

Abstract
Eukaryotic transcription-coupled nucleotide excision repair (TC-NER) is a pathway that removes DNA lesions capable of blocking RNA polymerase II (Pol II) transcription from the template strand. This process is initiated by lesion-arrested Pol II and the recruitment of Cockayne Syndrome B protein (CSB). In this review, we will focus on the lesion recognition steps of eukaryotic TC-NER and summarize the recent research progress toward understanding the structural basis of Pol II-mediated lesion recognition and Pol II-CSB interactions. We will discuss the roles of CSB in both TC-NER initiation and transcription elongation. Finally, we propose an updated model of tripartite lesion recognition and verification for TC-NER in which CSB ensures Pol II-mediated recognition of DNA lesions for TC-NER.
Keywords: DNA damage, Nucleotide excision repair, Transcription-coupled nucleotide excision repair, Cockayne syndrome, Lesion recognition, RNA polymerase II, Transcriptional arrest
Introduction
Nucleotide excision repair (NER) is the main repair pathway that removes diverse bulky and helix-distorting/destabilizing DNA damage (1–3). NER can be further divided into two subpathways: Transcription-coupled nucleotide excision repair (TC-NER) and global genome nucleotide excision repair (GG-NER)(Figure 1)(2, 3). TC-NER is responsible for the preferential repair of DNA lesions that block transcription, whereas GG-NER can occur anywhere in the genome (2).
Figure 1.
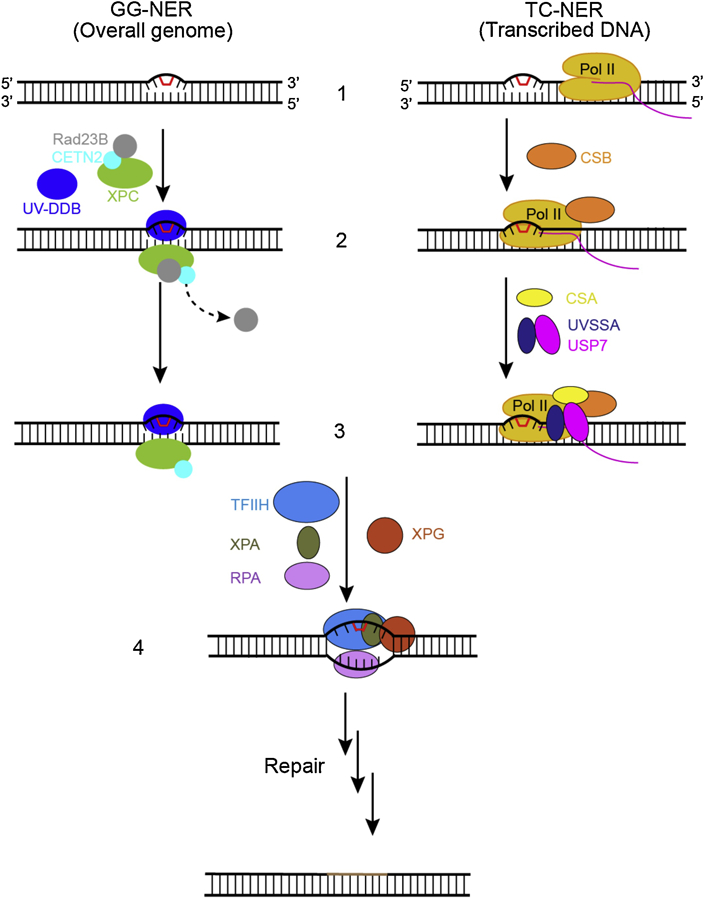
The recognition steps of two subpathways of nucleotide excision repair. In the global genome nucleotide excision repair subpathway (GG-NER), left, the damage sensor XPC, in complex with UV excision repair protein RAD23 homologue B (RAD23B) and centrin 2 (CETN2), which binds the non-damaged strand opposite the lesion with the help of the UV– DDB complex (step 2, left). Binding of the XPC complex to the damaged site results in RAD23B dissociation from the complex (step 3, left). In the transcription-coupled subpathway (TC-NER), damage recognition is initiated by the stalling of RNA polymerase II (Pol II). The stalled Pol II recruits CSB (step 2, right) and the Pol II–CSB complex serves as a platform to further recruitment of downstream repair factors such as CSA and UVSSA-USP7 (step 3, right). After damage recognition (step1–3), the TFIIH complex is recruited to the lesion in both GG-NER and TC-NER, along with XPA, RPA, and XPG (step 4). Once DNA lesion is verified, the damage-containing DNA short fragment is removed via dual incisions, new DNA fragment is synthesized, and NER reaction is completed through sealing the final nick by DNA ligase.
Transcription-coupled nucleotide excision repair (TC-NER), first discovered in mammalian cells (Chinese hamster ovary (CHO)) by Philip Hanawalt and colleagues in the mid-1980s (4, 5), is a highly conserved pathway that is reported in all three kingdoms of life (2, 6). Eukaryotic TC-NER is much more complicated than prokaryotic TC-NER (1–3, 7, 8). Yet, research over the last three decades has led to a significant increase in our understanding of the TC-NER pathway (2, 6, 9–17). Similar to GG-NER, TC-NER is composed of four steps: lesion recognition, lesion verification, incision steps (pre-incision repair complex assembly step and dual incision steps), and gap filling steps involved in DNA repair synthesis and ligation (2). The major difference between TC-NER and GG-NER is in the lesion recognition and verification steps (Figure 1).
In this review, we will focus on the recent progress that elucidated the structural basis of the early steps of lesion recognition and verification in eukaryotic TC-NER. A detailed review of GG-NER DNA lesion recognition can also be found in this issue by Schärer and colleagues (18). It is noted that some DNA lesions not only lead to local RNA Pol II stalling triggering TC-NER, but such lesions also lead to global inhibition of transcription during DNA repair (19, 20). The global effect of transcription inhibition and recovery after DNA repair is also a subject of intensive research, as reviewed by Marteijn and colleague in this issue (21).
Lesion Recognition in TC-NER
For TC-NER, the DNA lesion is initially sensed by the transcribing RNA polymerase (2). RNA polymerase arrested at a DNA lesion is proposed to be the signal that triggers TC-NER (2, 22–27). Damage detection in TC-NER is mainly through 1D-scanning, as RNA polymerase is a highly processive enzyme that travels along the DNA template several thousands or millions of base pair during transcription. This Pol II-mediated lesion recognition also provides an excellent DNA damage surveillance mechanism for the transcribed genome, a large portion of the whole genome (~50–90% of the whole genome, including protein-coding genes and non-coding regions) (28, 29).
There are several key mechanistic questions that are related to Pol II-mediated lesion recognition: What is the molecular basis for Pol II to sense structurally diverse DNA lesions? Is there a common mechanism for Pol II-mediated DNA lesion detection? What determines Pol II arrest versus bypass for a specific DNA lesion? Do DNA lesions induce dramatic conformational changes in Pol II? What is the difference, with respect to TC-NER, between a genuine DNA lesion-induced Pol II arrest and other obstruction-induced Pol II arrest (such as, intrinsic arrest DNA sequences, non-B structures, and nucleosome) (30)? How can cells distinguish among different forms of arrested Pol II complexes and commit only those encountering genuine DNA lesions to the TC-NER pathway? Given dozens of factors and the amount of time needed to complete repair and re-synthesis of the transcript, it is important that cells commit to this route only when absolutely necessary. Mammalian genes are generally much larger than those of prokaryotes and have a higher chance of transcription pausing by intrinsic arrest sequences or secondary DNA structures. What are the mechanisms for improving the fidelity of Pol II-mediated lesion recognition for TC-NER? In the following section, we will summarize the recent progress toward improving our understanding of these key questions related to the molecular basis of Pol II-mediated lesion recognition.
Structural Basis of Pol II Recognition of DNA Lesions
Over the last decade, substantial studies were carried out toward understanding how Pol II recognizes and processes DNA lesions. These biochemical and structural studies provide detailed mechanistic insight into the impact of DNA lesions on transcription elongation. Multiple outcomes can occur when RNA Pol II encounters a DNA lesion. For a small DNA lesion, RNA Pol II can bypass the lesion in either an error-free or error-prone manner (24, 31–37). The error-prone mode may lead to miscoded RNA products (31, 38, 39), also termed as transcriptional mutagenesis (24, 31, 39, 40). The mechanism of transcriptional bypass and mutagenesis is reviewed elsewhere (24, 27, 31, 37, 41). In contrast, Pol II can be completely blocked by a bulky DNA lesion (13, 24, 25, 34, 42). Here, we will focus on several recent structural studies in this area, particularly on the following four categories of DNA lesions: intrastrand DNA lesions; DNA major groove-related lesions; DNA minor groove-related lesions; and abasic sites (Figure 2).
Figure 2.
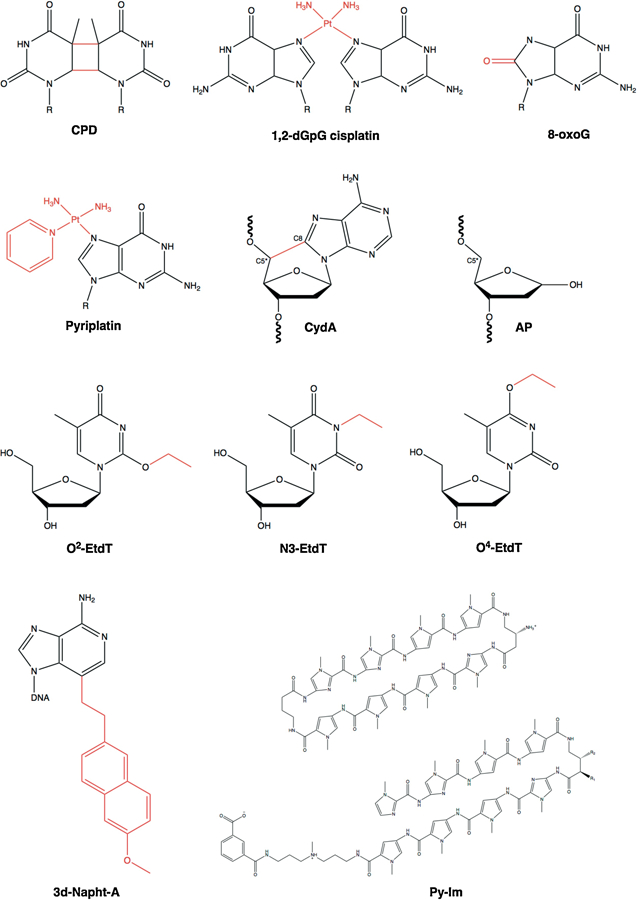
Chemical structures of DNA lesions: cyclobutane pyrimidine dimer (CPD), 1,2- dGpG cisplatin, 8-oxoguanine (8-oxoG), monofunctional pyriplatin, 8,5′-cyclo-2′-deoxyadenosine (CydA), abasic site (AP), O2-, N3- and O4-ethylthymideine (O2-, N3, and O4-EtdT), 3-deaza-3-methoxynaphtylethyl-adenosine (3d-Napht-dA), and non-covalent DNA binders Pyrrole-imidazole (Py-Im) polyamides. The DNA lesion is highlighted in red.
Intrastrand DNA lesions
Intrastrand DNA lesions represent a major class of DNA lesions that are recognized by NER. For example, UV irradiation leads to the formation of cyclobutane pyrimidine dimer (CPD), a common DNA lesion that crosslinks two adjacent pyrimidine nucleotides. In contrast, the anticancer drug cisplatin prefers to react with the N7-position of purines (G and A) and forms bulky 1,2-dGpG or 1,2-dApG intrastrand cross-links (account for ~90% of cisplatin-DNA adducts) (43, 44). One common characteristic shared by these DNA lesions is that the two covalently crosslinked nucleobases in the same strand lead to a significant distortion of the local DNA duplex structure (Figure 2), and both DNA lesions strongly block Pol II transcription (44–49).
Recent structural studies performed by the Cramer and Kashlev groups shed light on how the Pol II complex gets stalled by these DNA lesions (49–51). In particular, a structure capturing a CPD-stalled intermediate state showed that the CPD lesion is located above the bridge helix and does not reach into the +1 site, the canonical position for the template base after fully crossing over the bridge helix (PDB ID: 4A93) (Figure 3a, middle panel). This translocation intermediate state is very transient for an undamaged template (52). The captured intermediate state for the CPD lesion is attributed to the fact that intrastrand crosslinks greatly distorts the DNA template and restrains the flexibility of the phosphodiester backbone of the template strand during translocation. As a result, the CPD lesion gets stuck above the bridge helix. Since correct positioning of the template base is important for effective template-dependent NTP addition, the 3′-thymine of CPD cannot support template-dependent pairing for the incoming NTP. Instead, incorporation follows the so-called ‘A-rule’ where AMP is slowly incorporated in a non-template dependent manner (also see below for abasic site (AP) lesion-related transcription) (53). After slow translocation of the CPD lesion from +1 to the upstream −1 position, another AMP can be slowly incorporated, although UMP misincorporation opposite the 5′-thymine of CPD is also reported (49, 51). In contrast, the structure of the Pol II complex with a scaffold containing a 1,2-dGpG cisplatin-induced DNA lesion revealed that the lesion is accommodated at the downstream +2/+3 position (PDB ID: 2RZ7) (Figure 3b). A potential explanation for the failure to capture cisplatin at the +1/+2 intermediate is that the 1,2- dGpG cisplatin-DNA adduct is much bulkier than the CPD lesion. Therefore, the former imposes a stronger steric block for the damaged base to cross over the bridge helix and to be delivered into the active site in comparison with CPD lesion.
Figure 3.
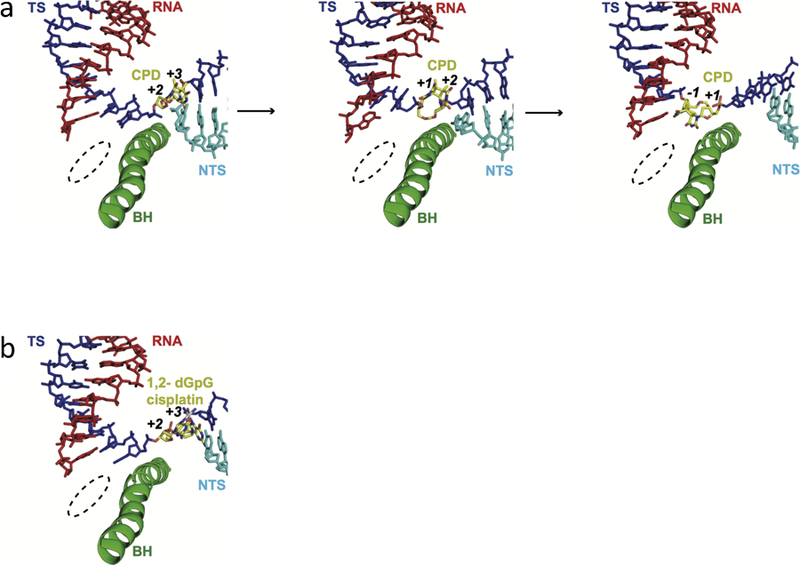
The structures of Pol II EC stalled at intrastrand crosslinks. a, CPD lesion-induced Pol II stall complex (PDB IDs: 2JA6, 4A93 and 2JA7). b, 1,2-dGpG cisplatin DNA lesion-induced Pol II stall complex (PDB ID: 2R7Z). RNA, template strand, non-template strand, and the bridge helix motif of RNA Pol II (Rpb1 810–840) are shown in red, blue, cyan, and green, respectively. The DNA lesion are highlighted and labeled in yellow. The template registers are also marked in the figure. The Pol II active site is indicated in dashed circle.
DNA major groove-related lesions
Reactive oxygen species (ROS) or free radicals from cellular metabolism can produce oxidatively-induced DNA lesions. 8-oxoguanine (8-oxoG) (Figure 2) is an abundant DNA lesion among more than 20 identified oxidatively-induced DNA lesions (54). Pol II can bypass the 8-oxoG lesion in either an error-free (cytosine incorporation) or error-prone (adenine incorporation) manner (55–57). The crystal structure of Pol II with the template strand containing a site-specific 8-oxoG showed that 8-oxoG adopts a canonical anti-conformation to pair with incoming CTP. In contrast, 8-oxoG uses the syn-conformation to form a Hoogsteen base pair with incoming ATP (Figure 4a) (58). Thus, the presence of an 8-carbonyl group reduces the stability of the 8-oxoG base in anti-anti pairing with C and increases the possibility of 8-oxoG in the syn-conformation pairing with A. Notably, the similar mechanism of adenine misincorporation at 8-oxoG is also observed in many DNA polymerases (59, 60). Whether 8-oxoG is the substrate for TC-NER is a subject of debate, partially because 8-oxoG does not induce strong Pol II arrest. Recently, it was reported that 8-oxoG in the ATM gene can be repaired in a transcription-coupled manner in vivo. This “non-canonical” transcription-coupled repair not only requires Pol II, CSB and UVSSA, but also requires hOGG1 and XPA, suggesting the participation of both base excision repair (BER) and NER in this process (61). It is interesting to note that hOGG1 can convert 8-oxoG to an abasic site, and AP endonuclease can subsequently generate a single-strand break. It is tempting to speculate that these BER intermediates of 8-oxoG can trigger Pol II arrest and the recruitment of TC-NER factors, such as CSB and UVSSA.
Figure 4.
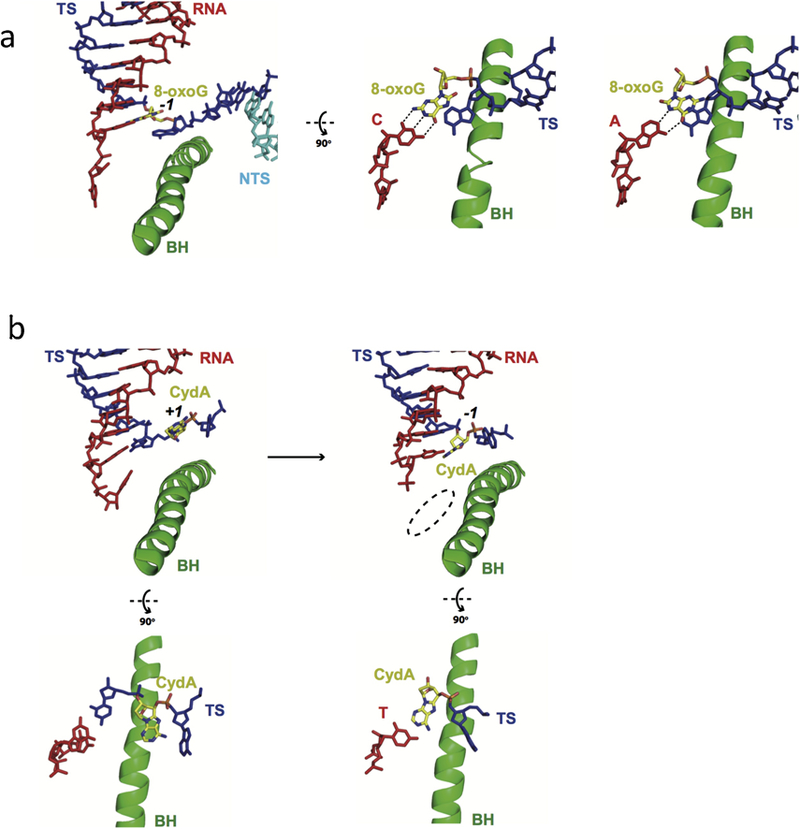
RNA Pol II recognition of oxidative DNA lesions. a. Pol II recognition of 8-oxoguanine (8-oxoG) lesion. Canonical Watson-Crick pair is formed between 8-oxo dG and C (PDB ID: 3I4M), but 8-oxo dG adopts syn-form when pairing with adenine in the Pol II-active site (PDB ID: 3I4N). b. Pol II EC stalled at an 8,5′-cyclo-2′-deoxyadenosine (CydA) site (PDB IDs: 4X6A and 4X67). Color codes are the same as Figure 3.
8,5′-Cyclopurine, such as 8,5′-cyclo-2′-deoxyadenosine (CydA) (Figure 2), is another type of oxidatively-induced DNA lesion caused by hydroxyl radical attack on DNA (62–64). Unlike 8-oxoG, 8,5′-cyclopurine DNA lesions strongly blocks RNA Pol II and interferes with gene transcription in mammalian cells (63–66). Interestingly, a portion of Pol II molecules can slowly bypass CydA and generate transcripts with 5′ A mutations opposite the 5′ nucleotide next to the lesion (63, 64, 66). To further understand this error-prone transcriptional bypass of CydA, in collaboration with the Kashlev and Brook labs, we systemically studied the molecular mechanism of CydA recognition and lesion bypass by Pol II in vitro (Figure 4b) (67). At the insertion step, the ‘A-rule’ is partially followed opposite the CydA lesion on the template DNA, although UMP incorporation is still predominant. The structure of the Pol II complex containing a CydA lesion at the +1 register revealed that the CydA lesion is accommodated above the bridge helix. The location of CydA lesion is remarkably similar to the previously discussed CPD lesion intermediate (51, 67) (Figure 4b, left). As a result, the CydA cannot form a canonical Watson-Crick base pair with 3′ UMP of the RNA primer. This provides a structural explanation to why UMP incorporation is inefficient. For the next extension step, the template base (5′) next to CydA is also accommodated above the bridge helix to keep the +1 position empty (Figure 4b, right). The steric restriction caused by the 8,5′-cyclo bond can lead to hindered loading of this template base into the active site (67). Thus, the ‘A-rule’ is also applied to this step, leading to 5′ A mutations in RNA transcripts.
Monofunctional pyriplatin (cis-diamminepyridinechloroplatinum(II)) and phenanthriplatin (cis-diamminephenanthridinechloroplatinum(II)) (Figures 2 and 5a) showed potent anticancer activities on several cancer cell lines that are resistant to cisplatin (68). These platinum compounds can form monofunctional bulky platinum-induced DNA lesions at dG and dA sites on DNA template, without distorting the DNA duplex structure. Intriguingly, unlike cisplatin, CPD, or CydA lesions, these monofunctional platinum-DNA lesions do not block the Pol II insertion step but strongly inhibit the extension step. To understand how these monofunctional platinum compounds are accommodated within the Pol II-active site and affect Pol II transcription, we solved the structure of Pol II stalled at a pyriplatin-DNA adduct (69) (Figure 5a). The structure provides insight into why these lesions can effectively support the insertion step (nucleotide incorporation opposite the damage site). In sharp contrast to the cisplatin-DNA adduct, pyriplatin-dG can fully cross over the bridge helix and be accommodated at the +1 position in the Pol II active site for efficient CMP incorporation. Indeed, a canonical Watson Crick base pair is formed between cytosine with pyriplatin-modified guanosine, in which the pyridine group extrudes toward the major groove. The structures also provide mechanistic insight into why RNA transcript extension is inhibited. We found that the platinum group of pyriplatin interacts with conserved residues of the bridge helix to stabilize the pre-translocation state. The presence of the bulky pyriplatin group also forms a strong steric barrier for the next nucleobase (5′) to cross over the bridge helix to reach the +1 position at the extension step. The phenanthriplatin-dG DNA lesion induced transcription stalling and bypass were also investigated in collaboration with the Lippard group (70). Similar to pyriplatin, the base of phenanthriplatin-dG at the +1 position does not affect CMP incorporation at the insertion step, which is almost the same as for the undamaged dG-containing template. The phenanthriplatin-dG lesion also significantly slows down transcription at the extension step, in which Pol II switches to an error-prone mode to slowly bypass the bulky lesion.
Figure 5.
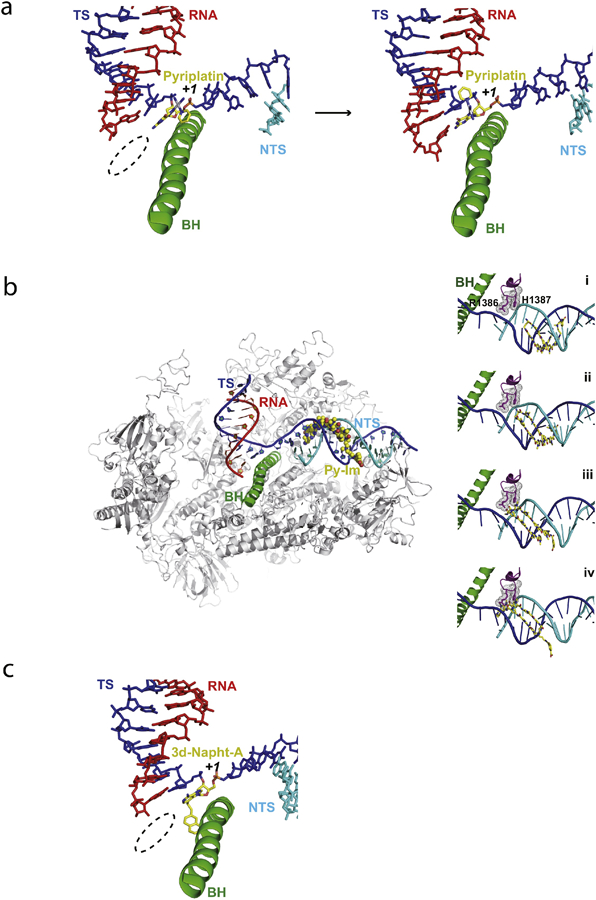
Pol II EC stalled by major groove or minor groove obstructions. a. Pol II EC stalled by a pyriplatin-DNA monofunctional adduct (PDB IDs: 3M4O and 3M3Y). b. Structural modeling of RNA Pol II transcriptional pausing by Py-Im polyamide. Two critical residues (R1386 and H1387) from the conserved Switch 1 region of Rpb1 play a key role in slowing down transcription progress through the polyamide-bound minor groove step by step. c. The Pol II EC adopts the post-translocation state with the modified adenine base (3d-Napht-dA) at +1 position, with 3-deaza-3-methoxynaphtylethyl group contacting the bridge helix in the Pol II active center from underneath (PDB ID: 5OT2). Color codes are the same as Figure 3.
DNA minor groove-related lesions and blockage
Pyrrole-imidazole (Py-Im) polyamides are synthetic small molecules that bind to specific DNA target sequences in the minor groove and with high affinity (~10 nM) (71, 72). Previous studies have suggested that cell-permeable Py-Im polyamides can modulate gene expression by preventing transcription factor binding and even causing Pol II subunit Rpb1 degradation (73–75). In collaboration with the Dervan lab, we investigated the effect of Py-Im polyamides on transcription elongation, and we found that Pol II elongation complex (EC) becomes persistently arrested by Py-Im polyamides (76). Strikingly, this Py-Im polyamide-induced Pol II arrest can not be rescued by TFIIS. This is in sharp contrast to the scenario where TFIIS can stimulate Pol II bypass of a nucleosomal protein barrier (~10 nM affinity for histones to DNA). Careful examination of Py-Im polyamide-induced Pol II stalling revealed that Pol II stalls a few base pairs upstream from the targeted Py-Im binding DNA sequence, indicating that Pol II can sense Py-Im before the small molecule reaches the Pol II active site or the downstream bubble opening edge. Structural modeling analyses revealed that two conserved residues, Arg1386 and His1387, in the Switch 1 region could play a key role in Pol II stalling at Py-Im binding sites (Figure 5b). The Switch regions serve as a hinge for mediating clamp closure of the RNAP active-center cleft when a DNA–RNA hybrid exists (77). These two critical residues from the Switch 1 region can insert into the DNA minor groove. Molecular modeling revealed that these residues might either directly interact with polyamide backbones (state ii) or sterically clash with the Py-Im polyamide (states iii and iv) during Pol II translocation toward to the Py-Im binding site (Figure 8). To directly verify the roles of these two residues, we generated a Pol II mutant with two site-specific substitutions (R1386A/H1387A). The transcription results with this Pol II mutant confirmed that these two residues are responsible for the early sensing or scanning of the DNA minor groove conformation, as two early stalling bands vanished with Pol II mutant.
Figure 8.
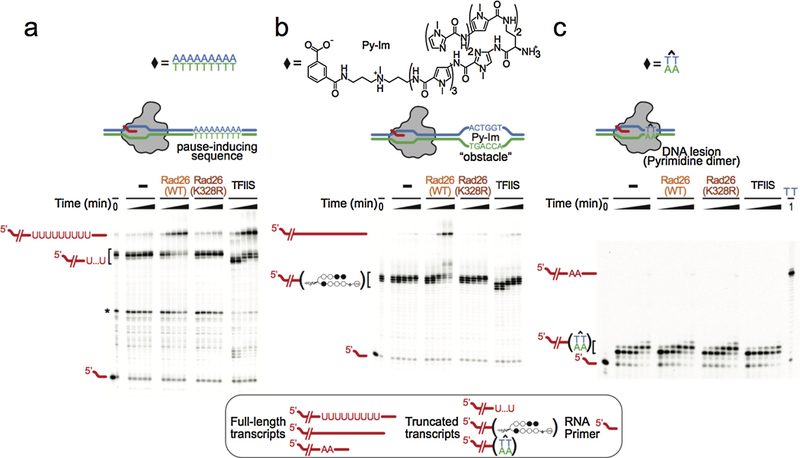
Rad26 helps Pol II discriminate among different transcription obstacles a-c.Transcription assays probing the ability of Rad26, Rad26 mutant, and TFIIS to discriminate among three representative transcription obstacles encountered by Pol II: A pause-inducing repetitive A-tract sequence (a); A sequence-specific DNA-binding polyamide (Py-Im) (b); and a TT cyclobutane pyrimidine dimer (CPD) DNA lesion (c). The asterisk in (a) represents a Pol II pausing site upstream of the A-tract sequence. Last lane in (c): full-length transcript in the absence of the CPD lesion.
Acylfulvenes (AFs) are a class of semisynthetic anticancer compounds produced as sesquiterpene derivatives of illudin S, a natural toxin isolated from the fungus Omphalotus illuden (109). These chemical agents alkylate DNA in the minor groove and interfere with DNA replication and transcription. The DNA alkylation process is also related to Pol II stalling triggering TR-NER (78–80). Recently, the Sturla and Cramer groups used several synthetic AF analogs to assess the effect of this type of alkylation on Pol II transcription elongation in vitro. Pol II was strongly blocked by the bulky AF analog, ‘3-deaza-3-methoxynaphtylethyl-adenosine (3d-Napht-dA)’, but it was able to bypass smaller analogs. The structure of Pol II containing 3d-Napht-dA showed that the damaged base can occupy the +1 position (Figure 5c). Interestingly, the 3-deaza-3-methoxynaphtylethyl group contacts the bridge helix mainly via van der Waals interactions between the second aromatic ring of 3d-Napht-A and the side chain methyl group of Thr831 on Rpb1. However, the ‘3d-Napht’ group can therefore severely impair trigger loop closure in the Pol II active site (81). The observed misincorporation could be explained by catalysis in a slow, trigger loop-independent, and low-fidelity fashion (82).
In addition to the parameters of size and chemical nature of DNA lesions, the position of nucleotide modifications also plays important roles in determining transcriptional outcomes. Recently, in collaboration with Yinsheng Wang’s group, we systematically investigated how Pol II recognizes and bypasses three regioisomeric alkylated DNA lesions (83). O2-, N3- and O4- ethylthymideine (O2-, N3, and O4-EtdT, see Figure 2) are alkylated by tobacco-specific metabolic activation and are at higher levels in smokers (Figure 10) (84–86). Although modifications happen on the same thymine, the different positions of the ethyl group (O2, N3, and O4) elicited distinct transcriptional responses of Pol II. O2- and N3-alkylation greatly blocked Pol II elongation, whereas O4-alkylation had limited blockage effect. Pol II blockage by O2- and N3-EtdT mainly occurred in both the insertion and extension steps. In contrast, for O4-EtdT, only weak pausing was found at the insertion step. Intriguingly, GMP was preferentially incorporated opposite O2- and O4-EtdT. Molecular modeling analyses indicated that guanine can form wobble base pairs with both O2- and O4-EtdT (Figure 6), but the canonical Watson-Crick pair of A:T could be impaired by the ethyl group. Hence, these wobble base pairs at the +1 template position could favor GMP binding and addition. After GMP incorporation, the newly formed rG:O2-EtdT or rG:O4-EtdT base pair needs to translocate into the −1 position so as to facilitate the subsequent nucleotide addition. For O4-EtdT, the ethyl group protruding into the major groove of DNA is well tolerated in the Pol II active site without any steric clash. Hence, transcriptional lesion bypass of the O4-EtdT lesion is very efficient. In sharp contrast, for O2-EtdT, the ethyl group points to the minor groove and severely clashes with P448 in Rpb1 during translocation. The steric clash of O2-EtdT and the P448-containing loop (or ‘Pro-gate’ loop) may explain why the minor groove O2-EtdT lesions significantly block Pol II elongation (83). Consistently, the orientation of the 3-deaza-3-methoxynaphtylethyl group of 3d-Napht at the +1 site is facing toward a secondary channel and away from the ‘Pro-gate’ loop, avoiding a steric clash (81).
Figure 10.
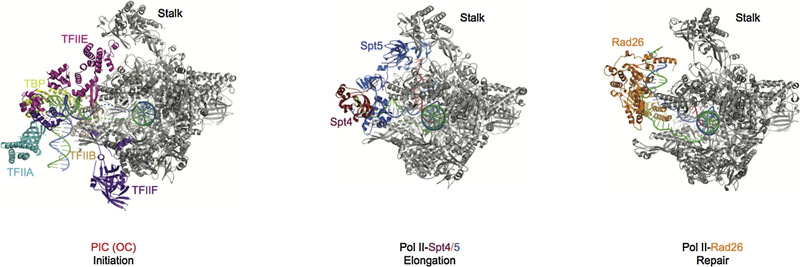
Comparison of three stages of Pol II complexes. The initiation open complex structure is shown on the left, PDB ID: 5FYW; The transcription elongation complex is shown in the middle, PDB ID: 5OIK; The transcription repair initiation complex is shown on the right, PDB ID: 5VVR. In the three different stages, TFIIE, Spt4/5, and CSB bind to similar positions on Pol II. Pol II is in gray and other factors are highlighted and labeled as indicated. Template and non-template DNA, and RNA are shown in blue, green, and red, respectively.
Figure 6.
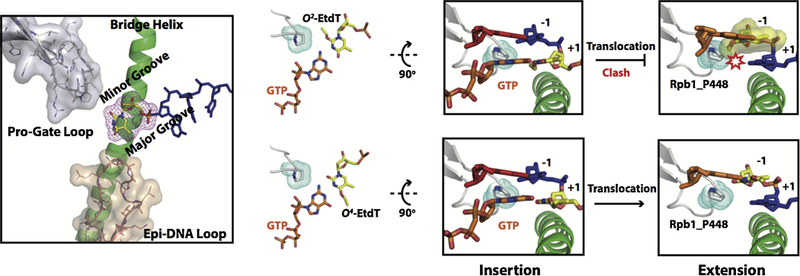
Structural modeling showed distinct effects of O4- and O2-EtdT lesions on transcriptional bypass. Briefly, the alkylation in the major groove (O4-EtdT) hardly affects translocation, whereas the minor-groove alkylation (O2-EtdT) has strong steric clash with the P448 residue on the ‘Pro-gate loop’ during translocation. Color codes are the same as Figure 3.
The abasic lesion
Abasic (AP) sites result from spontaneous depurination or intermediates of base excision repair and are among the most frequent DNA lesions in the steady-state (85, 87–89). The AP sites, if left unrepaired, are highly mutagenic for both DNA replication and transcription. For DNA replication, AP sites serve as strong blocks for replicative DNA polymerases, and A is predominately incorporated opposite the AP site (‘A-rule’) (53, 90–92). In contrast, several translesion DNA synthesis (TLS) DNA polymerases (mostly X and Y families) use different mechanisms (probably sequence-dependent) to bypass the AP sites (93–96). Similar to DNA replication, Pol II stalls at the AP site on the DNA template and then bypasses it slowly, following the ‘A-rule’ (97–99). Although many structures of DNA polymerases with AP-containing DNA have been solved, giving us significant insight into the molecular basis of ‘A-rule’ and translesion bypass for DNA polymerases, the precise molecular mechanism for Pol II transcriptional bypass of AP sites remains elusive.
Recently, in collaboration with the Kashlev lab, we took a combined structural and biochemical approach to systematically study the molecular mechanism of Pol II stalling on the AP site and translesion bypass in a step-wise manner (Figure 7) (100). We revealed that Pol II has two consecutive stalling events: the insertion opposite the AP site and the extension bypass of the AP site. Intriguingly, the nucleotide incorporation patterns for these two consecutive stalling steps are very distinct: AMP is preferentially favored in the first insertion step (A-rule), whereas the cognate nucleotide incorporation is favored in the next extension step (template-dependent manner). To elucidate the structural basis for these two consecutive stalling events, four structural snapshots of two consecutive stalled states at the insertion and extension steps were captured, respectively (Figure 7). The structures of EC-I (without NTP) and EC-II (with a bound ATP analog) represent the states of Pol II first encountering an AP site at the insertion step. Interestingly, while the upstream of RNA/DNA hybrid is already at a canonical post-translocation state, the AP site is located above the bridge helix as a half-translocation intermediate. The conserved residue Rpb1 Arg337 forms strong hydrogen bonds with the phosphate group of the AP site at the intermediate state. The EC-II structure revealed that AMPCPP binds to the addition site opposite the AP lesion despite the lack of a base pair. The driving force for ATP binding is likely due to base stacking. Indeed, the AMPCPP is sandwiched between the −1 base pair and Thr831 on the bridge helix. The trigger loop is in an open conformation that is consistent with slow nucleotide addition. To understand the second stalling event, we further solved the structures of EC-III (without cognate NTP) and EC-VI (with a bound cognate NTP analog). In the EC-III structure, we found significant flexibility for both AMP at the 3′-RNA terminus and 5′-template nucleotide adjacent to the AP lesion. This flexibility probably results from a lack of base-stacking interactions and base pair restraint from the AP site. As a result, the incoming cognate UTP fails to reach the canonical addition site and largely remains at the entrance site in the EC-VI structure. These structures provide a potential structural explanation as to why the extension step is substantially slow. The presence of the AP lesion alters the free energy landscape of Pol II translocation, as neither the 5′-template nucleotide nor the incoming cognate NTP is located at a canonical position that is poised for nucleotide addition. Both template and cognate NTP are needed to rotate toward the canonical positions to support template-dependent nucleotide addition. When the AP lesion is placed into the upstream RNA/DNA hybrid region and sandwiched by non-damaged base pairs, the AP site has a minimal effect on Pol II elongation.
Figure 7.
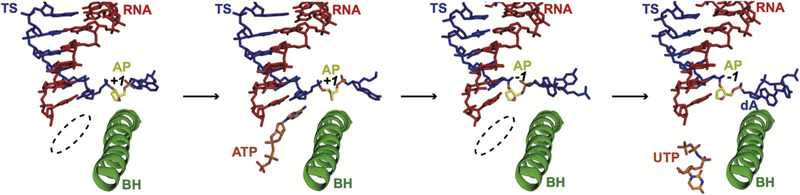
Structural analysis of transcription stalling by the AP site at insertion and extension steps with four structural snapshots (PDB IDs: 6BLO, 6BLP, 6BM2 and 6BM4). Color codes are the same as Figure 3.
Several general principles start to emerge from the comparison of Pol II arrested by different lesions. First, Pol II can be arrested through distinct mechanisms even for the DNA lesions induced by chemical-related agents. For example, Pol II gets arrested by the 1,2-dGpG cisplatin-DNA lesion because this lesion is a strong steric block to the crossover of the bridge helix. In sharp contrast, the pyriplatin-dG adduct (a monofunctional cisplatin derivative) can support effective CMP incorporation and form a Watson-Crick base pair with CMP. However, this lesion strongly blocks subsequent Pol II translocation (69). These findings highlight the importance of investigating Pol II arrest by different types of DNA lesions. Second, we found that Pol II can be trapped at a common translocation intermediate state by a subset of structurally-diverse DNA lesions, such as the CPD, CydA, and AP site (51, 67, 100). Quite intriguingly, all of these damaged DNA templates are all trapped above the bridge helix, representing a half-way intermediate to translocate into the +1 canonical template position (51, 67, 100). For CPD and AP site-arrested Pol II structures, it was found that Rpb1 R337 forms conserved hydrogen bonds with the template phosphodiester backbone that may further stabilize this intermediate state (51, 100). No much direct interactions were observed between the damaged bases and Pol II residues, indicating that these DNA lesions are not recognized through specific interactions. Instead, Pol II recognizes these DNA lesions through their failure to fully cross over the bridge helix to reach the +1 template position. This provides a structural explanation of a broad range of lesion-containing substrates for TC-NER. Third, a common theme starts to emerge for the structural basis of the “A-rule”. Pol II is able to slowly incorporate AMP opposite a subset of structural-diverse DNA lesions in a template-independent manner (‘A-rule’) with prolonged incubation. These DNA lesions are often trapped at the half-way intermediate position (above the bridge helix) rather than the canonical +1 site to form a base pair with incoming NTP. Finally, Pol II arrest can be induced by non-covalent obstructions, such as Py-Im and non-B DNA structures. It is unclear whether these Pol II arrest events can also trigger TC-NER or require additional factors to discriminate these forms of Pol II arrest from DNA lesion-induced Pol II arrest.
Function of CSB in Pol II-mediated DNA Lesion Recognition
CSB is among the first protein to be recruited to the arrested Pol II and has been considered as the master coordinator of TC-NER (2, 22, 101–109). CSB is well conserved from yeast to human and belongs to the Swi2/Snf2 family of nucleosome remodelers that all share the signature seven helicase motifs in their core ATPase domains (101, 102, 110, 111). CSB protein has DNA-dependent ATPase, DNA translocase, as well as chromatin remodeling activities (101, 111–115).
CSB recruits CSA to the sites of arrested Pol II (109). The CSA (ERCC8) protein has seven WD40 repeat motifs and beta-propeller architecture (116, 117) and is the substrate recognition subunit of a CRL4CSA E3 ubiquitin ligase complex (CSA-DDB1-Cul4A-Roc1) (117, 118). Recently, it was shown that CSA is also required for the recruitment of UVSSA-USP7 (119–122). Interestingly, USP7 can deubiquitylate CSB and prevent DNA damage-induced degradation of CSB (120, 122). On the other hand, CSA-dependent degradation of CSB is required for the recovery of global RNA synthesis after UV damage (123, 124). The ubiquitylation and deubiquitylation of CSB are tightly regulated during TC-NER and the recovery of RNA synthesis after UV damage. In addition, CSB, together with CSA, also recruits P300, XAB2, and HMGN1 to sites of stalled Pol II (109).
The exact role of CSB in Pol II-mediated lesion recognition is not very clear. Given that CSB is among the first proteins to be recruited to the arrested Pol II, we recently tested whether CSB has a role in discriminating Pol II arrested at a bulky lesion from other forms of Pol II arrests (such as Py-Im induced Pol II arrest) (125). We investigated the effect of Rad26, the Saccharomyces cerevisiae CSB ortholog, on Pol II stall/arrest in three representative scenarios: a non-damaged DNA containing an intrinsic pausing/arrest sequence (a long repetitive A sequence on the template strand, referred to as an A-tract), a non-covalent DNA binder (pyrrole-imidazole (Py-Im) polyamide), or a bulky covalent DNA lesion (CPD lesion) (Figure 8). We used an in vitro reconstituted transcription system with purified yeast Pol II and Rad26, where we assembled a Pol II elongation complex with a fully-matched transcriptional bubble upstream and placed a site-specific Pol II translocation barrier on the downstream template strand (A-tract sequence, Py-Im binding site, or CPD lesion). As shown in Figure 8, Pol II alone cannot distinguish among these three transcription barriers, as it becomes stalled in all three scenarios. Intriguingly, with the joint action of Pol II and Rad26, we found a distinct transcriptional readout for the CPD lesion compared with other non-covalent obstructions: in the presence of Rad26, Pol II is able to bypass the A-tract and Py-Im translocation barriers, whereas Pol II remains arrested at CPD lesions (Figure 8). The intact ATPase of Rad26 and (d)ATP is required for stimulating Pol II bypass of the A-tract and Py-Im translocation barriers. We also tested the effects of other transcription factors, such as TFIIS. In contrast to Rad26, TFIIS failed to distinguish Pol II arrested by the non-covalent Py-Im molecule from arrest at a CPD lesion (Figure 8). Furthermore, our recent unpublished data indicates Rad26 was also able to help Pol II bypass the nucleosome barrier in an ATP-dependent manner. Taken together, these biochemical results show that Rad26 can help to distinguish different Pol II arrest scenarios and facilitate Pol II transcriptional bypass through some arrest situations that do not require DNA repair.
Structure Basis of CSB in Pol II-mediated DNA Lesion Recognition
To further understand the structural basis of how CSB interacts with Pol II, in a joint-effort with the Leschziner lab, we recently solved yeast Pol II/Rad26 complex structure using single-particle cryo-EM. This Pol II/Rad26 complex structure revealed that Rad26 binds to the DNA upstream of the Pol II complex and sits between Pol II’s clamp (Rpb2 side) and stalk (Rpb4/7) regions (Figure 9a) (125). The protein-protein interface between Pol II and Rad26 is conserved from yeast to human, suggesting that what we learned from yeast Pol II-Rad26 is likely to also apply to mammalian Pol II-CSB. One of the most striking features is that the upstream DNA is bent about 80 degrees toward the stalk upon binding of Rad26 (Figure 9b). This unique structural feature may have two potential roles: first, it shields the binding site from other transcription factors (discussed in below); and second, the Pol II/Rad26 complex may also generate a new binding surface for the recruitment of other downstream repair factors.
Figure 9.

Pol II-CSB structure and upstream DNA bending. a, Atomic model for the Pol II– Rad26 complex. Cartoons of the structures highlighting their orientations. b, Superposition of the scaffolds from the Pol II–Rad26 and Pol II EC structures, with the latter shown in darker colors.
Several important structural insights could be obtained by comparing the structures of the Pol II initiation complex, Pol II elongation complex with Spt4/Spt5, and Pol II–Rad26 complex (repair complex) (Figure 10) (125–131). First, the core Pol IIs are in a similar conformation, except for minor changes in the stalk region. Second, the upstream DNA path changes dramatically among the three structures, whereas the downstream DNA path is essentially the same. Finally, and perhaps most importantly, the Pol II binding sites for initiation factors (such as TFIIE), elongation factors (Spt4/5), and repair factor (CSB/Rad26) are significantly overlapping, indicating the binding of these factors are mutually exclusive (Figure 10). Just like the case where Spt4/5 displaces TFIIE during the transition between transcription initiation and productive elongation, there is an intriguing possibility that the TC-NER factor Rad26 may also displace Spt4/5 when Pol II switches form the transcription elongation mode to the repair mode. This suggests an important functional interplay between the transcription-coupled repair factor Rad26 and other transcription factors during transcription and TCR through direct competition for binding to Pol II. Indeed, this explains early observations that Rad26 is able to antagonize the repression of TCR by Spt4/5 (132, 133).
A Unified Mode of Action of CSB
The structure of the Pol II-Rad26 complex revealed that Rad26 binds to the upstream region of Pol II and pulls the template strand away from Pol II in an ATP-dependent manner (125). As a result, this prevents Pol II backtracking and favors Pol II forward translocation, which is verified by biochemical assays. This suggests a mechanistic model for a unified mode of action of CSB in both TCR and transcription (Figure 11). During Pol II transcription elongation (on a non-damaged template), CSB can reduce Pol II dwell times by preventing backtracking or elemental pausing, and/or by stimulating Pol II forward translocation (from pre-translocation state to post-translocation state). Thus, CSB functions as a Pol II elongation factor, stimulating transcription elongation as reported (Figure 11) (106, 107, 134). Similarly, during transcription through templates containing less bulky DNA lesions, such as oxidized bases, CSB can rescue arrested Pol II by preventing extensive backtracking and increasing the chances of transcriptional bypass of these DNA lesions(134). For bulky DNA lesions that result in a strong blockage of translocation due to steric hindrance at the active site (such as CPD lesions)(49), while CSB can prevent extensive backtracking, CSB fails to promote efficient Pol II transcriptional bypass of the bulky DNA lesion. Therefore, the CPD-induced steric hindrance for Pol II transcription would persist even in the presence of CSB (Rad26), and Pol II-Rad26 complex would remain arrested at the DNA lesion and is subject to the downstream TCR process (2).
Figure 11.
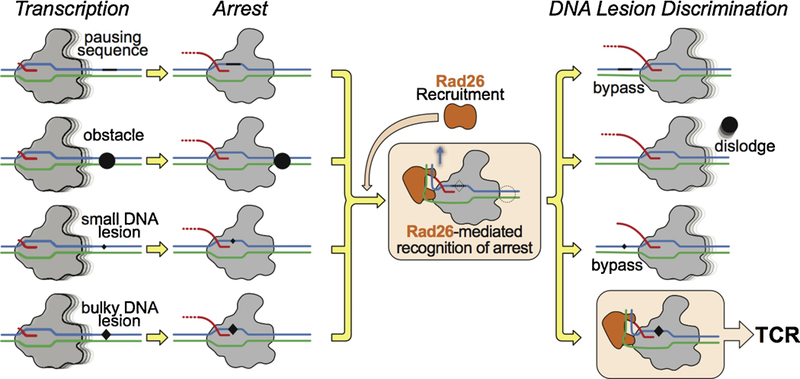
Model for lesion recognition by CSB. Rad26 recognizes a stalled Pol II and can reduce its dwell time by preventing backtracking, promoting Pol II forward translocation on non-damaged templates, and increasing the chances of transcriptional bypass through less bulky DNA lesions, all of which stimulate transcription elongation. However, Rad26 fails to promote efficient transcriptional bypass of bulky DNA lesions that lead to strong blockage of translocation (such as CPD lesions). The interaction between Rad26 and a Pol II persistently arrested at a bulky lesion would lead to the initiation of TCR.
Lesion Recognition and Verification during TCR
The structure of the Pol II-Rad26 complex also provides mechanistic insights into the roles of CSB in DNA lesion recognition and verification in eukaryotic TCR initiation. Based on the structural and biochemical data, we propose that CSB (Rad26) plays a central role in controlling (and improving) the fidelity of transcription-coupled DNA lesion recognition (step 1), thus reducing the rate of incorrect TCR initiation. The arrested Pol II-CSB (Rad26) complex presents new protein interaction interfaces that could facilitate loading other repair factors (2, 108, 135), such as TFIIH, downstream of the Pol II complex (Step 2). Based on the experimental data reported for TFIIH-dependent lesion verification in GG-NER at step 2, we propose that TFIIH has a similar mode of action in TCR (136–138). Indeed, it was reported recently that UVSSA can directly interact with the TFIIH p62 subunit (136). Therefore, it is conceivable that the Pol II-CSB complex could recruit TFIIH via UVSSA. Other factors may also be involved in TFIIH recruitment. TFIIH is loaded downstream of the Pol II-CSB complex and it uses its XPB and XPD helicases to verify whether a DNA lesion is present on the template strand. Pol II is either backtracked or displaced at this stage. The final step, the XPA-dependent lesion verification step, is expected to be the same for both GG-NER and TCR (step 3) (Figure 12) (137, 138). Taken together, there will be three sequential checkpoint steps for lesion recognition and verification for both GG-NER and TCR. Further experiments will be needed to test this model.
Figure 12.
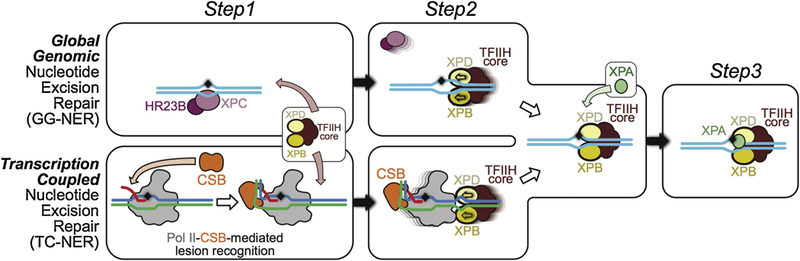
Three DNA lesion checkpoints for GG-NER and TC-NER. Check step 1: For GG-NER, XPC/HR23B detects base-pair disruption and helix distortion and binds to the DNA strand opposite to that carrying the lesion. This constitutes the initial lesion recognition. For TCR, CSB is recruited to a stalled Pol II to discriminate genuine DNA lesion-induced transcription arrest from other forms of transcriptional arrest, as diagrammed in panel a. At this step, CSB acts in conjunction with Pol II to mediate the initial recognition of DNA lesions that block transcription translocation. Check step 2: Core TFIIH is recruited to further verify the DNA lesion. In GG-NER, the XPD and XPB helicases in core TFIIH translocate the complex towards the lesion. This is the result of XPD tracking along the damage-containing strand in a 5´ to 3´ direction and XPB tracking along the opposite strand (non-damaged) in a 3´ to 5´ direction. In TCR, TFIIH is loaded downstream of the arrested Pol II-CSB complex, with XPD and XPB tracking the template and non-template strands, respectively. The XPD/XPB helicases in core TFIIH translocate towards the lesion, as is the case for GG-NER. As a result, Pol II-CSB is pushed upstream by TFIIH to expose the DNA lesion. Check step 3: XPA is recruited for a final validation of the TFIIH-recognized lesion and to ensure that only genuine NER lesions are subjected to dual incision by endonucleases ERCC1/XPF and XPG and downstream repair synthesis.
Conservation of the Pol II-CSB Complex
While there are some differences in TC-NER in yeast and human, the central themes and interactions among the repair factors are highly conserved (2, 10). Indeed, the conservation of the Pol II-CSB complex from yeast to humans is supported by the CryoEM structure. The core ATPase domain of Rad26 observed in our structure is highly conserved from yeast to human (101, 102, 111). Therefore, the structure of the human CSB core ATPase is expected to be very similar to that of Rad26 reported here. The structural conservation between mammalian and yeast Pol II elongation complex was also well-established (130, 139). Furthermore, and of particular relevance to the work presented here, most of the interaction interfaces we identified between Rad26 and DNA and Rad26 and Pol II are also highly conserved between yeast and human. Therefore, the Pol II-Rad26 structure presented here could serve as a model for studying Pol II-CSB interactions in human and other mammalian systems. Our study provides structural insight into the CSB-dependent TCR, at least the initiation stage of TCR, which is conserved from yeast to human. The difference of UV sensitivity for CSB deletion in mammalian cells versus Rad26 deletion in yeast strains is due to the existence of additional CSB/Rad26-independent pathways for repairing of UV lesions in yeast rather than a difference of mechanism of CSB-dependent TCR between yeast and human (2).
Conclusion and Future Perspectives
Recent structural studies in elucidating Pol II-mediated lesion recognition and the first structure of the Pol II-CSB (Rad26) complex have provided us important mechanistic insight into TC-NER lesion recognition. These studies reveal the molecular mechanisms of how Pol II becomes arrested by a variety of DNA lesions. In particular, the structure of CSB/Rad26 reveals molecular interactions between CSB/Rad26 and Pol II and provides novel mechanistic insight into the roles CSB plays in both TCR initiation and transcription elongation. We also revealed that CSB is critical for the fidelity of transcription-coupled lesion recognition. Several important mechanistic questions deserve future investigation: How does CSB displace other elongation factors when Pol II gets arrested? How are downstream repair factors recruited to the Pol II-CSB arrested complex for lesion verification? What is the fate of Pol II during lesion recognition and verification and at what stage is Pol II moved away (displaced, or backtracked, or ubiquitinylated and degraded) from the lesion site? When and how is CSB released from DNA lesion sites at a later stage? What are the roles of DNA damage induced post-translational modifications of Pol II, CSB and other repair factors in regulating TC-NER? What is the functional interplay between these TC-NER factors recruitment events and chromatin environments? Finding answers to these questions would not only provide a framework for us to understand the molecular mechanism of the TC-NER pathway, but also develop novel chemotherapeutic drugs targeting TC-NER in the future.
Acknowledgements
This work was supported by the National Institutes of Health (R01 GM102362) to D.W
Abbreviations
- 3d-Napht-dA
3-deaza-3-methoxynaphtylethyl-adenosine
- 8-oxoG
8-oxoguanine
- CydA
8,5′cyclo-2′-deoxyadenosine
- AP
Abasic site
- AF
Acylfulvene
- BER
Base excision repair
- CHO
Chinese hamster ovary
- CSB
Cockayne Syndrome B protein
- CPD
Cyclobutane pyrimidine dimer
- EC
Elongation complex
- GG-NER
Global genome nucleotide excision repair
- NER
Nucleotide excision repair
- O2-, N3-, and O4-EtdT
O2- N3- and O4-ethylthymideine
- Phenanthriplatin
cis-diamminephenanthridinechloroplatinum(II)
- Pol II
RNA polymerase II
- Py-Im
Pyrrole-imidazole polyamide
- Pyriplatin
cis-diamminepyridinechloroplatinum(II)
- ROS
Reactive oxygen species
- TC-NER
Transcription-coupled nucleotide excision repair
- TCR
Transcription-coupled repair
- TLS
Translesion DNA synthesis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Sancar A & Reardon JT (2004) Nucleotide excision repair in E. coli and man. Adv Protein Chem 69:43–71. [DOI] [PubMed] [Google Scholar]
- 2.Hanawalt PC & Spivak G (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9(12):958–970. [DOI] [PubMed] [Google Scholar]
- 3.Spivak G (2015) Nucleotide excision repair in humans. DNA Repair (Amst) 36:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohr VA, Smith CA, Okumoto DS, & Hanawalt PC (1985) DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40(2):359–369. [DOI] [PubMed] [Google Scholar]
- 5.Mellon I, Spivak G, & Hanawalt PC (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51(2):241–249. [DOI] [PubMed] [Google Scholar]
- 6.Spivak G (2016) Transcription-coupled repair: an update. Archives of toxicology [DOI] [PMC free article] [PubMed]
- 7.Selby CP (2017) Mfd Protein and Transcription-Repair Coupling in Escherichia coli. Photochemistry and photobiology 93(1):280–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pani B & Nudler E (2017) Mechanistic insights into transcription coupled DNA repair. DNA Repair (Amst) 56:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stantial N, Dumpe J, Pietrosimone K, Baltazar F, & Crowley DJ (2016) Transcription-coupled repair of UV damage in the halophilic archaea. DNA Repair (Amst) 41:63–68. [DOI] [PubMed] [Google Scholar]
- 10.Li S (2015) Transcription coupled nucleotide excision repair in the yeast Saccharomyces cerevisiae: The ambiguous role of Rad26. DNA Repair (Amst) 36:43–48. [DOI] [PubMed] [Google Scholar]
- 11.Spivak G & Ganesan AK (2014) The complex choreography of transcription-coupled repair. DNA Repair (Amst) 19:64–70. [DOI] [PubMed] [Google Scholar]
- 12.Fousteri M & Mullenders LH (2008) Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell research 18(1):73–84. [DOI] [PubMed] [Google Scholar]
- 13.Mullenders L (2015) DNA damage mediated transcription arrest: Step back to go forward. DNA Repair (Amst) 36:28–35. [DOI] [PubMed] [Google Scholar]
- 14.Svejstrup JQ (2010) The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci 35(6):333–338. [DOI] [PubMed] [Google Scholar]
- 15.Scharer OD (2013) Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5(10):a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vermeulen W & Fousteri M (2013) Mammalian transcription-coupled excision repair. Cold Spring Harb Perspect Biol 5(8):a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisker C, Kuper J, & Van Houten B (2013) Prokaryotic nucleotide excision repair. Cold Spring Harb Perspect Biol 5(3):a012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schärer O (2018) DNA Repair (Amst)
- 19.Epanchintsev A, et al. (2017) Cockayne’s Syndrome A and B Proteins Regulate Transcription Arrest after Genotoxic Stress by Promoting ATF3 Degradation. Mol Cell 68(6):1054–1066 e1056. [DOI] [PubMed] [Google Scholar]
- 20.Williamson L, et al. (2017) UV Irradiation Induces a Non-coding RNA that Functionally Opposes the Protein Encoded by the Same Gene. Cell 168(5):843–855 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geijer ME & Marteijn JA (2018) What happens at the lesion does not stay at the lesion: The effects of DNA damage in cis and trans on transcription and transcription-coupled nucleotide excision repair. DNA Repair (Amst):In press. [DOI] [PubMed]
- 22.Laine JP & Egly JM (2006) Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J 25(2):387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindsey-Boltz LA & Sancar A (2007) RNA polymerase: the most specific damage recognition protein in cellular responses to DNA damage? Proc Natl Acad Sci U S A 104(33):13213–13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saxowsky TT & Doetsch PW (2006) RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem Rev 106(2):474–488. [DOI] [PubMed] [Google Scholar]
- 25.Svejstrup JQ (2007) Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem Sci 32(4):165–171. [DOI] [PubMed] [Google Scholar]
- 26.Belotserkovskii BP, Mirkin SM, & Hanawalt PC (2013) DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev 113(11):8620–8637. [DOI] [PubMed] [Google Scholar]
- 27.Shin JH, Xu L, & Wang D (2017) Mechanism of transcription-coupled DNA modification recognition. Cell Biosci 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hangauer MJ, Vaughn IW, & McManus MT (2013) Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet 9(6):e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ransohoff JD, Wei Y, & Khavari PA (2018) The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 19(3):143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belotserkovskii BP, et al. (2010) Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci U S A 107(29):12816–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doetsch PW (2002) Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat Res 510(1–2):131–140. [DOI] [PubMed] [Google Scholar]
- 32.Scicchitano DA (2005) Transcription past DNA adducts derived from polycyclic aromatic hydrocarbons. Mutat Res 577(1–2):146–154. [DOI] [PubMed] [Google Scholar]
- 33.Scicchitano DA, Olesnicky EC, & Dimitri A (2004) Transcription and DNA adducts: what happens when the message gets cut off? DNA Repair (Amst) 3(12):1537–1548. [DOI] [PubMed] [Google Scholar]
- 34.Tornaletti S (2005) Transcription arrest at DNA damage sites. Mutat Res 577(1–2):131–145. [DOI] [PubMed] [Google Scholar]
- 35.Waters LS, et al. (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 73(1):134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, et al. (2015) Impact of template backbone heterogeneity on RNA polymerase II transcription. Nucleic Acids Res 43(4):2232–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, et al. (2014) Molecular basis of transcriptional fidelity and DNA lesion-induced transcriptional mutagenesis. DNA Repair (Amst) 19:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Zhou W, & Doetsch PW (1995) RNA polymerase bypass at sites of dihydrouracil: implications for transcriptional mutagenesis. Mol Cell Biol 15(12):6729–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bregeon D & Doetsch PW (2011) Transcriptional mutagenesis: causes and involvement in tumour development. Nat Rev Cancer 11(3):218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morreall JF, Petrova L, & Doetsch PW (2013) Transcriptional mutagenesis and its potential roles in the etiology of cancer and bacterial antibiotic resistance. J Cell Physiol 228(12):2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu L, et al. (2015) RNA polymerase II transcriptional fidelity control and its functional interplay with DNA modifications. Critical reviews in biochemistry and molecular biology 50(6):503–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tornaletti S & Hanawalt PC (1999) Effect of DNA lesions on transcription elongation. Biochimie 81(1–2):139–146. [DOI] [PubMed] [Google Scholar]
- 43.Wang D & Lippard SJ (2005) Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4(4):307–320. [DOI] [PubMed] [Google Scholar]
- 44.Todd RC & Lippard SJ (2009) Inhibition of transcription by platinum antitumor compounds. Metallomics 1(4):280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park H, et al. (2002) Crystal structure of a DNA decamer containing a cis-syn thymine dimer. Proc Natl Acad Sci U0 S A 99(25):15965–15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donahue BA, Yin S, Taylor JS, Reines D, & Hanawalt PC (1994) Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc Natl Acad Sci U S A 91(18):8502–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tornaletti S, Reines D, & Hanawalt PC (1999) Structural characterization of RNA polymerase II complexes arrested by a cyclobutane pyrimidine dimer in the transcribed strand of template DNA. J Biol Chem 274(34):24124–24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mei Kwei JS, et al. (2004) Blockage of RNA polymerase II at a cyclobutane pyrimidine dimer and 6–4 photoproduct. Biochem Biophys Res Commun 320(4):1133–1138. [DOI] [PubMed] [Google Scholar]
- 49.Brueckner F, Hennecke U, Carell T, & Cramer P (2007) CPD damage recognition by transcribing RNA polymerase II. Science 315(5813):859–862. [DOI] [PubMed] [Google Scholar]
- 50.Damsma GE, Alt A, Brueckner F, Carell T, & Cramer P (2007) Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat Struct Mol Biol 14(12):1127–1133. [DOI] [PubMed] [Google Scholar]
- 51.Walmacq C, et al. (2012) Mechanism of translesion transcription by RNA polymerase II and its role in cellular resistance to DNA damage. Mol Cell 46(1):18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silva DA, et al. (2014) Millisecond dynamics of RNA polymerase II translocation at atomic resolution. Proc Natl Acad Sci U S A 111(21):7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strauss BS (1991) The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays 13(2):79–84. [DOI] [PubMed] [Google Scholar]
- 54.Saxowsky TT, Meadows KL, Klungland A, & Doetsch PW (2008) 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proc Natl Acad Sci U S A 105(48):18877–18882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tornaletti S, Maeda LS, Kolodner RD, & Hanawalt PC (2004) Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 3(5):483–494. [DOI] [PubMed] [Google Scholar]
- 56.Kuraoka I, et al. (2007) RNA polymerase II bypasses 8-oxoguanine in the presence of transcription elongation factor TFIIS. DNA Repair (Amst) 6(6):841–851. [DOI] [PubMed] [Google Scholar]
- 57.Bregeon D, Doddridge ZA, You HJ, Weiss B, & Doetsch PW (2003) Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell 12(4):959–970. [DOI] [PubMed] [Google Scholar]
- 58.Damsma GE & Cramer P (2009) Molecular basis of transcriptional mutagenesis at 8-oxoguanine. J Biol Chem 284(46):31658–31663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brieba LG, et al. (2004) Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J 23(17):3452–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eoff RL, Irimia A, Angel KC, Egli M, & Guengerich FP (2007) Hydrogen bonding of 7,8-dihydro-8-oxodeoxyguanosine with a charged residue in the little finger domain determines miscoding events in Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem 282(27):19831–19843. [DOI] [PubMed] [Google Scholar]
- 61.Guo J, Hanawalt PC, & Spivak G (2013) Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoGuanine in human cells. Nucleic Acids Research 41(16):7700–7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y (2008) Bulky DNA lesions induced by reactive oxygen species. Chemical research in toxicology 21(2):276–281. [DOI] [PubMed] [Google Scholar]
- 63.Brooks PJ, et al. (2000) The oxidative DNA lesion 8,5’-(S)-cyclo-2’-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem 275(29):22355–22362. [DOI] [PubMed] [Google Scholar]
- 64.Marietta C & Brooks PJ (2007) Transcriptional bypass of bulky DNA lesions causes new mutant RNA transcripts in human cells. EMBO Rep 8(4):388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaruga P & Dizdaroglu M (2008) 8,5’-Cyclopurine-2’-deoxynucleosides in DNA: mechanisms of formation, measurement, repair and biological effects. DNA Repair (Amst) 7(9):1413–1425. [DOI] [PubMed] [Google Scholar]
- 66.You C, et al. (2012) A quantitative assay for assessing the effects of DNA lesions on transcription. Nat Chem Biol 8(10):817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walmacq C, et al. (2015) Mechanism of RNA polymerase II bypass of oxidative cyclopurine DNA lesions. Proc Natl Acad Sci U S A 112(5):E410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park GY, Wilson JJ, Song Y, & Lippard SJ (2012) Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc Natl Acad Sci U S A 109(30):11987–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang D, Zhu G, Huang X, & Lippard SJ (2010) X-ray structure and mechanism of RNA polymerase II stalled at an antineoplastic monofunctional platinum-DNA adduct. Proc Natl Acad Sci U S A 107(21):9584–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kellinger MW, Park GY, Chong J, Lippard SJ, & Wang D (2013) Effect of a monofunctional phenanthriplatin-DNA adduct on RNA polymerase II transcriptional fidelity and translesion synthesis. J Am Chem Soc 135(35):13054–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trauger JW, Baird EE, & Dervan PB (1996) Recognition of DNA by designed ligands at subnanomolar concentrations. Nature 382(6591):559–561. [DOI] [PubMed] [Google Scholar]
- 72.White S, Szewczyk JW, Turner JM, Baird EE, & Dervan PB (1998) Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands. Nature 391(6666):468–471. [DOI] [PubMed] [Google Scholar]
- 73.Nickols NG & Dervan PB (2007) Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci U S A 104(25):10418–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muzikar KA, Nickols NG, & Dervan PB (2009) Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc Natl Acad Sci U S A 106(39):16598–16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang F, et al. (2013) Antitumor activity of a pyrrole-imidazole polyamide. Proc Natl Acad Sci U S A 110(5):1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu L, et al. (2016) RNA polymerase II senses obstruction in the DNA minor groove via a conserved sensor motif. Proc Natl Acad Sci U S A 113(44):12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gnatt AL, Cramer P, Fu J, Bushnell DA, & Kornberg RD (2001) Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science 292(5523):1876–1882. [DOI] [PubMed] [Google Scholar]
- 78.Jaspers NG, et al. (2002) Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair (Amst) 1(12):1027–1038. [DOI] [PubMed] [Google Scholar]
- 79.Koeppel F, et al. (2004) Irofulven cytotoxicity depends on transcription-coupled nucleotide excision repair and is correlated with XPG expression in solid tumor cells. Clin Cancer Res 10(16):5604–5613. [DOI] [PubMed] [Google Scholar]
- 80.Otto C, et al. (2017) Modulation of Cytotoxicity by Transcription-Coupled Nucleotide Excision Repair Is Independent of the Requirement for Bioactivation of Acylfulvene. Chemical research in toxicology 30(3):769–776. [DOI] [PubMed] [Google Scholar]
- 81.Wang D, Bushnell DA, Westover KD, Kaplan CD, & Kornberg RD (2006) Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell 127(5):941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toulokhonov I, Zhang J, Palangat M, & Landick R (2007) A central role of the RNA polymerase trigger loop in active-site rearrangement during transcriptional pausing. Mol Cell 27(3):406–419. [DOI] [PubMed] [Google Scholar]
- 83.Xu L, et al. (2017) Mechanism of DNA alkylation-induced transcriptional stalling, lesion bypass, and mutagenesis. Proc Natl Acad Sci U S A 114(34):E7082–E7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Godschalk R, et al. (2002) Comparison of multiple DNA adduct types in tumor adjacent human lung tissue: effect of cigarette smoking. Carcinogenesis 23(12):2081–2086. [DOI] [PubMed] [Google Scholar]
- 85.Swenberg JA, et al. (2011) Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol Sci 120 Suppl 1:S130–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anna L, Kovacs K, Gyorffy E, Schoket B, & Nair J (2011) Smoking-related O4-ethylthymidine formation in human lung tissue and comparisons with bulky DNA adducts. Mutagenesis 26(4):523–527. [DOI] [PubMed] [Google Scholar]
- 87.Lindahl T (1993) Instability and decay of the primary structure of DNA. Naturen 362(6422):709–715. [DOI] [PubMed] [Google Scholar]
- 88.Nakamura J, et al. (1998) Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res 58(2):222–225. [PubMed] [Google Scholar]
- 89.Loeb LA & Preston BD (1986) Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet 20:201–230. [DOI] [PubMed] [Google Scholar]
- 90.Obeid S, et al. (2010) Replication through an abasic DNA lesion: structural basis for adenine selectivity. The EMBO journal 29(10):1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor JS (2002) New structural and mechanistic insight into the A-rule and the instructional and non-instructional behavior of DNA photoproducts and other lesions. Mutat Res 510(1–2):55–70. [DOI] [PubMed] [Google Scholar]
- 92.Pages V, Johnson RE, Prakash L, & Prakash S (2008) Mutational specificity and genetic control of replicative bypass of an abasic site in yeast. Proc Natl Acad Sci U S A 105(4):1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beard WA, Shock DD, Batra VK, Pedersen LC, & Wilson SH (2009) DNA polymerase beta substrate specificity: side chain modulation of the “A-rule”. Journal of Biological Chemistry 284(46):31680–31689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ling H, Boudsocq F, Woodgate R, & Yang W (2004) Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol Cell 13(5):751–762. [DOI] [PubMed] [Google Scholar]
- 95.Nair DT, Johnson RE, Prakash L, Prakash S, & Aggarwal AK (2009) DNA synthesis across an abasic lesion by human DNA polymerase iota. Structure 17(4):530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nair DT, Johnson RE, Prakash L, Prakash S, & Aggarwal AK (2011) DNA synthesis across an abasic lesion by yeast REV1 DNA polymerase. J Mol Biol 406(1):18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tornaletti S, Maeda LS, & Hanawalt PC (2006) Transcription arrest at an abasic site in the transcribed strand of template DNA. Chemical research in toxicology 19(9):1215–1220. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y, Sheppard TL, Tornaletti S, Maeda LS, & Hanawalt PC (2006) Transcriptional inhibition by an oxidized abasic site in DNA. Chemical research in toxicology 19(2):234–241. [DOI] [PubMed] [Google Scholar]
- 99.Clauson CL, Oestreich KJ, Austin JW, & Doetsch PW (2010) Abasic sites and strand breaks in DNA cause transcriptional mutagenesis in Escherichia coli. Proc Natl Acad Sci U S A 107(8):3657–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang W, Walmacq C, Chong J, Kashlev M, & Wang D (2018) Structural basis of transcriptional stalling and bypass of abasic DNA lesion by RNA polymerase II. Proc Natl Acad Sci U S A 115(11):E2538–E2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Troelstra C, et al. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 71(6):939–953. [DOI] [PubMed] [Google Scholar]
- 102.van Gool AJ, et al. (1994) RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. Embo J 13(22):5361–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Gool AJ, et al. (1997) The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J 16(19):5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tantin D, Kansal A, & Carey M (1997) Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol Cell Biol 17(12):6803–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Selby CP, Drapkin R, Reinberg D, & Sancar A (1997) RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res 25(4):787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Selby CP & Sancar A (1997) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci U S A 94(21):11205–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Selby CP & Sancar A (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J Biol Chem 272(3):1885–1890. [DOI] [PubMed] [Google Scholar]
- 108.Sarker AH, et al. (2005) Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell 20(2):187–198. [DOI] [PubMed] [Google Scholar]
- 109.Fousteri M, Vermeulen W, van Zeeland AA, & Mullenders LH (2006) Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell 23(4):471–482. [DOI] [PubMed] [Google Scholar]
- 110.Eisen JA, Sweder KS, & Hanawalt PC (1995) Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res 23(14):2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang L, et al. (2014) Regulation of the Rhp26ERCC6/CSB chromatin remodeler by a novel conserved leucine latch motif. Proc Natl Acad Sci U S A 111(52):18566–18571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Citterio E, et al. (2000) ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol 20(20):7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lake RJ, Geyko A, Hemashettar G, Zhao Y, & Fan HY (2010) UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell 37(2):235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lake RJ & Fan HY (2013) Structure, function and regulation of CSB: a multi-talented gymnast. Mech Ageing Dev 134(5–6):202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee JY, et al. (2017) NAP1L1 accelerates activation and decreases pausing to enhance nucleosome remodeling by CSB. Nucleic Acids Res 45(8):4696–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henning KA, et al. (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82(4):555–564. [DOI] [PubMed] [Google Scholar]
- 117.Fischer ES, et al. (2011) The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147(5):1024–1039. [DOI] [PubMed] [Google Scholar]
- 118.Groisman R, et al. (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113(3):357–367. [DOI] [PubMed] [Google Scholar]
- 119.Schwertman P, Vermeulen W, & Marteijn JA (2013) UVSSA and USP7, a new couple in transcription-coupled DNA repair. Chromosoma 122(4):275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schwertman P, et al. (2012) UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nature genetics 44(5):598–602. [DOI] [PubMed] [Google Scholar]
- 121.Fei J & Chen J (2012) KIAA1530 protein is recruited by Cockayne syndrome complementation group protein A (CSA) to participate in transcription-coupled repair (TCR). J Biol Chem 287(42):35118–35126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang X, et al. (2012) Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nature genetics 44(5):593–597. [DOI] [PubMed] [Google Scholar]
- 123.Jin J & Li J- M (2012) CRL Ubiquitin Ligases and DNA Damage Response. Frontiers in Oncology 2(29). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Groisman R, et al. (2006) CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes & development 20(11):1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xu J, et al. (2017) Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature 551(7682):653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hantsche M & Cramer P (2017) Conserved RNA polymerase II initiation complex structure. Current opinion in structural biology 47:17–22. [DOI] [PubMed] [Google Scholar]
- 127.Nogales E, Louder RK, & He Y (2016) Cryo-EM in the study of challenging systems: the human transcription pre-initiation complex. Current opinion in structural biology 40:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klein BJ, et al. (2011) RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc Natl Acad Sci U S A 108(2):546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Martinez-Rucobo FW, Sainsbury S, Cheung AC, & Cramer P (2011) Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J 30(7):1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bernecky C, Herzog F, Baumeister W, Plitzko JM, & Cramer P (2016) Structure of transcribing mammalian RNA polymerase II. Nature 529(7587):551–554. [DOI] [PubMed] [Google Scholar]
- 131.Ehara H, et al. (2017) Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 357(6354):921–924. [DOI] [PubMed] [Google Scholar]
- 132.Li W, Giles C, & Li S (2014) Insights into how Spt5 functions in transcription elongation and repressing transcription coupled DNA repair. Nucleic Acids Res 42(11):7069–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jansen LE, et al. (2000) Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. Embo J 19(23):6498–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Charlet-Berguerand N, et al. (2006) RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J 25(23):5481–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Laine JP & Egly JM (2006) When transcription and repair meet: a complex system. Trends Genet 22(8):430–436. [DOI] [PubMed] [Google Scholar]
- 136.Okuda M, Nakazawa Y, Guo C, Ogi T, & Nishimura Y (2017) Common TFIIH recruitment mechanism in global genome and transcription-coupled repair subpathways. Nucleic Acids Res 45(22):13043–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li C-L, et al. (2015) Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Molecular Cell 59(6):1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Marteijn Jurgen A, Hoeijmakers Jan HJ, & Vermeulen W (Check, Check …Triple Check: Multi-Step DNA Lesion Identification by Nucleotide Excision Repair. Molecular Cell 59(6):885–886. [DOI] [PubMed] [Google Scholar]
- 139.Liu X, Bushnell DA, & Kornberg RD (2013) RNA polymerase II transcription: structure and mechanism. Biochimica et biophysica acta 1829(1):2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]