

Abstract
There are 257 million persons worldwide with chronic hepatitis B virus (HBV) infection, a leading causes of liver cancer. Almost all adults with acute HBV infection have a rapid immune response to the virus resulting in life-long immunity, but there is no cure for individuals with chronic HBV infection, which they acquire during early life. The mechanisms that drive the progression of hepatitis B through distinct clinical phases to to end-stage liver disease are poorly understood. Likewise, it is not clear whether and how immune responses can be modulated to allow control and/or clearance of intrahepatic HBV DNA. We review the innate and adaptive immune responses to acute and chronic HBV infections and responses to antiviral therapy. Comparisons with hepatitis C virus infection provide insights into the reversibility of innate inflammatory responses and the potential for successful therapy to recover virus-specific memory immune responses.
Keywords: Hepatitis B virus, hepatitis C virus, immune memory, innate
Studies of the immune response to hepatitis B virus (HBV) antigens began >50 years ago, when Baruch Blumberg discovered HBV surface antigen (HBsAg), originally called Australia antigen, in sera from Australian aborigines 1. One important advance in our understanding of the immune response to HBV infection came with the observation that seroconversion from HBsAg+ to anti-HBs+ was a biomarker for immune control of the infection and protective immunity. Researchers began testing recombinant HBsAg in preventative vaccines, and countries that adopted universal immunization with HBsAg reported a decreased incidence of de novo infections,2 providing evidence for protective immunity.
Although we understand many features of acute self-limited hepatitis B and natural and vaccine-induced immunity, our understanding on the immune response to chronic HBV infection is lacking. Almost all cases of chronic HBV infection are established from perinatal or early childhood transmission. The geographic distribution and young age of the patients and the asymptomatic nature of early childhood infection have made it difficult to determine how infection during early life results in chronic infection. Due to the unpredictable timing of disease phases and disease activity flares, there have been few prospective studies of the immune response during chronic infection. Little is known about the mechanisms of immune activation and intrahepatic inflammation with increasing age.
We review the adaptive immune responses to HBV infection, highlighting similarities to and differences from hepatitis C virus (HCV) infection and identify areas for future research (Table 1). As the focus of drug development has moved from treatment of HCV infection to HBV infection, new immune cell targets and biomarkers for treatment are needed.
Table 1.
Current Knowledge and Knowledge Gaps related to innate and adaptive immune responses in HBV and HCV infection.
| What we know | What we do not know | |||
|---|---|---|---|---|
| HBV | HCV | HBV | HCV | |
| Innate Response |
– no ISG induction “stealth virus” |
– strong ISG induction | – mechanisms circumventing ISG-induction |
– relative role of ISGs in HCC and HCV-mediated interference with hepatic IFN system |
| – functional and phenotypical alterations of NK cells – IFN therapy leads to activation of immunity – DAA therapy leads to rapid downregulation of ISGs and normalization of NK cell phenotype |
– Responsible pathways and reversibility of NK cell alteration | |||
| Adaptive Response |
– viral clearance associated with multi-specific CD4+ and CD8+ T cell responses |
– relative role of cytolytic- versus non-cytolytic effector mechanisms |
||
| – enrichment of T cells at the site of infection | – impact of liver environment on function and phenotype of immune cells – nature of immune responses that drive disease progression |
|||
| – early loss of CD4+ T cell help leads to viral persistence | – mechanisms responsible for early CD4+ T cell failure | |||
| – T cell dysfunction and viral escape contribute to T cell failure |
– relative role of different mechanisms of T cell failure | |||
| – antiviral therapy leads at least to partial recovery of virus-specific T cell recovery |
– extent to which antiviral therapy and viral control lead to restoration of protective immunity |
|||
| – humoral immune responses detectable | – role of antibodies and B cells in different disease phases |
– contribution of antibodies to viral control |
||
ISG, interferon stimulated gene; HCC, hepatocellular carcinoma
Interestingly, there are shared features of the innate and adaptive immune responses to both infections. Chronic HBV and HCV infections are each associated with exhaustion and reduced antiviral function of virus-specific T cells, thought to result from chronic T-cell receptor stimulation by persisting viral antigens 3. Chronic HBV and HCV infections also share inflammation-induced changes in natural killer (NK) cell functions, such as increased cytotoxicity and decreased production of antiviral cytokines 4, which contribute to the regulation and deletion of T cells during HBV infection 5, 6. Interferon (IFN)-based and direct-activating antiviral (DAA) therapies have been used to treat patients with HBV or HCV infection. The intense study of HCV infection over the past decade has provided novel information on immune responses that may be applicable to and should be examined in the context of research on HBV infection. We review these parallels, and discuss the added complexities of the HBV lifecycle, immune response, and development of hepatitis, which make HBV infection more difficult to cure than HCV infection.
The Liver Immune System
HBV and HCV each infect cells of the liver. Based on its location and anatomical features, the liver’s immune system induces tolerance to antigens, which are delivered via the portal vein and sampled by sinusoidal cells and liver-resident macrophages 7. The liver’s immune tolerance is well known in the field of transplantation, because livers for transplantation do not require HLA matching with the recipient 8.
The liver contains specialized immune cell populations, such as memory T cells and NK cells that are not found in the blood 9–11 and innate-like T cells, such as mucosal-activated invariant T (MAIT) cells, which are shared by gut and liver 12, 13. Much of this immune system was only recently discovered and its role in the pathogenesis of viral hepatitis and use as a target for immunomodulator therapies has not been recognized. Likewise, the spatial organization of intrahepatic immune cell clusters is an area of active investigation. Some structures, such as B cell follicles, 14 are permanent whereas others, such as intrahepatic myeloid aggregates 15, form transiently to promote optimal proliferation of antiviral effector cells.
HBV and HCV each establish acute self-limited and chronic infection of the liver. However, they have different structures of genome organization and replication strategies, so it is not surprising that they use unique strategies to persist (Table 2). HBV, a double-stranded partially open DNA virus with multiple overlapping open reading frames, establishes a mini-chromosome as its transcriptional template and also integrates randomly into the host genome. It replicates within its own nucleocapsids in the cytoplasm of infected hepatocytes and does not induce IFN-mediated responses 16. In contrast, HCV is a single-stranded RNA virus with a single open reading frame. Its replication in the cytoplasm of infected cells is readily detected by intracellular pattern recognition receptors and results in the release of type I and III IFNs, which HCV escapes [reviewed in 17].
Table 2.
Comparison of HBV and HCV virology, immunology and treatment
| HBV | HCV | |
|---|---|---|
| Prevalence worldwide |
257 million people infected | 71 million people infected |
| Virology | ||
| Virus | 42 nm; enveloped nucleocapsid; partially double-stranded DNA genome |
50 nm; enveloped nucleocapsid; positive stranded RNA genome |
| Family | Hepadnaviridae | Flaviviridae; Hepacivirus Genus |
| Genotypes | 8 genotypes | 6 major genotypes; more than 50 subtypes; quasispecies in each infected patient |
| Mutation rate | low | high |
| Virus half-life | 2–3 days | 3 hours |
| Virus production | 1010 –1012 virions / day | 1012 virions / day |
| Acute Infection | ||
| Outcome | >90% self-limited infection | <30% self-limited infection |
| Immunity | Longterm protective immuity | ? |
| Chronic Infection | ||
| Cause | Mostly from vertical/perinatal transmission:mother-to-neonate, early childhood infection |
Mostly from horizontal transmission injection drug use, parenteral, sexual, nosocomial |
| Course | Distinct phases defined by ALT and viremia level HBeAg / antibody status |
Stable ALT levels and viremia |
| Spont resolution | About 1% per year (HBsAg loss) | No |
| Treatment | ||
| IFN-based | PegIFN; 25% HBeAg seroconversion 3–7% HBsAg loss |
PegIFN/ribavirin |
| Antiviral | Inhibition of transcriptase activity of viral polymerase; suppression of viral replication without elimination of cccDNA |
Inhibition of viral protease and polymerase; >95% cure |
| Cure | functional cure (cccDNA persists, reactivation possible) |
virological cure (virus completely cleared) |
The Immune Response During Acute, Self-limited Infection
Individuals infected with HBV or HCV can mount a success antiviral immune response. HBV is cleared by more than 95% of adults and HCV is cleared by about 30% of adults. Although takes several weeks for HBV to reach high viral titers in the blood, HCV viremia reaches high levels within days after infection (Fig. 1A, B). HBV persists as a stealth virus in infected cells 16, 18, 19, whereas HCV immediately induces a large number of IFN-stimulated genes (ISGs) and persists via elaborate strategies to escape the innate immune response (reviewed in 17).
Figure 1. Acute, Self-limited HBV and HCV infection and Chronic HBV Infection.
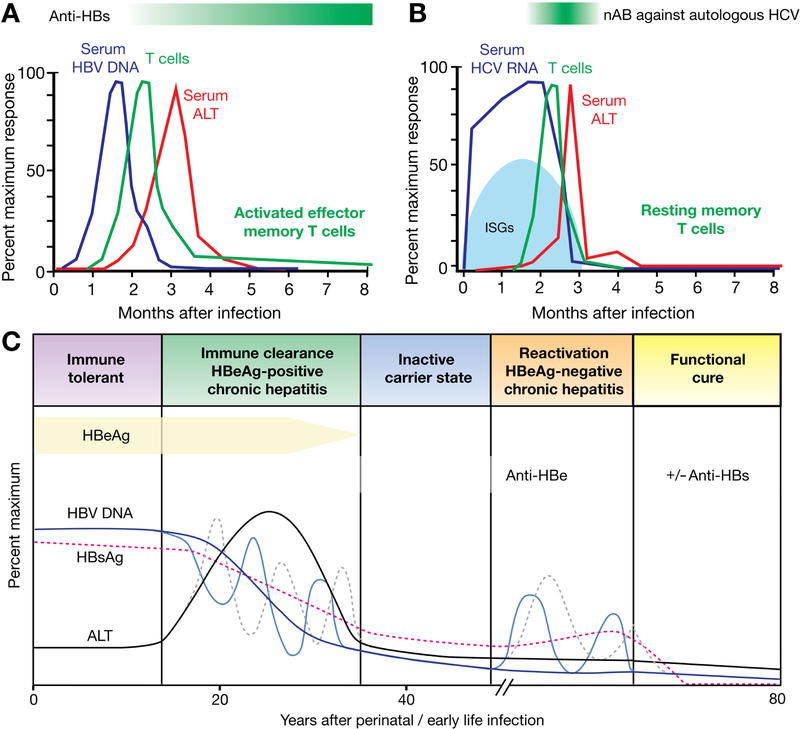
(A, B) Studies of acute, self-limited HBV and HCV infection provide information about successful immune responses. (A) HBV DNA is detectable within the first 2 months of infection and its peak precedes the onset of T-cell responses and the increase in serum level of ALT. (B) HCV RNA becomes detectable within a few weeks after infection and is associated with the induction of ISGs. T-cell responses occur after 8–12 weeks in most patients concomitant with a surge in serum ALT levels. Neutralizing antibodies (nAb) against HCV become detectable a few weeks later. (C) Phases of chronic HBV infection: the immune-tolerant phase is characterized by high levels of HBV DNA and HBsAg and a normal level of ALT. In the immune-clearance phase and the HBeAg+ chronic hepatitis phase, levels of HBV DNA and ALT decrease and the level of ALT increases, indicating liver injury. The transition between the first 2 phases of disease can be dynamic and associates with repeated increases and decreases in level of HBV DNA and activity of ALT (indicated by the dashed lines). This phase can ultimately lead to HBeAg seroconversion. The subsequent inactive carrier state is characterized by low levels of HBV and HBsAg and a normal level of ALT, but reactivation in the HBeAg-negative chronic hepatitis phase is associated with flares in levels of ALT and ongoing liver disease.
Virus-specific T cells can be detected in the blood about 8–10 weeks after HBV or HCV infection. This means that T cells respond when HBV viremia reaches high levels, but trail the appearance of HCV viremia by several weeks. In both infections, viremia starts to decrease shortly after antiviral T cells become detectable in the blood and the size of the CD8+ T cell infiltrate increases in the liver 20, 21. The ability to mount effective anti-virus immune responses is determined partly by genetic factors and is increased in individuals with specific HLA alleles. For example, HLA-B27 presents dominant epitopes from many viruses to CD8+ T cells, including HCV 22, 23.
In individuals with acute HBV infection, the decrease in viremia precedes the peak level of alanine aminotransferase (ALT), 20 indicating that antiviral functions of HBV-specific T cells occur before the initiation of severe liver damage. These antiviral functions are mediated by cytokines such as IFN gamma (IFNG) and tumor necrosis factor (TNF) 24, 25. This process is more efficient than perforin- and granzyme-mediated killing of infected cells, which requires direct interactions between each immune cell and its target cell 20.
Depletion of CD8+ T cells from chimpanzees interferes with clearance of HBV 26 and HCV 27. Depletion of CD4+ T cells prevents the effective of induction of CD8+ T cell-mediated responses and also prevents clearance of acute HBV 28 and HCV infection 29. HBV viremia typically decreases to undetectable levels with the onset of virus-specific T-cell responses. However, HCV viremia often is only temporarily controlled, and rebounds multiple times, due to the selection of viral quasispecies with mutations that allow them to escape detection by T cells 30, ultimately establishing chronic infection.
Humoral immune responses are detectable during acute HBV and HCV infections. During HBV infection, the seroconversion from HBeAg+ to anti-HBe+, and from HBsAg+ to anti-HBs+, are used as biomarkers on the road to recovery. Standard immunoassays for these serological markers can be used to monitor clearance of HCV infection, but the transient appearance of strain-specific antibodies has been associated with HCV clearance 31. Interestingly, HBV-specific antibodies persist for life, whereas HCV-specific antibodies disappear 10–20 years after recovery from acute hep atitis 32. This might result from continued antigen stimulation after resolution of acute HBV infection vs the complete antigen clearance after resolution of acute HCV infection.
Resolution of acute HBV and HCV infection therefore results in different outcomes. Self-limited acute HBV infection is effectively controlled by the immune response, resulting in the appearance of a cure, but the virus is never completely eliminated—small amounts of covalently closed circular DNA (cccDNA) and integrated HBV DNA persist. Trace amounts of HBV DNA below the limit of detection by quantitative commercial assays might sporadically induce responses by virus-specific T cells and antibodies 33. In contrast, self-limited acute HCV infection results in complete clearance of all viral RNA and viral antigens. This is consistent with the observation that HBV-specific memory T cells maintain an activated phenotype after resolution of acute HBV infection—they can be depleted from peripheral blood mononuclear cells with antibodies against the activation marker HLA-DR 33. In contrast, HCV-specific memory T cells have a resting phenotype 32.
This distinction may also have implications for protective immunity upon re-infection. Whereas spontaneous recovery from acute HBV infection results in life-long protective immunity, due to persistence of virus-specific T cells and antibodies, protective immunity after recovery from HCV infection is not always maintained. Complete immune protection with rapid clearance of a re-infecting virus has been observed in some but not all chimpanzees upon re-challenge 27, 34–36, and injection drug users who cleared HCV infection often develop hepatitis upon re-infection, 37 despite evidence for some limited strain-specific immune responses from prior exposure38. This concept is also relevant for strategies to develop a protective vaccine against HCV—the use of replicating viral vectors that are not completely cleared may be better suited to induce long-lasting T cell-based immunity than the use of protein vaccines39,40.
Establishment and Course of Chronic Infection
One of the keys to understanding chronic HBV infection lies in the immune response to of early childhood infection. After comparing cord blood from neonates of HBV-infected and uninfected mothers, Hong et al proposed that in utero exposure to HBV trains the immune response to tolerate the virus, resulting increased innate immune cell maturation and Th1 cell differentiation (trained immunity, a form of innate immune memory) 41. This was associated with an increased ability of cord blood immune cells to respond to bacterial infections in vitro. Perinatally acquired HBV has therefore been proposed to be a symbiont that confers advantages to the host. This observation is supported by the recent discovery that the entire hepadnaviral lineage, to which HBV belongs, co-evolved with mammals over at least 400 million years 42. Perinatal transmission is a perfect result of this co-evolution because it achieves a very high (>95%) rate of persistent infection without any immediate detriment to the host.
Several studies in mice have identified factors that may contribute to the age-dependent differences in immune responses against HBV. Although mice cannot become infected with HBV, because they do not express its entry receptor, it is possible to introduce a transgene that encodes replicating HBV. This can be done in Rag1–/– mice, which then secrete intact and infectious HBV in the absence of T and B cells 43. Adoptive transfer of naïve immune cells into adult transgenic mice results in T- and B-cell priming and seroconversion from HBsAg+ to anti-HBs+, whereas adoptive transfer of immune cells to young transgenic mice does not 44. This differential outcome has been attributed to age-dependent expression of costimulatory molecules on hepatic antigen-presenting cells 43. This was also associated with impaired responses of T-follicular helper cells, which are required for the optimal generation of virus-specific responses of CD8+ T cells and antibodies45. Young mice also have lower hepatic levels of CXCL13, a chemokine that is required for optimal induction of virus-specific B cells 44. Age-dependent changes in the gut microbiota might contribute to this differential immune response, because sterilization of gut microbiota in nontransgenic immunocompetent adult mice decreases HBV-specific immune responses in a model of acute hepatitis B 46.
In contrast to the chronic outcome of early childhood HBV infection, early childhood HCV infection is frequently cleared 47. Virus-specific factors are likely to be responsible for this difference in outcomes. In utero exposure to the secreted HBeAg has been implicated in the attenuation of HBe- and HBeAg-specific immune responses in neonates 48. Tian et al tried to model this prenatal exposure to HBV antigens by crossing female hemizygous HBV transgenic mice to naïve male mice. Offspring of these mice were exposed to HBeAg in utero, but are HBV negative after birth 49. When adult offspring of HBV-positive mothers is challenged by hydrodynamic injection of an HBV-encoding plasmid, their HBV-specific T-cell responses were impaired compared to those of adult offspring of HBV-negative mothers. This supports the concept of tolerance where abundant HBV antigens such as HBeAg and HBsAg functionally impair or delete T-cell clones 50. Accordingly, HBV-infected neonates and children typically have a non-inflammatory phase, with barely detectable HBV-specific T cells and normal levels of liver enzymes, despite high levels of circulating HBV DNA. Meanwhile, HBV integrates into the host genome, resulting in clonal expansion of hepatocytes and increased risk for cancer 51. The establishment of cccDNA as the template for HBV replication results in long-term HBV persistence.
Not only are the virus life cycle and immune response more complex for HBV than for HCV, but but so is the course of chronic infection. Although viremia and liver enzyme levels are relatively constant in patients with chronic HCV infection, there are wide variations in HBV titers and liver disease activity in patients with chronic infection (Fig. 1C). The non-inflammatory, immune-tolerant phase eventually transitions into an inflammatory phase with more severe liver disease and fluctuations in HBV DNA titer and ALT activity 52. Thereafter, many patients enter a phase of low virus replication, with loss of HBeAg and seroconversion to antiHBe+, reduced inflammation, and low HBV titers. Patients who enter this phase after the age of 40 years have a higher risk of developing cirrhosis and liver cancer, compared to patients who enter it earlier 53. Finally, a subset of patients loses HBeAg expression due to viral mutations. These patients develop active liver disease with incrased, fluctuating levels of liver enzymes, but lower levels of HBV DNA than during the HBeAg+ inflammatory phase. In general, HBeAg– patients with active live r inflammation progress more rapidly to fibrosis and cirrhosis than HBeAg+ patients 52.
Innate Immune Responses During Chronic HBV or HCV Infection
One of the most interesting immunological difference between HBV and HCV infection is the difference in interferon-mediated responses. Acute HBV infection does not induce a significant ISG-mediated response in liver 54; liver biopsies from patients with HBV infection do not have higher levels of ISG expression than those from patients without HBV infection 55. When liver biopsies of HBV-infected patients were exposed to toll-like receptor ligands, IFN signaling was not suppressed. This was corroborated by the observations that HBV does not affect the magnitude or breadth of ISG expression by other inducers of IFN and ISGs, such as poly(I:C), Sendai virus, or IFN alpha, 18 and that it does not protect HCV from the antiviral effects of IFN 19.
This differs from HCV, which induces strong IFN- and ISG-mediated responses 56, 57 (regulated by allelic variants near the IFNL4 gene 58) yet persists for decades despite the expression of hundreds of ISGs. Several mechanisms of viral interference with the hepatic IFN system have been identified and are reviewed elsewhere 59. Their relative roles require elucidation.
If HBV cannot be detected by the innate, IFN-mediated response, what leads to the inflammatory response in livers of patients with chronic infection? Co-culture experiments showed that macrophages were capable of sensing HBV if exposed to high HBV titers 18. When activated, the macrophages produced inflammatory cytokines, including IL1B, IL6, TNF, and CXCL10 18. These cytokines have direct antiviral effects 60 and activate other immune cells, such as NK 61, T, and B cells 62, 63. In mice with acute HBV infection, depletion of macrophages reduced recruitment of inflammatory cells and decreased liver injury 62.
To identify immune cells that promote the progression of chronic hepatitis B to liver cirrhosis, Vanwolleghem et al studied transcriptomes of peripheral blood samples from patients 64. Compared to blood mononuclear cells from patients in the noninflammatory disease phase, with high viral antigen levels and no liver disease, blood mononuclear cells from patients in the immunoactive phase highly upregulated expression of immunoglobulin genes. Overall, the results showed pronounced activity of B cells during the transition from the immunetolerant to the immunoactive phase, but no significant changes in gene expression patterns among T cells 64. This is consistent with a report from Park et al, who did not find differences in HBV-specific responses of T cells in patients during different phases of chronic HBV infection 65. Vanwolleghem et al reported induction of genes related to NK-cell activation during the immune active phase and HBeAg– hepatitis phase in patients 64. This is of interest because during chronic HBV and HCV infections, NK cells have increased cytotoxicity and decreased production of antiviral cytokines (reviewed in 4).
What Has IFN-based Therapy Taught Us About Innate Immune Responses?
IFN was part of the first treatment regimens for HBV and for HCV infection. IFN has direct antiviral effects against HBV. High doses of IFN alpha induce epigenetic changes in cccDNA-bound histones in cultured hepatoma cells, which inihibit HBV replication and reduce transcription of pregenomic RNA and subgenomic RNA from cccDNA 66, 67. IFN alpha also induces degradation of HBV cccDNA by upregulating expression of the deaminase APOBEC3A25 and inhibits HBV nucleocapsid formation 68. Whether it also accelerates the decay of nucleocapsids with pregenomic HBV RNA is unclear, as effects were shown by Xu et al 69 but not by Wieland et al 68. IFN’s direct antiviral effect is sufficient to reduce viral load and antigen levels in HBV-infected uPA/SCID mice with humanized livers, which lack immune cells 70.
Based on HBV’s susceptibility to IFN and the lack of endogenous IFN responses in HBV-infected patients, we would expect IFN-based therapies to be effective. However, for still unknown reasons, this is not the case. In contrast to the sharp 1–2 log10 decrease in HCV viremia within the first 48 hrs after initiation of pegylated IFN-based therapy 71, a decrease in HBV viremia is observed not earlier than 3–4 weeks into therapy 72, 73 and takes several months to reach its maximum 74. Less than 10% of HBV-infected patients convert to anti-HBs and most do so long after the end of therapy. Of note, IFN-based therapy does not increase the HBV-specific immune response. To the contrary, IFN treatment reduces numbers of CD8+ T cells (particularly late effector CD8+ T cells)74, whereas expression of immune inhibitory cell-surface markers (such as programmed cell death 1 [PDCD1 or PD2], LAG3, and CTLA4) 75 is maintained, resulting in impaired immune function74.
Loss of HBeAg and HBsAg and seroconversion to anti-HBe+ and anti-HBs+ is associated with reduced virus replication and long-term normalization of ALT levels 52. Loss of HBeAg and HBsAg and seroconversion can be associated with increased serum levels of IL12, IFNG, and IL2 76 and a temporary flares in levels of ALT, which suggests that the loss of HBeAg and HBsAg isimmune immune-mediated. Cytotoxic CD8+ T cells can be more readily expanded from blood of treatment responders than nonresponders 77. Changes in the inflammatory environment in the liver, which may take a long time to develop, might be a pre-requisite for off-treatment immune responses. This would be consistent with the observation that long-term (> 4 years) nucleos(t)ide analog therapy increases responses of HBeAg-negative patients to pegylated IFN add-on therapy 78. Strategies to enhance this response are an important area for further investigation.
Studies of patients with HCV infection receiving IFN-based therapy have provided unique insights into determinants of innate immune responsiveness 79. Pre-treatment levels of ISG expression associate with response to IFN alpha-based therapy; patients with high baseline levels of ISG expression cannot increase these levels with IFN-based therapy. Induction of negative feedback mechanisms, such as suppressors of cytokine signalling, likely contributes to resistance 80. In addition, genetic determinants of innate immune responses associate with the response to IFN alpha-based therapy. Rs1297960 and rRs368234815 are in linkage disequilibrium but rs368234815 has a higher predictive value in African Americans 81. Rs12979860 is located in the first intron of the IFNL4 gene 81 and rs368234815 regulates the transcription of the IFNL4 gene. The rs368234815 [DG] allele creates an open reading frame for expression of the IFNL4 protein, which induces expression of ISGs. In contrast, the rs368234815 [ΔG] haplotype results in a frameshift so that IFNL4 protein is not produced. This haplotype is associated with HCV clearance after acute infection and response to IFN-based therapy 82–84.
The negative correlation between pre-treatment levels of ISG expression and on-treatment increases in expression of ISGs is recapitulated in the response of innate immune cell populations that are sensitive to IFN alpha. For example, pre-treatment expression levels of the activating NK cell receptors NKp30, NKp46 and DNAM1 correlate inversely with the increase in theeexpression levels of these receptors during IFN-based treatment and the subsequent virologic response to treatment 85. NK cells from patients with a rapid first-phase decrease in HCV RNA have maximal responsiveness to type I IFN, based on high levels of phosphorylated STAT1 in NK cells, compared to that of patients with slow decrease in HCV RNA 86. This is consistent with greater NK cell cytotoxicity in treatment responders than nonresponders 87.
Although a virologic response to IFN-based therapy is durable in more than 97% of patients 88 it takes a long time until HCV is completely cleared. Small traces of HCV RNA can sporadically re-appear in the circulation within the first years after treatment along with a transient increase in HCV-specific T-cell responses 89. The persisting HCV RNA is replication-competent and infectious because it can transmit infection to chimpanzees 90 and on rare occasions result in relapse in the treated patients 91.
T-cell responses in Patients With Chronic HBV or HCV infection
Patients with HBV or HCV infection have low numbers of virus-specific T cells, compared to patients with other chronic infections, such as CMV or HIV3—may be related to the tolerogenic environment of the liver (for review, see 92). HBV-specific CD8+ and CD4+ T cells are more difficult to detect and expand than HCV-specific T cells 93. This could be due to reduced induction of HBV-specific T cells during perinatal infection or increased T-cell exhaustion. Increased T-cell exhaustion may be due to fact that HBV infection typically occurs earlier in life than HCV. This results in a longer period of antigen-specific stimulation followed by exhaustion and ultimately depletion of virus-specific T cells. HBV also has a lower rate of mutation than HCV (Table 2), and the HBV genome encodes several overlapping open reading frames, which reduces the likelihood of T-cell escape variants being compatible with viral fitness. This may result in a somewhat lower prevalence of viral escape variants in patients with HBV 94, 95 vs HCV infection 96, 97.
The ex vivo detectability of HBV-specific CD8+ T cells appears to be limited to individuals with low viral load and those, who have also cleared HBeAg 98–104. T-cell responses against HBV polymerase are the easiest to detect in recall assays whereas T-cell responses against HBsAg are almost completely absent 102, 105. This recapitulates the differential abundance of these proteins: HBsAg is expressed in subviral particles, which outnumber complete polymerase-containing virions by a factor of 100,0004.
In HBV and HCV infection, most virus-specific CD8+ T cells express multiple inhibitory receptors and are functionally impaired 102, 106–110. Dysregulated expression of transcription factors 104 and mitochondrial alterations have been identified as additional factors associated with the impaired function of HBV-specific T cells 111. Increased expression of the apoptosis-activating molecule Bim is thought to contribute to ultimate clonal deletion 112. However, studies from mice indicate that T-cell responses to individual viral epitopes can be polyclonal 113—not all virus-specific T cells are terminally exhausted. Likewise, HCV-specific CD8+ T cells are not a homogeneous population 114. Terminally exhausted CD127– PD1hi CD8+ T cells co-exist with CD127+PD1+ CD8+ T cells that target the same HCV epitope without viral escape mutation (Fig. 2). The CD127+PD1+ CD8+ T-cell subset has memory-like characteristics due to the expression of the memory transcription factor TCF1 and the expression of the IL7 receptor CD127, which allows them to maintain their capacity for self-renewal and act as a continuous source of effector cells that then undergo terminal exhaustion (Fig. 2B) 115. Memory-like cells can persist in the absence of antigen, shown by transfer of these cells from mice with chronic lymphocytic choriomeningitis virus infection to uninfected mice, 116 and give rise to a rapid, memory-like recall response. In this mouse model of chronic lymphocytic choriomeningitis virus infection, TCF1+ T cells have provide the proliferative burst after blockade of the inhibitory receptor PD1 117. Blockade of inhibitory receptors can increase the functional response of some HBV- and HCV-specific CD8+ T cells in vitro 65, 102, 109, so memory-like CD8+ T cells might be targeted to revive exhausted CD8+ T-cell populations in patients with viral hepatitis.
Figure 2. Phenotypic, Transcriptional, and Tunctional Properties of Virus-specific CD8+ T Cells.
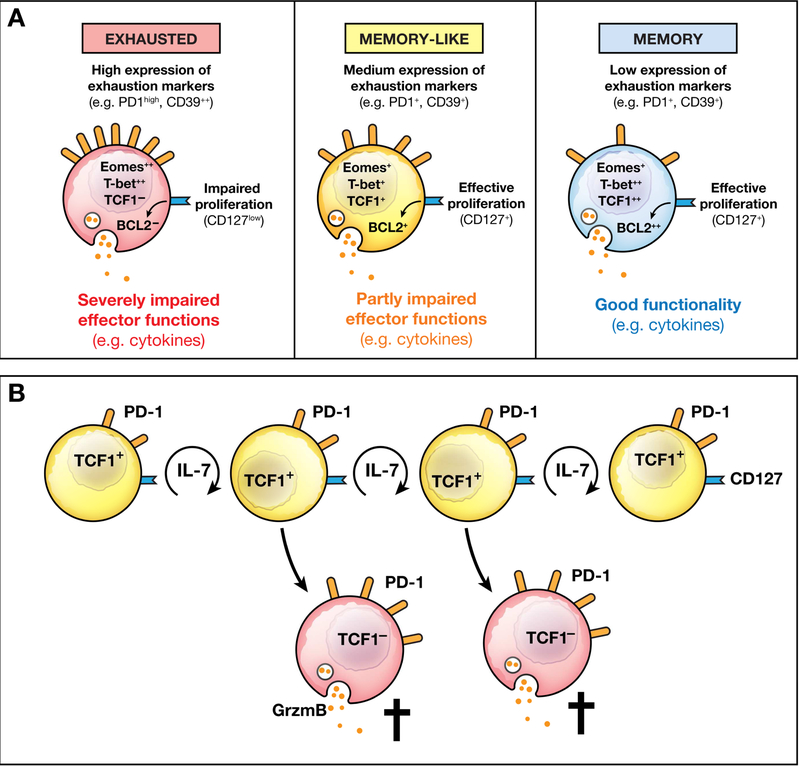
(A) Exhausted CD8+ T cells are characterized by high expression of exhaustion markers and the transcription factor eomesodermin and an impaired capacity to produce cytokines or proliferate. Memory-like CD8+ T cells have features of exhausted cells (PD1 expression) and memory cells (expression of the IL7 receptor CD127 and the transcription factor TCF1). Their functional properties are increased in comparison to exhausted cells but impaired in comparison to memory cells. Memory CD8+ T cells express low levels of exhaustion markers, have recall effector functions, and express the transcription factors T-box 21 (TBX21 or TBET) and HNF1 homeobox A (HNF1A or TCF1).(B) Virus-specific memory-like CD8+ T cells sustain the T-cell response during chronic infection (modified from 115 with permission).
Does Antiviral Therapy Restore Protective Immune Responses?
DAAs and nucleos(t)-ide analogues effectively inhibit viral replication in patients with HCV or HBV infection. Antiviral therapy decreases viral titers in blood and liver and reduces inflammatory liver disease in patients with HCV or HBV infection. Differences exist, however, in response kinetics. Whereas a sustained virologic response can be reached within 8 weeks for most patients treated for HCV infection and viral antigens are eventually complete cleared, HBV typically requires life-long therapy, and viral antigens remain despite suppression of viral replication. Studying immune responses to HCV and HBV can elucidate the relative roles of viral replication and persisting antigen on innate and adaptive immunity.
DAA-mediated clearance of HCV is accompanied by rapid downregulation of ISGs in the liver and blood, regardless of treatment outcome. Analysis of paired pre-treatment and end of treatment samples revealed that viral clearance is accompanied by decreased expression of type II and III IFNs, but surprisingly by increased expression of type I IFN (IFNA2) at the end of treatment 118. Restoration of type I IFN levels in the liver might therefore contribute to DAA-mediated HCV eradication and prevention of HCV re-infection. This is consistent with findings from Alao et al, who observed higher baseline expression of ISGs in patients who respond to DAA therapy compared to those with viral breakthrough 119. Successful DAA therapy also normalizes NK cell function and to restores their responsiveness to IFN alpha 120, 121, so innate immunity might contribute to HCV clearance during DAA therapy by preventing viral breakthrough. Nonetheless, even after HCV is cleared, the diversity of the NK cell repertoire remains altered. 122
DAA therapy does result in restoration of all parts of the innate immune response, as shown for cytokines and MAIT cells. MAIT cells are a population of innate-like T cells that is enriched at barrier sites such as liver and gut and activated by antigen (vitamin B metabolites displayed on non-classical MHC molecules) and by inflammatory cytokines (IL12 and IL18). Monocyte-derived IL18 is increased in the HCV-infected liver, resulting in MAIT cell activation and reduction of peripheral and intrahepatic MAIT cells 123. Effective DAA therapy decreases intrahepatic activation of monocytes and plasma levels of IL18, followed by a decrease in MAIT cell activation and an increase in intrahepatic MAIT cell frequency 123. The frequency of MAIT cells in peripheral blood is reduced for many months after the end of treatment 124 and their antigen-dependent effector function is not restored 123 124. Likewise, several cytokines do not normalize during the follow-up period 125. Collectively, these findings indicate that decades of HCV infection have lasting effects on parts of the innate immune system, even after treatment-induced viral clearance.
At the level of adaptive immune responses, a partial recovery of virus-specific T-cell responses, especially their proliferative function, has been reported in patients with HBV or HCV infection. In patients treated with nucleos(t)-ide analogues for HBV infection, this recovery was observed in assays that depend on in vitro stimulation 99, 126 and has been proposed to be transient 127. This could be the reason it is impossible to stop nucleos(t)-ide analogues treatment for most patients with chronic HBV infection. There is an association between the quantity and function of HBV-core and -pol specific CD8+ T cells and the ability to control HBV replication after nucleos(t)-ide analogue withdrawal 128. Functional HBV-specific CD8+ T cells were selectively enriched in the PD1+ population of T cells, supporting the existence of a persistent, memory-like population. The strongest recovery of HBV-specific CD8+ T-cell responses has been reported in patients with HBsAg seroconversion during nucleos(t)-ide analogue therapy. This includes the recovery of T-cell responses against HBsAg, which is typically not observed in patients with HBV chronic infection 103. As in patients with acute self-limited hepatitis, HBsAg clearance indicates effective immune control and is a prognostic marker for a good outcome of liver disease 129, 130. It is therefore tempting to speculate that viremia and the level of virus antigen contribute to HBV-specific T-cell suppression during chronic infection.
We have learned much about the fate of memory-like T cells in patients with HCV infection treated with DAAs. During and after DAA therapy, memory-like CD8+ T cells persist, whereas terminally exhausted T cells are rapidly lost (Fig. 3). This is similar to the immune contraction phase after acute viral infection, in which memory T-cell populations are maintained after successful antigen elimination while terminally differentiated effector T cells disappear. Memory-like HCV-specific CD8+ T cells therefore appear to be capable of antigen-independent survival, whereas terminally differentiated effector cells depend on continuous antigen stimulation.
Figure 3. Changes in HCV-specific T-cell Subsets During and After DAA Therapy.
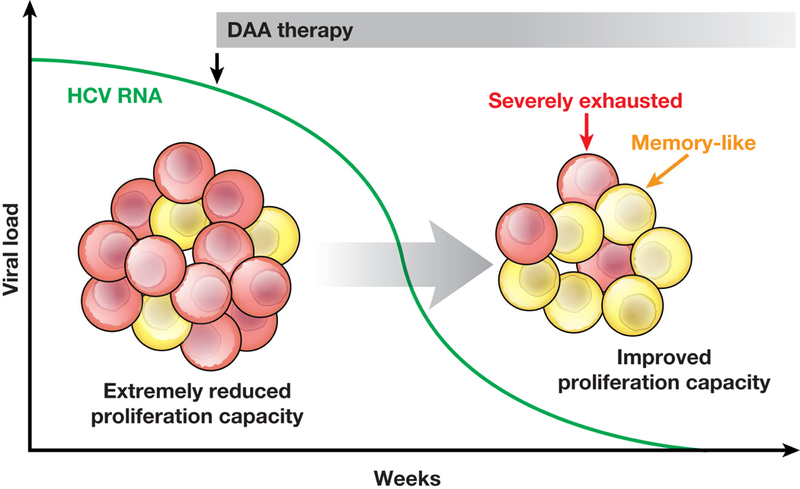
During chronic HCV infection, the 2 subsets of PD1+ HCV-specific CD8+ T cells are severely exhausted T cells and memory-like T cells. After successful DAA therapy, higher numbers of memory-like CD8+ T cells are maintained due to their antigen-independent proliferative capacity while severely exhausted, antigen-dependent CD8+ T cells decrease in number. This is reflected by an improved proliferative capacity of the overall HCV-specific CD8+ T-cell population.
Memory T cells are reactivated upon re-exposure viral antigen. However, even though HCV-specific T cells from patients who are successfully treated have increased proliferation and function, compared to exhausted HCV-specific effector cells, they do not reach the functional capacities of conventional memory T cells in in vitro assays 131. In a patient with a virologic relapse after DAA therapy, recall responses of HCV-specific CD8+ T cells were observed at the time of virological relapse and antigen re-exposure, but these did not mediate viral clearance and ultimately acquired a terminally exhausted phenotype 114. This finding is in agreement with results from a study in chimpanzees, which reported the persistence of HCV-specific CD8+ T cells after DAA-mediated HCV elimination but no protection from re-infection 132. Likewise, chronic infections are frequently observed in re-infected injection drug users. Antiviral therapy does not, therefore, fully restore immunity. The mechanisms of this effect are not well understood and could include quantitative and qualitative differences in the small population of memory-like T cells, a permanent and irreversible epigenetic imprinting of most virus-specific CD8+ T cells, 133 or indirect effects of cytokines or regulatory T cells134.
Future Directions and Opportunities
Research into HBV infection should aim to provide a better understanding of chronic disease and immune control. Host, viral, and environmental factors that affect the transition from non-inflammatory to inflammatory phases, and from HBeAg+ to anti-HBe+ phases, need to be identified and studied in order for us to understand mechanisms of pathogenesis and for identification of biomarkers. Patients with chronic HBV infection who spontaneously seroconvert to anti-HBs should be identified and assessed, as they represent a functional cure—the goal of treatment goal for chronic HBV infection. An ideal antiviral therapy would not just decrease HBV replication but also effectively reduce levels of HBsAg and eliminate or at least control cccDNA—it might need to synergize with the adaptive immune response to achieve this 135. Decrease in HBV replication, reduction of HBsAg levels and control of cccDNA might be achieved by directly increasing the immune response (such as with agonists of the toll like receptor, a therapeutic vaccine, or adoptive T-cell transfer) or through indirect activation (inhibition of HBsAg production).
Altough antiviral therapies can now completely eradicate HCV, even with reduction in treatment duration and costs, these regimens will likely not be affordable in countries where the prevalence of HCV and the incidence of new infections are highest. The development of a prophylactic vaccine that induces protective immunity is therefore necessary. A better understanding of the basic mechanisms of immune failure and its possible restoration by DAA therapy will be important to guide this attempt.
Acknowledgment:
This work was partially supported by the intramural research program of NIDDK, NIH. The work from R.T. is supported by the Deutsche Forschungsgemeinschaft (TRR 179 and SFB 1160 IMPATH)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The author state that no conflict of interest exists.
References
Author names in bold designate shared co-first authorship.
- 1.Blumberg BS. Hepatitis B - The hunt for a killer virus: Princeton University Press, 2002. [Google Scholar]
- 2.Chiang CJ, Yang YW, You SL, et al. Thirty-year outcomes of the national hepatitis B immunization program in Taiwan. JAMA 2013;310:974–6. [DOI] [PubMed] [Google Scholar]
- 3.Rehermann B Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med 2013;19:859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rehermann B Natural killer cells in viral hepatitis. Cell Mol Gastroenterol Hepatol 2015;1:578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peppa D, Gill US, Reynolds G, et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J Exp Med 2013;210:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boni C, Lampertico P, Talamona L, et al. Natural killer cell phenotype modulation and natural killer/T-cell interplay in nucleos(t)ide analogue-treated hepatitis e antigen-negative patients with chronic hepatitis B. Hepatology 2015;62:1697–709. [DOI] [PubMed] [Google Scholar]
- 7.Cantor HM, Dumont AE. Hepatic suppression of sensitization to antigen absorbed into the portal system. Nature 1967;215:744–5. [DOI] [PubMed] [Google Scholar]
- 8.Levitsky J, Feng S. Tolerance in clinical liver transplantation. Hum Immunol 2018;79:283–287. [DOI] [PubMed] [Google Scholar]
- 9.Pallett LJ, Davies J, Colbeck EJ, et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J Exp Med 2017;214:1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuff AO, Robertson FP, Stegmann KA, et al. Eomes high NK cells in human liver are long-lived and do not recirculate but can be replenished from the circulation. J Immunol 2016;197:4283–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stegmann KA, Robertson F, Hansi N, et al. CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci Rep 2016;6:26157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolte FJ, Rehermann B. Mucosal-associated invariant T cells in chronic inflammatory liver disease. Semin Liver Dis 2018;38:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ussher JE, Willberg CB, Klenerman P. MAIT cells and viruses. Immunol Cell Biol 2018. [DOI] [PMC free article] [PubMed]
- 14.Lefkowitch JH, Schiff ER, Davis GL, et al. Pathological diagnosis of chronic hepatitis C: a multicenter comparative study with chronic hepatitis B. The Hepatitis Interventional Therapy Group. Gastroenterology 1993;104:595–603. [DOI] [PubMed] [Google Scholar]
- 15.Huang LR, Wohlleber D, Reisinger F, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol 2013;14:574–83. [DOI] [PubMed] [Google Scholar]
- 16.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol 2005;79:9369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity 2014;40:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng X, Xia Y, Serti E, et al. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017;66:1779–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mutz P, Metz P, Lempp FA, et al. HBV bypasses the innate immune response and does not protect HCV from antiviral activity of interferon. Gastroenterology 2018;154:1791–1804 e22. [DOI] [PubMed] [Google Scholar]
- 20.Guidotti LG, Rochford R, Chung J, et al. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999;284:825–9. [DOI] [PubMed] [Google Scholar]
- 21.Shin EC, Park SH, Demino M, et al. Delayed induction, not impaired recruitment, of specific CD8(+) T cells causes the late onset of acute hepatitis C. Gastroenterology 2011;141:686–95, 695 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann-Haefelin C, Timm J, Schmidt J, et al. Protective effect of human leukocyte antigen B27 in hepatitis C virus infection requires the presence of a genotype-specific immunodominant CD8+ T-cell epitope. Hepatology 2010;51:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dazert E, Neumann-Haefelin C, Bressanelli S, et al. Loss of viral fitness and cross-recognition by CD8+ T cells limit HCV escape from a protective HLA-B27-restricted human immune response. J Clin Invest 2009;119:376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol 2001;19:65–91. [DOI] [PubMed] [Google Scholar]
- 25.Lucifora J, Xia Y, Reisinger F, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014;343:1221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thimme R, Wieland S, Steiger C, et al. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 2003;77:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoukry NH, Grakoui A, Houghton M, et al. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J Exp Med 2003;197:1645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asabe S, Wieland SF, Chattopadhyay PK, et al. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol 2009;83:9652–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grakoui A, Shoukry NH, Woollard DJ, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science 2003;302:659–62. [DOI] [PubMed] [Google Scholar]
- 30.Heller T, Rehermann B. Acute hepatitis C: a multifaceted disease. Semin Liver Dis 2005;25:7–17. [DOI] [PubMed] [Google Scholar]
- 31.Pestka JM, Zeisel MB, Blaser E, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 2007;104:6025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takaki A, Wiese M, Maertens G, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med 2000;6:578–82. [DOI] [PubMed] [Google Scholar]
- 33.Rehermann B, Ferrari C, Pasquinelli C, et al. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat Med 1996;2:1104–8. [DOI] [PubMed] [Google Scholar]
- 34.Major ME, Mihalik K, Puig M, et al. Previously infected and recovered chimpanzees exhibit rapid responses that control hepatitis C virus replication upon rechallenge. J Virol 2002;76:6586–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoukry NH, Sidney J, Sette A, et al. Conserved hierarchy of helper T cell responses in a chimpanzee during primary and secondary hepatitis C virus infections. J Immunol 2004;172:483–92. [DOI] [PubMed] [Google Scholar]
- 36.Lanford RE, Guerra B, Chavez D, et al. Cross-genotype immunity to hepatitis C virus. J Virol 2004;78:1575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacks-Davis R, Grebely J, Dore GJ, et al. Hepatitis C virus reinfection and spontaneous clearance of reinfection--the InC3 study. J Infect Dis 2015;212:1407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sugimoto K, Kaplan DE, Ikeda F, et al. Strain-specific T-cell suppression and protective immunity in patients with chronic hepatitis C virus infection. J Virol 2005;79:6976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen SG, Zak DE, Xu G, et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat Med 2018;24:130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fruh K, Picker L. CD8+ T cell programming by cytomegalovirus vectors: applications in prophylactic and therapeutic vaccination. Curr Opin Immunol 2017;47:52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong M, Sandalova E, Low D, et al. Trained immunity in newborn infants of HBV-infected mothers. Nat Commun 2015;6:6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lauber C, Seitz S, Mattei S, et al. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 2017;22:387–399 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Publicover J, Gaggar A, Jespersen JM, et al. An OX40/OX40L interaction directs successful immunity to hepatitis B virus. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed]
- 44.Publicover J, Gaggar A, Nishimura S, et al. Age-dependent hepatic lymphoid organization directs successful immunity to hepatitis B. J Clin Invest 2013;123:3728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Publicover J, Goodsell A, Nishimura S, et al. IL-21 is pivotal in determining age-dependent effectiveness of immune responses in a mouse model of human hepatitis B. J Clin Invest 2011;121:1154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou HH, Chien WH, Wu LL, et al. Age-related immune clearance of hepatitis B virus infection requires the establishment of gut microbiota. Proc Natl Acad Sci U S A 2015;112:2175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vogt M, Lang T, Frosner G, et al. Prevalence and clinical outcome of hepatitis C infection in children who underwent cardiac surgery before the implementation of blood-donor screening. N Engl J Med 1999;341:866–70. [DOI] [PubMed] [Google Scholar]
- 48.Milich DR, Jones JE, Hughes JL, et al. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci U S A 1990;87:6599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian Y, Kuo CF, Akbari O, et al. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 2016;44:1204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milich DR. The concept of immune tolerance in chronic hepatitis B virus infection Is alive and well. Gastroenterology 2016;151:801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mason WS, Gill US, Litwin S, et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 2016;151:986–998 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol 2011;8:275–84. [DOI] [PubMed] [Google Scholar]
- 53.Chen YC, Chu CM, Liaw YF. Age-specific prognosis following spontaneous hepatitis B e antigen seroconversion in chronic hepatitis B. Hepatology 2010;51:435–44. [DOI] [PubMed] [Google Scholar]
- 54.Wieland S, Thimme R, Purcell RH, et al. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A 2004;101:6669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suslov A, Boldanova T, Wang X, et al. Hepatitis B virus does not interfere with innate immune responses in the human liver. Gastroenterology 2018;154:1778–1790. [DOI] [PubMed] [Google Scholar]
- 56.Su AI, Pezacki JP, Wodicka L, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A 2002;99:15669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bigger CB, Brasky KM, Lanford RE. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J Virol 2001;75:7059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Brien TR, Yang H, Groover S, et al. Genetic factors that affect spontaneous clearance of hepatitis C or B virus, response to Tteatment, and disease progression. Gastroenterology 2018;in press. [DOI] [PubMed]
- 59.Horner SM, Gale M Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 2013;19:879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hosel M, Quasdorff M, Wiegmann K, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009;50:1773–82. [DOI] [PubMed] [Google Scholar]
- 61.Boltjes A, van Montfoort N, Biesta PJ, et al. Kupffer cells interact with hepatitis B surface antigen in vivo and in vitro, leading to proinflammatory cytokine production and natural killer cell function. J Infect Dis 2015;211:1268–78. [DOI] [PubMed] [Google Scholar]
- 62.Ando K, Moriyama T, Guidotti LG, et al. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J Exp Med 1993;178:1541–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kimura K, Moriwaki H, Nagaki M, et al. Pathogenic role of B cells in anti-CD40-induced necroinflammatory liver disease. Am J Pathol 2006;168:786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vanwolleghem T, Hou J, van Oord G, et al. Re-evaluation of hepatitis B virus clinical phases by systems biology identifies unappreciated roles for the innate immune response and B cells. Hepatology 2015;62:87–100. [DOI] [PubMed] [Google Scholar]
- 65.Park JJ, Wong DK, Wahed AS, et al. Hepatitis B virus-specific and global T-cell dysfunction in chronic hepatitis B. Gastroenterology 2016;150:684–695 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu F, Campagna M, Qi Y, et al. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 2013;9:e1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belloni L, Allweiss L, Guerrieri F, et al. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 2012;122:529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wieland SF, Eustaquio A, Whitten-Bauer C, et al. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc Natl Acad Sci U S A 2005;102:9913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu C, Guo H, Pan XB, et al. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 2010;84:9332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allweiss L, Volz T, Lutgehetmann M, et al. Immune cell responses are not required to induce substantial hepatitis B virus antigen decline during pegylated interferon-alpha administration. J Hepatol 2014;60:500–7. [DOI] [PubMed] [Google Scholar]
- 71.Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998;282:103–7. [DOI] [PubMed] [Google Scholar]
- 72.Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med 2005;352:2682–95. [DOI] [PubMed] [Google Scholar]
- 73.Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2004;351:1206–17. [DOI] [PubMed] [Google Scholar]
- 74.Micco L, Peppa D, Loggi E, et al. Differential boosting of innate and adaptive antiviral responses during pegylated-interferon-alpha therapy of chronic hepatitis B. J Hepatol 2013;58:225–33. [DOI] [PubMed] [Google Scholar]
- 75.Tan AT, Hoang LT, Chin D, et al. Reduction of HBV replication prolongs the early immunological response to IFNalpha therapy. J Hepatol 2014;60:54–61. [DOI] [PubMed] [Google Scholar]
- 76.Rossol S, Marinos G, Carucci P, et al. Interleukin-12 induction of Th1 cytokines is important for viral clearance in chronic hepatitis B. J Clin Invest 1997;99:3025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rehermann B, Lau D, Hoofnagle JH, et al. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J Clin Invest 1996;97:1655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ouzan D, Penaranda G, Joly H, et al. Add-on peg-interferon leads to loss of HBsAg in patients with HBeAg-negative chronic hepatitis and HBV DNA fully suppressed by long-term nucleotide analogs. J Clin Virol 2013;58:713–7. [DOI] [PubMed] [Google Scholar]
- 79.Heim MH. 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end. Nat Rev Immunol 2013;13:535–42. [DOI] [PubMed] [Google Scholar]
- 80.Sarasin-Filipowicz M, Wang X, Yan M, et al. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol 2009;29:4841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013;45:164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009;461:399–401. [DOI] [PubMed] [Google Scholar]
- 83.Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009;41:1105–9. [DOI] [PubMed] [Google Scholar]
- 84.Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009;461:798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Maria A, Fogli M, Mazza S, et al. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol 2007;37:445–55. [DOI] [PubMed] [Google Scholar]
- 86.Edlich B, Ahlenstiel G, Zabaleta Azpiroz A, et al. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology 2012;55:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahlenstiel G, Edlich B, Hogdal LJ, et al. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology 2011;141:1231–9, 1239 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Welker MW, Zeuzem S. Occult hepatitis C: how convincing are the current data? Hepatology 2009;49:665–75. [DOI] [PubMed] [Google Scholar]
- 89.Veerapu NS, Raghuraman S, Liang TJ, et al. Sporadic reappearance of minute amounts of hepatitis C virus RNA after successful therapy stimulates cellular immune responses. Gastroenterology 2011;140:676–685 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Veerapu NS, Park SH, Tully DC, et al. Trace amounts of sporadically reappearing HCV RNA can cause infection. J Clin Invest 2014;124:3469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hara K, Rivera MM, Koh C, et al. Sequence analysis of hepatitis C virus from patients with relapse after a sustained virological response: relapse or reinfection? J Infect Dis 2014;209:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol 2012;12:201–13. [DOI] [PubMed] [Google Scholar]
- 93.Rehermann B, Chang KM, McHutchinson J, et al. Differential cytotoxic T-lymphocyte responsiveness to the hepatitis B and C viruses in chronically infected patients. J Virol 1996;70:7092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rehermann B, Pasquinelli C, Mosier SM, et al. Hepatitis B virus (HBV) sequence variation of cytotoxic T lymphocyte epitopes is not common in patients with chronic HBV infection. J Clin Invest 1995;96:1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kefalakes H, Budeus B, Walker A, et al. Adaptation of the hepatitis B virus core protein to CD8(+) T-cell selection pressure. Hepatology 2015;62:47–56. [DOI] [PubMed] [Google Scholar]
- 96.Chang KM, Rehermann B, McHutchison JG, et al. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J Clin Invest 1997;100:2376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Timm J, Li B, Daniels MG, et al. Human leukocyte antigen-associated sequence polymorphisms in hepatitis C virus reveal reproducible immune responses and constraints on viral evolution. Hepatology 2007;46:339–49. [DOI] [PubMed] [Google Scholar]
- 98.Maini MK, Boni C, Lee CK, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med 2000;191:1269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boni C, Penna A, Ogg GS, et al. Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology 2001;33:963–71. [DOI] [PubMed] [Google Scholar]
- 100.Reignat S, Webster GJ, Brown D, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med 2002;195:1089–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Webster GJ, Reignat S, Brown D, et al. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol 2004;78:5707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boni C, Fisicaro P, Valdatta C, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol 2007;81:4215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boni C, Laccabue D, Lampertico P, et al. Restored function of HBV-specific T cells after long-term effective therapy with nucleos(t)ide analogues. Gastroenterology 2012;143:963–73 e9. [DOI] [PubMed] [Google Scholar]
- 104.Kurktschiev PD, Raziorrouh B, Schraut W, et al. Dysfunctional CD8+ T cells in hepatitis B and C are characterized by a lack of antigen-specific T-bet induction. J Exp Med 2014;211:2047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bertoletti A, Ferrari C. Adaptive immunity in HBV infection. J Hepatol 2016;64:S71–S83. [DOI] [PubMed] [Google Scholar]
- 106.Fisicaro P, Valdatta C, Massari M, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010;138:682–93, 693 e1–4. [DOI] [PubMed] [Google Scholar]
- 107.Schurich A, Khanna P, Lopes AR, et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011;53:1494–503. [DOI] [PubMed] [Google Scholar]
- 108.Nebbia G, Peppa D, Schurich A, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One 2012;7:e47648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raziorrouh B, Schraut W, Gerlach T, et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology 2010;52:1934–47. [DOI] [PubMed] [Google Scholar]
- 110.Bengsch B, Martin B, Thimme R. Restoration of HBV-specific CD8+ T cell function by PD-1 blockade in inactive carrier patients is linked to T cell differentiation. J Hepatol 2014;61:1212–9. [DOI] [PubMed] [Google Scholar]
- 111.Fisicaro P, Barili V, Montanini B, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med 2017;23:327–336. [DOI] [PubMed] [Google Scholar]
- 112.Lopes AR, Kellam P, Das A, et al. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. J Clin Invest 2008;118:1835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ishikawa T, Kono D, Chung J, et al. Polyclonality and multispecificity of the CTL response to a single viral epitope. J Immunol 1998;161:5842–50. [PubMed] [Google Scholar]
- 114.Wieland D, Kemming J, Schuch A, et al. TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat Commun 2017;8:15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Utzschneider DT, Charmoy M, Chennupati V, et al. T Cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 2016;45:415–27. [DOI] [PubMed] [Google Scholar]
- 116.Utzschneider DT, Legat A, Fuertes Marraco SA, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol 2013;14:603–10. [DOI] [PubMed] [Google Scholar]
- 117.Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016;537:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meissner EG, Wu D, Osinusi A, et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J Clin Invest 2014;124:3352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alao H, Cam M, Keembiyehetty C, et al. Baseline Intrahepatic and Peripheral Innate Immunity are Associated with Hepatitis C Virus Clearance During DAA Therapy. Hepatology 2018. [DOI] [PMC free article] [PubMed]
- 120.Serti E, Chepa-Lotrea X, Kim YJ, et al. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology 2015;149:190–200 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Serti E, Park H, Keane M, et al. Rapid decrease in hepatitis C viremia by direct acting antivirals improves the natural killer cell response to IFNalpha. Gut 2017;66:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Strunz B, Hengst J, Deterding K, et al. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat Commun 2018;9:2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bolte FJ, O’Keefe AC, Webb LM, et al. Intra-hepatic depletion of mucosal-associated invariant T cells in hepatitis C virus-Induced liver inflammation. Gastroenterology 2017;153:1392–1403 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hengst J, Strunz B, Deterding K, et al. Nonreversible MAIT cell-dysfunction in chronic hepatitis C virus infection despite successful interferon-free therapy. Eur J Immunol 2016;46:2204–10. [DOI] [PubMed] [Google Scholar]
- 125.Hengst J, Falk CS, Schlaphoff V, et al. Direct-acting antiviral-induced hepatitis C virus clearance does not completely restore the altered cytokine and chemokine milieu in patients with chronic hepatitis C. J Infect Dis 2016;214:1965–1974. [DOI] [PubMed] [Google Scholar]
- 126.Boni C, Bertoletti A, Penna A, et al. Lamivudine treatment can restore T cell responsiveness in chronic hepatitis B. J Clin Invest 1998;102:968–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Boni C, Penna A, Bertoletti A, et al. Transient restoration of anti-viral T cell responses induced by lamivudine therapy in chronic hepatitis B. J Hepatol 2003;39:595–605. [DOI] [PubMed] [Google Scholar]
- 128.Rivino L, Le Bert N, Gill US, et al. Hepatitis B virus-specific T cells associate with viral control upon nucleos(t)ide-analogue therapy discontinuation. J Clin Invest 2018;128:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim GA, Lim YS, An J, et al. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: clinical outcomes and durability. Gut 2014;63:1325–32. [DOI] [PubMed] [Google Scholar]
- 130.Yuen MF, Wong DK, Fung J, et al. HBsAg seroclearance in chronic hepatitis B in Asian patients: replicative level and risk of hepatocellular carcinoma. Gastroenterology 2008;135:1192–9. [DOI] [PubMed] [Google Scholar]
- 131.Wieland D, Hofmann M, Thimme R. Overcoming CD8+ T-cell exhaustion in viral hepatitis: lessons from the mouse model and clinical perspectives. Dig Dis 2017;35:334–338. [DOI] [PubMed] [Google Scholar]
- 132.Callendret B, Eccleston HB, Satterfield W, et al. Persistent hepatitis C viral replication despite priming of functional CD8+ T cells by combined therapy with a vaccine and a direct-acting antiviral. Hepatology 2016;63:1442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016;354:1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Knolle PA, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 2014;146:1193–207. [DOI] [PubMed] [Google Scholar]
- 135.Protzer U, Gehring A. New drug development for curing hepatitis B: immune-based Gastroenterology 2018;in press.