI] [PubMed] [Google Scholar]
- 16.Uchio N, Taira K, Ikenaga C, et al. Granulomatous myositis induced by anti–PD-1 monoclonal antibodies. Neurol Neuroimmunol Neuroinflamm 2018;5:e464. [Google Scholar]
- 17.Liao B, Shroff S, Kamiya-Matsuoka C, Tummala S. Atypical neurological complications of ipilimumab therapy in patients with metastatic melanoma. Neuro Oncol 2014;16:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohan A, Peter J. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975;292:344–347. [DOI] [PubMed] [Google Scholar]
- 19.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975;292:403–407. [DOI] [PubMed] [Google Scholar]
- 20.Hoogendijk JE, Amato AA, Lecky BR, et al. ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:377–345. [DOI] [PubMed] [Google Scholar]
- 21.Rose MR, Ibm E, Group W. 188th ENMC international workshop: inclusion body myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 2013;23:1044–1055. [DOI] [PubMed] [Google Scholar]
- 22.Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2153–2165. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi Y, Sakurai Y, Mannen T, Shimizu J. Rapidly progressive polymyositis with elevated antiacetylcholine receptor antibody activity. Intern Med 2000;39:1108–1110. [DOI] [PubMed] [Google Scholar]
- 24.Maekawa R, Shibuya H, Hideyama T, Shiio Y. A case of myasthenia gravis with invasive thymoma associated with diffuse panbronchiolitis, alopecia, dysgeusia, cholangitis and myositis. Clin Neurol 2014;54:703–708. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki S, Baba A, Kaida K, et al. Cardiac involvements in myasthenia gravis associated with anti-Kv1.4 antibodies. Eur J Neurol 2014;21:223–230. [DOI] [PubMed] [Google Scholar]
- 26.Hida A, Yamashita T, Hosono Y, et al. Anti-TIF1-γ antibody and cancer-associated myositis: a clinicohistopathologic study. Neurology 2016;87:300–308. [DOI] [PubMed] [Google Scholar]
- 27.Gilhus NE. Myasthenia gravis. N Engl J Med 2016;375:2570–2581. [DOI] [PubMed] [Google Scholar]
- 28.Stefanou MI, Komorowski L, Kade S, Bornemann A, Ziemann U, Synofzik M. A case of late-onset, thymoma-associated myasthenia gravis with ryanodine receptor and titin antibodies and concomitant granulomatous myositis. BMC Neurol 2016;16:172–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Meulen MF, Bronner IM, Hoogendijk JE, et al. Polymyositis: an overdiagnosed entity. Neurology 2003;61:316–321. [DOI] [PubMed] [Google Scholar]
- 30.Ikenaga C, Kubota A, Kadoya M, et al. Clinicopathologic features of myositis patients with CD8-MHC-1 complex pathology. Neurology 2017;89:1060–1068. [DOI] [PubMed] [Google Scholar]
- 31.Vilela VS, Prieto-González S, Milisenda JC, Selva-O Callaghan A, Grau JM. Polymyositis, a very uncommon isolated disease: clinical and histological re-evaluation after long-term follow-up. Rheumatol Int 2015;35:915–920. [DOI] [PubMed] [Google Scholar]
- 32.Zamecnik J, Vesely D, Jakubicka B, et al. Muscle lymphocytic infiltrates in thymoma-associated myasthenia gravis are phenotypically different from those in polymyositis. Neuromuscul Disord 2007;17:935–942. [DOI] [PubMed] [Google Scholar]
- 33.Evoli A, Minisci C, Di Schino C, et al. Thymoma in patients with MG: characteristics and long-term outcome. Neurology 2002;59:1844–1850. [DOI] [PubMed] [Google Scholar]
- 34.Fukuhara M, Higuchi M, Owada Y, et al. Clinical and pathological aspects of microscopic thymoma with myasthenia gravis and review of published reports. J Thorac Dis 2017;9:1592–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology 1993;43:1167–1172. [DOI] [PubMed] [Google Scholar]
- 36.Shelly S, Agmon-levin N, Altman A, Shoenfeld Y. Thymoma and autoimmunity. Cell Mol Immunol 2011;8:199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med 2016;375:1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murata K, Dalakas MC. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol 1999;155:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016;39:98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chuang WY, Ströbel P, Gold R, et al. A CTLA4high genotype is associated with myasthenia gravis in thymoma patients. Ann Neurol 2005;58:644–648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigators.